

Abstract
Abnormally high concentrations of Zn2+, Cu2+, and Fe3+ are present along with amyloid-β (Aβ) in the senile plaques in Alzheimer disease, where Al3+ is also detected. Aβ aggregation is the key pathogenic event in Alzheimer disease, where Aβ oligomers are the major culprits. The fundamental mechanism of these metal ions on Aβ remains elusive. Here, we employ 4,4′-Bis(1-anilinonaphthalene 8-sulfonate) and tyrosine fluorescence, CD, stopped flow fluorescence, guanidine hydrochloride denaturation, and photo-induced cross-linking to elucidate the effect of Zn2+, Cu2+, Fe3+, and Al3+ on Aβ at the early stage of the aggregation. Furthermore, thioflavin T assay, dot blotting, and transmission electron microscopy are utilized to examine Aβ aggregation. Our results show that Al3+ and Zn2+, but not Cu2+ and Fe3+, induce larger hydrophobic exposures of Aβ conformation, resulting in its significant destabilization at the early stage. The metal ion binding induces Aβ conformational changes with micromolar binding affinities and millisecond binding kinetics. Cu2+ and Zn2+ induce similar assembly of transiently appearing Aβ oligomers at the early state. During the aggregation, we found that Zn2+ exclusively promotes the annular protofibril formation without undergoing a nucleation process, whereas Cu2+ and Fe3+ inhibit fibril formation by prolonging the nucleation phases. Al3+ also inhibits fibril formation; however, the annular oligomers co-exist in the aggregation pathway. In conclusion, Zn2+, Cu2+, Fe3+, and Al3+ adopt distinct folding and aggregation mechanisms to affect Aβ, where Aβ destabilization promotes annular protofibril formation. Our study facilitates the understanding of annular Aβ oligomer formation upon metal ion binding.
Keywords: Alzheimer Disease, Amyloid, Kinetics, Metals, Protein Folding
Introduction
The brain deposition of amyloid plaques composed of Aβ2 is the pathological hallmark of AD (1, 2). Aβ is generated from sequential cleavages of amyloid precursor protein by β- and γ-secretases (3, 4). The predominant Aβ isoforms are Aβ40 and Aβ42, which differ in two residues at the C terminus, where Aβ42 is less abundant but more neurotoxic (5–8). Aβ is a natively unfolded protein prone to aggregating into cross-β-amyloid fibrils through a nucleation-dependent polymerization pathway (9). Aβ aggregation is considered the major culprit in AD, in which the Aβ oligomers, but not fibrils, better correlate with cognitive impairment and synaptic dysfunction (10). Aβ oligomers are referred to various different metastable intermediates found in the aggregation, including low molecular weight oligomers, spherical oligomers, Aβ-derived diffusible ligands, globulomers, annular protofibrils, Aβ56*, and curvilinear protofibrils (11–18).
Specific metal ions have been observed in the lesions of the disease. Analysis of the autopsy of AD patients shows abnormally high levels of specific metal ions present in the senile plaques (Cu2+, 25 μg/g, ∼393 μm; Zn2+, 69 μg/g, ∼1055 μm; and Fe3+, 52 μg/g, ∼940 μm) (19, 20). The levels of Cu2+, Zn2+, and Fe3+ in the AD neuropil are also significantly elevated (19), where Zn2+ is elevated from 346 to 786 μm, Cu2+ from 69 to 304 μm, and Fe3+ from 338 to 695 μm (19, 20). Al3+ has also been detected in amyloid fibers in cores of the senile plaques (21). Moreover, the imbalance of cellular Zn2+ and/or Cu2+ homeostasis modulates AD pathology (22, 23), and dietary Cu2+ and Al3+ are risk factors for AD (24–25). These facts indicate that the elevation of the metal ions is relevant to AD pathology.
Aβ is able to bind to the metal ions (22, 23). Aβ ion coordination, binding affinity, and induced aggregation have been studied intensively in various conditions; however, the results and mechanisms remain inconclusive. Aβ-Cu2+ coordination involves three intramolecular histidines (i.e. His-6, His-13, and His-14) in Aβ (26–28), with the fourth coordinate being either the amino group of the N terminus (29), an oxygen from Tyr-10 (30), or an oxygen from Glu-3 (29). The Aβ-Zn2+ complex is reportedly more complicated. A similar coordination with Cu2+ has been proposed for Zn2+ using the three histidines and the N terminus (31, 32). Both intermolecular Aβ-Cu2+ and Aβ-Zn2+ coordination have been reported via histidine bridges (28, 30–33). Fe3+ has also been shown to interact with histidines (37). Dissociation constants for the binding affinity of ions and Aβ ranging from attomolar to 11 μm for Cu2+ and 2–300 μm for Zn2+ have been reported (38–40).
Zn2+ and Cu2+ have been shown to accelerate Aβ deposition (41) but form amorphous aggregates (42–48). Al3+ and Fe3+ promote Aβ fibrils (43, 49, 50) or oligomer formation (23). Zn2+, especially at lower concentrations, and Cu2+ show protective effects toward Aβ mediated toxicity (42, 51). In contrast, histidine-bridged Aβ-Cu2+ dimers are neurotoxic (33). In addition, the metal chelators, clioquinol CQ and its analogous PBT2, are able to reverse ion-induced Aβ aggregation, reduce plaque load, and reverse cognition deficits in the transgenic AD mice (52, 53). The PBT2 is currently under phase II clinical trials (54).
The involvement of the metal ions with Aβ in AD and the potential development of metal chelating therapy indicate the importance of elucidating fundamental mechanisms of their effect on Aβ. Here, we systematically examine the metal ion effects, especially of Zn2+, Cu2+, Fe3+, and Al3+, on early and aggregated stages of full-length Aβ40. By employing different spectroscopic methods, including far-UV CD, tyrosine fluorescence, and 4,4′-Bis(1-anilinonaphthalene 8-sulfonate) (Bis-ANS) fluorescence, we monitor the conformational changes of Aβ and its ion binding affinity at the early stage. The binding kinetics and conformational stability are further examined by stopped flow machinery and guanidine hydrochloride denaturation. Photo-induced cross-linking of unmodified proteins (PICUP), thioflavin T (ThT) assay, dot blotting, and transmission electron microscopy (TEM) are also employed to monitor the oligomerization and fibrillization during aggregation. A mechanism for the metal ion effects on Aβ stability, oligomerization, and aggregation is then proposed.
EXPERIMENTAL PROCEDURES
Materials
GdnHCl, ThT, ammonium persulfate, and Tris (2,2′-bipyridyl)dichlororuthenium (II)) were purchased from Sigma-Aldrich (St. Louis, MO). Tris and NaCl were from Amresco (Solon, OH). CaCl2·2H2O was from J. T. Backer (Phillipsburg, NJ). AlCl3·6H2O, CuCl2, FeCl3·6H2O, and ZnCl2 were from Riedel-de Haeen Inc. (Sigma-Aldrich, St. Louis, MO). All metal ions were prepared in double-distilled Milli-Q water.
Aβ Preparation
Aβ40 peptide was synthesized using Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry and purified by reversed phase high performance liquid chromatography, as described previously (7). The molecular mass was identified by MALDI-TOF mass spectrometry (UltraFlex II; Bruker BioSciences, Billerica, MA). To prepare the Aβ stock, lyophilized peptide was freshly dissolved in Buffer A (10 mm Tris-HCl, pH 7.4) containing 8 m GdnHCl and refolded into Buffer A at a concentration of ∼1 mg/ml. Then the stock was then centrifuged at 17,000 × g at 4 °C for 15 min. The supernatant was collected and quantified by absorbance at 280 nm (ϵ = 1,280 cm−1 m−1) (55) and used as a stock solution to prepare Aβ at 25 μm for all experiments.
Fluorescence Spectroscopy
Fluorescence emission spectra were obtained using a FluoroMax-3 spectrofluorometer (Horiba Jobin Yvon). The emission spectra of Bis-ANS at 5 μm were collected from 450 to 550 nm with excitation at 400 nm. The emission spectra of tyrosine were collected from 290 to 360 nm with excitation at 270 nm. Both fluorescent experiments were performed at 25 °C. Temperature was controlled by a circulating water bath. The buffer backgrounds were subtracted.
Far-UV CD
Far-UV CD spectra were collected from 250 to 202 nm at 25 °C using a Jasco J-815 spectropolarimeter (Jasco Inc., Easton, MD). A circular quartz cell with a path length of 1 mm was used. Six scans were performed and averaged for each condition.
Equilibrium Binding of Metal Ion and Aβ by Titration
The 25 μm Aβ solution was titrated with different metal ion stock solutions: ZnCl2, CuCl2, and FeCl3 at 2 mm or AlCl3 at 20 mm. Less than 10% of the solution volume was increased after titration. Three different signals were collected, and the dilution factor was corrected. The monitored signals were 490 nm for Bis-ANS fluorescence, 305 nm for tyrosine fluorescence, and 216 nm for CD. The final titration signal was used as unity for normalization. The normalized data were plotted against the metal ion concentration. The amount of aluminum atom in the solution was confirmed by atomic absorption spectroscopy. Data fitting was performed by using an equation describing single protein ligand binding (56),
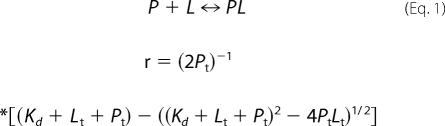 |
where γ is the fraction of the observed signal changes representing the bound protein fraction, Pt is the total Aβ concentration, Lt is the total ligand concentration, and Kd is the dissociation constant. In addition, the data were fitted to an equation describing one-protein and two-ligand binding,
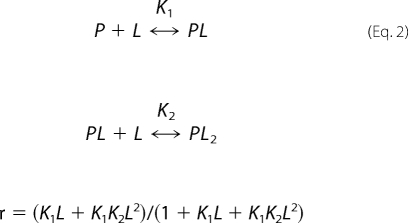 |
where γ is the fraction of the observed signal change, K1 and K2 are the association constants for the first and second ligand binding, and L is the free ligand concentration. Because the free ligand concentration cannot be faithfully determined in our experimental conditions, we used the total ligand concentration as L for the fitting.
Stopped Flow Experiments for Aβ and Metal Ion Binding Kinetics
The kinetics of Aβ and metal ion binding was examined using a stopped flow module (Bio-Logic, Claix, France) attached to a Jasco J-815 spectropolarimeter. The stopped flow module was composed of a multimixer SFM-400, a motor power supply MPS-250, and a photomultiplier system PMS-250. A fluorescence cuvette, FC-15, with an optical path of 1.5 mm was used. Excitation at 270 nm was used to monitor the tyrosine fluorescence of Aβ at 12.5 μm. A single mixing reaction using a volume ratio of 9:1 for the metal ions and Aβ was performed at 25 °C. The final metal ion concentrations were 1.25 mm; hence the metal-to-Aβ ratio was 100 to 1. Here, Aβ was prepared in 100 mm Tris-HCl, pH 7.4, to avoid metal ion-induced acidity. In this buffer system, the reactions were in neutral pH. The flow rate was fixed at 11 ml/s, resulting in a dead time of 4.7 ms. The data were collected every 0.5 ms for the first 2 s, every 20 ms in the range of 2–60 s, and every 0.5 s for 60–100 s. The data were fitted to multi-exponential equations using Bio-kine 32 V4.51 (Bio-Logic, Claix, France) with either single or double exponential equations with a linear base line,
where a and b are the slope and offset for the linear base line, and c and k are the amplitudes and rate constants for the exponential phases, respectively.
GdnHCl Denaturation
The denaturation study was performed, as described previously (58), at 25 °C. Briefly, the titration was performed by titrating an unfolded Aβ solution in Buffer A containing ∼6.2 m GdnHCl into Aβ solution in Buffer A containing <0.1 m GdnHCl. The desired metal ion concentrations, 25 μm of Aβ, and 5 μm of Bis-ANS were present in both solutions. The duration for each titration was ∼30 s, and each set of denaturation experiment was less than 1 h. The Bis-ANS fluorescence emission at 490 nm was collected, averaged, and normalized. The normalized emission intensity versus GdnHCl concentration was plotted.
PICUP
The experiment was performed as described previously (59). Here, we prepared the Aβ stock by urea instead of GdnHCl to facilitate running of SDS-PAGE. Briefly, Aβ samples at 25 μm prepared in Buffer A with different ion concentrations, as indicated, were immediately subjected to photo-induced cross-linking. A 90% Aβ solution was mixed with 5% each of 1 mm Tris(2,2′-bipyridyl)dichlororuthenium(II) and 20 mm ammonium persulfate. After mixing, the samples were exposed to a blue light LED in a closed chamber with a manual switch for 10 s. The cross-linking reaction was stopped by adding the SDS-PAGE sample buffer, and the samples were run on Tris-Tricine SDS-PAGE. All of the actions were performed without delay. The gel was subjected to Western blotting with anti-Aβ antibody 6E10 (Chemicon Inc., Billerica, MA) recognizing Aβ residues 1–17.
ThT Assay
Aβ (25 μm) in Buffer A with different metal ion concentrations and 25 μm ThT were incubated in an ELISA plate and monitored by a microplate reader (SpectraMax M5; Molecule Devices) at 25 °C. The samples were in quiescence during the incubation, except for 10 s of mixing prior to the measurement. The ThT emission was measured at 485 nm, whereas excitation was at 442 nm. The signals were collected automatically every 1 h for the first 100 h, after which they were collected in a longer intervals.
Dot Blotting
To detect the oligomers during the aggregation, 2 μl of the samples from the ThT assay were dotted onto a nitrocellulose membrane at the indicated incubation time and recognized using an anti-Aβ oligomer antibody, A11 (Invitrogen).
TEM
The samples were placed on glow-discharged, 400-mesh Formvar carbon-coated copper grids (EMS Inc., Hatfield, PA) for 3 min, rinsed, and negatively stained with 2% uranyl acetate. The samples were examined with a Tecnai G2 Spirit TWIN TEM (FEI, Hillsboro, OR) with an accelerating voltage of 75 kV.
Sedimentation Assay
The aggregated products of 25 μm Zn2+ or 100 μm Al3+ and 25 μm Aβ were incubated in Buffer A and subjected to centrifugation at 10,000 × g at 4 °C for 20 min. The supernatant and pellet were collected and analyzed by dot blotting using A11 and 6E10 antibodies and TEM. The pellet was resuspended in the original volume of Buffer A.
RESULTS
Al3+ and Zn2+ Increase Hydrophobic Exposure of Aβ Conformation, whereas Cu2+ Decreases
To reveal the effect of metal ions on Aβ, the ion-induced Aβ structural changes were monitored by three spectroscopic techniques: Bis-ANS fluorescence, tyrosine fluorescence, and far-UV CD spectra (Fig. 1). Bis-ANS is able to report the hydrophobic clusters exposed on protein surfaces and to probe Aβ conformation at the early stage, where the binding sites are at the flanking regions of the protease-resistant segment of Aβ (58, 60, 61). The experiments were performed with Aβ at 25 μm in the presence of Zn2+, Cu2+, Fe3+, and Al3+ above their saturated concentrations, as determined by the metal ion binding experiment described below. The Aβ spectra with and without 200 μm of Zn2+, Cu2+, and Fe3+ and 500 μm of Al3+ are shown in Fig. 1A and supplemental Fig. S1. In the presence of Al3+ and Zn2+, the Bis-ANS emission of Aβ40 showed ∼30- and 15-fold enhancement, respectively, but decreased ∼2.5-fold in the presence of Cu2+. The enhancement of the emission intensity indicates a larger extent of hydrophobic clusters exposed on the protein surface. Fe3+ did not induce significant changes on the hydrophobic surfaces of Aβ. Apart from the intensity difference, all of the spectra were blue-shifted from 500 nm to a range of ∼480–495 nm, indicating Bis-ANS encountering the hydrophobic environment.
FIGURE 1.

Conformational changes of Aβ in the presence of the metal ions. Aβ in the absence (●) and presence of 200 μm Zn2+ (▿), 200 μm Cu2+ ( ), 200 μm Fe3+ (○), or 500 μm Al3+ (□) is shown. A, Bis-ANS fluorescence spectra of Aβ with and without the metal ions. The inset shows the lower fluorescence range. In comparison with Aβ without ions, Aβs in the presence of Zn2+ and Al3+ contained more hydrophobic exposed protein surfaces but was more compact in Cu2+. Aβ in Fe3+ did not differ significantly. B, tyrosine fluorescence spectra of Aβ with and without the metal ions. Zn2+ and Al3+ both increased the quantum yield of Aβ, but Cu2+ and Fe3+ decreased the emission. C, far-UV CD spectra of Aβ with and without the metal ions. All of the ions altered the residual structures of Aβ to a lesser degree of the mean residue ellipticity.
), 200 μm Fe3+ (○), or 500 μm Al3+ (□) is shown. A, Bis-ANS fluorescence spectra of Aβ with and without the metal ions. The inset shows the lower fluorescence range. In comparison with Aβ without ions, Aβs in the presence of Zn2+ and Al3+ contained more hydrophobic exposed protein surfaces but was more compact in Cu2+. Aβ in Fe3+ did not differ significantly. B, tyrosine fluorescence spectra of Aβ with and without the metal ions. Zn2+ and Al3+ both increased the quantum yield of Aβ, but Cu2+ and Fe3+ decreased the emission. C, far-UV CD spectra of Aβ with and without the metal ions. All of the ions altered the residual structures of Aβ to a lesser degree of the mean residue ellipticity.
In addition to extrinsic fluorescence, we used intrinsic tyrosine fluorescence at residue 10 of Aβ to report local conformational changes. In the absence of metal ions, the tyrosine emission had a maximum at ∼302 nm while excited at 270 nm. We found that the tyrosine emissions were significantly quenched in the presence of Fe3+ and Cu2+ but enhanced in the presence of Al3+ and Zn2+ (Fig. 1B). The base lines of the spectra moved upward with concentration dependence to Al3+ and Zn2+ but not to Fe3+ and Cu2+ (supplemental Fig. S2). The increase in base line may be due to the formation of other unknown fluorescence species. Furthermore, far-UV CD spectra were employed to examine the secondary structure changes of Aβ (Fig. 1C). In the absence of the metal ions, a random coil-dominant spectrum was observed, as expected (62). However, all four metal ions further decreased ellipticity, especially with Zn2+ and Al3+, revealing that the metal ions reduced the content of residual secondary structures existing in Aβ conformation. Together, our results showed that Zn2+ and Al3+ increased the hydrophobic exposed protein surfaces, induced higher tyrosine emission, and reduced most residual secondary structures of Aβ. In contrast, Cu2+ and Fe3+ induced less or did not alter hydrophobic exposed surfaces, decreased tyrosine emission, and reduced residual secondary structures of Aβ.
Ion Binding Stoichiometry and Affinity to Aβ
To understand the conformational changes, binding stoichiometry, and affinity of Aβ and the metal ions, titration methods monitored by Bis-ANS fluorescence, tyrosine fluorescence, and far-UV CD were employed (Fig. 2). Aβ solutions at 25 μm were used, and the signals were normalized as described under “Experimental Procedures.” The fractions of Aβ bound with the metals were plotted against the metal ion to Aβ ratio. In general, the results showed that the three reporting signals changed coordinately but with some degree of differences. In the presence of Zn2+, three signals showed closed changes, and the data could be fitted to a single protein ligand interaction with the apparent dissociation constants, Kd = 3.9, 12, and 14 μm for Bis-ANS, tyrosine, and CD signals, respectively. In the presence of Cu2+, both CD and tyrosine signals changed concomitantly and can be described by a single ligand binding with Kd = 6.3 and 4.9 μm. However, the Bis-ANS signal was biphasic and delayed, suggesting that the hydrophobic exposed cluster of Aβ in the presence of Cu2+ may be less sensitive to the changes of tyrosine environment and secondary structures.
FIGURE 2.

Binding equilibrium of Zn2+ (A), Cu2+ (B), Fe3+ (C), and Al3+ (D) with Aβ. Conformational changes of Aβ were monitored by Bis-ANS fluorescence (□), tyrosine fluorescence (○), and circular dichroism () spectra. The signals at 490 nm for Bis-ANS fluorescence, 305 nm for the tyrosine fluorescence, and 216 nm for CD were collected, normalized, and plotted against the ratio of metal ion to Aβ concentration. Standard deviation of the data and residuals of the fits are shown. The solid lines are fits describing protein ligand interactions, and the dashed lines illustrate the data traces.
In the presence of Fe3+, the three signals changed in a similar fashion; however, their standard deviations became larger. The data could be fitted to a single protein and ligand binding (data not shown) but were best fitted to the equation describing one-protein and two-ligand binding, assuming total ligand concentration as the free ligands. The Kd1 and Kd2 were 59 and 44 μm for Bis-ANS, 185 and 55 μm for tyrosine, and 606 and 8 μm for CD signals. In the presence of Al3+, Bis-ANS signal changed faster than tyrosine signal, and tyrosine signal changed faster than CD signal. The Bis-ANS and tyrosine signals were best fitted to one-protein and two-ligand binding (Kd1 and Kd2 were 108 and 61 μm for Bis-ANS and 322 and 85 μm for tyrosine), where CD signal was biphasic. Thus, the result suggested that in the presence of Al3+, the hydrophobic exposed surface was most sensitive to Aβ structural changes than the tyrosine environment, and the secondary structural changes were the least sensitive. The fitting suggested that the stoichiometry of Zn2+ or Cu2+ to Aβ is 1, and that of Fe3+ and Al3+ to Aβ may be 2. However, the exact concentration of Al3+ ion in the solution was unknown because of the formation of aluminum hydroxide complexes in neural pH (63). Furthermore, higher concentration of Al3+ (i.e. above 200 μm in Buffer A)-induced acidity (supplemental Table S1).
Millisecond Binding Kinetics of the Metal Ions to Aβ
The stopped flow apparatus monitoring total quantum yield of Aβ tyrosine fluorescence was employed to study the binding kinetics of the metal ions and Aβ (Fig. 3). The final metal ion concentration was 100 times more than Aβ concentration. The signal was monitored from the instrumental dead time, ∼5 ms, up to 100 s. Aβ mixed with water had no signal change through time, indicating no conformational changes. When Aβ mixed with the metal ions, the signals showed exponential increases for Zn2+ and Al3+, whereas the signal decayed exponentially for Fe3+. The signal for Cu2+ had no significant change after 5 ms; however, the signal was already drastically decreased in the burst phase. The offset of Zn2+ was elevated, similar to the base-line elevation in the steady state spectra (supplemental Fig. S2A). The offset changes for Al3+ and Fe3+ were smaller and may be neglected. Overall, the amplitude changes of kinetic signals were consistent with the changes observed in the steady state spectra (Fig. 1B), where Zn2+ and Al3+ increased the fluorescence, but Fe3+ and Cu2+ quenched the fluorescence.
FIGURE 3.

Binding kinetics of Zn2+, Cu2+, Fe3+, and Al3+ with Aβ. Intrinsic tyrosine fluorescence of Aβ, 12.5 μm, in the absence (●) or presence of Zn2+ (▿), Cu2+ (), Fe3+ (○), or Al3+ (□) was monitored with excitation at 270 nm. Aβ was mixed to a 9:1 volume ratio with H2O as a control or 12.5 mm metal ion stocks to reach the final ion concentration of 1.25 mm. Every 100th collected data point is shown in the plot for clarity. Exponential fits for Zn2+, Fe3+, and Al3+ are shown as solid lines, and the residuals are shown above. Linear fits are used to illustrate the data of the control and Cu2+.
The data were fitted to exponential equations with a linear base line as described under “Experimental Procedures,” assuming pseudo-first-order reactions occurred. Fe3+ and Al3+ were best fitted to two exponential equations, whereas Zn2+ was best fitted to a single exponential equation. The number of kinetic events reflected the binding stoichiometry monitored from the titration data. The rate constants and amplitudes are described in supplemental Table S2, and the linear time scale of the data is shown in supplemental Fig. S3. In contrast to Zn2+, Fe3+, and Al3+, Cu2+ had nearly no signal change after the dead time, indicating that the events were completed in the burst phase. A very small increase observed at ∼50 s can be attributed to diffusion. Therefore, the order of binding from the fastest to the slowest was Cu2+ (submilliseconds) > Al3+ (k1 = 6.067 s−1, τ = 0.114 s) > Fe3+ (k1 = 3.455 s−1, τ = 0.20 s) > Zn2+ (k1 = 0.124 s−1, τ = 5.553 s). The observed offset differences could be due to submillisecond binding events or environmental effects altering the tyrosine quantum yield. In the case of Zn2+, because the fitting precisely described the events from 0.005 to 100 s, and the elevation was also observed in the steady state spectra, we suggested that the elevation is not due to a binding event occurred in the burst phase.
Effects of the Metal Ions on the Stability of Aβ
The possible effects of the metal ions to Aβ stability were then investigated. Although Aβ is considered to be intrinsically disordered (62), solution NMR and limited proteolysis studies have shown the existence of residual Aβ structures (64, 65). Previously, we have shown that Bis-ANS fluorescence can distinguish “native” Aβ, Aβ in native buffer, unfolded Aβ, and Aβ in high concentrations of denaturant. The chemical denaturation coupled with data fitting is able to provide equilibrium folding mechanisms for Aβ40 and Aβ42 (58). Here, GdnHCl denaturation was employed to examine the stability changes of Aβ in the presence of the metal ions (Fig. 4). In the native buffer, Bis-ANS binding to Aβ showed a significantly enhanced emission and blue-shifted spectrum in comparison with that in the presence of unfolded Aβ in 3 m GdnHCl (supplemental Fig. S4A). Tyrosine and CD spectra also showed different spectra between Aβ in native and unfolded conditions (supplemental Fig. S4, B and C). The Bis-ANS signals of Aβ in the absence and presence of the metal ions were monitored in different GdnHCl concentrations, and the data were averaged and normalized. In the absence of the metal ions, a tilted pretransition, <0.5 m GdnHCl, followed by a single transition between 0.5 and 1.5 m GdnHCl, and a post-transition >1.5 m GdnHCl were observed, indicating Aβ adopted an apparent two-state equilibrium mechanism (N⇆U), where a “native” and an unfolded ensemble were present at the equilibrium (58). In the presence of 25 μm Zn2+, 25 μm Cu2+, 50 μm Fe3+, or 100 μm Al3+, the concentrations at the transition midpoints from the titration study, we found that Cu2+ and Fe3+ did not significantly alter the midpoint of the transition [GdnHCl]½ at ∼1 m. On the contrary, Zn2+ and Al3+ shifted [GdnHCl]½ to ∼0.4 and ∼0.6 m, respectively. The pretransitions were lost in the presence of Zn2+ and Al3+. The results demonstrated that Zn2+ and Al3+ drastically destabilized Aβ. In addition, we performed reversed titration to titrate unfolded Aβ with Aβ in the native buffer. The denaturation was reversible in all conditions (supplemental Fig. S5A). Alternatively, we monitored the denaturation by tyrosine fluorescence (supplemental Fig. S5B). The results also showed that Aβs in the presence of Zn2+ and Al3+ shifted the midpoints to lower GdnHCl concentrations. Other ions that do not bind to Aβ, including Fe2+, Ca2+, Mg2+, and Na+, had no effect on Aβ stability (data not shown).
FIGURE 4.

GdnHCl denaturation of Aβ in the absence and presence of the metal ions. The effect of the metal ions on Aβ stability was examined by GdnHCl denaturation and monitored by Bis-ANS fluorescence. Aβ in the absence (●) and presence of 25 μm Zn2+ (▿), 25 μm Cu2+ (), 50 μm Fe3+ (○), or 100 μm Al3+ (□) are shown. Zn2+ and Al3+ significantly destabilized Aβ, whereas Cu2+ and Fe3+ did not change the stability significantly.
Effect of Metal Ions on Aβ Oligomerization
We employed PICUP assay to detect transiently appearing Aβ oligomers in the presence of the metal ions at the early stage (Fig. 5). PICUP was developed to photo-cross-link the short-lived metastable assembly in the fast equilibrium at the early stage (59). Here, we examined the effect in the presence of 25 μm Zn2+, 25 μm Cu2+, 50 μm Fe3+, and 100 μm Al3+. Before PICUP, Aβs were predominantly monomers, as examined by SDS-PAGE (data not shown). After PICUP, Aβ in the absence of the metal ions showed a dominant monomeric species and a minor dimeric species. No additional oligomer was induced in the presence of Fe3+ and Al3+. However, in the presence of Zn2+ and Cu2+, trimeric species were induced. Therefore, our results showed that Zn2+ and Cu2+, but not Fe3+ and Al3+, are capable of inducing higher assembly of Aβ oligomers that transiently appear at the early stage.
FIGURE 5.

Cu2+ and Zn2+ triggered Aβ oligomerization examined by PICUP assay. Photo-induced cross-linked Aβs with and without 25 μm Zn2+, 25 μm Cu2+, 50 μm Fe3+, and 100 μm Al3+ were run on SDS-PAGE. Aside from the dominant monomeric species, in the presence of Cu2+ and Zn2+, trimeric species were induced. The others showed only minor dimer populations.
Effect of the Metal Ions on Aβ Oligomerization and Fibrillization
To investigate the kinetics of Aβ oligomerization and fibrillization, ThT assay was first applied to monitor the cross-β-sheet formation during aggregation (Fig. 6). ThT is commonly used as a fluorescence probe in reporting the cross-β structures in amyloid fibrils (66). Aβ fibrillization is believed to undergo a nucleation-dependent pathway, where an oligomer nucleus is formed followed by an elongation event to mature fibrils (9). Five concentrations of the metal ions in different ranges were used, as indicated, for a better visualization of the effect. Without metal ions, Aβ followed the classic pattern of amyloid fibril formation to nucleate at a lag time of 20 h and elongated to reach a steady state after 60 h. In the presence of metal ions, concentration-dependent effects on Aβ fibrillization were observed. In the presence of Zn2+, the lag time was completely diminished, even in the lowest ion concentration (5 μm), and ThT intensity increased readily without lag time (Fig. 6A). A plateau was seen after 20 h with a lower ThT intensity in comparison with Aβ alone. Apart from Zn2+, the other three metal ions had similar inhibition effects, albeit occurring at different concentrations of ions. At 5 μm, Cu2+ prolonged the lag phase significantly to more than 60 h and decreased ThT fluorescence (Fig. 6B). At 50 μm, Cu2+ completely abolished the aggregation. Fe3+ and Al3+ above 50 μm both prolonged the lag phase to ∼40 h, where Al3+ has a strong inhibition effect above 250 μm (Fig. 6, C and D). A weaker inhibition effect from Al3+ was observed when the pH was neutralized by performing the experiment in Tris buffer at pH 7.85 (supplemental Fig. S6A). Thus, the strong inhibition may be primarily due to the acidic pH.
FIGURE 6.

Oligomerization and fibrillization of Aβ in the presence of the metal ions monitored by ThT and dot blotting. Aβ in the absence (black line) and presence of various concentrations of Zn2+ (A), Cu2+ (B), Fe3+ (C), and Al3+ (D) are shown. From lowest to highest, the ion concentrations are labeled with blue, green, orange, pink, and red lines. The concentrations examined were 5, 12.5, 25, 50, and 200 μm for Zn2+ and Cu2+; 25, 50, 100, 150, and 200 μm for Fe3+; and 25, 50, 100, 250, and 500 μm for Al3+. Time courses of Aβ in the presence of different metal ions were examined by dot blotting probed by anti-amyloid oligomer antibody, A11. The results are shown at the lower portions of each panel. Zn2+ accelerated Aβ aggregation without a lag phase but promoted oligomers formation with a Zn2+ concentration dependence. Cu2+, Fe3+, and Al3+ prolonged the lag phase with ion concentration dependence, where Al3+ at lower concentrations also promoted oligomer formation.
The corresponding oligomer appearance was further examined by dot blotting probed by anti-amyloid oligomer antibody, A11. A11 is an amyloid oligomer-specific antibody recognizing common epitopes in various amyloid proteins (13). Aβ incubated with different metal ion concentrations were spotted on the nitrocellulose membrane at different times during the aggregation. In the absence of metal ions, no significant A11-positive oligomer appeared. In the presence of Zn2+, starting from 25 to 200 μm, A11-positive signal was significantly enhanced and appeared at the first time point (Fig. 6A). For Cu2+ and Fe3+, no obvious A11-positive signal was detected (Fig. 6, B and C). Al3+ induced A11-positive signals in a time-dependent manner; however, the signals disappeared in the presence of 500 μm Al3+ (Fig. 6D). The disappearance of A11 signals was not caused by the acidic pH because the signal was also significantly weakened at higher concentrations of Al3+ when the solution was kept neutral (supplemental Fig. 6B).
Morphology of the Aggregates
We further employed TEM to observe the morphology of the Aβ aggregates in the presence of various metal ion concentrations (Fig. 7). In the absence of the metal ions, Aβ formed large amounts of mature fibrils. In the presence of Zn2+, no fibrils were found. Instead, massive amounts of heterogeneous oligomers mixed with several amorphous aggregates were observed. In contrast, with increased Cu2+ concentration, the appearance of fibrils decreased; however, nonfibril, amorphous aggregates increased. Some short and fragmented fibrils were also observed at 25 μm of Cu2+. A similar phenomenon was seen in the presence of higher Fe3+ concentration, although without observing the short fibrils. A decrease in fibrils was also observed in the presence of Al3+. However, at 100 μm Al3+, fibrils and oligomers were observed to co-exist. At 500 μm Al3+, the solution became acidic, and the aggregates were mainly amorphous.
FIGURE 7.

The morphology of the Aβ aggregates in the presence of different metal ions. The end point products of Aβ after aggregation were monitored by TEM in the indicated metal ion concentrations. Zn2+ promoted oligomer formation in a concentration-dependent manner, whereas others in general reduced fibril formation and increased amorphous aggregates. Oligomers and fibrils were observed to co-exist in the presence of 100 μm Al3+. The scale bars are 200 nm.
We further examined the secondary structural content of the aggregates by far-UV CD spectra (supplemental Fig. S7). The spectra of Aβ with 50 μm of Fe3+ showed similar β-sheet-dominant spectra to Aβ fibrils alone, indicating fibril formation in 50 μm Fe3+. In the presence of 100 μm Al3+, the ellipticity was reduced. The aggregates formed in 25 μm of Cu2+ and Zn2+ were significantly different from β-sheet-rich fibrils, where the spectra of Aβ in the presence of Zn2+ had a helix-like double minima spectra. However, the minima were not at the classic wavelength for α-helices. This result is consistent with the data obtained from ThT assay and TEM images.
To further characterize the Zn2+- and Al3+-induced oligomers, sedimentation was employed to separate the aggregates in soluble and insoluble fractions. The fractions were then analyzed by dot blotting and TEM (Fig. 8). At 10,000 × g for 20 min, the A11 signals for both Zn2+ and Al3+ appeared mostly in the pellets, whereas half of the amount of the total protein remained in the supernatants for Zn2+ and little protein for Al3+ as demonstrated by 6E10 immunoblotting (Fig. 8A). TEM analysis (Fig. 8B) showed large amounts of heterogeneous, ring-shaped, and pore-like oligomers in the pellet of Aβ-Zn2+ aggregates with an average diameter of ∼23 nm and a pore size of ∼12 nm. Weakly stained small oligomers that were difficult to count were also present. Fewer oligomers remained in the supernatant, and the species appeared to have a rougher surface. Close examination showed no obvious spherical oligomers, only mostly pore-like conformers. In the aggregates of Aβ-Al3+, we found fibrils and oligomers co-existed in the pellet. Most oligomers were small (<10 nm) and had smooth surface; however, some large oligomers (∼25 nm) without pores were also present, especially in close proximity with the fibrils. A few rugged oligomers were observed in the supernatant, but no fibrils. Similar results were observed for dot blotting and TEM by examining the aggregates of Aβ in the presence of 250 μm Al3+ starting form Tris buffer, pH 7.85 (supplemental Fig. S6, C and D). Pore-like oligomers and little curvilinear protofibrils were mostly observed in the supernatant, whereas fibrils and oligomers were observed to co-exist in the pellet. Those ring-shaped, pore-like oligomers most resembled the annular protofibrils described in the literature (67).
FIGURE 8.

The distribution of Zn2+- and Al3+-induced oligomers. The end point products of Aβ in the presence of 25 μm Zn2+ or 100 μm Al3+ were subjected to centrifugation. The oligomers were mostly precipitated in the pellets, as revealed by dot blotting (A) and TEM images (B). Supernatant and pellet were denoted as S and P. Annular protofibrils were observed predominantly. The scale bars for all images are 100 nm, except for the inset, which is 20 nm.
DISCUSSION
To understand the involvement of specific metal ions in Aβ, we examined the influence of the metal ions on Aβ initial conformation and stability and the mechanism of the effect of these factors on its aggregation process. All four ions, i.e. Zn2+, Cu2+, Fe3+, and Al3+, reduced residual secondary structures of Aβ upon formation of metal-Aβ complex, but only Zn2+ and Al3+ induced larger exposure of the hydrophobic protein surfaces. Upon ion binding, a structural change was observed, and the binding occurred in submillisecond or millisecond time scale, in which Cu2+ binds faster than Al3+, Fe3+, and Zn2+. The binding stoichiometries were suggested to be 1 for Zn2+ and Cu2+ and 2 for Fe3+ and Al3+. GdnHCl denaturation showed that Zn2+ and Al3+, but not Cu2+ and Fe3+, destabilized Aβ. Further examination on the metal ion effect on the aggregation process revealed that Zn2+ preferentially facilitates annular protofibril formation without a nucleation phase, whereas other metal ions inhibit fibril formation, likely by partitioning Aβ into fibril or amorphous aggregate/protofibril formation pathways. The results are consistent with previous reports on the metal ion effects on Aβ aggregation, where Zn2+ and Cu2+ accelerate Aβ aggregation to form nonfibrillar aggregates but not ThT-positive fibrils (41–48), and Al3+ promotes Aβ42 fibrils or oligomers formation (49, 68).
The fact that both Zn2+ and Al3+ induced more hydrophobic exposure and destabilization of initial Aβ conformation, resulting in annular protofibrils formation indicates that Aβ conformation and stability are key factors in the formation of specific aggregates such as annular protofibrils. Although Cu2+ induced slightly more compact Aβ conformation, the stability was not significantly different, suggesting that the Cu2+-induced retardation of fibril formation may not be due to the stability change of Aβ. For Aβ aggregation in the presence of Cu2+, Fe3+, and Al3+, the partition between amorphous aggregates/protofibrils and fibrils seems to correlate with to their binding affinities, where the inhibition is more profound in higher concentration of Fe3+ and Al3+. Although Zn2+ and Cu2+ induced similar trends of transiently appearing Aβ oligomers at the early stage, their aggregations were distinct, as reflected by ThT intensity, A11 dot blotting, and aggregate morphology. Our results demonstrated that the four metal ions adopt different mechanisms to influence Aβ, yet similarities in partitioning pathways or stability influence on annular protofibril formation are present.
Aβ is intrinsically disordered; however, residual structures that contain a turn connected by two loose β-strands are revealed by solution NMR (64) and limited proteolysis (65). The structure of the N terminus of Aβ from residues 1 to 10 is considered to be flexible and disordered. The region is not incorporated in the structural assembly of fibrils (69) and oligomers (70). The two histidinyl residues, His-13 and His-14, involved in the metal ion coordination are located at the edge of the first β-strand in fibrils (69) and are involved in a solvent accessible turn formed between His-13 to Gln-15 in disc-shaped Aβ42 oligomers (70). Our results showing Aβ structural changes concomitantly upon metal ion binding demonstrated that the ion binding at the N terminus is able to affect the overall residual conformation of Aβ. Several familial AD mutants containing single mutations at the N terminus such as English mutant, H6R (71), and Tottori mutant D7N (72), as well as the N-terminal truncated pyroglutamate Aβ may possess different ion binding properties and result in altered aggregation pathways.
It is particularly interesting that Zn2+ induced preferentially ring-shaped, pore-like annular protofibrils without an apparent nucleation process and that Al3+ induced similar oligomers. Recently, heterogeneous Aβ oligomers have been found to exist in immunological distinct structural states (67, 73). These Aβ oligomers have been characterized as prefibrillar oligomers, fibrillar oligomers, and annular protofibrils. The annular protofibrils are considered to be assembled from the prefibrillar oligomers, where the conversion is accelerated in the presence of lipid vesicles and other artificial conditions. The annular protofibrils are less toxic and membrane-permeable in comparison with prefibrillar oligomers, but they share common structural epitopes with the bacterial toxin formed by β-barrel structures (67). In this study, the Zn2+- or Al3+-induced Aβ oligomers were morphologically similar to the annular protofibrils and also shared a common epitope recognized by A11 antibody. The extent to which the Zn2+- or Al3+-induced Aβ oligomers resemble those annular protofibrils or others remains uno oknown. The structural conversion, membrane permeability, and toxicity of those annular protofibrils need further structural and functional characterizations. However, our far-UV CD spectra of Zn2+-induced oligomers showed no enrichment of the β-conformation, which suggests that the oligomers are not fibrillar oligomers and should be different from the β-barrel toxins.
In physiological conditions, less than 0.5 μm of extracellular Zn2+ is present in brain; however, a high concentration of Zn2+ was found in the glutamergic neurons during neuronal activity especially in cerebrocortex or amygdala (22, 74). The presynapic Zn2+ is concentrated by the zinc transporter protein, ZnT3, to achieve ∼300 μm in the cleft (22, 75). The in vivo evidence shows that the ZnT3 knock-out transgenic amyloid precursor protein mice with a reduction of Zn2+ in the hippocampus did not develop obvious amyloid plaques in comparison with those with normal ZnT3 level (34). In AD patients, a decrease of metallothionein 3 that regulates the uptake of synaptic Zn2+ has been observed (35), and the release of synaptic Zn2+ during activity is critical for Aβ oligomer synaptic targeting (36). These studies show that synaptic Zn2+ and Zn2+ homeostasis play crucial roles in AD pathology. According to our results, we suspect that the Zn2+-induced oligomer formation resulting in heterogeneous Aβ annular protofibrils may occur at presynapses during synaptic activities, thereby triggering oligomer formation and leading to concentrated Aβ oligomers at the synaptic clefts. This hypothesis may also contribute to the mechanism of rapid cognition restoration in the metal chelating therapy (53). In summary, our fundamental folding and aggregation studies facilitate the understanding of the clinically related metal ions in AD to Aβ and provide a rationalized mechanism for annular protofibril formation. The study may contribute to potential pathogenic implication in AD.
Supplementary Material
Acknowledgments
We thank the specialists of Academia Sinica, Tai-Lang Lin (Institute of Cellular and Organismic Biology) for assisting TEM imaging, and Cheng-Ying Yu (Institute of Chemistry) for assisting atomic absorption operation.
This work was supported by Grants 98-2320-B-001-020-MY3, 96-2113-M-001-030-MY2, and 97-2320-B-010-027-MY3 from the Genomics Research Center (Academia Sinica, Taiwan, National Science Council, Taiwan) and National Health Research Institute Grant NHRI-EX98-9816NC.

The on-line version of this article (available at http://www.jbc.org) contains supplemental text, Tables S1 and S2, and Figs. S1–S7.
- Aβ
- amyloid-β
- AD
- Alzheimer disease
- Bis-ANS
- 4,4′-Bis(1-anilinonaphthalene 8-sulfonate)
- GdnHCl
- guanidine hydrochloride
- PICUP
- photo-induced cross-linking of unmodified proteins
- ThT
- thioflavin T
- TEM
- transmission electron microscopy
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
REFERENCES
- 1. Masters C. L., Simms G., Weinman N. A., Multhaup G., McDonald B. L., Beyreuther K. (1985) Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mucke L. (2009) Nature 461, 895–897 [DOI] [PubMed] [Google Scholar]
- 3. Kang J., Lemaire H. G., Unterbeck A., Salbaum J. M., Masters C. L., Grzeschik K. H., Multhaup G., Beyreuther K., Müller-Hill B. (1987) Nature 325, 733–736 [DOI] [PubMed] [Google Scholar]
- 4. Thinakaran G., Koo E. H. (2008) J. Biol. Chem. 283, 29615–29619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bibl M., Esselmann H., Mollenhauer B., Weniger G., Welge V., Liess M., Lewczuk P., Otto M., Schulz J. B., Trenkwalder C., Kornhuber J., Wiltfang J. (2007) J. Neurochem. 103, 467–474 [DOI] [PubMed] [Google Scholar]
- 6. Schoonenboom N. S., Mulder C., Van Kamp G. J., Mehta S. P., Scheltens P., Blankenstein M. A., Mehta P. D. (2005) Ann. Neurol. 58, 139–142 [DOI] [PubMed] [Google Scholar]
- 7. Burdick D., Soreghan B., Kwon M., Kosmoski J., Knauer M., Henschen A., Yates J., Cotman C., Glabe C. (1992) J. Biol. Chem. 267, 546–554 [PubMed] [Google Scholar]
- 8. Jarrett J. T., Berger E. P., Lansbury P. T., Jr. (1993) Biochemistry 32, 4693–4697 [DOI] [PubMed] [Google Scholar]
- 9. Roychaudhuri R., Yang M., Hoshi M. M., Teplow D. B. (2009) J. Biol. Chem. 284, 4749–4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haass C., Selkoe D. J. (2007) Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 11. Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., Rowan M. J., Selkoe D. J. (2002) Nature 416, 535–539 [DOI] [PubMed] [Google Scholar]
- 12. Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., Ashe K. H. (2006) Nature 440, 352–357 [DOI] [PubMed] [Google Scholar]
- 13. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003) Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 14. Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., Morgan T. E., Rozovsky I., Trommer B., Viola K. L., Wals P., Zhang C., Finch C. E., Krafft G. A., Klein W. L. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hoshi M., Sato M., Matsumoto S., Noguchi A., Yasutake K., Yoshida N., Sato K. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 6370–6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barghorn S., Nimmrich V., Striebinger A., Krantz C., Keller P., Janson B., Bahr M., Schmidt M., Bitner R. S., Harlan J., Barlow E., Ebert U., Hillen H. (2005) J. Neurochem. 95, 834–847 [DOI] [PubMed] [Google Scholar]
- 17. Harper J. D., Wong S. S., Lieber C. M., Lansbury P. T. (1997) Chem. Biol. 4, 119–125 [DOI] [PubMed] [Google Scholar]
- 18. Lashuel H. A., Lansbury P. T., Jr. (2006) Q. Rev. Biophys. 39, 167–201 [DOI] [PubMed] [Google Scholar]
- 19. Lovell M. A., Robertson J. D., Teesdale W. J., Campbell J. L., Markesbery W. R. (1998) J. Neurol. Sci. 158, 47–52 [DOI] [PubMed] [Google Scholar]
- 20. Frederickson C. J., Koh J. Y., Bush A. I. (2005) Nat. Rev. Neurosci. 6, 449–462 [DOI] [PubMed] [Google Scholar]
- 21. Yumoto S., Kakimi S., Ohsaki A., Ishikawa A. (2009) J. Inorg. Biochem. 103, 1579–1584 [DOI] [PubMed] [Google Scholar]
- 22. Bush A. I. (2003) Trends Neurosci. 26, 207–214 [DOI] [PubMed] [Google Scholar]
- 23. Zatta P., Drago D., Bolognin S., Sensi S. L. (2009) Trends Pharmacol. Sci. 30, 346–355 [DOI] [PubMed] [Google Scholar]
- 24. Sparks D. L., Schreurs B. G. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 11065–11069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frisardi V., Solfrizzi V., Capurso C., Kehoe P. G., Imbimbo B. P., Santamato A., Dellegrazie F., Seripa D., Pilotto A., Capurso A., Panza F. (2010) J. Alzheimers Dis. 20, 17–30 [DOI] [PubMed] [Google Scholar]
- 26. Ma Q. F., Hu J., Wu W. H., Liu H. D., Du J. T., Fu Y., Wu Y. W., Lei P., Zhao Y. F., Li Y. M. (2006) Biopolymers 83, 20–31 [DOI] [PubMed] [Google Scholar]
- 27. Syme C. D., Nadal R. C., Rigby S. E., Viles J. H. (2004) J. Biol. Chem. 279, 18169–18177 [DOI] [PubMed] [Google Scholar]
- 28. Minicozzi V., Stellato F., Comai M., Serra M. D., Potrich C., Meyer-Klaucke W., Morante S. (2008) J. Biol. Chem. 283, 10784–10792 [DOI] [PubMed] [Google Scholar]
- 29. Karr J. W., Akintoye H., Kaupp L. J., Szalai V. A. (2005) Biochemistry 44, 5478–5487 [DOI] [PubMed] [Google Scholar]
- 30. Stellato F., Menestrina G., Serra M. D., Potrich C., Tomazzolli R., Meyer-Klaucke W., Morante S. (2006) Eur. Biophys. J. 35, 340–351 [DOI] [PubMed] [Google Scholar]
- 31. Danielsson J., Pierattelli R., Banci L., Gräslund A. (2007) FEBS J. 274, 46–59 [DOI] [PubMed] [Google Scholar]
- 32. Syme C. D., Viles J. H. (2006) Biochim. Biophys. Acta 1764, 246–256 [DOI] [PubMed] [Google Scholar]
- 33. Smith D. P., Smith D. G., Curtain C. C., Boas J. F., Pilbrow J. R., Ciccotosto G. D., Lau T. L., Tew D. J., Perez K., Wade J. D., Bush A. I., Drew S. C., Separovic F., Masters C. L., Cappai R., Barnham K. J. (2006) J. Biol. Chem. 281, 15145–15154 [DOI] [PubMed] [Google Scholar]
- 34. Lee J. Y., Cole T. B., Palmiter R. D., Suh S. W., Koh J. Y. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 7705–7710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu W. H., Lukiw W. J., Bergeron C., Niznik H. B., Fraser P. E. (2001) Brain Res. 894, 37–45 [DOI] [PubMed] [Google Scholar]
- 36. Deshpande A., Kawai H., Metherate R., Glabe C. G., Busciglio J. (2009) J. Neurosci. 29, 4004–4015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nair N. G., Perry G., Smith M. A., Reddy V. P. (2010) J. Alzheimers Dis. 20, 57–66 [DOI] [PubMed] [Google Scholar]
- 38. Garzon-Rodriguez W., Yatsimirsky A. K., Glabe C. G. (1999) Bioorg. Med. Chem. Lett. 9, 2243–2248 [DOI] [PubMed] [Google Scholar]
- 39. Atwood C. S., Scarpa R. C., Huang X., Moir R. D., Jones W. D., Fairlie D. P., Tanzi R. E., Bush A. I. (2000) J. Neurochem. 75, 1219–1233 [DOI] [PubMed] [Google Scholar]
- 40. Tõugu V., Karafin A., Palumaa P. (2008) J. Neurochem. 104, 1249–1259 [DOI] [PubMed] [Google Scholar]
- 41. Bush A. I., Pettingell W. H., Multhaup G. d., Paradis M., Vonsattel J. P., Gusella J. F., Beyreuther K., Masters C. L., Tanzi R. E. (1994) Science 265, 1464–1467 [DOI] [PubMed] [Google Scholar]
- 42. Yoshiike Y., Tanemura K., Murayama O., Akagi T., Murayama M., Sato S., Sun X., Tanaka N., Takashima A. (2001) J. Biol. Chem. 276, 32293–32299 [DOI] [PubMed] [Google Scholar]
- 43. Ha C., Ryu J., Park C. B. (2007) Biochemistry 46, 6118–6125 [DOI] [PubMed] [Google Scholar]
- 44. Noy D., Solomonov I., Sinkevich O., Arad T., Kjaer K., Sagi I. (2008) J. Am. Chem. Soc. 130, 1376–1383 [DOI] [PubMed] [Google Scholar]
- 45. Ryu J., Girigoswami K., Ha C., Ku S. H., Park C. B. (2008) Biochemistry 47, 5328–5335 [DOI] [PubMed] [Google Scholar]
- 46. Smith D. P., Ciccotosto G. D., Tew D. J., Fodero-Tavoletti M. T., Johanssen T., Masters C. L., Barnham K. J., Cappai R. (2007) Biochemistry 46, 2881–2891 [DOI] [PubMed] [Google Scholar]
- 47. Garai K., Sahoo B., Kaushalya S. K., Desai R., Maiti S. (2007) Biochemistry 46, 10655–10663 [DOI] [PubMed] [Google Scholar]
- 48. Tõugu V., Karafin A., Zovo K., Chung R. S., Howells C., West A. K., Palumaa P. (2009) J. Neurochem. 110, 1784–1795 [DOI] [PubMed] [Google Scholar]
- 49. Ricchelli F., Drago D., Filippi B., Tognon G., Zatta P. (2005) Cell Mol. Life Sci. 62, 1724–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kawahara M., Kato M., Kuroda Y. (2001) Brain Res. Bull. 55, 211–217 [DOI] [PubMed] [Google Scholar]
- 51. Lovell M. A., Xie C., Markesbery W. R. (1999) Brain Res. 823, 88–95 [DOI] [PubMed] [Google Scholar]
- 52. Cherny R. A., Atwood C. S., Xilinas M. E., Gray D. N., Jones W. D., McLean C. A., Barnham K. J., Volitakis I., Fraser F. W., Kim Y., Huang X., Goldstein L. E., Moir R. D., Lim J. T., Beyreuther K., Zheng H., Tanzi R. E., Masters C. L., Bush A. I. (2001) Neuron 30, 665–676 [DOI] [PubMed] [Google Scholar]
- 53. Adlard P. A., Cherny R. A., Finkelstein D. I., Gautier E., Robb E., Cortes M., Volitakis I., Liu X., Smith J. P., Perez K., Laughton K., Li Q. X., Charman S. A., Nicolazzo J. A., Wilkins S., Deleva K., Lynch T., Kok G., Ritchie C. W., Tanzi R. E., Cappai R., Masters C. L., Barnham K. J., Bush A. I. (2008) Neuron 59, 43–55 [DOI] [PubMed] [Google Scholar]
- 54. Faux N. G., Ritchie C. W., Gunn A., Rembach A., Tsatsanis A., Bedo J., Harrison J., Lannfelt L., Blennow K., Zetterberg H., Ingelsson M., Masters C. L., Tanzi R. E., Cummings J. L., Herd C. M., Bush A. I. (2010) J. Alzheimers Dis. 20, 509–516 [DOI] [PubMed] [Google Scholar]
- 55. Edelhoch H. (1967) Biochemistry 6, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 56. Chen Y. R., Clark A. C. (2004) Protein Sci. 13, 2196–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Deleted in proof.
- 58. Chen Y. R., Glabe C. G. (2006) J. Biol. Chem. 281, 24414–24422 [DOI] [PubMed] [Google Scholar]
- 59. Bitan G., Lomakin A., Teplow D. B. (2001) J. Biol. Chem. 276, 35176–35184 [DOI] [PubMed] [Google Scholar]
- 60. Brand L., Gohlke J. R. (1972) Annu. Rev. Biochem. 41, 843–868 [DOI] [PubMed] [Google Scholar]
- 61. LeVine H., 3rd. (2002) Arch. Biochem. Biophys. 404, 106–115 [DOI] [PubMed] [Google Scholar]
- 62. Riek R., Güntert P., Döbeli H., Wipf B., Wüthrich K. (2001) Eur. J. Biochem. 268, 5930–5936 [DOI] [PubMed] [Google Scholar]
- 63. Vasudevaraju P., Govindaraju M., Palanisamy A. P., Sambamurti K., Rao K. S. (2008) Indian J. Med. Res. 128, 545–556 [PubMed] [Google Scholar]
- 64. Hou L., Shao H., Zhang Y., Li H., Menon N. K., Neuhaus E. B., Brewer J. M., Byeon I. J., Ray D. G., Vitek M. P., Iwashita T., Makula R. A., Przybyla A. B., Zagorski M. G. (2004) J. Am. Chem. Soc. 126, 1992–2005 [DOI] [PubMed] [Google Scholar]
- 65. Lazo N. D., Grant M. A., Condron M. C., Rigby A. C., Teplow D. B. (2005) Protein Sci. 14, 1581–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. LeVine H., 3rd. (1993) Protein Sci. 2, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kayed R., Pensalfini A., Margol L., Sokolov Y., Sarsoza F., Head E., Hall J., Glabe C. (2009) J. Biol. Chem. 284, 4230–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Drago D., Bettella M., Bolognin S., Cendron L., Scancar J., Milacic R., Ricchelli F., Casini A., Messori L., Tognon G., Zatta P. (2008) Int. J. Biochem. Cell Biol. 40, 731–746 [DOI] [PubMed] [Google Scholar]
- 69. Tycko R. (2006) Q. Rev. Biophys. 39, 1–55 [DOI] [PubMed] [Google Scholar]
- 70. Ahmed M., Davis J., Aucoin D., Sato T., Ahuja S., Aimoto S., Elliott J. I., Van Nostrand W. E., Smith S. O. (2010) Nat. Struct. Mol. Biol. 17, 561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Janssen J. C., Beck J. A., Campbell T. A., Dickinson A., Fox N. C., Harvey R. J., Houlden H., Rossor M. N., Collinge J. (2003) Neurology 60, 235–239 [DOI] [PubMed] [Google Scholar]
- 72. Wakutani Y., Watanabe K., Adachi Y., Wada-Isoe K., Urakami K., Ninomiya H., Saido T. C., Hashimoto T., Iwatsubo T., Nakashima K. (2004) J. Neurol. Neurosurg. Psychiatry 75, 1039–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wu J. W., Breydo L., Isas J. M., Lee J., Kuznetsov Y. G., Langen R., Glabe C. (2010) J. Biol. Chem. 285, 6071–6079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Frederickson C. J., Bush A. I. (2001) BioMetals 14, 353–366 [DOI] [PubMed] [Google Scholar]
- 75. Palmiter R. D., Cole T. B., Quaife C. J., Findley S. D. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 14934–14939 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.