

Abstract
The 57-residue small hydrophilic endoplasmic reticulum-associated protein (SHERP) shows highly specific, stage-regulated expression in the non-replicative vector-transmitted stages of the kinetoplastid parasite, Leishmania major, the causative agent of human cutaneous leishmaniasis. Previous studies have demonstrated that SHERP localizes as a peripheral membrane protein on the cytosolic face of the endoplasmic reticulum and on outer mitochondrial membranes, whereas its high copy number suggests a critical function in vivo. However, the absence of defined domains or identifiable orthologues, together with lack of a clear phenotype in transgenic parasites lacking SHERP, has limited functional understanding of this protein. Here, we use a combination of biophysical and biochemical methods to demonstrate that SHERP can be induced to adopt a globular fold in the presence of anionic lipids or SDS. Cross-linking and binding studies suggest that SHERP has the potential to form a complex with the vacuolar type H+-ATPase. Taken together, these results suggest that SHERP may function in modulating cellular processes related to membrane organization and/or acidification during vector transmission of infective Leishmania.
Keywords: Circular Dichroism (CD); Lipid; NMR; Parasite; Protein Cross-linking; Protein Folding; Vacuolar ATPase; Leishmania major; SHERP; SRCD, Synchrotron Radiation Circular Dichroism
Introduction
Leishmaniasis, caused by the digenetic kinetoplastid parasite Leishmania, is one of the world's neglected diseases. According to the World Health Organization, an estimated 12 million people are currently infected, with up to 350 million people at risk in 88 countries (1). Transmitted by blood-feeding female sand flies, more than 20 species of Leishmania are pathogenic in humans. Infection results in a range of clinical conditions, with the visceral form of leishmaniasis usually fatal if untreated (reviewed in Ref. 2). There are no licensed vaccines for protection against infection, and postexposure therapies are often ineffective, with unacceptable side effects. Co-infection with Leishmania and HIV presents an additional complication, with parasite infection known to accelerate the onset of AIDS (reviewed in Ref. 3). Clearly, an improved understanding of the molecular mechanisms leading to pathogenesis in humans is of key importance in developing new therapeutic options for tackling these debilitating diseases.
Leishmania cycles between extracellular life in the alimentary tract of its sand fly vector and intracellular maintenance within the acidic phagolysosomes of mammalian host macrophages. Following uptake during sand fly blood-feeding, dividing parasites reach a stationary growth phase prior to differentiation, with the end point of that process being the production of highly motile metacyclic organisms that are preadapted for survival following inoculation into the host (4, 5). This differentiation is characterized by modifications to the parasite surface glycocalyx, principally to the major lipid-anchored glycoconjugate, lipophosphoglycan, that confer complement resistance (6–9). At the same time, other cellular processes, such as translation, are down-regulated.
In contrast, the 6.2-kDa small hydrophilic endoplasmic reticulum-associated protein (SHERP)7 is exclusively expressed in metacyclic parasites, the only highly expressed Leishmania protein identified to date that is specific to these insect stage organisms (10). SHERP is hydrophilic with an acidic pI and shares no sequence identity with any other known protein, in common with up to 40% of the proteins encoded by the Leishmania genomes (11, 12). Although SHERP has an unusual dual localization at the cytosolic face of the ER and the mitochondrial outer membrane, in vivo cross-linking and fractionation experiments have shown that it is a peripheral membrane protein of as yet unknown function (10). Its unusually high level of stage-specific expression (estimated at 100,000 molecules/cell), localization, and partial biochemical characterization suggest that SHERP may have a vital function in metacyclic parasites during transmission to the mammalian host.
To gain an improved insight into the functional properties of SHERP, we have used a combined biophysical and biochemical approach to characterize the structure and interactions of this protein both in vitro and in vivo. Using synchrotron radiation circular dichroism (SRCD) and NMR spectroscopic methods, we show that SHERP adopts a globular fold when bound to either anionic phospholipids or SDS detergent. Cross-linking and binding studies suggest that SHERP can form a complex with a vacuolar type H+-ATPase (V-ATPase). From these data, we postulate that an anionic environment or surface is a key molecular recognition template for SHERP and may facilitate its ability to form a complex with V-ATPase. Furthermore, formation of a membrane-localized SHERP·V-ATPase complex could in turn modulate acidification processes that may prove important for the differentiation of Leishmania within its sand fly vector.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Reagents
Escherichia coli strains XL1-Blue and BL21(λDE3) were obtained from Stratagene (La Jolla, CA). Thermus thermophilus strain AH8 was a gift from Professor Ken Yokoyama (Tokyo Institute of Technology, Yokohama, Japan). Unless otherwise noted, chemicals were purchased from Sigma-Aldrich. Oligonucleotides were synthesized by Sigma-Genosys. Other suppliers were as follows: Deep Vent DNA polymerase and T4 DNA ligase, New England Biolabs (Hitchin, UK); restriction enzymes, Roche Applied Science and New England Biolabs; M199 medium, fetal calf serum, and DNA molecular weight markers, Invitrogen; isopropyl β-d-thiogalactoside, Genesys (London, UK); Taq PCR Ready-To-Go beads, protein molecular weight standards, and Coomassie Brilliant Blue R250, Amersham Biosciences; reagents for production of Luria-Bertani (LB) medium, Merck; Spectra 9-CN medium, Spectra Gases Ltd. (Cambridge, UK); Centricon protein concentration devices, Millipore (Watford, UK); UV-activated cross-linking reagent sulfo-N-hydroxysuccinimidyl-2-(6-[biotinamido]-2-(p-azidobenzamido)-hexanoamido) ethyl-1,3′-dithioproprionate (sulfo-SBED), ImmunoPure immobilized monomeric avidin, and Ultralink immobilized streptavidin, Pierce. Synthetic phospholipids were purchased from Avanti Polar Lipids (Alabaster, AL) as lyophilized solids, and Triton X-100 and n-dodecyl-β-d-maltopyranoside were from Anatrace, Inc. (Maumee, OH).
Cloning, Expression, and Purification of Recombinant L. major SHERP
Plasmid DA-PET15/3 (10), which contains the L. major ORF of SHERP1, was used as template in PCR amplification with primers LmN_F (5′-CCCCCCCCATGGTTCATCATCATCATCATCATTCTAGAGCTAGTTCGTACACAATGGACCAGGAGACAAG-3′) and LmN_R (5′-GGGAAAGGATCCTTACGAGCCACCGC-3′). The resulting PCR product contains NcoI and BamHI restriction sites for cloning into the isopropyl β-d-thiogalactoside-inducible expression vector pET-28a(+). The resulting construct, pTLmCSHERP, was transformed into heat shock-competent E. coli XL1-Blue cells and selected on L-agar containing 30 μg/ml kanamycin. Plasmids were purified from single colonies using the QIAprep spin minikit and subsequently screened for the presence of the SHERP insert by PCR amplification screening using Taq Ready-To-Go PCR beads with the T7 promoter and LmN_R primers. The DNA sequence of the SHERP insert in PCR-positive plasmids was subsequently confirmed by the Advanced Biotechnology Centre (Imperial College London).
Plasmid pTLmCSHERP was next transformed into E. coli strain BL21(λDE3), and expression trials indicated that recombinant SHERP could be overproduced as a soluble protein. An optimized purification protocol was developed, which was initiated by inoculating LB medium containing 30 μg/ml kanamycin with a 1:100 dilution of an overnight culture of E. coli BL21(λDE3)(pTLmCSHERP). Cells were grown at 37 °C with shaking to A600 of 0.4–0.8. Isopropyl β-d-thiogalactoside was added to a final concentration of 1 mm, and the cells were left at 37 °C for a further 4 h prior to harvesting by centrifugation at 10,000 × g and 4 °C for 25 min. Supernatants were discarded, and the cell pellets stored at −20 °C until required. Frozen cells were thawed at room temperature and resuspended using 1.5% of the original culture volume in a buffer containing 50 mm K2HPO4/KH2PO4, 300 mm NaCl, 5 mm imidazole, and 0.5 mm PMSF, pH 7.0. Lysozyme was then added to produce a final concentration of 0.75 mg/ml, and the cells were incubated at room temperature for 25 min prior to lysis by sonication with three 30-s bursts, with 30-s intervals on ice using an XL 2020 sonicator (Labcaire Systems Ltd., Avon, UK) at 60% intensity. Insoluble materials were pelleted by centrifugation at 15,000 × g and 4 °C for 25 min. The supernatant was decanted and passed through a 0.45-μm filter (Millipore, Watford, UK) and then applied to prepared TALON resin (BD Biosciences) in a Bio-Rad disposable plastic column, and the flow-through was retained. SHERP was eluted using 50 mm K2HPO4/KH2PO4, 300 mm NaCl, and 300 mm imidazole, pH 7.0. Elution fractions were analyzed by SDS-PAGE, and SHERP fractions were pooled prior to concentration using a Centri-Prep YM-3 centrifugal filter unit (Millipore, Watford, UK) with a 3-kDa cut-off limit, at 3000 × g and 4 °C.
SRCD Spectroscopy and Data Analysis
Lyophilized samples of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-(phospho-rac-(1-glycerol)) (sodium salt) (DOPG), 1,2-dioleoyl-sn-glycero-3-phospho-l-serine, and/or 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were dissolved in methanol. Methanol was subsequently evaporated from aliquots of phospholipids using a rotary evaporator. The lipid layer was resuspended in 100 mm sodium phosphate buffer at pH 7.4 and bath-sonicated for 30 min. Small unilamellar vesicles were produced by passaging the samples through an extruder with a pore size of 50 nm. The concentration of SHERP was determined by measuring its absorbance at 280 nm and using an experimentally determined extinction coefficient of 2689 m−1 cm−1, based upon quantitative amino acid analyses (carried out at the Protein & Nucleic Acid Chemistry Facility, University of Cambridge, UK). SHERP and small unilamellar vesicle samples were mixed and allowed to equilibrate for 10 min at final concentrations of 2.1 mg/ml SHERP, 1.4 mg/ml DOPC/DOPE (3:1 lipid ratio), or 1.6 mg/ml DOPC/DOPG (1:1 lipid ratio). SDS solutions were prepared at room temperature by dissolving weighed quantities of SDS powder into a 100 mm sodium phosphate buffer, pH 7.4. Prior to data collection, samples were degassed to remove any dissolved nitrogen or oxygen and centrifuged at 13,000 × g for 2 min to remove any insoluble material and/or large vesicles. SHERP concentrations ranged from 1.6 to 2.2 mg/ml in the lipid experiments.
SRCD spectra were acquired on Beamline CD12 at the Synchrotron Radiation Source (Daresbury, UK) and/or at Beamline UV1 at the Institute for Synchrotron Studies (Århus, Denmark). Samples were measured in either Suprasil quartz circular demountable cells or calcium fluoride cells (13) with path lengths ranging from 4.2 to 21.6 μm. All samples were in 100 mm sodium phosphate buffer, pH 7.4. For samples containing buffer only (no detergent or lipid) or in buffer containing 50 mm SDS, the concentration of SHERP was 5.6 mg/ml. SHERP concentrations ranged from 1.6 to 2.2 mg/ml in the lipid experiments. Typically, spectra were acquired at 4 °C, over the wavelength range from 280 to 168 nm with an interval of 1.0 nm and an averaging (dwell) time of 1 s (CD12) or 3 s (UV1). The low wavelength cut-off values of the data were established from measurements of the high tension signal (a measure of pseudoabsorption) as described previously (14). Three repeats of each sample and base-line spectrum (which contained all of the components present in the sample cell except the protein) were acquired. The beamlines were calibrated at the beginning of each beam injection using camphorsulfonic acid, as described previously (15). Small unilamellar vesicle stability was verified by comparisons of the SRCD and high tension spectra obtained at the beginning and end of spectral acquisition, with changes suggesting vesicle instability or aggregation.
SRCD spectra were processed using CDtool software (16). Base-line spectra were averaged and then subtracted from the averaged sample spectra. The resulting spectra were zeroed between 263 and 270 nm and smoothed with a Savitsky-Golay filter (16). A mean residue weight of 112 was used in the calculations.
The secondary structures were calculated using the DICHROWEB server (17, 18) with the CDSSTR (19) and CONTINLL (20) algorithms and the SELMAT (21) version of SELCON3 (22), all with the reference data set SP175 (23). Because the helical type of secondary structure is the most accurately defined by these algorithms, and because “other” is the dominant secondary structure in this protein, the analyses reported focus primarily on these types of secondary structure. The uncertainty (±) values reported are one S.D. between average calculated secondary structure values produced from all the algorithms. The normalized root mean square deviation value (24), which is a reflection of the difference between the experimentally derived values for the mean residue ellipticity and those calculated by the fitting algorithm, is reported for the CONTINLL method. Values of ≤0.10 indicate that the calculated secondary structure corresponds well to the experimental data.
Nuclear Magnetic Resonance Spectroscopy
All data were acquired on a Bruker DRX 500 MHz (1H frequency) NR spectrometer equipped with a cryoprobe with the exception of the three-dimensional 1H-13C NOESY experiment, which was acquired with a Varian INOVA spectrometer operating at 800 MHz (1H frequency). Spectra collected at 277 K were acquired using 0.5 mm SHERP in 10 mm HEPES, pH 7.4. All spectra used for assignment and collection of structural restraints were acquired at 298 K using 0.5 mm SHERP in 10 mm HEPES, pH 7.4, and 50 mm SDS in a 90%/10% D2O mixture. 15N,13C-labeled samples of recombinant L. major SHERP were produced in Spectra 9-CN medium, a minimal medium containing 0.07% 15NH4Cl and 0.2% 13C6-glucose following the optimized purification protocol described above. All NMR spectra for assignment data were recorded hybrid 15N,13C-labeled samples with unlabeled detergent. Sequence-specific backbone 1HN, 15N, 13C′, 13Cα, and 13Cβ were determined using standard triple resonance methods (for a review, see Ref. 25). Hα and Hβ assignments were obtained using HBHA(CBCACO)NH (25). The side chain assignments were completed using HCCH total correlation spectroscopy and (H)CC(CO)NH total correlation spectroscopy (25). Three-dimensional 1H-15N/13C NOESY-HSQC (mixing time 100 ms at 500 and 800 MHz) experiments provided the distance restraints used in the final structure calculation. Heteronuclear 1H-15N NOE data with minimal water saturation were acquired using the pulse sequence described previously (26). 1H-15N residual dipolar couplings were measured in strained polyacrylamide gel according to published methods (27).
The ARIA protocol (28) was used for completion of the NOE assignment and structure calculation. Dihedral angle restraints derived from TALOS (29) and 50 1H-15N residual dipolar coupling measurements were also implemented. The frequency window tolerance for assigning NOEs was ±0.04 and ±0.06 ppm for direct and indirect proton dimensions and ±0.7 and ±1.2 ppm for nitrogen and carbon dimensions, respectively. The ARIA parameters, p, Tv, and Nv, were set to default values. A slow cooling step was invoked using 72,000 dynamic steps of 0.003 ps (30). The 10 lowest energy structures had no NOE violations greater than 0.5 Å and no dihedral angle violations greater than 5°. For the SDS titration, increasing SDS concentrations up to 40 mm were added in aliquots to a solution of 0.5 mm 15N-labeled SHERP in 10 mm HEPES, pH 7.4. 2D 1H/15N HSQC spectra were recorded at each point.
In Vitro Cross-linking with SHERP
Purified recombinant SHERP-labeled with sulfo-SBED (SHERP-SBED in PBS) was generated following the manufacturer's protocol (Pierce). The L. major strain null for the LmcDNA16 locus encoding SHERP and HASP proteins (ΔcDNA16::HYG/ΔcDNA16::PAC) (31) was maintained and subcultured as described previously (31). Procyclic and metacyclic promastigote stages were monitored by their distinct morphology and changing agglutination with peanut lectin at days 2–3 and 6–8, respectively (31).
For cross-linking, L. major promastigotes taken after 6–8 days in culture were centrifuged at 300 rpm for 10 min, washed three times with 0.25 m sucrose in 10 mm Tris, pH 7.4, and then resuspended in 1 cell volume equivalent of lysis buffer (0.2 m Tris-HCl, pH 8.0, 6 mm MgCl2, 1 mm EDTA, 1 mm DTT, 100 μg/ml leupeptin, 500 μg/ml Pefabloc, 5 μg/ml pepstatin, 198 μg/ml 1,10-phenanthroline, 25 μg/ml E64). Cells were lysed on ice by sonication with three 10-s bursts with 1-min intervals on ice using an XL 2020 sonicator (Labcaire Systems Ltd., Avon, UK) at 60% intensity. Cells were split into two equal portions in polypropylene tubes and placed in the dark. To one portion 7 μg of SHERP-SBED per 1 × 109 cells were added, and both portions were then left to incubate for 1 h on ice. Both samples were then exposed for 8 min to a 125-watt mercury lamp placed 5 cm from each open polypropylene tube. SDS was added to a final concentration of 1% (w/v), and the cells were centrifuged at 5000 × g for 5 min. The supernatant was then removed, and aliquots were analyzed by SDS-PAGE and blotting to probe for successful biotinylation using HRP-conjugated streptavidin. An ImmunoPure immobilized monomeric avidin column (Pierce) was then equilibrated with PBS followed by biotin blocking and elution buffer (2 mm d-biotin in PBS). Biotin was removed from reversible binding sites with 12 ml of regeneration buffer (0.1 m glycine, pH 2.8) followed by further PBS washing. The cell supernatant samples were adjusted to a final volume of 2 ml and applied to the column. A total of six 2-ml wash steps using PBS were carried out, followed by six 2-ml elution steps using the biotin blocking and elution buffer. 2-ml fractions were collected and analyzed using SDS-PAGE followed by Coomassie or silver staining. Elution fractions from the avidin column were then added to the washed streptavidin beads and allowed to incubate for 1 h at room temperature. The mixture was then centrifuged at 5000 × g for 1 min, the supernatant was removed, and the beads were washed four times using PBS. 25 μl of SDS-PAGE sample buffer was added to the washed beads prior to boiling for 5 min and analysis by SDS-PAGE. Gels were visualized using either colloidal Coomassie or silver staining. Fractions containing proteins isolated under this protocol were digested using trypsin, and the peptide fragments were analyzed by MALDI-TOF by Philip Nugent in the Technology Facility at the University of York.
Surface Plasmon Resonance Studies
The affinity of SHERP for T. thermophilus V-ATPase was studied using surface plasmon resonance (SPR) methods. V-ATPase was purified from the T. thermophilus strain AH8 in which the atpA gene encoding the A subunit had been modified to contain a His tag for metal affinity purification of the complex (32). The purification protocol was carried out as described previously (32) with the following minor modifications. First, the concentration of Triton X-100 was reduced from 1 to 0.5% in the buffer used in the metal affinity purification step. In addition, a Mono Q column (GE Healthcare) was used for ion exchange chromatography, and the concentration of Triton X-100 was reduced from 0.05 to 0.03%. All nine subunits were clearly present based upon SDS-PAGE analysis as reported previously (32), and the sample was exchanged by spin column into a buffer containing 10 mm Tris-HCl (pH 8.0), 100 mm NaCl, and 0.03% n-dodecyl-β-d-maltopyranoside.
SPR studies were carried out using a BIAcore 2000 instrument with a dextran-coated CM5 sensor chip. Two flow cells were activated according to the manufacturer's instructions. To one flow cell, V-ATPase at 0.2 mg/ml in 10 mm sodium acetate buffer, pH 5.0, was immobilized to a CM5 chip by amine coupling. Unreacted groups on both flow cells were capped with 100 mm ethanolamine. Both flow cells were washed and demonstrated insignificant levels of nonspecific binding using bovine serum albumin. Prior to injecting SHERP, cells were washed with HBS (10 mm HEPES, 150 mm NaCl, 0.01% (w/v) P20 detergent), and injections of 0.5 m NaCl and 20 mm CHAPS were used to stabilize the base line. SHERP in HBS was injected into both flow cells at six concentrations (0.5, 1, 2, 5, 10, and 15 μm) at a flow rate of 5 μl/min. Binding curves obtained from the flow cell containing the immobilized V-ATPase were corrected by subtraction of low level SHERP binding to the null cell. Sensorgrams were analyzed with BIAevaluation 3.x software using a simple 1:1 binding model shown below.
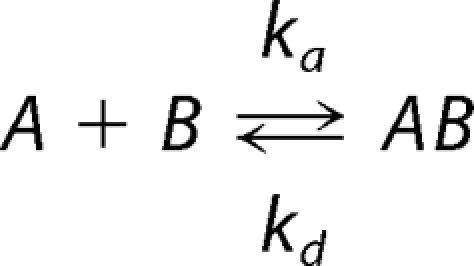 |
where KD = kd/ka, and KA = ka/kd.
RESULTS
Induced Folding of SHERP by Anionic Reagents
The SHERP open reading frame from L. major was expressed and purified to homogeneity as a recombinant protein containing the 57 residues that comprise SHERP, followed by a 10-residue non-hydrolyzable linker (33) to improve stability and a 6-residue polyhistidine linker for affinity purification (Fig. 1). Secondary structure analyses using SRCD were carried out on purified recombinant L. major SHERP in the presence and absence of lipids. The use of SRCD spectroscopy as opposed to conventional CD measurements was important in these studies for a number of reasons: 1) SRCD beamlines have a detector geometry that reduces the apparent light scattering, which can be problematic for membrane samples (24); 2) the higher signal-to-noise levels enable the use of smaller amounts of protein; 3) because lipid and detergents absorb light, although they do not produce a CD signal, the higher light flux of the synchrotron permits the use of higher lipid/protein ratios; and 4) the data obtained at lower wavelengths (∼170 nm compared with 190 nm) can provide more accurate information on secondary structures (34), especially for proteins that contain a large amount of disordered structure because the signature spectral characteristics for disordered structures occur below 200 nm. Wavelengths below 200 nm are not easily or accurately achievable for lipid-containing samples in conventional CD instruments.
FIGURE 1.

The expressed sequence of recombinant L. major SHERP. The translated open reading frame of SHERP sequence is shown in green, the linker region in red, and the histidine tag in yellow.
SHERP is highly soluble in aqueous solution in the absence of lipids or detergents; however, its SRCD spectrum (Fig. 2) is that typically found for a mostly disordered protein, with a large negative peak at ∼200 nm. Secondary structure analyses on the SHERP spectrum suggest that in the absence of lipid or detergents, the protein has little defined secondary structure (Table 1), with only 10% of the protein being helical and >40% being “other” (i.e. not one of the canonical helical, sheet, or turn types of secondary structures; it is usually considered to be unordered or disordered). This experimental observation is in contrast to a previously published protein sequence-based secondary structure prediction, based on computational analysis, which suggested that SHERP would be a mainly helical structure, containing a possible turn in the middle of its sequence (10).
FIGURE 2.

Induced folding of SHERP by anionic lipid or detergent shown monitored by SRCD spectroscopy. SRCD spectra of SHERP are shown in aqueous solution (black), in DOPC/DOPE (red; 1.4 mg/ml DOPC/DOPE; 3:1 lipid ratio), in DOPC/DOPG (blue; 1.6 mg/ml DOPC/DOPG; 1:1 lipid ratio), and in 50 mm SDS (green). Anionic lipid (DOPG) or detergent (SDS) induces SHERP to adopt a mainly helical structure.
TABLE 1.
Calculated secondary structures of L. major SHERP samples based on SRCD data
| Sample addition | Helixa | Otherb | NRMSDc |
|---|---|---|---|
| % | % | ||
| None | 12 ± 5 | 44 ± 2 | 0.053 |
| DOPC/DOPE | 13 ± 5 | 43 ± 2 | 0.049 |
| DOPC/DOPG | 48 ± 3 | 30 ± 4 | 0.053 |
| SDS | 64 ± 4 | 22 ± 4 | 0.037 |
a Values are shown ± S.D. between values calculated by three different algorithms (see “Experimental Procedures” for explanation).
b Unordered or disordered secondary structure.
c Normalized root mean square deviation value, a goodness of fit parameter between experimental data and calculated secondary structure (24).
Given that SHERP was previously localized near ER and mitochondrial membranes (10), the effect of phospholipids on the folding and structure of SHERP was examined. The addition of small unilamellar vesicles composed of either neutral (zwitterionic) phospholipids, DOPC, or a mixture of DOPC and DOPE produced no appreciable spectral changes or corresponding alterations in calculated secondary structure composition (Fig. 2 and Table 1) compared with SHERP alone in aqueous solution. However, significant spectral changes (Fig. 2) were observed upon the addition of vesicles containing equimolar amounts of DOPC and the anionic phospholipid DOPG. Analyses of these spectra revealed a marked reduction in the amount of “other” structure coinciding with a considerable increase in the amount of helical component, displaying a maximum value of ∼50% in the presence of DOPC/DOPG vesicles (Table 1). In addition, SRCD spectra of SHERP collected in the presence of a phosphatidylcholine/phosphatidylserine mixture (data not shown) were very similar to those for the DOPC/DOPG mixture.
The observed induced folding of recombinant L. major SHERP was characterized in further detail using NMR spectroscopy. NMR spectra of SHERP alone in aqueous buffer were consistent with SHERP adopting an unfolded or disordered structure (Fig. 3). The addition of the short chain phospholipid 1,2-dihexanol-sn-glycero-3-phospho-l-serine to SHERP yielded NMR spectra with severely broadened line widths due to the large size of the complex (data not shown). To improve the spectral characteristics, we examined whether SHERP could also be induced to fold in detergents that result in smaller complexes that are more compatible with NMR measurements. Although the neutral detergent β-octyl glucoside detergent did not induce significant structural changes in SHERP (data not shown), the amphipathic, anionic detergent SDS produced a SRCD spectrum characteristic of a mostly helical structure similar to that found for the protein in the anionic lipids (Fig. 2). Analyses of the spectra obtained for SHERP in the presence of SDS or the DOPC/DOPG mixture both indicate a helical content of ∼50–60% (Table 1). Taken together, these results suggest that an anionic environment is key to driving the structural transition of SHERP from a predominantly unordered structure to one possessing significant α-helical content. Comparison of the measured secondary structures of the construct in the presence of these partner molecules with that predicted based on sequence methods (10) produces similar results; the C-terminal 16-residue addition, including the His tag, is not predicted to form an ordered structure, but 39 of the 73 total residues are, which corresponds to a structure that would be 53% helical (35). Interestingly, sequence-based disorder predictions (36) only identify the termini as disordered and so do not suggest that it would be disordered in the absence of a binding partner.
FIGURE 3.

Two-dimensional 1H-15N HSQC NMR spectrum of SHERP. Data shown were collected at 35 °C in the absence (red) and presence (black) of 50 mm SDS. Residue assignments are shown with blue labels. Increased dispersion of amide resonances in the presence of SDS indicates folding of SHERP into a distinct structure.
The Solution Structure of SHERP
The high resolution solution structure of SHERP in the presence of SDS was solved using heteronuclear multidimensional NMR spectroscopy. Initially, 15N-labeled SHERP was analyzed by two-dimensional NMR methods. In the absence of SDS, the 1H-15N HSQC spectrum of SHERP showed a narrow 1H chemical shift dispersion clustered mainly between 8.0 and 8.6 ppm (Fig. 3), suggesting that SHERP is unstructured and highly dynamic in solution. Spectra were also collected at 4 °C in an attempt to maximize the likelihood of stabilizing the structure (supplemental Figs. S1 and S2). The observation of the three Gly cross-peaks at low temperatures compared with higher temperatures is probably due to the reduced amide exchange with water. It is unlikely that the spectral dispersion of these three Gly residues is due to residual structure because the rest of the amides do not show evidence for this, and the heteronuclear NOE (Fig. 3) confirms the highly dynamic nature of an unfolded polypeptide. These observations are also consistent with the SRCD spectrum of SHERP at 4 °C (Fig. 2), collected in the absence of SDS or lipids, that shows that SHERP is a mostly disordered protein. Upon titration with SDS, the spectrum displays a wide dispersion of the amide resonances, indicative of a fully folded protein with regular secondary structure (Fig. 3); the saturation point for the titration of SDS with L. major SHERP was obtained at a ratio of 80:1 (SDS/SHERP).
The backbone assignment of the 15N/13C-labeled L. major SHERP was carried out using standard triple resonance methodology (25). Of the 73 residues that comprise this recombinant form of SHERP, peaks corresponding to 66 residues were assigned, which include 56 of 57 residues encoded by the SHERP open reading frame and the 10 residues present in the non-hydrolyzable linker. No assignment was made of the N-terminal methionine or the 6 residues of the C-terminal polyhistidine tag, although the latter did show some increase in order most likely arising from electrostatic interactions with the SDS micelle. Analysis of chemical shift values for SHERP using the TALOS program (29) revealed that 46 of the 66 residues were predicted to adopt a helical conformation.
Using a combination of manual and automated NMR assignment methods for analysis of NOESY spectra (28), a family of high resolution structures for L. major SHERP in the presence of SDS micelles was calculated (Fig. 4a), with excellent agreement with experimental data and structural quality (Table 2). All areas of secondary structure are very well defined; the average pairwise root mean square deviation for the water-refined final structures is 0.19 ± 0.05 Å for the backbone atoms and 0.65 ± 0.17 Å for the heavy atoms of residues present in secondary structure. Steady-state heteronuclear NOE data show that L. major SHERP in the presence of SDS possesses flexible N and C termini (Fig. 5).
FIGURE 4.

The solution structure of SHERP incorporated into SDS micelles. a, the final family of 10 high resolution structures for L. major SHERP determined in the presence of SDS micelles. b and c, ribbon diagram and electrostatic surface representation of SHERP in two 90° orientations. The N terminus of SHERP (N) is shown in a and b. Only the coordinates for residues corresponding to the L. major encoded SHERP sequence are shown. Images were produced using PyMOL (DeLano Scientific LLC).
TABLE 2.
NMR and refinement statistics for L. major SHERP in SDS
| Parameter | Value |
|---|---|
| No. of experimental restraints | |
| Total NOE-derived | 883 |
| Ambiguous | 415 |
| Unambiguous | 468 |
| TALOS (f/y) | 114 |
| Root mean square deviation from experimental restraints | |
| Distance (Å) | 0.04 ± 0.003 |
| Dihedral angle (degrees) | 1.0 ± 0..02 |
| Root mean square deviation from idealized covalent geometry | |
| Bonds (Å) | 0.0035 ± 0.0001 |
| Angles (degrees) | 0.71 ± 0.008 |
| Energies (kcal mol−1) | |
| ENOE | 84.8 ± 22.5 |
| Ebond | 12.8 ± 0.3 |
| Eangle | 138 ± 3.5 |
| Evdw | −583 ± 4.5 |
| Coordinate root mean square deviation (Å) | |
| Backbone atoms in secondary structure | 0.19 ± 0.03 |
| Heavy atoms in secondary structure | 0.65 ± 0.17 |
| Ramachandran plot | |
| Residues in favored regions (%) | 77 |
| Residues in allowed regions (%) | 20 |
| Residues in outlier regions (%) | 3a |
a The outliers include residue Asp2, which is so near to the N terminus that it shows some evidence of flexibility, and Asp29, which lies at the apex of the helical hairpin.
FIGURE 5.

Steady-state heteronuclear 15N-1H NOE values for SHERP in a 1:100 molar ratio with SDS. Rigid secondary structural elements are characterized by a high heteronuclear NOE, whereas lower values indicate significant flexibility on the pico- to nanosecond time scale. Black circles show the experimentally determined values, and the red error bars represent one S.D. value. A secondary structure prediction using chemical shift values is shown above for comparison.
As shown in Fig. 4b, the structure for monomeric L. major SHERP in the presence of SDS adopts a helix-turn-helix motif. The flexible N-terminal region is followed by the first helix, which stretches from Gln3 to Leu27, and then a hairpin turn followed by a second helix from residue Gly32 to Leu53 and finally the flexible C-terminal region. These regions of secondary structure correlate well with the backbone dynamics calculated from the heteronuclear NOE values, where high heteronuclear NOE values (indicating increased rigidity; Fig. 5) correspond to regions of low flexibility (small root mean square deviation values). Visualization of the calculated surface electrostatic potential of this structure for SHERP (Fig. 4c) shows it to be amphipathic with positively charged residues, such as lysine and arginine, being present on opposite faces to negatively charged aspartate and glutamate residues.
SHERP Can Form a Complex with Vacuolar ATPase
Earlier localization studies identified SHERP as a peripheral membrane protein (10), and the in vitro studies described above indicate that SHERP can be induced to fold in the presence of anionic phospholipids or SDS. Although these findings raise the possibility that SHERP may form specific interactions with anionic phospholipids in vivo, the protein may also form protein-protein interactions with other binding partners in a membrane environment, as suggested previously (10). To explore this hypothesis further, L. major SHERP was labeled with the chemical cross-linking reagent sulfo-SBED to yield SHERP-SBED. This form of the protein was then incubated with a lysed culture of metacyclic promastigotes of a genetically modified strain of L. major (ΔcDNA16::HYG/ΔcDNA16::PAC) lacking both alleles of the LmcDNA16 locus that codes for the SHERP genes (31). SHERP-SBED-cross-linked complexes, which contain a biotin affinity label, were isolated by streptavidin affinity chromatography. Fractions from column elutions and washes were collected and analyzed by SDS-PAGE (supplemental Fig. S3). Eight samples (five from the wash and three from the elution) were excised from these gels and then submitted for MALDI-TOF mass spectrometry analysis.
Five proteins (supplemental Table S1) were identified with statistically significant levels of confidence: one protein from the column elution step, subunit B of V-ATPase, and four proteins from the column wash (peroxidoxin, paraflagellar rod protein, the heat shock protein Hsp70, and the SA subunit of the 40 S ribosome). The four proteins in the wash fractions probably represent false positive results, derived from proteins present at high abundance in the parasite lysates (as shown in proteomic analyses (e.g. see Ref. 37)) that have nonspecifically bound to the affinity column. Such proteins would be expected to be present in this fraction, and their identification represents a robust control. In comparison, subunit B from the vacuolar or V-ATPase was isolated from the column elution step, suggesting that it was indeed captured as a biotin-labeled cross-linked complex with SHERP. V-ATPase is a membrane protein complex known to be involved in the acidification of intracellular compartments in eukaryotes; subunit B is one of the two large components that form the extracellular catalytic rotor (reviewed in Refs. 38–40). Given that vacuolar acidification is known to occur during parasite differentiation (discussed below), V-ATPase was thus considered to be a potential genuine binding partner for SHERP.
To further study the interaction of L. major SHERP with V-ATPase, we have used surface plasmon resonance to obtain the binding affinity of SHERP for the V-ATPase homologue from T. thermophilus, a well characterized membrane protein complex with subunits that can be purified for in vitro studies (32). Amino acid sequence alignment of subunit B from T. thermophilus and L. major demonstrates sufficient levels of identity and similarity (56 and 72%, respectively (supplemental Fig. S4)) to support the use of T. thermophilus V-ATPase as a good model system to assess complex formation with SHERP. Sensorgrams (supplemental Fig. S5) of L. major SHERP binding to T. thermophilus V-ATPase complex immobilized to a CM5 dextran chip show rapid association and dissociation phases. A concentration-independent elevated base line is also observed following the dissociation phase that may indicate a small but constant amount of irreversible binding of SHERP to the chip surface containing immobilized V-ATPase. However, the observation of a rapid dissociation phase suggests that SHERP mainly interacts with V-ATPase in a specific and reversible manner. Non-linear regression using the global fitting Bioevaluation 3.x software yielded a KD of 2.0 ± 0.1 μm, indicative of the formation of a functionally stable complex between SHERP and the T. thermophilus V-ATPase.
DISCUSSION
Identification of a Molecular Recognition Template for SHERP Folding
In this study, both biophysical and biochemical approaches have been used to gain more information about SHERP and its molecular interactions, in order to advance our understanding of the role of this unusual small molecule. We produced a new recombinant form of SHERP, ∼8 kDa, containing a 6-residue C-terminal polyhistidine tag and small non-hydrolyzable linker, to enable rapid purification and to improve its stability against proteolytic degradation. SHERP is a highly soluble protein, which was shown here to exist in a predominantly disordered state in aqueous solution using SRCD (Fig. 2 and Table 1) and NMR (Fig. 3 and supplemental Figs. S1 and S2). Based upon its hydrophilicity and apparent lack of membrane spanning or anchoring motifs, together with in vivo cross-linking data, it has been previously suggested that SHERP is a peripheral membrane protein that localizes on the cytosolic side of membranes, perhaps in association with another integral or peripheral membrane protein (10).
To explore this hypothesis further, we initiated studies to assess whether SHERP could interact with phospholipids. A key but unexpected finding was that this protein can be induced to fold by vesicles containing anionic phospholipids. SRCD spectra (Fig. 2 and Table 1) of SHERP in the presence of vesicles containing DOPG or 1,2-dioleoyl-sn-glycero-3-phospho-l-serine clearly show that the helical content of the protein increases significantly. This effect could also be mimicked using the anionic detergent SDS and was observed using both SRCD (Fig. 2 and Table 1) and heteronuclear NMR (Fig. 3) methods. Furthermore, the observations that SHERP does not fold in the presence of neutral phospholipids or neutral detergents suggest that the molecular recognition template that triggers SHERP to fold in vivo is dominated by a positively charged electrostatic surface. This hypothesis is further supported by the amphipathic nature of the globular fold of SHERP, determined in the presence of SDS using multidimensional heteronuclear NMR methods. As shown in Fig. 4b, SHERP adopts a well defined amphipathic helix-turn-helix fold from residue 3 to 53, consistent with earlier bioinformatic predictions (10) and with the helical propensity observed in the SRCD spectra. A clear distribution of electrostatically positive and negative regions on opposite faces of the folded structure of SHERP is also evident, as seen in Fig. 4c.
The ability of anionic phospholipids to induce protein folding is a phenomenon that has been well characterized in proteins such as α-synuclein (41, 42) and human plasma apolipoprotein A-I (43). These proteins adopt amphipathic helical folds with a membrane recognition motif characterized by 11-residue repeating units (44, 45). Molecular “stripes” of neutral or negative or positively charged residues are visible along the distinct faces of the helices. The charge distribution observed in these α-helical structures was first used to propose a mode of binding to the lipid surface by apolipoproteins (44) where positively charged side chains interact with the negatively charged phospholipid headgroup (46). In this model, the hydrophobic interaction of helices penetrates to a depth of ∼3 Å into the lipid surface, an effect often described as “snorkeling,” with larger assemblies forming a “carpet” of extended helices, an observation initially supported by NMR studies of apolipoprotein C-II in dodecyl phosphocholine (47). More recently, the three-dimensional NMR structure of the complex apolipoprotein C-III with SDS clearly shows that this protein wraps itself around an SDS micelle as a “necklace” of extended helices (48). The “snorkeling” interaction with phospholipids was also proposed for α-synuclein (49) and supported by EPR spin labeling studies (50, 51). However, there is considerable debate about the conformation of membrane-bound α-synuclein. Current data indicate that a bent helical or helix-turn-helix-type fold is present when α-synuclein is in complex with detergent micelles (52, 53), although at very high concentrations of SDS (where SDS is more rodlike in structure), alternative binding modes have been observed, including an extended elongated helical fold (54). Several different binding modes have also been proposed to occur in biological membranes, possibly due to differences in composition and curvature. These include the bent (helix-turn-helix) helix fold (55) and an elongated extended helix (51, 53), both of which would be embedded in a membrane/vesicular surface in vivo. In addition, a recent NMR study suggests that α-synuclein may assemble into α-helical bundles that could enable the protein to penetrate deeper into a membranous bilayer (56).
SHERP, in comparison, lacks the strong repetitive character seen in the 11-residue repeats of apolipoproteins (57, 58) and α-synuclein (45). However, as shown in Fig. 4c, the electrostatic distribution of charge surrounding its helices clearly shows that SHERP possesses the characteristic amphipathic properties typical of other helical proteins that bind anionic phospholipids (44). Thus, in a similar manner to apolipoproteins and α-synuclein, the amphipathic helical fold of SHERP could allow it to be surface-localized on membrane surfaces when sufficient concentrations of anionic phospholipids are present. It is also worth noting that the hairpin turn observed in SHERP differs from the other helix-forming sequences in this molecule in that it contains a glycine residue (Gly31) that can act together with nearby residues to favor hairpin formation. In the α-synuclein complex with SDS (59), a similar turn is observed in its three-dimensional structure (52, 53).
The NMR-determined structure of SHERP in the presence of SDS does not discount the possibility that this protein could adopt alternative conformations and/or binding modes in vivo. For example, it has been argued that SDS micelles are limited in their capability to mimic a membrane bilayer due to their small size and high curvature (50, 51). The SRCD data reported here shows a higher helical content (16%) for SHERP in the presence of SDS compared with vesicles containing anionic phospholipids (Table 1 and Fig. 2), suggesting the presence of some conformational differences. Indeed, a similar increase in helical content has been reported previously for α-synuclein under analogous conditions using CD (59), and EPR and FRET studies indicate that α-synuclein adopts an extended helical fold on a membrane surface (51, 53). It is also important to note that the interhelical interactions of SHERP observed in the NMR data in SDS reported here could arise from interactions between independent monomeric units. Such interactions could permit SHERP to “carpet” or “cluster” on membrane surfaces (49, 60) or even form membrane-penetrating α-helical bundles (56) as described previously for α-synuclein. Given the plethora of biophysical approaches to study membrane-protein interactions, future studies can examine whether the fold and/or oligomeric state of SHERP differ significantly in the presence of membrane bilayers from what is reported here.
Potential Functional Roles of SHERP
Metacyclogenesis in Leishmania represents the end point of parasite development in the sand fly vector, when parasites cease cell division and are preadapted for inoculation into and survival within the mammalian host (4). Generation of metacyclic parasites can be induced by low pH and nutrient depletion in vitro, whereas reduced tetrahydrobiopterin levels may also act as a signal for parasite differentiation in vivo (61, 62). These differentiated parasites display distinctive morphological and biochemical features; they have a small cell body and relatively long flagellum and are highly motile and resistant to human complement, thereby facilitating parasite survival in the host following transmission. Complement resistance is associated with presence of an extensive glycocalyx composed chiefly of the complex lipid-anchored glycoconjugate, lipophosphoglycan. Modification of lipophosphoglycan during metacyclogenesis facilitates parasite detachment from the mid-gut in the sand fly and is essential for vector transmission (reviewed in Ref. 8). The identity and function of other parasite factors required in later stages of development and their role in metacyclic transmission are poorly understood.
SHERP, together with HASPB, were originally identified as two of the few differentially regulated genes in L. major, expressing high RNA and protein levels predominantly in metacyclic parasites (10, 63–65), both in culture and, more recently, in metacyclics dissected from sand flies (66). Both proteins are encoded within the LmcDNA16 gene locus on chromosome 23 (63). Homozygous null mutants deleted for the whole diploid locus, generated by targeted gene deletion, are still infective in vitro (in cultured macrophages) and in vivo (in a susceptible BALB/c mouse model) (31). These in vivo experiments involved direct needle inoculation of cultured parasites, however, rather than their introduction by sand fly bite, experimental conditions that more closely mimic in vivo transmission. Recent data have shown that the null mutants described above, that are also more sensitive to complement lysis than wild type parasites, are compromised in their development in the sand fly vector (66). Genetic complementation of the null phenotype confirms that the LmcDNA16 locus is essential for metacyclogenesis in vivo.
Previous work using indirect immunofluorescent imaging, immunogold electron microscopy, and cell fractionation demonstrated that SHERP associates with the ER and outer mitochondrial membranes as a peripheral membrane protein (10). Additional experiments to investigate potential SHERP-cytoskeletal interactions or its influence on organellar morphology showed no significant effects. Overall, these data have given limited insight into the functional role of SHERP in metacyclic Leishmania.
The amphipathic nature of SHERP, its localization at intracellular membranes (10), and its ability to interact with anionic phospholipids suggest that one function of SHERP may be to bind and possibly recruit anionic lipids during metacyclogenesis. Characterization of total membrane composition in Leishmania promastigotes (but not specifically metacyclic parasites) has identified the neutral phospholipids (phosphatidylcholine and phosphatidylethanolamine) as principal components, with negatively charged phospholipids (phosphatidylinositol, phosphatidylglycine, and phosphatidylserine) also present but in considerably smaller quantities (67–69). The structural similarities between SHERP and apolipoproteins or α-synuclein suggest that SHERP may not only embed itself in a membrane surface but possibly penetrate into the bilayer. These modes of membrane interaction suggest that SHERP might be required for cellular reorganization or signaling processes occurring during parasite differentiation.
The identification of V-ATPase as another potential binding partner of SHERP raises the possibility that this protein may have some role in modulating V-ATPase-dependent cellular processes in vivo. These molecular complexes are membrane-localized ATP-driven proton pumps that are primarily responsible for acidification in a variety of intracellular endosomal/lysosomal-type compartments and secretory vesicles as well as for proton transport across the plasma membrane in eukaryotes. In general, eukaryotic V-ATPases contain 14 subunits of varying stoichiometries that assemble as V1, a peripheral membrane complex containing the catalytic ATPase activity and V0, an integral membrane motor complex (39, 70). This architecture is also shared by the intact T. thermophilus V-ATPase formed from nine subunits, which principally differs from eukaryotic V-ATPases in its V0 rather than the V1 subcomplex assembly (71). A key feature of V-ATPase is that dynamic and reversible disassembly of the V1 from the V0 complex regulates its activity, and this has been shown to be important for a wide range of cellular processes related to acidification (38, 72, 73). Data presented here indicate that SHERP may have the potential to form a functionally stable complex with the L. major V-ATPase in vivo, presumably through interactions with the V1 subcomplex. If this occurs, SHERP could either prevent dissociation of the V-ATPase complex from a membrane, perhaps enhancing organelle acidification, or inhibit the catalytic activity V1 subcomplex through interactions with subunit B or other subunits in a manner analogous to that observed for the eukaryote-specific subunit H regulatory protein (74).
Optimum expression of SHERP occurs in metacyclic parasites that have a short extracellular life span within the sand fly prior to transmission and phagocytosis by host macrophages. Within these phagocytic cells, metacyclic Leishmania are maintained within the phagosome, an acidic lysosomal/endocytic compartment, prior to differentiation into amotile amastigotes (75). In addition, autophagy has been identified as a key process for Leishmania differentiation and parasite virulence (76–78). This catabolic process, common in eukaryotic cells, appears to be essential for the extensive cellular remodeling and size changes associated with progression through the stages of the parasite life cycle. In particular, metacyclogenesis has been shown to be dependent on late endosome function and autophagy, with the multivesicular body-like network found in multiplicative promastigotes maturing into a lysosomal-like structure of high lytic capacity and low pH in metacyclic cells (76). Given the localization of SHERP to mitochondrial and ER membranes (both of which can be processed by autophagic digestion) and its potential interaction with the V-ATPase in vitro, perhaps this small protein plays a regulatory role in the vacuolar acidification associated with parasite autophagy in the vector.
In summary, we have developed a strategy using a range of biophysical and biochemical studies to elucidate structural properties and identify molecular interactions of the unusual Leishmania protein, SHERP. These studies have established that the structure of SHERP is highly disordered in solution, whereas in the presence of anionic phospholipids or detergent, it adopts a highly helical fold with properties that could enable it to embed or even penetrate into a membrane bilayer. Our data suggest that the molecular recognition template that induces this structural transformation in SHERP is an anionic surface and that anionic phospholipids and/or V-ATPase may be among the potential binding partners of SHERP in vivo. Taken together, these observations suggest a number of potential roles for SHERP that can now be assessed in the context of the whole organism.
Supplementary Material
Acknowledgments
Access to the CD1 beamline at the Institute for Synchrotron Studies is acknowledged under the European Union Integrated Infrastructure Initiative (I3), Integrated Activity on Synchrotron and Free Electron Laser Science (IA-SFS), contract RII3-CT-2004-506008. NMR spectra were recorded at the Medical Research Council Biomedical NMR Centre (Mill Hill, UK). We thank Dr. G. Kelly and Dr. T. A. Frankiel for assistance in data recording.
This work was supported in part by grants from the United Kingdom Biotechnology and Biological Science Research Council (to K. A. B., D. F. S., and B. A. W.); Wellcome Trust Program Grant 077503 (to D. F. S.) and Program Grant 079819 and Equipment Grant 085464 (to S. J. M.); and SRCD beamtime grants (to B. A. W. and K. A. B.).
The atomic coordinates and structure factors (code 2X43) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S5.
- SHERP
- small hydrophilic endoplasmic reticulum-associated protein
- SRCD
- synchrotron radiation circular dichroism
- V-ATPase
- vacuolar type H+-ATPase
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPE
- 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- DOPG
- 1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]
- SBED
- N-hydroxysuccinimidyl-2-(6-[biotinamido]-2-(p-azidobenzamido)-hexanoamido) ethyl-1,3′-dithioproprionate
- ER
- endoplasmic reticulum.
REFERENCES
- 1. Desjeux P. (2004) Nat. Rev. Microbiol. 2, 692–693 [DOI] [PubMed] [Google Scholar]
- 2. Murray H. W., Berman J. D., Davies C. R., Saravia N. G. (2005) Lancet 366, 1561–1577 [DOI] [PubMed] [Google Scholar]
- 3. Chappuis F., Sundar S., Hailu A., Ghalib H., Rijal S., Peeling R. W., Alvar J., Boelaert M. (2007) Nat. Rev. Microbiol. 5, 873–882 [DOI] [PubMed] [Google Scholar]
- 4. Sacks D. L., Perkins P. V. (1984) Science 223, 1417–1419 [DOI] [PubMed] [Google Scholar]
- 5. Bates P. A. (2007) Int. J. Parasitol. 37, 1097–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sacks D. L., Hieny S., Sher A. (1985) J. Immunol. 135, 564–569 [PubMed] [Google Scholar]
- 7. Beverley S. M., Turco S. J. (1998) Trends Microbiol. 6, 35–40 [DOI] [PubMed] [Google Scholar]
- 8. Sacks D., Kamhawi S. (2001) Annu. Rev. Microbiol 55, 453–483 [DOI] [PubMed] [Google Scholar]
- 9. Descoteaux A., Turco S. J. (2002) Microbes Infect. 4, 975–981 [DOI] [PubMed] [Google Scholar]
- 10. Knuepfer E., Stierhof Y. D., McKean P. G., Smith D. F. (2001) Biochem. J. 356, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ivens A. C., Peacock C. S., Worthey E. A., Murphy L., Aggarwal G., Berriman M., Sisk E., Rajandream M. A., Adlem E., Aert R., Anupama A., Apostolou Z., Attipoe P., Bason N., Bauser C., Beck A., Beverley S. M., Bianchettin G., Borzym K., Bothe G., Bruschi C. V., Collins M., Cadag E., Ciarloni L., Clayton C., Coulson R. M., Cronin A., Cruz A. K., Davies R. M., De Gaudenzi J., Dobson D. E., Duesterhoeft A., Fazelina G., Fosker N., Frasch A. C., Fraser A., Fuchs M., Gabel C., Goble A., Goffeau A., Harris D., Hertz-Fowler C., Hilbert H., Horn D., Huang Y., Klages S., Knights A., Kube M., Larke N., Litvin L., Lord A., Louie T., Marra M., Masuy D., Matthews K., Michaeli S., Mottram J. C., Müller-Auer S., Munden H., Nelson S., Norbertczak H., Oliver K., O'neil S., Pentony M., Pohl T. M., Price C., Purnelle B., Quail M. A., Rabbinowitsch E., Reinhardt R., Rieger M., Rinta J., Robben J., Robertson L., Ruiz J. C., Rutter S., Saunders D., Schäfer M., Schein J., Schwartz D. C., Seeger K., Seyler A., Sharp S., Shin H., Sivam D., Squares R., Squares S., Tosato V., Vogt C., Volckaert G., Wambutt R., Warren T., Wedler H., Woodward J., Zhou S., Zimmermann W., Smith D. F., Blackwell J. M., Stuart K. D., Barrell B., Myler P. J. (2005) Science 309, 436–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peacock C. S., Seeger K., Harris D., Murphy L., Ruiz J. C., Quail M. A., Peters N., Adlem E., Tivey A., Aslett M., Kerhornou A., Ivens A., Fraser A., Rajandream M. A., Carver T., Norbertczak H., Chillingworth T., Hance Z., Jagels K., Moule S., Ormond D., Rutter S., Squares R., Whitehead S., Rabbinowitsch E., Arrowsmith C., White B., Thurston S., Bringaud F., Baldauf S. L., Faulconbridge A., Jeffares D., Depledge D. P., Oyola S. O., Hilley J. D., Brito L. O., Tosi L. R., Barrell B., Cruz A. K., Mottram J. C., Smith D. F., Berriman M. (2007) Nat. Genet. 39, 839–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wien F., Wallace B. A. (2005) Appl. Spectrosc. 59, 1109–1113 [DOI] [PubMed] [Google Scholar]
- 14. Miles A. J., Wallace B. A. (2006) Chem. Soc. Rev. 35, 39–51 [DOI] [PubMed] [Google Scholar]
- 15. Miles A. J., Wien F., Lees J. G., Rodger A., Janes R. W., Wallace B. A. (2003) Spectroscopy 17, 653–661 [Google Scholar]
- 16. Lees J. G., Smith B. R., Wien F., Miles A. J., Wallace B. A. (2004) Anal. Biochem. 332, 285–289 [DOI] [PubMed] [Google Scholar]
- 17. Whitmore L., Wallace B. A. (2004) Nucleic Acids Res. 32, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Whitmore L., Wallace B. A. (2008) Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 19. Compton L. A., Johnson W. C., Jr. (1986) Anal. Biochem. 155, 155–167 [DOI] [PubMed] [Google Scholar]
- 20. van Stokkum I. H., Spoelder H. J., Bloemendal M., van Grondelle R., Groen F. C. (1990) Anal. Biochem. 191, 110–118 [DOI] [PubMed] [Google Scholar]
- 21. Lees J. G., Miles A. J., Janes R. W., Wallace B. A. (2006) BMC Bioinformatics 7, 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sreerama N., Venyaminov S. Y., Woody R. W. (1999) Protein Sci. 8, 370–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lees J. G., Miles A. J., Wien F., Wallace B. A. (2006) Bioinformatics 22, 1955–1962 [DOI] [PubMed] [Google Scholar]
- 24. Mao D., Wallace B. A. (1984) Biochemistry 23, 2667–2673 [DOI] [PubMed] [Google Scholar]
- 25. Sattler M., Schleucher J., Griesinger C. (1999) Prog. NMR Spectrosc. 34, 93–158 [Google Scholar]
- 26. Farrow N. A., Zhang O., Forman-Kay J. D., Kay L. E. (1994) J. Biomol. NMR 4, 727–734 [DOI] [PubMed] [Google Scholar]
- 27. Meier S., Häussinger D., Grzesiek S. (2002) J. Biomol. NMR 24, 351–356 [DOI] [PubMed] [Google Scholar]
- 28. Linge J. P., Habeck M., Rieping W., Nilges M. (2003) Bioinformatics 19, 315–316 [DOI] [PubMed] [Google Scholar]
- 29. Cornilescu G., Delaglio F., Bax A. (1999) J. Biomol. NMR 13, 289–302 [DOI] [PubMed] [Google Scholar]
- 30. Fossi M., Oschkinat H., Nilges M., Ball L. J. (2005) J. Magn. Reson. 175, 92–102 [DOI] [PubMed] [Google Scholar]
- 31. McKean P. G., Denny P. W., Knuepfer E., Keen J. K., Smith D. F. (2001) Cell Microbiol. 3, 511–523 [DOI] [PubMed] [Google Scholar]
- 32. Yokoyama K., Nagata K., Imamura H., Ohkuma S., Yoshida M., Tamakoshi M. (2003) J. Biol. Chem. 278, 42686–42691 [DOI] [PubMed] [Google Scholar]
- 33. Urwin P. E., McPherson M. J., Atkinson H. J. (1998) Planta 204, 472–479 [DOI] [PubMed] [Google Scholar]
- 34. Wallace B. A., Lees J. G., Orry A. J., Lobley A., Janes R. W. (2003) Protein Sci. 12, 875–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cole C., Barber J. D., Bartonm G. J. (2008) Nucleic. Acids Res. 36, W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ward J. J., Sodhi J. S., McGuffin L. J., Buxton B. F., Jones D. T. (2004) J. Mol. Biol. 337, 635–645 [DOI] [PubMed] [Google Scholar]
- 37. Leifso K., Cohen-Freue G., Dogra N., Murray A., McMaster W. R. (2007) Mol. Biochem. Parasitol. 152, 35–46 [DOI] [PubMed] [Google Scholar]
- 38. Forgac M. (2007) Nat. Rev. Mol. Cell Biol. 8, 917–929 [DOI] [PubMed] [Google Scholar]
- 39. Qi J., Wang Y., Forgac M. (2007) J. Bioenerg. Biomembr. 39, 423–426 [DOI] [PubMed] [Google Scholar]
- 40. Marshansky V., Futai M. (2008) Curr. Opin. Cell Biol. 20, 415–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Davidson W. S., Jonas A., Clayton D. F., George J. M. (1998) J. Biol. Chem. 273, 9443–9449 [DOI] [PubMed] [Google Scholar]
- 42. Jo E., McLaurin J., Yip C. M., St George-Hyslop P., Fraser P. E. (2000) J. Biol. Chem. 275, 34328–34334 [DOI] [PubMed] [Google Scholar]
- 43. Boucher J., Ramsamy T. A., Braschi S., Sahoo D., Neville T. A., Sparks D. L. (2004) J. Lipid Res. 45, 849–858 [DOI] [PubMed] [Google Scholar]
- 44. Segrest J. P., Garber D. W., Brouillette C. G., Harvey S. C., Anantharamaiah G. M. (1994) Adv. Protein Chem. 45, 303–369 [DOI] [PubMed] [Google Scholar]
- 45. Bussell R., Jr., Eliezer D. (2003) J. Mol. Biol. 329, 763–778 [DOI] [PubMed] [Google Scholar]
- 46. Wang G., Sparrow J. T., Cushley R. J. (1997) Biochemistry 36, 13657–13666 [DOI] [PubMed] [Google Scholar]
- 47. MacRaild C. A., Howlett G. J., Gooley P. R. (2004) Biochemistry 43, 8084–8093 [DOI] [PubMed] [Google Scholar]
- 48. Gangabadage C. S., Zdunek J., Tessari M., Nilsson S., Olivecrona G., Wijmenga S. S. (2008) J. Biol. Chem. 283, 17416–17427 [DOI] [PubMed] [Google Scholar]
- 49. Ramakrishnan M., Jensen P. H., Marsh D. (2003) Biochemistry 42, 12919–12926 [DOI] [PubMed] [Google Scholar]
- 50. Jao C. C., Der-Sarkissian A., Chen J., Langen R. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 8331–8336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jao C. C., Hegde B. G., Chen J., Haworth I. S., Langen R. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 19666–19671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ulmer T. S., Bax A., Cole N. B., Nussbaum R. L. (2005) J. Biol. Chem. 280, 9595–9603 [DOI] [PubMed] [Google Scholar]
- 53. Trexler A. J., Rhoades E. (2009) Biochemistry 48, 2304–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ferreon A. C., Deniz A. A. (2007) Biochemistry 46, 4499–4509 [DOI] [PubMed] [Google Scholar]
- 55. Drescher M., Godschalk F., Veldhuis G., van Rooijen B. D., Subramaniam V., Huber M. (2008) Chembiochem 9, 2411–2416 [DOI] [PubMed] [Google Scholar]
- 56. Bodner C. R., Dobson C. M., Bax A. (2009) J. Mol. Biol. 390, 775–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. George J. M., Jin H., Woods W. S., Clayton D. F. (1995) Neuron 15, 361–372 [DOI] [PubMed] [Google Scholar]
- 58. Weinreb P. H., Zhen W., Poon A. W., Conway K. A., Lansbury P. T., Jr. (1996) Biochemistry 35, 13709–13715 [DOI] [PubMed] [Google Scholar]
- 59. Chandra S., Chen X., Rizo J., Jahn R., Südhof T. C. (2003) J. Biol. Chem. 278, 15313–15318 [DOI] [PubMed] [Google Scholar]
- 60. Pandey A. P., Haque F., Rochet J. C., Hovis J. S. (2009) Biophys. J. 96, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cunningham M. L., Titus R. G., Turco S. J., Beverley S. M. (2001) Science 292, 285–287 [DOI] [PubMed] [Google Scholar]
- 62. Bates P. A. (2008) Curr. Opin. Microbiol. 11, 340–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Flinn H. M., Smith D. F. (1992) Nucleic Acids Res. 20, 755–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Flinn H. M., Rangarajan D., Smith D. F. (1994) Mol. Biochem. Parasitol. 65, 259–270 [DOI] [PubMed] [Google Scholar]
- 65. McKean P. G., Delahay R., Pimenta P. F., Smith D. F. (1997) Mol. Biochem. Parasitol. 85, 221–231 [DOI] [PubMed] [Google Scholar]
- 66. Sádlová J., Price H. P., Smith B. A., Votýpka J., Volf P., Smith D. F. (2010) Cell. Microbiol. 12, 1765–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Beach D. H., Holz G. G., Jr., Anekwe G. E. (1979) J. Parasitol. 65, 201–216 [PubMed] [Google Scholar]
- 68. Wassef M. K., Fioretti T. B., Dwyer D. M. (1985) Lipids 20, 108–115 [DOI] [PubMed] [Google Scholar]
- 69. Rakotomanga M., Blanc S., Gaudin K., Chaminade P., Loiseau P. M. (2007) Antimicrob. Agents Chemother. 51, 1425–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kane P. M. (2007) J. Bioenerg. Biomembr. 39, 415–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lau W. C., Rubinstein J. L. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 1367–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kane P. M. (2006) Microbiol. Mol. Biol. Rev. 70, 177–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Trombetta E. S., Ebersold M., Garrett W., Pypaert M., Mellman I. (2003) Science 299, 1400–1403 [DOI] [PubMed] [Google Scholar]
- 74. Diab H., Ohira M., Liu M., Cobb E., Kane P. M. (2009) J. Biol. Chem. 284, 13316–13325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Alexander J., Russell D. G. (1992) Adv. Parasitol. 31, 175–254 [DOI] [PubMed] [Google Scholar]
- 76. Besteiro S., Williams R. A., Morrison L. S., Coombs G. H., Mottram J. C. (2006) J. Biol. Chem. 281, 11384–11396 [DOI] [PubMed] [Google Scholar]
- 77. Williams R. A., Tetley L., Mottram J. C., Coombs G. H. (2006) Mol. Microbiol. 61, 655–674 [DOI] [PubMed] [Google Scholar]
- 78. Besteiro S., Williams R. A., Coombs G. H., Mottram J. C. (2007) Int. J. Parasitol. 37, 1063–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.