

Abstract
1-Aminocyclopropane-1-carboxylic acid (ACC) deaminase (ACCD) is a pyridoxal 5′-phosphate (PLP) dependent enzyme that cleaves the cyclopropane ring of ACC, to give α-ketobutyric acid and ammonia as products. The cleavage of the Cα-Cβ bond of an amino acid substrate is a rare event in PLP-dependent enzyme catalysis. Potential chemical mechanisms involving nucleophile- or acid-catalyzed cyclopropane ring opening have been proposed for the unusual transformation catalyzed by ACCD, but the actual mode of cyclopropane ring cleavage remains obscure. In this report, we aim to elucidate the mechanistic features of ACCD catalysis by investigating the kinetic properties of ACCD from Pseudomonas sp. ACP and several of its mutant enzymes. Our studies suggest that the pKa of the conserved active site residue, Tyr294, is lowered by a hydrogen bonding interaction with a second conserved residue, Tyr268. This allows Tyr294 to deprotonate the incoming amino group of ACC in order to initiate the aldimine exchange reaction between ACC and the PLP coenzyme, and also likely helps to activate Tyr294 for a role as nucleophile to attack and cleave the cyclopropane ring of the substrate. In addition, solvent kinetic isotope effect, proton inventory, and 13C-KIE studies of the wild type enzyme suggest that the Cα-Cβ bond cleavage step in the chemical mechanism is at least partially rate limiting under kcat/Km conditions, and is likely preceded in the mechanism by a partially rate limiting step involving the conversion of a stable gem-diamine intermediate into a reactive external aldimine intermediate that is poised for cyclopropane ring cleavage. When viewed within the context of previous mechanistic and structural studies of ACCD enzymes, our studies are most consistent with a mode of cyclopropane ring cleavage involving nucleophilic catalysis by Tyr294.
1-Aminocyclopropane-1-carboxylate deaminase (ACCD) is a pyridoxal-5′-phosphate (PLP)-dependent enzyme expressed in certain soil bacteria and fungi that catalyzes the conversion of 1-aminocyclopropane-1-carboxylic acid (ACC, 1) to α-ketobutyrate (α-KB, 2) and ammonia (Scheme 1).1 In plants, ACC is the immediate biosynthetic precursor to ethylene, an important plant hormone that regulates fruit ripening, seed germination, leaf senescence, responses to environmental stress, and many other physiological events.2 In the soil-dwelling organisms that express ACCD, the function of ACCD may be to salvage useful sources of carbon and nitrogen for metabolic needs. Bacterial ACCDs have been incorporated into transgenic plants to regulate ethylene biosynthesis, and ACCD expression in plant tissues can lead to significant delays in fruit ripening, helping to increase the shelf life of fruits and vegetables.3
Scheme 1.

The reaction catalyzed by ACC deaminase
The catalytic cycles of most PLP-dependent enzymes begin with the formation of an external aldimine (such as 4, Scheme 2) between the amino group of the substrate and the PLP coenzyme.4–6 In the vast majority of PLP-dependent enzymes, a Cα anion species (such as 5) is then generated either by the deprotonation of the Cα-H or by decarboxylation of the α-carboxyl group of the substrate. In this respect, the reaction catalyzed by ACCD is unusual, because the amino acid substrate, ACC, contains no Cα proton and the carboxylate group is retained in the α-KB product. In ACCD, the Cα-anion equivalent could be generated by cleavage of one of the two Cα-Cβ bonds of ACC. However, Cα-Cβ bond cleavage is a rare event in PLP-dependent enzyme catalysis and has been observed in only one other enzyme, serine hydroxymethyltransferase (SHMTase).7,8 In this enzyme, the Cα-Cβ bond of the serine-PLP external aldimine is believed to be cleaved either by a retro-aldol type reaction or as a result of direct nucleophilic attack by the tetrahydrofolate co-substrate on the Cβ atom of the serine substrate.
Scheme 2.

Putative chemical mechanisms for ACCD involving nucleophile- and base-catalyzed ring scission (paths A and B, respectively).
Several possible mechanisms can be envisioned for the Cα-Cβ bond cleavage event catalyzed by ACCD.9 As shown in Scheme 2 route A, following conversion of the internal aldimine (3) to the ACC-PLP-external aldimine (4), an active-site nucleophile could attack and trigger the cleavage of the ACC cyclopropane ring10,11 to yield the Cα anion equivalent (or quinonoid, 5). This mechanism is conceivable because cyclopropane rings bearing electron withdrawing substituents are known to be susceptible toward nucleophilic addition reactions.12–15 Removal of a proton at Cβ of the quinonoid (5)9,16 by an active site base (B1) could then eliminate the nucleophile to afford the vinylglycyl-PLP quinonoid intermediate (6). Support for the intermediacy of 6 in the catalytic cycle of ACCD is derived from mechanistic studies with D-vinylglycine (7, Scheme 2), which can also be converted to α-KB (2) and ammonia by ACCD.9 Subsequent Cγ protonation of 6 by a separate active site residue (B2)16 could give the PLP-aminocrotonate species (8). Aldimine exchange and hydrolysis of (9) could then regenerate the internal aldimine (3) and yield the reaction products, α-KB (2) and ammonia. A variety of stereochemical studies have suggested that the Cβ-Cα bond cleavage is pro-S stereospecific,10 that Cβ deprotonation (5 → 6, pro-R dominant) and protonation (9 → 2, pro-R dominant) are mediated by a single base (B1), and that both B1 and B2 are located on the si face of the alkene moiety of 6.16 A second potential mechanism that has been proposed for the ACC ring scission step involves deprotonation of one of the two Cβ-methylene carbons of the ACC-PLP-external aldimine (4) to generate the extended quinonoid intermediate (6) directly (Scheme 2, route B).9 However, the pKa of ~ 46 for the ring hydrogens of cyclopropane17 seems prohibitively high for this mechanism to be feasible.
The X-ray crystal structures of ACCD enzymes from both yeast and bacterial sources have been reported,18–21 and together with the stereochemical studies discussed above,9,10,16 have provided important insight into the putative roles of several active site amino acid residues in the ACCD-catalyzed reaction. Specifically, the phenoxyl side chain of Tyr294 (Pseudomonas sp ACP numbering) is positioned on the si face of the ACC-PLP adduct (4) in the active site, and is located only 3 Å from the pro-S Cβ of the ACC cyclopropane ring (Figure 1). As such, it appears to be positioned appropriately to serve as a nucleophile to attack the pro-S Cβ and cleave the pro-S Cβ-Cα bond of the ACC cyclopropane ring. In support of this proposed catalytic role, the Y294F mutant is completely inactive and no consumption of ACC was detected by 1H-NMR spectroscopy after a 24 h incubation period.20 A putative hydrogen bonding interaction between Tyr268 and Tyr294 (2.5 Å) could lower the pKa of Tyr294, enhancing its role as a catalytic nucleophile. Ser78 is also located close to the ACC-PLP adduct in the active site, where it form a hydrogen bond with the carboxylate group of ACC (2.8 Å) and could help mediate proton transfers during turnover.22 Finally, Glu295 appears to form an ion pair interaction with the pyridinium group of PLP. Such interactions are common in PLP enzymes and are generally believed to raise the pKa of the pyridine ring in order to maximize the electrophilic properties of the coenzyme.
Figure 1.
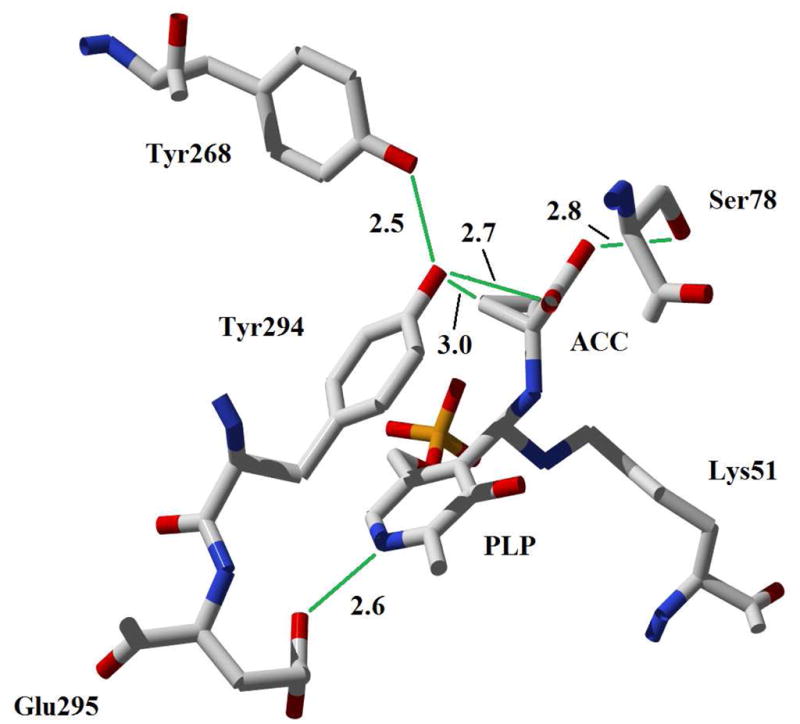
Active site of wt ACCD from Pseudomonas sp. ACP in complex with ACC (1). Distances are indicated in Å.
An unusual feature of the wt ACCD-ACC co-crystal structure is the apparent accumulation of a gem-diamine species (10, Scheme 3) in the active site, where the amino groups of ACC and the Schiff-base forming lysine residue (Lys51) are both covalently linked to the C4′ atom of the coenzyme (Figure 1).20,21 In most PLP-dependent enzymes, the gem-diamine is a transient species during the aldimine exchange reaction (see 3 → 4, Scheme 2). The observation of this unusual gem-diamine species in the wt ACCD-ACC co-crystal structure prompted us to consider an alternative mechanistic possibility for ACCD, in which Tyr294 acts as an active site acid to facilitate the cleavage of the cyclopropane ring directly from the gem-diamine intermediate (Scheme 3).21 After formation of the gem-diamine intermediate (10), a “push” by the electron pair of the ACC amine and a “pull” by the concomitant protonation at the pro-S Cβ of ACC by Tyr294, could facilitate the cleavage of the Cα-Cβ bond to generate 11. Stereospecific Cβ deprotonation of 11 could give 12, and the aldimine exchange reaction could release the aminocrotonate (9) and regenerate the internal aldimine (3). Subsequent tautomerization of 9 and hydrolysis of the resulting imine would eliminate ammonia to give α-KB (2). Although this proposal represents an unprecedented catalytic cycle for a PLP-dependent enzyme in that the electron sink properties of the coenzyme are not used, it provides a rationale for the accumulation of the gem-diamine (10) in the wt ACCD-ACC co-crystal structure. Furthermore, the acid-catalyzed electrophilic cleavage of cyclopropanes is well-documented in model studies.23–27
Scheme 3.

Putative chemical mechanism for ACCD involving acid-catalyzed ring scission
In this study, we investigate the kinetic and spectroscopic properties of wt ACCD from Pseudomonas sp ACP and several of its active site mutants in an attempt to distinguish among the possible chemical mechanisms for ACCD catalysis involving either nucleophile or acid-catalyzed cyclopropane ring opening. Kinetic-pH profiles are used to assess the involvement of acid-base chemistry in catalysis and to help define the protonation states of groups that are important to substrate binding and catalysis. Pre-steady state stopped-flow absorption spectroscopy of wt ACCD and its mutants is used to provide insight into the PLP-linked intermediates that form during turnover. Finally, solvent kinetic isotope effects, proton inventory studies, viscosity variation experiments, and 13C-KIE studies of the wt enzyme help to define the sequence of chemical events involved in catalysis and to characterize the nature of the steps that limit steady state turnover.
MATERIALS AND METHODS
General
Wild type (wt) ACCD from Pseudomonas sp. ACP, and its Y268F and Y294F mutants were expressed in E. coli as N-His6-fusion proteins as described previously.21 The wt ACCD gene in the pET28b(+) construct (Stratagene, La Jolla, CA) was used as a template to generate the Glu295→Asp295 mutation using the QuikChange site-directed mutagenesis kit (Amersham, Arlington Heights, IL) with the forward primer, 5′-GCTGACCGATCCCGTCTACGATGGCAAATCGATGCACGGCA, and the reverse primer, 5′-TGCCGTGCATCGATTTGCCATCGTAGACGGGATCGGTCAGC (mutation in bold). The mutation was confirmed by DNA sequencing performed by the Institute of Cellular and Molecular Biology Core Labs of the University of Texas at Austin. The E295D protein was expressed and purified in a similar fashion to the other enzymes used in this study.21 L-Lactic dehydrogenase from rabbit muscle, β-nicotinamide adenine dinucleotide (NADH), ACC (1), and all buffer components used in this study were purchased from Sigma-Aldrich (St. Louis, Mo). All protein concentrations were determined by the Bradford method using bovine serum albumin as the standard.28 All curve fitting was performed with GraFit 5 (Erithacus Software, Horley, Surrey, UK).
pH-Dependence of the steady state kinetic parameters
Steady state kinetic measurements were made with a continuous coupled enzyme assay where the α-KB (2) produced by ACCD turnover is reduced with NADH to 2-hydroxybutanoic acid by lactate dehydrogenase (LDH).9,22 A typical 500 μL reaction contained 100 units of LDH, 200 μM NADH, 1.68 μM wt ACCD, and variable concentrations of ACC (0.1 – 50 mM) in assay buffer (25 mM MOPS, 25 mM MES, 50 mM HMP) adjusted to the appropriate pH with 10 M HCl or 10 M NaOH. After adjusting the buffer pH, the final ionic strength was raised to 100 mM by the addition of NaCl. Upon addition of ACC to initiate the reaction, the rate of NADH consumption at 340 nm was monitored (ε340 = 6220 M−1cm−1) at 25 °C. For Y268F, E295D, and wt ACCD in D2O, the concentration of enzyme was increased to 15.4 μM, 4.95 μM, and 3.36 μM, respectively. For assays performed in D2O, the assay buffer components were mixed directly in D2O (99.9 atom %D, Sigma-Aldrich) and were adjusted to the appropriate pD (pD = pH meter reading + 0.4) with 10 M HCl or 10 M NaOH. This solution was used to make the NADH, LDH, ACC, and enzyme stock solutions. The final mole fraction of protium in these reaction mixtures was calculated to be < 5%.
The choice of ACC concentrations used depended on the enzyme and the Km value at the particular pH being assayed, but in all cases at least 3–4 different substrate concentrations both above and below the Km value were used. The initial velocity at each substrate concentration was measured in triplicate and the substrate concentration dependence of the initial velocities were fit with the Michaelis-Menten equation for the determination of kcat and Km. The kcat and kcat/Km values determined at each pH [kcat,app and (kcat/Km)app] were normalized by the enzyme concentration, and the log of these kinetic constants were plotted versus pH and fit with Eq 1–3. The log (kcat/Km)app data were fit with Eq 1 or 2, which describe inverse bell-shaped profiles with slopes of +1 and −1 at low and high pH, respectively. In these equations, kcat/Km is the pH-independent value of kcat/Km, pK1 and pK2 (Eq 1) are the apparent pKa values of titrable groups in the free enzyme and free substrate, and pK′ (in Eq 2) is the average of two closely spaced apparent pKa values. Equation 3 describes a profile that increases to a maximum value (kcat) at high pH with a slope of +1 and the apparent dissociation constant, pK3.
| Eq 1 |
| Eq 2 |
| Eq 3 |
Spectrophotometric pH titrations of the internal aldimine
To monitor the pH dependence of the tautomerization between the enolimine and ketoenamine forms of the internal aldimine, enzymes (stored in 50 mM Na2HPO4, 10% glycerol, pH 7.5) were diluted to 50 μM in 100 mM buffer (specified below) and, following a 1 min incubation, the absorbance of each sample was recorded from 200 – 900 nm on a Beckman DU 650 spectrophotometer at 25 °C. The buffers used were HEPES (pH 6.8–7.5), EPPS (pH 7.3–8.2), TAPS (pH 8.0–9.0), and CAPSO (pH 9.2–10.2). The spectra were normalized by setting the average absorbance values from 850–900 nm to zero.
Viscosity variation experiments
Viscosity variation experiments were performed under pseudo-first order conditions with wt ACCD using glycerol as the viscogen. The reactions contained 1.2 μM ACCD, 30 mM ACC, and variable concentrations of glycerol (0, 10, 16, 20, 24, 30, and 32% w/v) in 25 mM MOPS, 25 mM MES, 50 mM HMP, pH 7.5, 25 °C. The coupled LDH reporter assay described above was used to detect the formation of the α-KB product. Each reaction was performed in triplicate and the dependence of the initial velocity on the relative viscosity of the solution was determined by fitting the data to Eq 4, where ko, kη, and ηrel are the initial velocity in buffer with no added viscogen, the initial velocity in buffer of relative viscosity η, and the relative viscosity of the solution, respectively. The slope of the plot, S, is unity for a reaction that is completely diffusion controlled, zero for a reaction that is not diffusion controlled, and is 1 > S > 0 for a reaction that is partially diffusion controlled. The relative viscosities of the solutions containing variable concentrations of glycerol were taken from St. Maurice et al.29
| Eq 4 |
Proton inventory study of wt ACCD
For these studies, the production of α-KB was monitored directly by following the increase in absorbance at 329 nm (ε329 = 0.0176 mM−1 cm−1 for α-KB). The reactions were performed at pL = 8.0, where the kcat in both 100% H2O and 100% D2O is relatively independent of pL. TAPS buffer (50 mM) was prepared with 0, 20, 40, 60, 80, or 100% (v/v) D2O/H2O. The pL was adjusted to 8.0 using 10 M and 1 M solutions of NaOH, and the ionic strength was adjusted to 100 mM with NaCl. In each separate D2O/H2O water mixture, 10 μL of the wt ACCD stock (in H2O, 1.19 mM) and 30 μL of a 500 mM ACC solution (dissolved in the appropriate D2O/H2O mixture) were added to a total reaction volume of 500 μL, giving wt ACCD and ACC in 23.8 μM and 30 mM final concentrations, respectively. The mole fraction of deuterium for each water mixture was corrected to account for the different molar volumes of H2O and D2O,30 and also for solvent exchangeable protons derived from TAPS, NaOH, ACC, and the components of the stock enzyme solution. The enzyme was pre-incubated in each water mixture for approximately 30 min prior to the addition of ACC to start the reaction. Following a 4 min incubation period, the production of α-KB was monitored over 4 min period by the increase in A329. Under these conditions, the formation of α-KB was a linear function of time for all reactions. In each water mixture, replicate measurements of the initial velocity were made under pseudo-first order substrate concentrations (as an estimate of kcat), and the data were fitted with several different forms of the Gross-Butler equation (Eq 5), where ko and kn are the kcat values in buffers of deuterium atom fraction 0 and n, respectively, and ΦTS and Φr are fractionation factors for exchangeable hydrogen sites in the transition and reactant states, respectively.30
| Eq 5 |
13C-KIE studies of the wt ACCD-catalyzed reaction
Carbon-13 kinetic isotope effects on kcat/Km for the wt ACCD-catalyzed reaction were measured by a competition experiment using the methodology developed by Singleton,31 wherein the 13C-enrichemnt at each carbon atom of the substrate (R/RO) is measured by 13C-NMR spectroscopy and is used along with the fractional conversion of reactants (F) to calculate the 13(kcat/Km) at each carbon atom via Eq 6.31–35 For our purposes, RO is the relative enrichment of 13C (defined relative to the carboxyl carbon of ACC) at a specific carbon atom of an ACC sample prior to the reaction, R is the relative enrichment of 13C at the same carbon atom in residual ACC recovered from a large-scale ACCD reaction mixture, and F is the fraction of reaction. Detailed descriptions of the large-scale reaction mixtures, purification protocols for residual ACC, 13C-NMR spectroscopy conditions, and the measurement of the fraction of reaction are all given in the supporting information, along with descriptions for calculating the standard error associated with the KIE measurement.31
| Eq 6 |
Stopped-flow absorption measurements
Pre-steady state absorbance changes in the PLP chromophore were measured with a HI-TECH Scientific SF-61 Double Mixing Stopped-Flow System equipped with a diode array detector for monitoring time-dependent changes in the PLP spectrum. The pH of all stock solutions was adjusted to the appropriate value immediately prior to conducting the experiments at 25 °C. The ionic strength of the final reaction mixtures was 150 mM. All concentrations given below correspond to those after mixing in the stopped flow. For wt ACCD, the reactions contained 100 μM enzyme and 50 mM ACC in 100 mM EPPS (pH 7.5). For Y268F, the reactions contained 96.5 μM enzyme and 50 mM ACC in 100 mM TAPS (pH 8.8). For E295D, reactions contained 60 μM enzyme and 50 mM ACC and were performed in 100 mM MOPS (pH 7.5). For Y294F, reactions contained 50 μM enzyme, 50 mM ACC, and were performed in 100 mM MOPS (pH 7.5). The pre-steady state kinetic data were fit with exponential equations (of the form shown by Eq 7) to estimate the amplitudes and rates associated with each observable kinetic phase. In this equation At is the absorbance at a particular wavelength at time, t, after mixing, An and kn are the observed amplitude and first order rate constant, respectively, of kinetic phase, n, and C is an offset that accounts for absorbance at t = 0.
| Eq 7 |
RESULTS
Steady state kinetic studies
The pH-dependence of the steady state kinetic parameters (kcat and kcat/Km) were determined for wt ACCD, E295D, and Y268F over the pH range of 6.5 – 9.8 (Figure 2 and Table S1). The initial velocity data were fit with Eqs 1–3 to extract estimates for the apparent pKa of groups that contribute to substrate binding and catalysis, and estimates for the pH-independent values of the kinetic constants (Table 1). The kcat/Km profiles for each enzyme were fit with both Eqs 1 and 2, and the fit that gave the lowest mean square residual is reported. For wt ACCD and for E295D in H2O, the fits to Eq 1 give similar values for pK1 (6.9 – 7.3) and pK2 (8.7 – 9.3) as well as similar pH optima (8.0 and 8.1 for wt ACCD and E295D, respectively). For the wt ACCD reaction in D2O, the apparent pK1 and pK2 values in the kcat/Km profile are both shifted to more basic pL, an expected result in 100% D2O.30 For Y268F, the best fits were obtained with Eq 2, which contains only a single apparent dissociation constant, pK′ = 8.7. This pK′ parameter is the average of two closely-spaced apparent pKa values. The relatively large errors associated with the kcat/Km parameter in each of the fits are likely due to the close separation of the apparent pKa values governing the shape of the profiles.
Figure 2.

pH-Dependence of the steady state kinetic parameters for wild type ACCD and its mutants (pL = pH or pD, pD = pH meter reading + 0.4). White circles: wt ACCD in H2O; blue circles: wt ACCD in D2O; red: E295D in H2O; green: Y268F in H2O. The values of kcat and Km at each pH for each enzyme were determined by non-linear regression of the initial velocity data using the Michaelis-Menten equation, and are shown in Table S1.
Table 1.
Summary of pH-dependence of the steady state kinetic parameters for wt ACCD and its mutants. See Materials & Methods for definitions of Eqs 1–3. The standard errors in the last digit of each parameter estimate were obtained from the non-linear fits to Eqs 1–3 and are shown in parentheses. MSR is the mean square residual of the non-linear fit.
| Enzyme | Equation | kcat/Km (M−1s−1) | pK1 | pK2 | pK′ | kcat (s−1) | pK3 | MSR |
|---|---|---|---|---|---|---|---|---|
| wt (H2O) | 1 | 1.5(0.7) × 104 | 7.3(2) | 8.7(2) | - | 1.32(8)† | - | 0.0134 |
| wt (D2O) | 1 | 9(5) × 103 | 7.9(3) | 9.8(3) | - | 0.22(2)† | - | 0.0112 |
| E295D | 1 | 2(1) × 103 | 6.9(2) | 9.3(2) | - | 0.132(4)† | - | 0.0076 |
| Y268F | 2 | 3.3(2) × 101 | - | - | 8.7(3) | - | 7.79(3) | .0029 |
| 3 | - | - | - | - | 0.028(1) | 7.8(0.03) | .0011 |
Determined by averaging the kcat values at each pH
Inspection of the kcat–pH profiles reveals that this parameter is relatively independent of pH in the wt and E295D enzymes across the pH range tested. This indicates that if acid/base catalysis is involved in the rate limiting step, the catalytic acid/base functional groups are not accessible to protonation/deprotonation in the enzyme-substrate/intermediate complexes. The kcat parameter for E295D is reduced roughly 10-fold relative to wt ACCD across the pH range studied and this accounts for the majority of the reduction in catalytic efficiency for the E295D mutant. Similar modest reductions in kcat and kcat/Km have been observed in other PLP-dependent enzymes when the conserved Glu and Asp residues that interact with the pyridine N atom of the coenzyme are interchanged.36–39 These data suggest an important catalytic function for Glu295. In contrast to wt ACCD and E295D, kcat increases at high pH for the Y268F mutant, and fits of the data to Eq 3 suggest that an active site group with a pKa ~ 7.8 needs to be deprotonated for catalysis. In this mutant, kcat does not appear to level off at lower pH, suggesting that while there may be two forms of the Y268F:ACC Michaelis complex (one protonated and one unprotonated), only the unprotonated enzyme form is active. These results, along with the ~ 50 fold decrease in kcat for the Y268F mutant, suggest that Tyr268 also plays an important role in catalysis.
pH-Dependence of internal aldimine absorption
The pH-dependence of the electronic absorption of the internal aldimine was studied for the wt ACCD, Y268F, Y294F, and E295D enzymes. Protonated internal aldimines typically exhibit absorption bands near 330 and 420 nm, which correspond to the enolimine (13) and ketoenamine (14) tautomers of the protonated Schiff base (Scheme 4).40 In contrast, deprotonated Schiff bases (15) typically exhibit an absorption maximum near 380 nm.41,42 In the resting state of each enzyme, absorption maxima are observed near 330 and 420 nm, consistent with the presence of both enolimine (13) and ketoenamine (14) forms of the internal aldimine (Figure 3 and Figure S1), which coexist in many PLP enzymes at physiological pH.38,43–48 As the pH is increased, the ketoenamine tautomer (14) is converted to the enolimine tautomer (13). Importantly, there is no red shift in the λmax of the ~ 330 nm peak at high pH in any of the enzymes, suggesting that the internal aldimine is not deprotonated (to 15) as the pH is increased. Thus, the internal aldimine of ACCD and its mutants is likely protonated in the free enzyme form across the pH range assayed in this study. Correspondingly, the pK1, pK2, and pK3 values determined from the kinetic-pH profiles likely correspond to other groups – either to active site amino acid side chains or to ionizable groups on the substrate. The pH-dependence of the internal aldimine absorption may result from an enzyme conformational change that serves to alter the distribution between the ketoenamine and enolimine tautomers, but further investigation will be required to substantiate this claim.
Scheme 4.

Common protonation states and tautomeric forms of internal Schiff bases
Figure 3.

UV-visible absorption spectra of the low (blue spectra) and high (fuscia spectra) pH forms of the internal aldimine (3) of wt ACCD. The pH-dependence of the absorption at 419 nm is shown in the inset to the panel. Similar pH-dependent absorption changes were also seen with the E295D, Y268F, and Y294F mutant enzymes (see Figure S1).
Solvent kinetic isotope and viscosity effects on the wt ACCD-catalyzed reaction
To assess the involvement of solvent exchangeable protons in the reaction catalyzed by wt ACCD, we performed solvent KIE and viscosity variation studies of the wt enzyme. From the kinetic data reported in Table 1, significant solvent kinetic isotope effects (D2Okcat = kcat,H2O/kcat,D2O = 5.7 and D2Okcat/Km = (kcat/Km)H2O/(kcat/Km)D2O = 2.5) were calculated for the wt enzyme using the data collected at the pL optimum in each solvent (8.1 in H2O and 8.8 in D2O). This result clearly suggests that solvent exchangeable protons play an important role in ACCD catalysis, and that bonds involving solvent exchangeable protons are likely breaking in the step(s) that limit steady state turnover. The larger magnitude of D2Okcat relative to D2Okcat/Km suggests that there may be solvent kinetic isotope effects on steps following the first irreversible step in the mechanism, which is assumed to be the ACC cyclopropane ring cleavage step. Inspection of both of the putative mechanisms for ACCD catalysis (Schemes 2 and 3) shows that there are several possible steps following the Cα-Cβ bond cleavage that could potentially exhibit solvent KIEs.
Because the relative viscosity of 100% D2O solutions is higher than that of 100% H2O solutions,49 it is possible that the apparent solvent KIEs may actually be due to the variation in solvent viscosity when H2O is replaced with D2O. It is known that increasing the solvent viscosity can lower the diffusion coefficient of solutes which, in turn, can slow the rates of bimolecular association and dissociation processes.50–52 In the case of enzyme catalysis, viscosity effects are often manifested on steps in the kinetic mechanism involving substrate/product binding or release, or when large-scale unimolecular enzyme conformational changes occur.52–58 To assess the dependence of the wt ACCD-catalyzed reaction on solvent viscosity, the effects of glycerol concentration on the reaction rate under pseudo-first order conditions was analyzed (Figure S2). Fitting the kinetic data to Eq 4 gives a slope that is not significantly different than zero (S = 0.05 ± 0.05), demonstrating that kcat is not limited by product dissociation or large-scale enzyme conformational changes under these conditions. These results indicate that the D2Okcat is a legitimate solvent KIE, and is not the result of increasing the relative viscosity of the solvent.
Proton inventory for the wt ACCD-catalyzed reaction
A proton inventory study was carried out to further explore the large solvent KIE measured on kcat for the wt ACCD reaction.30 The kcat versus pL profiles for the wt ACCD reaction indicated that kcat is relatively independent of pL over the entire pL range assayed (Figure 2). Hence, we chose to perform the proton inventory studies at pL = 8.0 with 30 mM ACC, which is well above the Km for ACC in both 100% H2O and 100% D2O at pL = 8.0 (see Table S1). The non-linear fits of the proton inventory data to several different forms of Eq 5 are shown in Figure 4 along with the residual plots. The definitions of the various forms of Eq 5 used to fit the data and a summary of the fitted parameters are listed in Table 2. The best fits, as evidenced by the residual plots (Figure 4) and lack of fit F-tests (Table S2), were obtained with models where multiple transition state protons are moving in the step(s) that limit turnover under pseudo-first order conditions (the T2 and T1S models). The simple linear model (T1) did not exhibit any significant lack of fit at the 95% confidence interval (Table S2). However, from the residual plot for the fit to this model (Figure 4), there appears to be systematic deviations between the fitted curve and the mean value of the data points. Because of this, the T2 and T1S models appear to be more appropriate models for fitting the proton inventory data.
Figure 4.

Proton inventory data for wt ACCD. See Table 2 for the definition of each model (T1, T2, and T1S) and for the values of the fitted parameters.
Table 2.
Summary of fits of the proton inventory data to various forms of the Gross-Butler equation (Eq 5).
| Model | Equation (kn/ko =) | ko | Φ1 | Φ2 | ΦS | MSR |
|---|---|---|---|---|---|---|
| T1 | (1 − n + n*Φ1) | 0.60(1) | 0.19(1) | - | - | 0.00043 |
| T2 | (1 − n + n*Φ1) (1 − n + n*Φ2) | 0.61(1) | 0.33(7) | 0.7(1) | - | 0.00024 |
| T1S | (1 − n + n*Φ1) ΦSn | 0.61(1) | 0.30(4) | - | 0.7(1) | 0.00023 |
The slightly bowl-shaped curvature of the proton inventory plots suggests that D2Okcat is composed of at least two, normal solvent KIEs.30 In both the T2 and T1S models, one exchangeable site (Φ1) is predicted to be responsible for the majority of the solvent KIE on kcat (Φ1 = 0.3 – 0.33, KIE = 3.0 – 3.3), while a second site (Φ2) or sites (ΦS) contributes a smaller normal effect (KIE ~ 1.4 – 1.5). Based on the measured differences in D2Okcat (5.7) and D2Okcat/Km (2.5) discussed above, our hypothesis is that one of the two normal solvent KIEs is expressed on a step prior to or concomitant with cyclopropane ring cleavage (thus contributing to both D2O kcat and D2Okcat/Km), while the other normal solvent KIE is expressed on a step (or steps) following cyclopropane ring cleavage (and is expressed only on kcat).
13C Kinetic isotope effects
To determine if the step in the ACCD-catalyzed reaction involving the cleavage of the cyclopropane ring of ACC contributes to steady state rate limitation under kcat/Km conditions, a 13C-KIE study of the wt ACCD-catalyzed reaction was conducted using the method developed by Singleton, which allows for the simultaneous determination of 13(kcat/Km) at each carbon atom of the substrate.31 As the wt ACCD-catalyzed reaction approaches completion, any residual ACC starting material in the reaction mixture will be enriched in 13C at the Cα and pro-S Cβ positions if there is a 13C-KIE on kcat/Km. This is because ACC molecules containing 13C at the scissile pro-S Cβ–Cα bond will react more slowly than ACC molecules containing 12C at the scissile bond. The relative 13C enrichment of unreacted and residual samples of ACC (RO and R, respectively) is measured by 13C-NMR spectroscopy and is used to calculate the enrichment ratio (R/RO) along with the fraction of reaction (F) to calculate the KIE at each carbon atom via Eq 6.35
A typical 13C-NMR spectrum for ACC acquired under the NMR conditions reported in the supporting information is shown in Figure S3. The 13C peak integrations from each of the replicate NMR spectra of each ACC sample, and the replicate measurements of the fraction of reaction are shown in Tables S3 and S4, respectively. The R/RO and F observables and their standard errors calculated from this data for each of the two replicate large-scale reactions are listed in Table S5, along with the calculated KIEs on Cα and the pro-S Cβ. Equation 6 was used to calculate the KIE at Cα. However, the 13C-NMR signals for the pro-R and pro-S β-carbons of ACC are indistinguishable, yielding an observed, combined enrichment, Rβ = Rpro-S + Rpro-R, versus the reference. If the initial enrichment at each β-carbon is the same, then the values of Rpro-S and Rpro-R prior to reaction are both given by ROpro-S = ROpro-R = ROβ/2, where ROβ is the observed, combined enrichment prior to reaction. Next, if we assume that only the pro-S β-carbon exhibits a non-unit isotope effect, then the observed, combined enrichment at some fraction of reaction F will be given by Rβ = Rpro-S + ROpro-R (or Rβ = Rpro-S + ROβ/2) because Rpro-R remains equal to its initial value. Therefore, the ratio of interest for computing the isotope effect associated with the pro-S β-carbon alone, i.e., Rpro-S/ROpro-S, can be calculated as 2Rβ/ROβ − 1:
| Eq 8 |
The calculated 13(kcat/Km) on the Cα and pro-S Cβ atoms of ACC from the two replicate measurements were then averaged together to give 13C-KIEs of 1.015 ± 0.005 on Cα and of 1.017 ± 0.017 on pro-S Cβ, respectively. Unfortunately, while 13(kcat/Km)α appeared to be significantly larger than unity in both reactions, the average 13(kcat/Km)β was within error of unity. Nevertheless, the 1.5% isotope effect at Cα compares favorably with 13kcat/Km isotope effects measured for other enzyme-catalyzed reactions,59–64 and suggest that the Cα-Cβ bond cleavage step in ACCD catalysis is at least partially rate limiting under kcat/Km conditions.
Multiwavelength stopped-flow studies
In an attempt to identify and characterize the PLP-linked intermediates that form during turnover under saturating ACC concentrations, absorption spectra for wt ACCD and its E295D, Y268F, and Y294F mutants were collected by a stopped-flow apparatus using a diode array detector (Figure 5). In the fast reaction phase for the wt enzyme, the ketoenamine tautomer of the internal aldimine (λmax = 419 nm) is converted into a new species (λmax = 329 nm) with an apparent rate constant, k1 = 260 s−1 (Figure S4, Table S6). This species is most likely either the enolimine form of an external aldimine or the gem-diamine species that is observed in the crystal structure21 and which also has an absorption maximum in the 330 nm region.65 Following the fast phase, there is a slower phase occurring at a catalytically competent observed rate (k2 = 2.3 s−1, Figure S4), during which the 329 nm absorption band decreases in intensity and broadens slightly.
Figure 5.

Multiwavelength stopped-flow studies of wt ACCD and its mutants. The times in each panel indicate the time after mixing with 50 mM ACC. For each enzyme, time-dependent absorption changes in the ~ 430 nm region were also fit with the appropriate exponential expression (Eq 10) to extract estimates for rates and amplitudes of each observable kinetic phase (see Figure S4 and Table S6).
In contrast to the wt enzyme, the λmax of both of the internal aldimine peaks (λmax = 331 and 421 nm) in the E295D mutant shift slightly upon mixing with ACC to values of 329 and ~ 428 nm, respectively, at an observed first order rate constant of ~ k1 = 150 s−1 (Figure S4). The spectral shifts are associated with a slight decrease in the absorption intensity in the ~ 420 nm region and a corresponding increase in the ~ 330 nm region. Similar rapid spectral shifts have been observed in other PLP-dependent enzymes, and are usually attributed to the formation of an external aldimine intermediate, whose absorption properties often differ slightly from the internal aldimine.66 In the Y268F mutant, a species with a λmax at 431 nm forms upon mixing with ACC at an observed rate of k1 = 70 s−1. As in the E295D mutant, the λmax of this intermediate is red-shifted relative to the λmax of the ketoenamine tautomer of the internal aldimine (419 nm, see Figure S1), suggesting that the external aldimine is likely accumulating upon ACC binding to Y268F. The absorbance changes at 431 nm are triphasic and, interestingly, the intensity of the 431 nm absorption band increases during the fast phase (k1 ~ 70 s−1), decreases over the next ~ 200 msec (k2 ~ 15 s−1), and then increases again over the next several seconds in a slow phase (k3 ~ 0.5 s−1, Figure S4). All three kinetic phases are fast enough to be kinetically competent (see Tables S1 and S6). In the catalytically inactive Y294F mutant,20 the ketoenamine form of an external aldimine (λmax = 431 nm) also appears to accumulate upon ACC binding. Support for this assignment is provided by the crystal structure of the yeast Y295F enzyme (analogous to the Pseudomonas sp. ACP Y294F mutant) solved in the presence of ACC, where an external aldimine species was detected.19 Like the Y268F reaction, the absorbance changes in the Y294F mutant at 431 nm upon ACC binding were multiphasic. However, in the case of Y294F, the 431 nm absorption increased in the fast phase (k1 = 6.1 s−1), then continued to increase in an intermediate phase (k2 = 0.5 s−1) before decaying in a slow phase (k3 = 0.1 s−1, Figure S4).
DISCUSSION
Based on the kinetic characterization of wt ACCD and its mutants reported in this study, a working model for the pH-dependence of the steady state kinetic parameters and the protonation states of the important active site residues can be proposed (Scheme 5). The group responsible for pK1 is assigned to the phenoxyl group or Tyr294, which likely needs to be deprotonated for ACC binding. Support for this conclusion is derived from the similarity of pK1 in the wt and E295D mutant enzymes and from the increase in pK1 of ~ 1.5 units in the Y268F enzyme. In this model, the pKa of Tyr294 is lowered by its hydrogen bonding interaction with Tyr268 (2.5 Å, Figure 1), which allows ACC to bind and Tyr294 to deprotonate the amino group of ACC, facilitating the aldimine exchange reaction. A role for Tyr294 in ACC deprotonation was first proposed on the basis of structural studies by Ose et al.19 and is also supported by our stopped-flow studies of the Y294F mutant, in which the rate of the aldimine exchange reaction to form an external aldimine is significantly reduced.
Scheme 5.

Revised chemical mechanism for ACCD. Putative hydrogen bonds are shown with dashed lines.
The pK2 values determined from the kcat/Km-pH profiles are relatively invariant, suggesting that this apparent ionization constant likely corresponds either to the amino group of ACC or the side chain of Ser78. The pKa of cyclopropylamine is ~ 8.7 in aqueous solution,67 which is very similar to the kinetic pK2 values (~ 9) measured in our studies. A protonated, positively-charged ACC amino group could engage in favorable electrostatic interactions with the deprotonated, negatively-charged Tyr294 to facilitate ACC binding. Alternatively, if Ser78 is responsible for pK2, Ser78 would likely need to be protonated to form a hydrogen bond to the ACC carboxylate group (see Figure 1). The pKa of the hydroxyl side chain of Ser78 may be lowered by hydrogen bonding interactions with the backbone amide groups of Gln80 and Thr81 and/or through a putative helix-dipole interaction. Similar helix-dipole interactions have been shown to lower the pKa of Cys residues in model peptides68 and in enzyme active sites69,70 by nearly two units. The lowering of the pKa of Ser78 could also serve a catalytic function, as this residue has been implicated in acid/base chemistry during turnover, perhaps serving to mediate proton transfers at C3 of the substrate-PLP adduct following cyclopropane ring scission.22 Finally, spectroscopic studies of the internal aldimines of wt ACCD, Y268F, Y294F, and E295D demonstrate that the Schiff base remains protonated across the pH range studied, suggesting that the deprotonation of PLP is not responsible for pK2.
Analysis of the kinetic properties of the ACCD mutant enzymes provides insight into the chemical mechanism for cyclopropane ring cleavage. PLP species consistent with the ketoenamine tautomer of an external aldimine (4, Scheme 5) form in the pre-steady state fast phases of the E295D and Y268F-catalyzed reactions. Since the external aldimine is an essential intermediate in the nucleophile-induced ring scission, but is not required in the acid-catalyzed ring cleavage, the detection of 4 suggests that the acid-catalyzed strategy shown in Scheme 3 is probably not employed in the mutant enzymes. Stronger evidence for a nucleophile-induced ring scission is provided by the kcat-pH profile for the Y268F mutant, where an active site group in the Michaelis complex with a pKa of ~ 7.8 must be deprotonated for activity. The most likely candidate for this important catalytic group is the side chain of Tyr294. This conclusion is supported by the conservation of the Tyr268-Tyr294 pair in ACCD homologues, the close proximity of the phenoxyl groups of Tyr294 and Tyr268 (2.5 Å), the position of the Tyr294 phenoxyl group near the pro-S Cβ of ACC in the co-crystal structure (3.0 Å), and the complete inactivity of the Y294F mutant.18,19,21 The 10-fold reduction in kcat for the E295D mutant and the reported inactivity of the E295Q mutant19 are also most consistent with the nucleophile-induced ring cleavage mechanism. The weakened ion pair interaction between the carboxylate group of Glu-295 and the pyridinium nitrogen of PLP in the E295D mutant may raise the energy of the transition state for the ring-scission step if the liberated cyclopropane ring electrons can not be delocalized efficiently into the coenzyme.
Interestingly, a clearly definable ketoenamine tautomer of an external aldimine species cannot be detected in the pre-steady state of the wt ACCD reaction. Instead, a species consistent with either an enolimine tautomer of an internal/external aldimine or a gem-diamine (10, Scheme 5) accumulates rapidly at an observed rate constant of ~ 260 s−1 under saturating concentrations of ACC. Although the observed PLP intermediate is spectroscopically indistinguishable from the enolimine tautomer of the internal aldimine resting state, the attack of the incoming ACC amino group on the C4′ atom of PLP to initiate the aldimine exchange is expected to be fast in the wt enzyme, in light of the observations that the Y268F and E295D mutant enzymes quickly catalyze the aldimine exchange reaction. Thus, the observable species in the stopped-flow studies of the wt enzyme is tentatively assigned as the gem-diamine that is observed in the X-ray crystal structure of the wt ACCD enzyme in the presence of ACC.20,21
According to our model for substrate binding and catalysis (Scheme 5), the side chain of Tyr294 is protonated in the gem-diamine state of the wt ACCD-ACC complex, allowing it to hydrogen bond with the ACC carboxylate group (2.7 Å). It is possible that deprotonation of Tyr294 in the Michaelis complex and the collapse of the gem-diamine into the external aldimine (10 → 4) is energetically demanding, allowing the gem-diamine species to accumulate in the pre-steady state. A similar gem-diamine intermediate also accumulates in incubations of wt ACCD with 1-aminocyclopropane-1-phosphonate (ACP) which, despite its similar stereoelectronic properties to ACC, is not turned over into any products.21,65 Interestingly, the internuclear distance between the oxygen atom of the Tyr294 side chain and the phosphonate oxygen of ACP in the gem-diamine form of the ACP-ACCD complex (2.3 Å) is shortened relative to the distance between Tyr294 and the ACC carboxylate group in the gem-diamine form of the ACC-ACCD complex (2.7 Å).21 It is tempting to speculate that this shortened hydrogen bonding distance could help to trap the ACP-ACCD complex in an unreactive gem-diamine state, perhaps accounting for the slow, tight-binding inhibition properties of ACP.65 At present, it is still unclear why the aldimine exchange reaction in wt ACCD is halted in the gem-diamine state, while both the Y268F and E295D mutants can apparently move quickly through this intermediate to generate the external aldimine rapidly in the pre-steady state. Based on this unusual observation and the fact that the ACP inhibitor also forms a gem-diamine complex when bound to wt ACCD, it is possible that the gem-diamine species detected in the wt ACCD-catalyzed reaction is an off-pathway species.
The proposed mechanism for ACCD catalysis involving nucleophilic addition of Tyr294 to the cyclopropane ring of ACC (Scheme 5) can be rationalized on the basis of the solvent and 13C kinetic isotope effects determined in this study. The interpretation of the KIEs depends on whether or not the putative gem-diamine species (10) is a catalytically competent intermediate. If 10 is a species on the reaction pathway, the steps leading from gem-diamine decay to the external aldimine (10 → 4) and from the external aldimine to the quinonoid species (4 → 5) may both be partially rate limiting during steady state turnover under kcat/Km conditions. Here, the normal D2O kcat/Km of 2.5 could be a result of the decay of the gem-diamine species. Deprotonation of Tyr294 and/or protonation of the amino side chain of Lys51 during its elimination from the gem-diamine intermediate could potentially generate the normal solvent KIE on kcat/Km. The 13(kcat/Km) isotope effect of 1.5% on Cα of ACC would arise from the partially rate limiting conversion of the reactive external aldimine intermediate to the quinoniod (4 → 5). The magnitude of 13(kcat/Km) on the wt ACCD-catalyzed reaction is modest when compared to the ~ 4–6% KIEs measured for enzymatic PLP-dependent decarboxylation reactions when the decarboxylation step is fully rate limiting.62, 63 The modest 13(kcat/Km) on the wt ACCD-catalyzed reaction would be consistent with a stepwise mechanism in which the 13C-KIE is attenuated by a preceding and partially rate limiting gem-diamine decay step.
Alternatively, if the gem-diamine species (10) that accumulates in the wt ACCD-catalyzed reaction is an off-pathway intermediate, the solvent and 13C-KIEs could be reporting on the same step in the mechanism. Here, the external aldimine would be generated rapidly upon ACC binding to the wt enzyme (Scheme 6), but may only represent a minor fraction of the total enzyme present in the reaction mixtures with ACC (with the major enzyme form being a non-productive gem-diamine state). As such, the external aldimine could elude detection by stopped-flow absorption spectroscopy. In this concerted mechanism, Tyr294 deprotonation could occur concomitantly with nucleophilic attack and Cβ-Cα bond cleavage. The magnitude of the solvent KIE for wt ACCD (D2O kcat/Km = 2.5) is consistent with solvent KIE values measured for enzymatic reactions where deprotonation of oxygen nucleophiles is concerted with nucleophilic attack.71 In this scenario, the 1.5% 13(kcat/Km) on the ACCD-catalyzed reaction could indicate an early (or late) transition state for C-C bond cleavage, rather than a stepwise mechanism where the C-C bond cleavage step is only partially rate limiting. According to the Hammond Postulate, an early transition state for C-C bond cleavage would be expected for the exothermic cyclopropane ring opening event.
Scheme 6.

Putative mechanism for ACCD catalysis involving concerted deprotonation and nucleophilic addition.
Following cleavage of the ACC cyclopropane ring, transient protonation at C4′ of PLP to form the ketimine species (16) may help to activate the Cβ proton (at C3) by lowering the anionic character at Cα (Scheme 5), in analogy to the reaction catalyzed by cystathionine-γ-synthase.72 The tyrosine nucleophile could then be eliminated from 16 by deprotonation of the pro-R proton at C3, yielding the β,γ-unsaturated ketimine intermediate (17). Based on stereochemical,9,16 structural,18–21 and biochemical studies,22 Ser78 is the most likely candidate for catalyzing this deprotonation. Most notably, the catalytic efficiency of the S78A mutant is significantly reduced, Ser78 is covalently modified by the electrophilic ACC analogue, 2-methylene-ACC, and Ser78 is required for Cα-deprotonation of ACCD-bound D-aminoacid-PLP adducts.22 Protonation at Cγ of the quinonoid species (17) to give the PLP-aminocrotonate species (8), followed by aldimine exchange (8 → 3), and hydrolysis of the aminocrotonate (9 → 2) would complete the catalytic cycle. The groups responsible for mediating these transformations are not clear, but they likely involve some level of participation by Tyr268, Tyr294, Ser78, Lys51, and the PLP. Since these steps follow the irreversible cyclopropane ring cleavage step, one or more of them is likely responsible for the additional portion of the normal solvent KIE that is measured only on kcat. At this point, the irreversibility of the ACCD-catalyzed reaction and our inability to conclusively detect species 5, 6, and 8 in the wt ACCD reaction using stopped-flow spectroscopy has made characterization of the late stages of the reaction mechanism difficult.
Taken together, the accumulated evidence strongly favors a mechanism for ACCD catalysis involving nucleophilic cleavage of the cyclopropane ring. However, the unusual stability of the gem-diamine intermediate in the wt ACCD-ACC complex prompted us to propose an alternative mechanism (Scheme 3) in which Tyr294 acts as a general acid to facilitate the opening of the cyclopropane ring of ACC.20 Precedence for a similar mode of electrophilic cyclopropane ring cleavage has been provided by Jencks and co-workers in their model studies of the reactivity of phenylcyclopropanols.23 Though this mechanism cannot be rigorously excluded at the moment, it is at odds with several experimental observations. First, the E295D and Y268F enzymes accumulate an external aldimine intermediate in the pre-steady state, which is not predicted to form along the putative reaction coordinate for the acid-catalyzed ring opening mechanism. Second, solvent proton incorporation into Cγ of the α-KB product is not fully stereospecific,16 which is unlikely if Tyr294 is serving as a general acid to initiate ring cleavage from the gem-diamine intermediate. Third, the E295Q mutant is inactive19 and the activity of the E295D mutant is substantially decreased. These observations suggest that electron delocalization into the pyridinium ring (a hallmark of normal PLP-catalysis that would not be required for turnover in the acid-catalyzed mechanism) is an important feature of wt ACCD catalysis. Finally, in addition to the deamination of ACC, wt ACCD catalyzes a variety of Cα-deprotonation and Cβ-elimination reactions with other amino acid substrates.9,11,22 These reactions most likely proceed via quinonoid species that are common to many other PLP-dependent enzyme mechanisms. Thus, the wt ACCD enzyme is at least capable of accommodating these typical PLP intermediates, which would not be employed in the acid-catalyzed ring opening mechanism.
Supplementary Material
Table 3.
Summary of 13kcat/Km data for the wt ACCD reaction. R/Ro and its standard error (ΔR/Ro) were calculated from the NMR data in Table S3. F and its standard error (ΔF) were calculated from the LDH reporter assay data in Table S4. 13(kca/Km)Cα and 13(kcat/Km)Cβ were then calculated from R/Ro and F using eqs 6 and 8, respectively, and the standard errors in the final digit of the KIE measurements (in parentheses) were calculated using Eqs S2–S4. The arithmetic average (and its standard error) of the two replicate measurements for each 13C-KIE was then calculated to determine the reported KIE. *The R/Ro of unity leads to errors of 0 in Eqs S2 and S3. For calculation of the average value of 13(kcat/Km)Cβ, we assumed a relative error of 0.5% on 13(kca/Km)Cβ for reaction 1 (the average relative error of the other KIE measurements).
| Reaction | R/Ro (Cα) | ΔR/Ro (Cα) | R/Ro (Cβ) | ΔR/Ro (Cβ) | F | ΔF | 13(kcat/Km)Cα | 13(kcat/Km)Cβ |
|---|---|---|---|---|---|---|---|---|
| 1 | 1.031 | 0.017 | 1.000* | 0.005 | 0.906 | 0.006 | 1.013(7) | 1.000(5)* |
| 2 | 1.030 | 0.010 | 1.031 | 0.006 | 0.834 | 0.006 | 1.017(6) | 1.034(3) |
| Average | 1.015(5) | 1.017(17) | ||||||
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health (GM040541).
The authors thank Dr. Mark Ruszczycky and Professor Christian P. Whitman for their critical reading of the manuscript, Professor Paul Cook for his helpful discussions, Dr. Zhihua Tao for constructing some of the mutant enzymes employed in this study, Steve Sorey for his assistance with the NMR experiments, and a Fellowship from the Graduate School of University of Texas at Austin to C.J.T.
ABBREVIATIONS
- α-KB
2-keto-1-butanoic acid
- ACC
1-aminocyclopropane-1-carboxylic acid
- ACCD
1-aminocyclopropane-1-carboxylate deaminase
- ACP
1-aminocyclopropane-1-phosphonate
- CAPSO
3-(cyclohexylamino)-2-hydroxy-1-propanesulfonic acid
- EPPS
4-(2-hydroxyethyl)-1-piperazinepropane-sulfonic acid
- HMP
4-hydroxyl-N-methylpiperidine
- HEPES
4-(2-hydroxyethyl)-1-piperizineethanesulfonic acid
- KIE
kinetic isotope effect
- LDH
L-lactate dehydrogenase
- MES
2-(N-morpholino)ethanesulfonic acid
- MOPS
3-(N-morpholino)propanesulfonic acid
- MSR
mean square residual of the fit
- NADH
nicotinamide adenine dinucleotide (reduced form)
- NMR
nuclear magnetic resonance
- PLP
pyridoxal-5′-phosphate
- SHMTase
serine hydroxymethyltransferase
- TAPS
N-tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid
- wt
wild type
Footnotes
Supporting Information Available. Steady state kinetic data, error analysis for the proton inventory studies, raw data used for the calculation of the 13C-KIEs, and non-linear fits of the stopped-flow data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Honma M, Shimomura T. Metabolism of 1-aminocyclopropane-1-carboxylic acid. Agric Biol Chem. 1978;42:1825–1831. [Google Scholar]
- 2.Kende H. Ethylene biosynthesis. Annu Rev Plant Physiol Plant Mol Biol. 1993;44:283–307. [Google Scholar]
- 3.Klee HJ, Hayford MB, Kretzmer KA, Barry GF, Kishore GM. Control of ethylene synthesis by expression of a bacterial enzyme in transgenic tomato plants. Plant Cell. 1991;3:1187–1193. doi: 10.1105/tpc.3.11.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 5.Dunathan HC. Conformation and reaction specificity in pyridoxal phosphate enzymes. Proc Natl Acad Sci USA. 1966;55:712–716. doi: 10.1073/pnas.55.4.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toney MD. Reaction specificity in pyridoxal phosphate enzymes. Arch Biochem Biophys. 2005;433:279–287. doi: 10.1016/j.abb.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 7.Matthews RB, Drummond JT. Providing one-carbon units for biological methylations: mechanistic studies on serine hydroxymethyltransferase, methylenetetrahydrofolate reductase, and methyltetrahydrofolate-homocysteine methyltransferase. Chem Rev. 1990;90:1275–1290. [Google Scholar]
- 8.Schirch V, Szebenyi DME. Serine hydroxymethyltransferase revisited. Curr Opin Chem Biol. 2005;9:482–487. doi: 10.1016/j.cbpa.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 9.Walsh CT, Pascal RA, Jr, Johnston M, Raines R, Dikshit D, Krantz A, Honma M. Mechanistic studies on the pyridoxal phosphate enzyme 1-aminocyclopropane-1-carboxylate deaminse from Pseudomonas sp. Biochemistry. 1981;20:7509–7519. doi: 10.1021/bi00529a028. [DOI] [PubMed] [Google Scholar]
- 10.Hill RK, Prakash SR, Wiesendanger R, Angst W, Martinoni B, Arigoni D, Liu H-w, Walsh CT. Stereochemistry of the enzymatic ring opening of 1-aminocyclopropanecarboxylic acid. J Am Chem Soc. 1984;106:795–796. [Google Scholar]
- 11.Li K, Du W, Que NLS, Liu H-w. Mechanistic studies of 1-aminocyclopropane-1-carboxylate deaminase: unique covalent catalysis by coenzyme B6. J Am Chem Soc. 1996;118:8763–8764. [Google Scholar]
- 12.Stewart JM, Westberg HH. Nucleophilic ring-opening additions to 1, 1-disubstituted cyclopropanes. J Org Chem. 1965;30:1951–1955. [Google Scholar]
- 13.Yates P, Helferty PH, Mahler P. Nucleophilic and acid-catalyzed cleavage of the cyclopropane rings of β, γ - unsaturated α-spirocyclopropyl ketones. Can J Chem. 1983;61:71–85. [Google Scholar]
- 14.Danishefsky S. Electrophilic cyclopropanes in organic synthesis. Acc Chem Res. 1979;12:66–72. [Google Scholar]
- 15.Cleland WW. The use of pH studies to determine chemical mechanisms of enzyme-catalyzed reactions. Methods Enzymol. 1982;87:390–405. doi: 10.1016/s0076-6879(82)87024-9. [DOI] [PubMed] [Google Scholar]
- 16.Liu H-w, Auchus R, Walsh CT. Stereochemical studies on the reactions catalyzed by the PLP-dependent enzyme 1-aminocyclopropane-1-carboxylate deaminase. J Am Chem Soc. 1984;106:5335–5348. [Google Scholar]
- 17.Battiste MA, Coxon JM. Acidity and basicity of cyclopropanes. In: Rappoport Z, editor. The chemistry of the cyclopropyl group. Part 1. John Wiley & Sons; Chichester: 1987. pp. 255–305. [Google Scholar]
- 18.Yao M, Ose T, Sugimoto H, Horiuchi A, Nakagawa A, Wakatsuki S, Yokoi S, Murakami T, Honma M, Tanaka I. Crystal structure of 1-aminocyclopropane-1-carboxylate deaminase from Hansenula saturnus. J Biol Chem. 2000;275:34557–34565. doi: 10.1074/jbc.M004681200. [DOI] [PubMed] [Google Scholar]
- 19.Ose T, Fujino A, Yao M, Watanabe N, Honma M, Tanaka I. Reaction intermediate structures of 1-aminocyclopropane-1-carboxylate deaminase: Insight into PLP-dependent cyclopropane ring-opening reaction. J Biol Chem. 2003;278:41069–41076. doi: 10.1074/jbc.M305865200. [DOI] [PubMed] [Google Scholar]
- 20.Karthikeyan S, Zhao Z, Kao CL, Zhou Q, Tao Z, Zhang H, Liu H-w. Structural analysis of 1-aminocyclopropane-1-carboxylate deaminase: Observation of an aminyl intermediate and identification of Tyr 294 as the active-site nucleophile. Angew Chem Int Ed. 2004;43:3425–3429. doi: 10.1002/anie.200453353. [DOI] [PubMed] [Google Scholar]
- 21.Karthikeyan S, Zhou Q, Zhao Z, Kao CL, Tao Z, Robinson H, Liu H-w, Zhang H. Structural analysis of Pseudomonas 1-aminocyclopropane-1-carboxylate deaminase complexes: Insight into the mechanism of a unique pyridoxal-5′-phosphate dependent cyclopropane ring-opening reaction. Biochemistry. 2004;43:13328–13339. doi: 10.1021/bi048878g. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Z, Chen H, Li K, Du W, He S, Liu H-w. Reaction of 1-amino-2-methylenecyclopropane-1-carboxylate with 1-aminocyclopropane-1-carboxylate deaminase: Analysis and mechanistic implications. Biochemistry. 2003;42:2089–2103. doi: 10.1021/bi020567n. [DOI] [PubMed] [Google Scholar]
- 23.Thibblin A, Jencks WP. Unstable carbanions. General acid catalysis of the cleavage of 1-phenylcyclopropanol and 1-phenyl-2-arylcyclopropanol anions. J Am Chem Soc. 1979;101:4963–4973. [Google Scholar]
- 24.Wiberg KB, Kass SR. Electrophilic cleavage of cyclopropanes. Acetolysis of alkylcyclopropanes. J Am Chem Soc. 1985;107:988–995. [Google Scholar]
- 25.Baird RL, Aboderin AA. Concerning the role of protonated cyclopropane intermediates in solvolytic reactions. The solvolysis of cyclopropane in deuteriosulfuric acid. J Am Chem Soc. 1964;86:252–255. [Google Scholar]
- 26.McKinney MA, Smith SH, Hempelman S, Gearen MM, Pearson L. Proteolytic cleavage of cyclopropanes: The structure of the transition state in the protonation of arylcyclopropanes. Tetra Lett. 1971:3657–3660. [Google Scholar]
- 27.McKinney MA, So EC. Protolytic cleavage of cyclopropanes. The two mechanisms for the acid-catalyzed cleavage of 1-phenylcyclopropylmethyl ether. J Org Chem. 1972;37:2818–2822. [Google Scholar]
- 28.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 29.St Maurice M, Bearne SL. Kinetics and thermodynamics of mandelate racemase catalysis. Biochemistry. 2002;41:4048–4058. doi: 10.1021/bi016044h. [DOI] [PubMed] [Google Scholar]
- 30.Schowen KB, Schowen RL. Solvent isotope effects on enzyme systems. Methods Enzymol. 1982;87:551–606. [PubMed] [Google Scholar]
- 31.Singleton DA, Thomas AA. High-precision simultaneous determination of multiple small kinetic isotope effects at natural abundance. J Am Chem Soc. 1995;117:9357–9358. [Google Scholar]
- 32.Northrop DB. Steady-state analysis of kinetic isotope effects in enzymic reactions. Biochemistry. 1975;14:2644–2651. doi: 10.1021/bi00683a013. [DOI] [PubMed] [Google Scholar]
- 33.Simon H, Palm D. Isotope effects in organic chemistry and biochemistry. Angew Chem Int Ed Engl. 1966;5:920–933. [Google Scholar]
- 34.Cleland WW. The use of isotope effects to determine enzyme mechanisms. Arch Biochem Biophys. 2005;433:2–12. doi: 10.1016/j.abb.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 35.Bigeleisen J, Wolfsberg M. Adv Chem Phys. 1958;1:15. [Google Scholar]
- 36.Yano T, Kuramitsu S, Tanase S, Morino Y, Kagamiyama H. Role of Asp222 in the catalytic mechanism of Escherichia coli aspartate aminotransferase: the amino acid residue which enhances the function of the enzyme-bound coenzyme pyridoxal 5′-phosphate. Biochemistry. 1992;31:5878–5887. doi: 10.1021/bi00140a025. [DOI] [PubMed] [Google Scholar]
- 37.Yano T, Hinoue Y, Chen VJ, Metzler DE, Miyahara I, Hirotsu K, Kagamiyama H. Role of an active site residue analyzed by combination of mutagenesis and coenzyme analog. J Mol Biol. 1993;234:1218–1229. doi: 10.1006/jmbi.1993.1672. [DOI] [PubMed] [Google Scholar]
- 38.Gong J, Hunter GA, Ferreira GC. Aspartate-279 in aminolevulinate synthase affects enzyme catalysis through enhancing the function of the pyridoxal 5′-phosphate cofactor. Biochemistry. 1998;37:3509–3517. doi: 10.1021/bi9719298. [DOI] [PubMed] [Google Scholar]
- 39.Momany C, Levdikov V, Blagova L, Lima S, Phillips RS. Three-dimensional structure of kynureninase from Pseudomonas fluorescens. Biochemistry. 2004;43:1193–1203. doi: 10.1021/bi035744e. [DOI] [PubMed] [Google Scholar]
- 40.Morozov YV. Spectroscopic properties, electronic structure, and photochemical behavior of vitamin B6 and analogs. In: Dolphin D, Poulson R, Avramovic O, editors. Vitamine B6 pyridoxal phosphate: chemiscal, biochemical, and medical aspects. Part A. John Wiley & Sons; New York: 1986. pp. 131–222. [Google Scholar]
- 41.Eliot AC, Kirsch JF. Modulation of the internal aldimine pKa’s of 1-aminocyclopropane-1-carboxylate synthase and aspartate aminotransferase by specific active site residues. Biochemistry. 2002;41:3836–3842. doi: 10.1021/bi016084l. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Feng L, Kirsch JF. Kinetic and spectroscopic investigations of wild-type and mutant forms of apple 1-aminocyclopropane-1-carboxylate synthase. Biochemistry. 1997;36:15477–15488. doi: 10.1021/bi971625l. [DOI] [PubMed] [Google Scholar]
- 43.Zhou X, Toney MD. pH Studies on the mechanism of the pyridoxal phosphate-dependent dialkylglycine decarboxylase. Biochemistry. 1999;38:311–320. doi: 10.1021/bi981455s. [DOI] [PubMed] [Google Scholar]
- 44.Osterman AL, Brooks HB, Rizo J, Phillips MA. Role of Arg-277 in the binding of pyridoxal 5′-phosphate to trypanosoma brucei ornithine decarboxylase. Biochemistry. 1997;36:4558–4567. doi: 10.1021/bi962916h. [DOI] [PubMed] [Google Scholar]
- 45.Hayashi H, Mizuguchi H, Kagamiyama H. Rat liver aromatic L-amino acid decarboxylase: Spectroscopic and kinetic analysis of the coenzyme and reaction intermediates. Biochemistry. 1993;32:812–818. doi: 10.1021/bi00054a011. [DOI] [PubMed] [Google Scholar]
- 46.Metzler CM, Viswanath R, Metzler DE. Equilibria and absorption spectra of tryptophanase. J Biol Chem. 1991;266:9374–9381. [PubMed] [Google Scholar]
- 47.Karsten WE, Ohshiro T, Izumi Y, Cook PF. Reaction of serine-glyoxylate aminotransferase with the alternative substrate ketomalonate indicates rate-limiting protonation of a quinonoid intermediate. Biochemistry. 2005;44:15930–15936. doi: 10.1021/bi051407p. [DOI] [PubMed] [Google Scholar]
- 48.Karsten WE, Ohshiro T, Izumi Y, Cook PF. Initial velocity, spectral, and pH studies of the serine-glyoxylate aminotransferase from Hyphomicrobium methylovorum. Arch Biochem Biophys. 2001;388:267–275. doi: 10.1006/abbi.2001.2294. [DOI] [PubMed] [Google Scholar]
- 49.Grinnell J, Fornwalt HJ. The viscosity of deuterium oxide and its mixtures with water at 25°. J Chem Phys. 1936;4:30–33. [Google Scholar]
- 50.Cerjan C, Barnett RE. Viscosity dependence of a putative diffusion-limited reaction. J Phys Chem. 1972;76:1192–1195. [Google Scholar]
- 51.Nakatani H, Dunford HB. Meaning of diffusion-controlled association rate constants in enzymology. J Phys Chem. 1979;83:2662–2665. [Google Scholar]
- 52.Brouwer AC, Kirsch JF. Investigation of diffusion-limited tates viscosity variation. Biochemistry. 1982;21:1302–1307. doi: 10.1021/bi00535a030. [DOI] [PubMed] [Google Scholar]
- 53.Hardy LW, Kirsch JF. Diffusion-limited component of reactions catalyzed by Bacillus cereus β-lactamase. Biochemistry. 1984;23:1275–1282. doi: 10.1021/bi00301a040. [DOI] [PubMed] [Google Scholar]
- 54.Kurz LC, Weitkamp E, Frieden C. Adenosine deaminase: viscosity studies and the mechanism of binding of substrate and of ground- and transition-state analogue inhibitors? Biochemistry. 1987;26:3027–3032. doi: 10.1021/bi00385a012. [DOI] [PubMed] [Google Scholar]
- 55.Adams JA, Taylor SS. Energetic limits of phosphotransfer in the catalytic subunit of cAMP dependent protein kinase as measured by viscosity experiments? Biochemistry. 1992;31:8516–8522. doi: 10.1021/bi00151a019. [DOI] [PubMed] [Google Scholar]
- 56.Wood BM, Chan KK, Amyes TL, Richard JP, Gerlt JA. Mechanism of the orotidine 5′-monophosphate decarboxylase-catalyzed reaction: Effect of solvent viscosity on kinetic constants. Biochemistry. 2009;48:5510. doi: 10.1021/bi9006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raber ML, Freeman MF, Townsend CA. Dissection of the stepwise mechanism to β-lactam formation and elucidation of a rate-determining conformational change in β-lactam synthetase. J Biol Chem. 2009;284:207–217. doi: 10.1074/jbc.M805390200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Francis K, Gadda G. Probing the chemical steps of nitroalkane oxidation catalyzed by 2 nitropropane dioxygenase with solvent viscosity, pH, and substrate kinetic isotope effects. Biochemistry. 2006;45:13889–13898. doi: 10.1021/bi060566l. [DOI] [PubMed] [Google Scholar]
- 59.Scharschmidt M, Fisher MA, Cleland WW. Variation of transition-state structure as a function of the nucleotide in reactions catalyzed by dehydrogenases. 1. Liver alcohol dehydrogenase with benzyl alcohol and yeast aldehyde dehydrogenase with benzaldehyde. Biochemistry. 1984;23:5471–5478. doi: 10.1021/bi00318a015. [DOI] [PubMed] [Google Scholar]
- 60.Hess RA, Hengge AC, Cleland WW. Isotope effects on enzyme-catalyzed acyl transfer from p-nitrophenyl acetate: Concerted mechanisms and increased hyperconjugation in the transition state. J Am Chem Soc. 1998;120:2703–2709. [Google Scholar]
- 61.Lee JK, Bain AD, Berti PJ. Probing the transition states of four glucoside hydrolyses with 13C kinetic isotope effects measured at natural abundance by NMR spectroscopy. J Am Chem Soc. 2004;126:3769–3776. doi: 10.1021/ja0394028. [DOI] [PubMed] [Google Scholar]
- 62.Swanson T, Brooks HB, Osterman AL, O’Leary MH, Phillips MA. Carbon-13 isotope effect studies of Trypanosoma brucei ornithine decarboxylase. Biochemistry. 1998;37:14943–14947. doi: 10.1021/bi981154i. [DOI] [PubMed] [Google Scholar]
- 63.O’Leary MH. Transition-state structures in enzyme-catalyzed decarboxylations. Acc Chem Res. 1988;21:450–455. [Google Scholar]
- 64.O’Leary MH, Richards DT, Hendrickson DW. Carbon isotope effects on the enzymic decarboxylation of glutamic acid. J Am Chem Soc. 1970;92:4435–4440. doi: 10.1021/ja00717a048. [DOI] [PubMed] [Google Scholar]
- 65.Erion MD, Walsh CT. 1-Aminocyclopropanephosphonate: Time-dependent inactivation of 1-aminocyclopropanecarboxylate deaminase and Bacillus stearothermophilus alanine racemase by slow dissociation behavior. Biochemistry. 1987;26:3417–3425. doi: 10.1021/bi00386a025. [DOI] [PubMed] [Google Scholar]
- 66.Woehl EU, Tai CH, Dunn MF, Cook PF. Formation of the α-aminoacrylate intermediate limits the overall reaction catalyzed by O-acetylserine sulfhydrylase. Biochemistry. 1996;35:4776–4783. doi: 10.1021/bi952938o. [DOI] [PubMed] [Google Scholar]
- 67.Tidwell TT. Conjugative and substitutive properties of the cyclopropyl group. In: Rappoport Z, editor. The chemistry of the cyclopropyl group. Vol. 1. John Wiley & Sons; New York: 1987. pp. 565–632. [Google Scholar]
- 68.Kortemme T, Creighton TE. Ionization of cysteine residues at the termini of model α-helical peptides. Relevance to unusual thiol pKa values in proteins of the thioredoxin family. J Mol Biol. 1995;253:799–812. doi: 10.1006/jmbi.1995.0592. [DOI] [PubMed] [Google Scholar]
- 69.Davies C, Heath RJ, White SW, Rock CO. The 1.8 Å crystal structure and active-site architecture of β-ketoacyl-acyl carrier protein synthase III (FabH) from Escherichia coli. Structure. 2000;8:185–195. doi: 10.1016/s0969-2126(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 70.Snook CF, Tipton PA, Beamer LJ. Crystal structure of GDP-mannose dehydrogenase: A key enzyme of alginate biosynthesis in P. aeruginosa. Biochemistry. 2003;42:4658–4668. doi: 10.1021/bi027328k. [DOI] [PubMed] [Google Scholar]
- 71.Anderson VE, Ruszczycky MW, Harris ME. Activation of oxygen nucleophiles in enzyme catalysis. Chem Rev. 2006;106:3236–3251. doi: 10.1021/cr050281z. [DOI] [PubMed] [Google Scholar]
- 72.Brzovic P, Holbrook EL, Greene RC, Dunn MF. Reaction mechanism of Escherichia coli cystathionine γ-synthase: Direct evidence for a pyridoxamine derivative of vinylglyoxylate as a key intermediate in pyridoxal phosphate dependent γ-elimination and γ-replacement reactions. Biochemistry. 1990;29:442–451. doi: 10.1021/bi00454a020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.