

Abstract
Osteogenesis imperfecta (OI) is a heterogeneous genetic disorder characterized by bone fragility and susceptibility to fractures after minimal trauma. After mutations in all known OI genes had been excluded by Sanger sequencing, we applied next-generation sequencing to analyze the exome of a single individual who has a severe form of the disease and whose parents are second cousins. A total of 26,922 variations from the human reference genome sequence were subjected to several filtering steps. In addition, we extracted the genotypes of all dbSNP130-annotated SNPs from the exome sequencing data and used these 299,494 genotypes as markers for the genome-wide identification of homozygous regions. A single homozygous truncating mutation, affecting SERPINF1 on chromosome 17p13.3, that was embedded into a homozygous stretch of 2.99 Mb remained. The mutation was also homozygous in the affected brother of the index patient. Subsequently, we identified homozygosity for two different truncating SERPINF1 mutations in two unrelated patients with OI and parental consanguinity. All four individuals with SERPINF1 mutations have severe OI. Fractures of long bones and severe vertebral compression fractures with resulting deformities were observed as early as the first year of life in these individuals. Collagen analyses with cultured dermal fibroblasts displayed no evidence for impaired collagen folding, posttranslational modification, or secretion. SERPINF1 encodes pigment epithelium-derived factor (PEDF), a secreted glycoprotein of the serpin superfamily. PEDF is a multifunctional protein and one of the strongest inhibitors of angiogenesis currently known in humans. Our data provide genetic evidence for PEDF involvement in human bone homeostasis.
Main Text
Osteogenesis imperfecta (OI; MIM 166200, 166210, 610854, 259420, 166220, 610967, 610968, 610682, 610915, and 259440 for type I to IX of the disease) is a genetic disorder characterized by bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality.1,2 Typical extraskeletal manifestations, which affect a variable number of OI patients, are dentinogenesis imperfecta, hearing loss, and blue sclera. Most individuals with OI have autosomal-dominantly inherited mutations in COL1A1 (MIM 120150) or COL1A2 (MIM 120160), the two genes that encode the α chains of the major bone matrix protein collagen type 1. Mutations in these genes result in quantitative and/or qualitative defects in type 1 collagen production by osteoblasts.3–6
In a minority of OI patients, the disorder is inherited in an autosomal-recessive manner. Recently, mutations in six different genes have been associated with these OI forms. CRTAP (MIM 605497), LEPRE1 (MIM 610339), and PPIB (MIM 123841) encode components of the prolyl 3-hydroxylation complex.7–14 This protein complex is located in the rough endoplasmatic reticulum and, among other functions, modifies the Pro986 residue of the type 1 collagen α1 chains.12,15 SERPINH1 (MIM 600943) and FKBP10 (MIM 607063) encode collagen chaperones.16–20 HSP47, the gene product of SERPINH1, may monitor the final integrity of the triple helix that is formed out of two α1 chains and one α2 chain.17 SP7 or OSX (MIM 606633) encodes a transcription factor that, based on the function of its murine homolog, is assumed to regulate the differentiation of preosteoblasts to osteoblasts.21,22 In a substantial number of COL1A1 and COL1A2 mutation-negative OI cases, the underlying molecular defect is unknown.17
The current standard therapy for moderate and severe forms of OI (types II, III, and IV in the Sillence classification23) is cyclic intravenous application of bisphophonates.1 These compounds are synthetic pyrophosphate analogs that deposit on the bone surface, where they inhibit osteoclasts.24,25 Bisphosphonate treatment, ideally starting in early childhood, increases bone mineral density. There is evidence that it also reduces fracture rates, chronic bone pain, and immobility in OI patients.26
Here, we report the identification of a gene that is mutated in a severe form of OI. The gene was identified in an affected male individual who has a similarly affected older brother. The parents come from the United Arab Emirates and are second cousins, suggesting autosomal-recessive inheritance of the disease. Mutations in all genes known to cause autosomal-dominant and -recessive OI (see above) had been excluded by direct Sanger sequencing of genomic DNA. We therefore decided to follow a genome-wide sequencing approach to identify the cause of the disease in this patient. The study was approved by the Medical Ethics Committee of the Radboud University Nijmegen Medical Centre. Written informed consent to participate in the study and to publish clinical data and radiographs was obtained for the index patient and for all other OI patients described in this paper.
We sequenced the exome (∼18,600 genes) of the index patient by using the SureSelect human exome kit (Agilent, Santa Clara, CA, USA) and one-quarter of a SOLiD 4 system sequencing slide (Life Technologies, Carlsbad, CA, USA). We obtained 3.7 Gb of mappable sequence data. Color space reads were mapped to the hg18 reference genome with the SOLiD bioscope software version 1.2, which utilizes an iterative mapping approach. In total, 71% of bases came from the targeted exome, resulting in a mean coverage of 65-fold (Table S1, available online). Single nucleotide variants were subsequently called by the DiBayes algorithm with high call stringency. Ninety-two percent of the targeted exons were covered more than ten times. Small insertions and deletions were detected with the SOLiD Small InDel Tool. Called SNP variants and indels were then combined and annotated with a custom analysis pipeline.
Initial quality filtering (>5 variant reads and >15% variant reads) resulted in the identification of 13,487 genetic variants, including 6,298 nonsynonymous changes, in the coding regions or the canonical dinucleotides of the splice sites (Table S2). We applied a prioritization scheme to identify the pathogenic mutation, similar to what was done in recent studies.27–29 For a recessive disease, it is possible that pathogenic mutations are listed as benign polymorphisms in dbSNP or in our internal variant database because individuals without the respective phenotype can be heterozygous carriers. However, given the rare incidence of autosomal-recessive OI, we considered such a scenario to be unlikely. We therefore excluded known dbSNP130 variants as well as variants from our in-house variant database, reducing the number of candidates by more than 98% to a total of 318 variants.
For further analysis, we applied an autosomal-recessive disease model and assumed that the causal mutation was inherited from a common ancestor because of parental consanguinity. Under this assumption, only 17 autosomal candidate genes with homozygous variants in the index patient remained. To prioritize these candidate genes, we developed an algorithm for unbiased identification of homozygous regions in the genome: we extracted the genotypes of all dbSNP130-annotated autosomal SNPs that had a read coverage of at least 15-fold from the exome sequencing data of the index patient. This resulted in a total of 299,494 genotypes, which increased the number of potential genetic markers for the identification of homozygous regions in the genome more than 17-fold. Variants displaying an identical SNP allele in ≥95% of all reads were considered to be homozygous, SNPs with 30%–70% variation reads were considered to be heterozygous, and SNPs with <30% or with 70%–95% variation reads were considered ambiguous. A genomic region was identified as a homozygous stretch if at least 500 consecutive SNP markers were called as homozygous; there was a maximum tolerance of two heterozygous SNPs per 500 markers (to account for possible sequencing or mapping errors). In the dataset of the index patient, this algorithm identified seven homozygous regions on chromosomes 1, 6, 11, and 17 (Table S3 and Figure S1). Only three of the 17 homozygous variants in the index patient's exome were located in one of these homozygous regions: two missense variants on chromosome 1 (p.Gln361Arg [c.1082A>G] in LPHN2 [RefSeq accession number NM_012302.2] and p.Thr1799Met [c.5396C>T] in INADL [MIM 603199; NM_176877.2]) and a stop mutation (p.Tyr232X [c.696C>G]) affecting SERPINF1 (MIM 172860; NM_002615.4) on chromosome 17p13.3 (Table S4). The stop mutation was embedded into a homozygous stretch spanning 2.99 Mb (Figure 1D).
Figure 1.
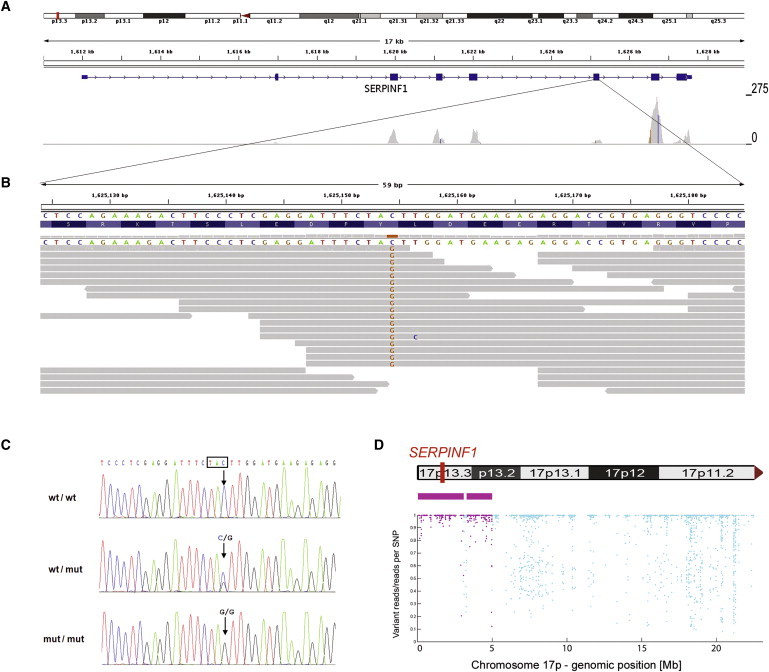
Identification of a Homozygous Mutation in SERPINF1 by Exome Sequencing
(A) Overview of SERPINF1 coverage by exome sequencing. Upper: An ideogram illustrating the SERPINF1 location in the chromosomal band 17p13.3 (red bar). Middle: The genomic structure and localization of SERPINF1 in more detail. Lower: The per-base coverage, with an axis to the right ranging from 0 to 275-fold coverage.
(B) Detailed view of individual sequencing reads overlapping the stop mutation in exon 6. All 18 reads at the genomic position g.1,625,154 (hg18) display the homozygous stop mutation c.696C>G.
(C) Validation of the SERPINF1 mutation in exon 6 by Sanger sequencing of genomic DNA. Electropherograms of a control individual, of the index patient, and of his father are shown. The position of the mutation is marked with an arrow. The affected codon is framed in black. The control individual carries the wild-type sequence (wt) in both copies of the gene. The patient's father is a heterozygous carrier of the mutation (genotype wt/mut). The patient is homozygous for the c.696C>G mutation (mut/mut).
(D) Upper: Contains an ideogram of the p-arm of chromosome 17 illustrating the localization of SERPINF1. Lower : The homozygous regions on chromosome 17 at the genomic positions g.97,058-3,091,589 and g.3,299,188-4,983,922 are recognizable. Each dot represents one of the SNP markers that were included in the analysis. SNPs within a region that was called as homozygous are colored in purple. The position along the y axis indicates the ratio of the number of reads with the nonreference allele to the total number of reads; the value is 1.0 if only reference or nonreference reads are present at a given SNP position (y axis, ratio of variant reads/all reads per SNP position; x axis, genomic position along the p-arm of chromosome 17).
We regarded SERPINF1 as the best candidate for further evaluation. Validation by Sanger sequencing confirmed that the c.696C>G (p.Tyr232X) mutation was indeed present in a homozygous state in the index patient (Figure 1C). We also tested DNA samples of the patient's parents, his affected brother (patient 2), and his two healthy sisters. Only the affected brother carried the mutation in a homozygous state, whereas both parents and both sisters were heterozygous carriers, compatible with autosomal-recessive inheritance of the disorder in the family (Figure S2A). The mutation was not detected on 460 control chromosomes from individuals with the same regional ancestry as the index patient. These results confirmed that the identified mutation is not a rare polymorphism in the United Arab Emirates population. No other SERPINF1 nonsense variant was observed in more than 120 in-house exomes, indicating the rare nature of loss-of-function mutations in this gene.
As a next step, we performed Sanger sequencing of all seven coding SERPINF1 exons on genomic DNA samples obtained from two unrelated Turkish patients (patients 3 and 4), an Iraqi patient, and an Italian patient, all clinically diagnosed with OI. These four patients had no detectable COL1A1 or COL1A2 mutations. The Italian patient had moderate OI (type IV in the Sillence classification23), whereas the other patients suffered from a severe form of OI, comparable to the course of the disease in the Arabian index family. Only the Turkish patients were born to consanguineous parents. In these two patients, we identified truncating SERPINF1 mutations: patient 3 was homozygous for the insertion c.324_325dupCT (p.Tyr109SerfsX5) in exon 4, resulting in a frameshift with a premature stop codon (Figure 2 and Figure S3A). Patient 4 carried the homozygous stop mutation c.1132C>T (p.Gln378X) in exon 8 (Figure 2 and Figure S3B). The patients' parents (as obligate carriers) and two healthy sisters of patient 3 were heterozygous for the respective mutation (Figures S2B and S2C). The insertion mutation c.324_325dupCT was excluded in 460 Turkish control chromosomes, and the stop mutation c.1132C>T was not present on 272 Turkish control alleles, demonstrating that neither of these two sequence changes is a rare polymorphism. The Iraqi and the Italian patient were negative for mutations in the coding region of SERPINF1.
Figure 2.
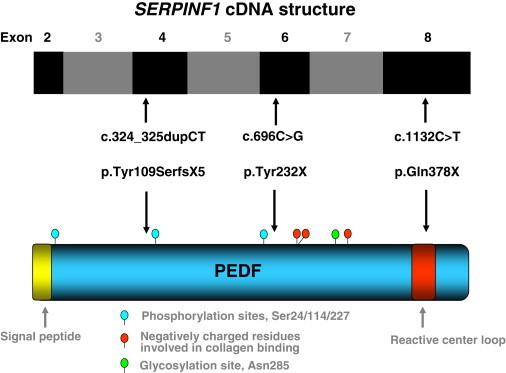
SERPINF1 and PEDF Scheme
Schematic representation of pigment epithelium-derived factor (PEDF, NP_002606) and of the coding exons of the SERPINF1 cDNA (NM_002615.4). The locations of the exons are aligned relative to the regions of the PEDF each exon encodes. The positions of the three truncating mutations identified in OI patients are indicated by arrows. Several important protein domains described in the literature are depicted.36
SERPINF1 encodes pigment-epithelium-derived factor (PEDF), a 50 kDa secreted glycoprotein of the serpin superfamily. The p.Tyr232X and p.Tyr109SerfsX5 mutations can be predicted to cause null alleles because of nonsense-mediated mRNA decay (NMD). We found experimental evidence for NMD of the mutated transcript in peripheral blood by direct sequencing of a PCR-amplified cDNA fragment encompassing the frameshift mutation p.Tyr109SerfsX5 (Figure S4). In the father of patient 3, the cDNA sequence of the mutated allele was only detectable as a faint background signal behind the wild-type sequence, whereas Sanger sequencing on genomic DNA derived from this obligate mutation carrier displayed equally strong signals for the mutated and the wild-type allele. This observation suggests that the number of mutated transcripts serving as templates for the preceding RT-PCR reaction is strongly reduced in comparison to the number of transcripts with the wild-type sequence. The c.1132C>T (p.Gln378X) mutation is located in the last coding exon of SERPINF1, 123 bp upstream of the regular stop codon (Figure 2). The corresponding transcript should therefore escape the NMD machinery.30 However, the C terminus of PEDF (amino acids 384–415) represents one of the four highly conserved regions of the protein.31 Extensive mutagenesis studies with cDNA expression vectors of human PEDF revealed that any deletion of the C terminus, which encompasses more than the four C-terminal amino acids (amino acids 415–418), abolishes secretion of the protein by transiently transfected mammalian cells.32 Moreover, the premature stop codon identified in patient 3 resides within the reactive center loop (RCL) of PEDF (Figure 2). An in vitro deletion of a segment of the RCL (Pro373–Ala380) as well as substitutions of Gly376 and Leu377 by alanine resulted in abrogation of PEDF secretion, suggesting that the RCL is important for the interaction of PEDF with the secretory apparatus.32 The p.Gln378X mutation therefore most likely represents a functional null allele. This interpretation is supported by the observation that the phenotypic consequences of the p.Gln378X mutation are indistinguishable from those of the p.Tyr232X and p.Tyr109SerfsX5 mutations (see below).
The four affected children with SERPINF1 mutations (referred to as patients 1–4) exhibited similar clinical symptoms and disease severity, currently classified as OI type III. There were no intrauterine fractures reported, and birth length and weight were normal. Dentinogenesis imperfecta was not present. Formal hearing tests were not performed, but there were no clinical signs of hearing impairment. The sclerae were grayish. No ophthalmological problems were seen. The serum levels showed moderately elevated levels for alkaline phosphatase and procollagen-1-C-peptide and moderately increased values for the osteoclast marker deoxypyridinoline (Table 1). Compared to other OI patients, these biochemical findings are not uncommon and can also be found in patients with COL1A1 or COL1A2 mutations. All four patients had fractures of long bones and severe vertebral compression fractures, and resulting deformities were seen as early as in their first year of life. Some of the fractured bones required surgical corrections of deformities and insertions of telescopic rods. Figures 3 and 4 show a selection of radiographs of patients 3 and 4. The radiological findings were typical for patients with severe forms of OI. There were no distinctive radiological abnormalities that would allow a further diagnostic subclassification. In Figure S5, we present whole-body DXA scans taken of patients 2–4 during their last bone-mineral-density measurement. These DXA scans give an impression of the degree of skeletal deformity.
Table 1.
Clinical Features of Patients with Recessive OI and Mutations in SERPINF1
| Findings | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| OI typea | III | III | III | III |
| Age at first visit (years) | 1 8/12 | 11 4/12 | 6/12 | 1 1/12 |
| Age at last visit (years) | 3 2/12 | 16 3/12 | 8 7/12 | 5 11/12 |
| Age at start of bisphosphonate treatment | 1 8/12 | 11 4/12 | 7/12 | 1 1/12 |
| Birth length and birth weight | normal | normal | normal | normal |
| Confirmed prenatal fractures | no | no | no | no |
| Age at first fracture (months) | 4 | unknown | 6 | 6 |
| Color of sclera | grayish | grayish | grayish | grayish |
| Dentinogenesis imperfecta | no | no | no | no |
| Hypermobility of joints | no | no | no | no |
| Hearing impairment | no | no | no | no |
| Old fractures of extremitiesb | yes | yes | yes | yes |
| Vertebral fracturesb | multiple | multiple | multiple | multiple |
| Bowing of upper extremitiesb | moderate | severe | mild | moderate |
| Bowing of lower extremitiesb | severe | severe | moderate | moderate |
| Shortening of upper extremitiesb | moderate | severe | mild | mild |
| Shortening of lower extremitiesb | moderate | severe | moderate | mild |
| Weight at first visit kg/BMI (SD) | 9/−0.5 | 40/+3.2 | 7.5/−1.4 | 11.9/+1.8 |
| Weight at last visit kg/BMI (SD) | 9.9/1.1 | 45/+3.5 | 17/−1.4 | 14/−1.5 |
| Length at first visit cm/(SD) | not done | 98/−8.8 | 67/0.2 | 78.3/−0.7 |
| Length at last visit cm/(SD) | 76/−5.6 | 101/−10.2 | 110/−3.8 | 101.5/−2.8 |
| Retarded gross motor functions | yes | yes | yes | yes |
| Mobility at first visit (BAMFc) | 2 | 1 | 0 | 4 |
| Mobility at last visit (BAMFc) | 4 | 2 | 6 | 6 |
| Intelligence | normal | normal | normal | normal |
| Calcium levelb | normal | normal | normal | normal |
| Alkaline phosphatase at first visit [U/l] | 353 | 283 | 295 | 348 |
| Alkaline phosphatase at last visit [U/l] | 328 | 154 | 269 | 344 |
| Procollagen-1-C-peptide (marker for osteoblastic activity) [μg/l]b | 720 | 430 | 200 | 375 |
| Deoxypyridinoline/creatinine (marker for osteoclastic activity) [nM/mM] at first visit | not done | 86.5 | 118.9 | not done |
| Deoxypyridinoline/creatinine (marker for osteoclastic activity) [nM/mM] at last visit | 64.9 | 60.1 | 63.1 | 65.3 |
| First available bone density, DXA whole-body measurement (Z score) | −6.1 (lumbar spine) | −3.7 | −2.7 | −2.0 |
Figure 3.

Radiological Features of Patient 3
(A and B) Thorax and pelvis of patient 3 at the age of 6 months showing no signs of rip or clavicular fractures, only a slightly decreased vertebral height, a small pelvis and moderately delayed development of the hips.
(C and D) Radiographs of the right and left leg when the patient was 1.6 years old show severe bowing of both femurs, multiple old and new fractures, and a pseudarthrosis in the right femur. In the epiphysis, lines resulting from intravenous bisphosphonate treatment are visible. The tibiae display no severe deformities.
Figure 4.
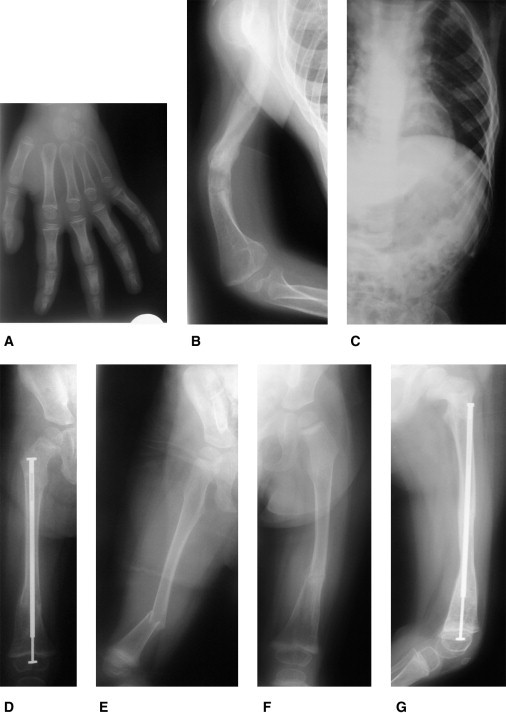
Radiological Features of Patient 4
(A) Hand radiogram at the age of 6 years shows no signs of fractures or skeletal dysplasia and only a mild delay of bone age.
(B) The right arm at the age of 5.4 years shows severe deformities of the humerus with multiple old fractures and callus formation in the diaphysis.
(C) Left hemithorax at the age of 4.3 years displays no deformities or fractures of rips.
(D) Right leg at the age of 2.9 years after surgical rodding with an intramedullar telescopic rod because of a fracture.
(E) Fractured right leg 3 months earlier. Also visible are the epiphyseal lines resulting from cyclic intravenous bisphosphonate treatment.
(F) Left leg with a fracture after minimal trauma at the age of 2.2 years.
(G) Left leg a few weeks later after surgical treatment with an intramedullar telescopic rod.
In all patients, bone mineral density was severely reduced. All four patients developed short stature during early childhood and were wheelchair bound. Three patients had received early medical treatment and physiotherapeutic training and were able to stand with assistance or to walk a few steps with assistive devices. The fourth patient, who was 11 years old at first presentation and severely adipose with a BMI of 44.1 kg/m2, was completely immobile and dependent on external support for all activities of daily life. The clinical findings are summarized in Table 1. There were no hints of an involvement of other organ systems, and abdominal ultrasound scans gave normal results. All patients had received a cyclic intravenous bisphosphonate treatment with the compound neridronate prior to this study, according to the Italian regime for children with OI.33 Upon treatment, there was vertebral reshaping and an increase in vertebral height, an observation that has been reported previously by several OI investigators.1 As an example of treatment outcome, the changes of vertebral size during treatment of patient 3 are presented in Figure S6. This assumed therapeutic effect was similar in all four patients. The parents of the patients, all heterozygous carriers of the disease-causing mutations, were healthy and had no overt signs of reduced bone mineral density and no history of previous fractures. DXA measurements have not been performed in the parents.
To analyze the effect of PEDF-truncating mutations on posttranslational modification of collagens, we performed a standard biochemical analysis of type I, III, and V collagens extracted from cultured dermal fibroblasts of patient 3.34 SDS-PAGE showed normal migration of collagens (Figure S7), which suggests that altered posttranslational modification of collagens is unlikely to play a role in the disease pathogenesis. Abnormal type I collagen migration in SDS-PAGE analyses can be observed in autosomal-recessive OI caused by CRTAP, LEPRE1, and PPIB mutations, as well as in most patients with autosomal-dominant OI resulting from COL1A1 or COL1A2 mutations.2,9,10,13,14 In order to investigate whether the secretion of triple helical collagen type I was impaired, pulse-chase experiments were performed. As shown in Figure S8, collagen secretion in fibroblasts of patient 3 was normal. However, subtle impairments of type I collagen secretion might remain unrecognized with this experimental design and therefore cannot be excluded. Delayed collagen secretion has been observed in fibroblasts from OI patients with LEPRE1, SERPINH1, and FKBP10 mutations, and it is also a frequent effect of COL1A1 or COL1A2 glycine substitutions impairing triple helix formation.10,16,17,35
The human genome encodes 36 serpin proteins, most of which are protease inhibitors.36 On the basis of the sequence similarities of the proteins and exon-intron organization of the genes, serpins have been organized into 16 clades. PEDF falls into the F1 clade. HSP47, which has been associated with autosomal-recessive OI in humans and in dachshunds, is categorized as clade H1 (and thus encoded by SERPINH1).17,18 The closest homologs of PEDF are alpha-2 antiplasmin (encoded by SERPINF2 [MIM 613168]) and complement component 1 inhibitor (encoded by SERPING1[ MIM 606860]).37 Unlike these two proteins, PEDF and HSP47 are not thought to exhibit protease inhibitory activity.38,39 PEDF contains two conserved regions with significant homology to other serpins.31 One of these regions is the C terminus (amino acids 384–415). Among 16 human serpins with strong sequence similarities to the PEDF C terminus, HSP47 displays the greatest homology (47% sequence identity and 88% sequence homology).31
PEDF was initially isolated from conditioned medium of fetal human retinal pigment epithelium cells and characterized as a factor that can induce the differentiation of retinoblastoma cells into cells with several characteristics of normal neurons.40–42 With the discovery that the protein is one of the strongest inhibitors of angiogenesis in humans known to date, utilizing this property for therapeutic applications became a main focus of PEDF research.43,44 PEDF is thought to counteract the effect of vascular endothelial growth factor (VEGF).45,46 VEGF-A is the molecular target for the neutralizing humanized antibody bevacizumab (Avastatin, FDA-approved for the treatment of certain types of malignant tumors) and its fragment ranibizumab (Lucentis, a FDA-approved medication for wet macular degeneration).47 Consequently, the first clinical FDA trials with PEDF are dedicated to the treatment of the wet form of age-related macular dengeneration as an ocular disease resulting from choroidal neovascularization. These clinical trials with intravitreal administration of virally delivered PEDF are currently in Phase II.46,48,49 In a mouse model of ischemia-induced retinal angiogenesis, systemically administered purified PEDF was able to completely eliminate aberrant neovascularization.50
There is a growing list of other cellular functions of PEDF, including immunomodulation, protection against oxidative stress, and expansion of the neural stem cell niche.36,51 The underlying molecular mechanisms have only been partially elucidated. PEDF is expressed in a wide range of fetal and adult tissues, including bone marrow, lung, brain, liver, and eye.52 An intracellular function, for example as a chaperone, has not been reported so far. As a secreted protein, PEDF accumulates in the extracellular matrix and binds, among others, to collagen type I.53,54
It is somewhat unexpected that loss of PEDF leads to severe OI in humans because the murine knockout model has no overt bone phenotype but exhibits vascular and epithelial abnormalities. Serpinf1−/− mice lacking exons 3–6 of the gene are viable and fertile and display increased microvessel density in the retina, kidney, pancreas, and prostate. On gross examination, the pancreas appeared enlarged. At 3 months of age, the mice developed prostatic hyperplasia.55 However, impaired organ functions were not reported, as is also the case in our OI patients with SERPINF1 mutations. Clarifying whether the OI phenotype is indeed restricted to humans with loss of PEDF will require detailed analyses of bone homeostasis and bone mineral density in Serpinf1−/− mice. Of note, experimental data demonstrating absence of mRNA and protein expression have not been provided with the original publication.55 Therefore, it remains possible that, e.g., splicing between Serpinf1 exon 2 and exon 7 is able to produce a form of the protein with a residual function that is essential to bone.
Expression analyses performed with bone tissue from wild-type mice as well as recent in vitro experiments with murine cell systems strongly support a function of PEDF in bone formation and remodeling. PEDF is expressed in osteoblasts, chondrocytes, and,to a lesser extent, osteoclasts. The protein was detected in areas of active bone remodeling. In growing mouse bone, strong positive immunostaining for PEDF was observed predominantly in the bone matrix of the trabeculae of the primary spongiosa and the periosteum; adjacent osteoblasts displayed only faint staining.56,57 PEDF is also expressed in avascular layers of the epiphyseal growth plate, as opposed to the expression pattern of VEGF, which is found in vascularized layers.56,58 Cell-culture experiments provided evidence that PEDF inhibits osteoclast differentiation and hence bone resorption via osteoprotegerin (OPG) and RANKL (receptor activator of NF-kB ligand) in a dose-dependent manner.59 This inhibitory effect was confirmed in vivo with a mouse model system for inflammatory bone destruction. Receptor activator of NF-kB (RANK), its ligand RANKL, and the decoy receptor OPG are central regulators of osteoclast development and function.
In light of these data and our collagen studies with patient fibroblasts, it is intriguing to speculate that the OI phenotype observed in patients with SERPINF1 mutations is the result of a pathomechanism that is primarily independent of alterations in type I collagen synthesis or intracellular processing. The published immunostaining pattern is consistent with a scenario in which PEDF is rapidly secreted by osteoblasts and subsequently binds to the newly laid type I collagen in the extracellular matrix.56 The balance between PEDF and VEGF expression in regions of active bone formation is thought to regulate neovascularisation at these sites, a process that is necessary for the provision of osteoblast and osteoclast precursor cells.45,57 Loss of PEDF could disturb this balance. As a result, there could be an excess of osteoclast precursor cells in areas of active bone remodeling and at the epiphyseal plate. In the absence of PEDF secretion by osteoblasts, osteoclast precursor cells could differentiate into osteoclasts without restraints, leading to excessive bone resorption. Dissecting the pathomechanism of this OI form will require further studies.
In conclusion, we report the identification of pathogenic germline mutations in human SERPINF1. The fact that four patients who have the uniform diagnosis of OI type III and descend from consanguineous families from two populations are homozygous for three different truncating SERPINF1 mutations establishes loss of PEDF as the molecular cause of the observed phenotype. The power of exome sequencing in combination with homozygosity analysis as a strategy for unraveling the cause of autosomal-recessive disorders in consanguineous families is highlighted by the fact that finding the mutated gene required only a single affected individual. Because we identified mutations in two out of four additional COL1A1- and COL1A2-negative families, there might be a substantial number of other autosomal-recessively inherited OI cases caused by SERPINF1 mutations. The data presented here provide in vivo evidence for an involvement of PEDF in human bone homeostasis. This well-characterized protein could be a promising candidate for the development of a drug for the treatment of OI patients with inborn loss of PEDF and possibly also for the treatment of other low bone-mass phenotypes. Deciphering the molecular pathway that leads to reduced bone mineral density in patients with SERPINF1 mutations might unveil additional targets for therapeutic interventions in this severe form of OI.
Acknowledgments
We are grateful to our patients and their families for participating in this study. We would like to thank Angelika Schwarze for support in collagen biochemical analysis, Angelika Stabrey for preparing Figure 3, Figure 4, and Figure S5, Bärbel Tutlewski for performing the DXA scans, and Karin Boss for critically reading the manuscript. C.N. was supported by the “Köln Fortune” program of the Cologne Medical Faculty. This work was partially supported by the German Federal Ministry of Education and Research (BMBF) by grant number 01GM0880 (SKELNET) to B.W. The next-generation sequencing platforms and bioinformatics pipeline have been funded in part by the Netherlands Organization for Health Research and Development (ZonMW grants 917-66-36 and 911-08-025 to J.V). Part of this work was funded by the EU-project TECHGENE (grant Health-F5-2009-223143 to J.A.V.) and by the AnEUploidy project (grant LSHG-CT-2006-37627 to A.H. and J.A.V.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
dbSNP, build 130, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi?build_id=130
IGV browser, http://www.broadinstitute.org/igv
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
University of California-Santa Cruz Genome Bioinformatics, http://www.genome.ucsc.edu
References
- 1.Rauch F., Glorieux F.H. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. doi: 10.1016/S0140-6736(04)16051-0. [DOI] [PubMed] [Google Scholar]
- 2.Basel D., Steiner R.D. Osteogenesis imperfecta: Recent findings shed new light on this once well-understood condition. Genet. Med. 2009;11:375–385. doi: 10.1097/GIM.0b013e3181a1ff7b. [DOI] [PubMed] [Google Scholar]
- 3.Barsh G.S., Byers P.H. Reduced secretion of structurally abnormal type I procollagen in a form of osteogenesis imperfecta. Proc. Natl. Acad. Sci. USA. 1981;78:5142–5146. doi: 10.1073/pnas.78.8.5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byers P.H., Tsipouras P., Bonadio J.F., Starman B.J., Schwartz R.C. Perinatal lethal osteogenesis imperfecta (OI type II): A biochemically heterogeneous disorder usually due to new mutations in the genes for type I collagen. Am. J. Hum. Genet. 1988;42:237–248. [PMC free article] [PubMed] [Google Scholar]
- 5.Chu M.L., Williams C.J., Pepe G., Hirsch J.L., Prockop D.J., Ramirez F. Internal deletion in a collagen gene in a perinatal lethal form of osteogenesis imperfecta. Nature. 1983;304:78–80. doi: 10.1038/304078a0. [DOI] [PubMed] [Google Scholar]
- 6.Williams C.J., Prockop D.J. Synthesis and processing of a type I procollagen containing shortened pro-alpha 1(I) chains by fibroblasts from a patient with osteogenesis imperfecta. J. Biol. Chem. 1983;258:5915–5921. [PubMed] [Google Scholar]
- 7.Baldridge D., Schwarze U., Morello R., Lennington J., Bertin T.K., Pace J.M., Pepin M.G., Weis M., Eyre D.R., Walsh J. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 2008;29:1435–1442. doi: 10.1002/humu.20799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnes A.M., Carter E.M., Cabral W.A., Weis M., Chang W., Makareeva E., Leikin S., Rotimi C.N., Eyre D.R., Raggio C.L., Marini J.C. Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N. Engl. J. Med. 2010;362:521–528. doi: 10.1056/NEJMoa0907705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes A.M., Chang W., Morello R., Cabral W.A., Weis M., Eyre D.R., Leikin S., Makareeva E., Kuznetsova N., Uveges T.E. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N. Engl. J. Med. 2006;355:2757–2764. doi: 10.1056/NEJMoa063804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cabral W.A., Chang W., Barnes A.M., Weis M., Scott M.A., Leikin S., Makareeva E., Kuznetsova N.V., Rosenbaum K.N., Tifft C.J. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007;39:359–365. doi: 10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang W., Barnes A.M., Cabral W.A., Bodurtha J.N., Marini J.C. Prolyl 3-hydroxylase 1 and CRTAP are mutually stabilizing in the endoplasmic reticulum collagen prolyl 3-hydroxylation complex. Hum. Mol. Genet. 2010;19:223–234. doi: 10.1093/hmg/ddp481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marini J.C., Cabral W.A., Barnes A.M. Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res. 2010;339:59–70. doi: 10.1007/s00441-009-0872-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 14.van Dijk F.S., Nesbitt I.M., Zwikstra E.H., Nikkels P.G., Piersma S.R., Fratantoni S.A., Jimenez C.R., Huizer M., Morsman A.C., Cobben J.M. PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 2009;85:521–527. doi: 10.1016/j.ajhg.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishikawa Y., Wirz J., Vranka J.A., Nagata K., Bächinger H.P. Biochemical characterization of the prolyl 3-hydroxylase 1.cartilage-associated protein.cyclophilin B complex. J. Biol. Chem. 2009;284:17641–17647. doi: 10.1074/jbc.M109.007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alanay Y., Avaygan H., Camacho N., Utine G.E., Boduroglu K., Aktas D., Alikasifoglu M., Tuncbilek E., Orhan D., Bakar F.T. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christiansen H.E., Schwarze U., Pyott S.M., AlSwaid A., Al Balwi M., Alrasheed S., Pepin M.G., Weis M.A., Eyre D.R., Byers P.H. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:389–398. doi: 10.1016/j.ajhg.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drögemüller C., Becker D., Brunner A., Haase B., Kircher P., Seeliger F., Fehr M., Baumann U., Lindblad-Toh K., Leeb T. A missense mutation in the SERPINH1 gene in Dachshunds with osteogenesis imperfecta. PLoS Genet. 2009;5:e1000579. doi: 10.1371/journal.pgen.1000579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tasab M., Batten M.R., Bulleid N.J. Hsp47: A molecular chaperone that interacts with and stabilizes correctly-folded procollagen. EMBO J. 2000;19:2204–2211. doi: 10.1093/emboj/19.10.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishikawa Y., Vranka J., Wirz J., Nagata K., Bächinger H.P. The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J. Biol. Chem. 2008;283:31584–31590. doi: 10.1074/jbc.M802535200. [DOI] [PubMed] [Google Scholar]
- 21.Lapunzina P., Aglan M., Temtamy S., Caparrós-Martín J.A., Valencia M., Letón R., Martínez-Glez V., Elhossini R., Amr K., Vilaboa N., Ruiz-Perez V.L. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;87:110–114. doi: 10.1016/j.ajhg.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakashima K., Zhou X., Kunkel G., Zhang Z., Deng J.M., Behringer R.R., de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 23.Sillence D.O., Senn A., Danks D.M. Genetic heterogeneity in osteogenesis imperfecta. J. Med. Genet. 1979;16:101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fisher J.E., Rogers M.J., Halasy J.M., Luckman S.P., Hughes D.E., Masarachia P.J., Wesolowski G., Russell R.G., Rodan G.A., Reszka A.A. Alendronate mechanism of action: Geranylgeraniol, an intermediate in the mevalonate pathway, prevents inhibition of osteoclast formation, bone resorption, and kinase activation in vitro. Proc. Natl. Acad. Sci. USA. 1999;96:133–138. doi: 10.1073/pnas.96.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marini J.C. Bone: Use of bisphosphonates in children-proceed with caution. Nat Rev Endocrinol. 2009;5:241–243. doi: 10.1038/nrendo.2009.58. [DOI] [PubMed] [Google Scholar]
- 26.Phillipi C.A., Remmington T., Steiner R.D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst. Rev. 2008;4:CD005088. doi: 10.1002/14651858.CD005088.pub2. [DOI] [PubMed] [Google Scholar]
- 27.Gilissen C., Arts H.H., Hoischen A., Spruijt L., Mans D.A., Arts P., van Lier B., Steehouwer M., van Reeuwijk J., Kant S.G. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 2010;87:418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoischen A., van Bon B.W., Gilissen C., Arts P., van Lier B., Steehouwer M., de Vries P., de Reuver R., Wieskamp N., Mortier G. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 29.Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 30.Nagy E., Maquat L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998;23:198–199. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- 31.Tombran-Tink J., Aparicio S., Xu X., Tink A.R., Lara N., Sawant S., Barnstable C.J., Zhang S.S. PEDF and the serpins: Phylogeny, sequence conservation, and functional domains. J. Struct. Biol. 2005;151:130–150. doi: 10.1016/j.jsb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Shao H., Schvartz I., Shaltiel S. Secretion of pigment epithelium-derived factor. Mutagenic study. Eur. J. Biochem. 2003;270:822–831. doi: 10.1046/j.1432-1033.2003.03374.x. [DOI] [PubMed] [Google Scholar]
- 33.Gatti D., Antoniazzi F., Prizzi R., Braga V., Rossini M., Tatò L., Viapiana O., Adami S. Intravenous neridronate in children with osteogenesis imperfecta: A randomized controlled study. J. Bone Miner. Res. 2005;20:758–763. doi: 10.1359/JBMR.041232. [DOI] [PubMed] [Google Scholar]
- 34.Steinmann B., Rao V.H., Vogel A., Bruckner P., Gitzelmann R., Byers P.H. Cysteine in the triple-helical domain of one allelic product of the alpha 1(I) gene of type I collagen produces a lethal form of osteogenesis imperfecta. J. Biol. Chem. 1984;259:11129–11138. [PubMed] [Google Scholar]
- 35.Raghunath M., Bruckner P., Steinmann B. Delayed triple helix formation of mutant collagen from patients with osteogenesis imperfecta. J. Mol. Biol. 1994;236:940–949. doi: 10.1006/jmbi.1994.1199. [DOI] [PubMed] [Google Scholar]
- 36.Filleur S., Nelius T., de Riese W., Kennedy R.C. Characterization of PEDF: A multi-functional serpin family protein. J. Cell. Biochem. 2009;106:769–775. doi: 10.1002/jcb.22072. [DOI] [PubMed] [Google Scholar]
- 37.Xu X., Zhang S.S., Barnstable C.J., Tombran-Tink J. Molecular phylogeny of the antiangiogenic and neurotrophic serpin, pigment epithelium derived factor in vertebrates. BMC Genomics. 2006;7:248. doi: 10.1186/1471-2164-7-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Becerra S.P., Sagasti A., Spinella P., Notario V. Pigment epithelium-derived factor behaves like a noninhibitory serpin. Neurotrophic activity does not require the serpin reactive loop. J. Biol. Chem. 1995;270:25992–25999. doi: 10.1074/jbc.270.43.25992. [DOI] [PubMed] [Google Scholar]
- 39.Law R.H., Zhang Q., McGowan S., Buckle A.M., Silverman G.A., Wong W., Rosado C.J., Langendorf C.G., Pike R.N., Bird P.I., Whisstock J.C. An overview of the serpin superfamily. Genome Biol. 2006;7:216. doi: 10.1186/gb-2006-7-5-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tombran-Tink J., Chader G.G., Johnson L.V. PEDF: A pigment epithelium-derived factor with potent neuronal differentiative activity. Exp. Eye Res. 1991;53:411–414. doi: 10.1016/0014-4835(91)90248-d. [DOI] [PubMed] [Google Scholar]
- 41.Steele F.R., Chader G.J., Johnson L.V., Tombran-Tink J. Pigment epithelium-derived factor: Neurotrophic activity and identification as a member of the serine protease inhibitor gene family. Proc. Natl. Acad. Sci. USA. 1993;90:1526–1530. doi: 10.1073/pnas.90.4.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bouck N. PEDF: Anti-angiogenic guardian of ocular function. Trends Mol. Med. 2002;8:330–334. doi: 10.1016/s1471-4914(02)02362-6. [DOI] [PubMed] [Google Scholar]
- 43.Dawson D.W., Volpert O.V., Gillis P., Crawford S.E., Xu H., Benedict W., Bouck N.P. Pigment epithelium-derived factor: A potent inhibitor of angiogenesis. Science. 1999;285:245–248. doi: 10.1126/science.285.5425.245. [DOI] [PubMed] [Google Scholar]
- 44.Tombran-Tink J., Barnstable C.J. Therapeutic prospects for PEDF: More than a promising angiogenesis inhibitor. Trends Mol. Med. 2003;9:244–250. doi: 10.1016/s1471-4914(03)00074-1. [DOI] [PubMed] [Google Scholar]
- 45.Broadhead M.L., Akiyama T., Choong P.F., Dass C.R. The pathophysiological role of PEDF in bone diseases. Curr. Mol. Med. 2010;10:296–301. doi: 10.2174/156652410791065345. [DOI] [PubMed] [Google Scholar]
- 46.Tombran-Tink J. PEDF in angiogenic eye diseases. Curr. Mol. Med. 2010;10:267–278. doi: 10.2174/156652410791065336. [DOI] [PubMed] [Google Scholar]
- 47.Ferrara N., Kerbel R.S. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 48.Rasmussen H., Chu K.W., Campochiaro P., Gehlbach P.L., Haller J.A., Handa J.T., Nguyen Q.D., Sung J.U. Clinical protocol. An open-label, phase I, single administration, dose-escalation study of ADGVPEDF.11D (ADPEDF) in neovascular age-related macular degeneration (AMD) Hum. Gene Ther. 2001;12:2029–2032. [PubMed] [Google Scholar]
- 49.Campochiaro P.A., Nguyen Q.D., Shah S.M., Klein M.L., Holz E., Frank R.N., Saperstein D.A., Gupta A., Stout J.T., Macko J. Adenoviral vector-delivered pigment epithelium-derived factor for neovascular age-related macular degeneration: Results of a phase I clinical trial. Hum. Gene Ther. 2006;17:167–176. doi: 10.1089/hum.2006.17.167. [DOI] [PubMed] [Google Scholar]
- 50.Stellmach V., Crawford S.E., Zhou W., Bouck N. Prevention of ischemia-induced retinopathy by the natural ocular antiangiogenic agent pigment epithelium-derived factor. Proc. Natl. Acad. Sci. USA. 2001;98:2593–2597. doi: 10.1073/pnas.031252398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramírez-Castillejo C., Sánchez-Sánchez F., Andreu-Agulló C., Ferrón S.R., Aroca-Aguilar J.D., Sánchez P., Mira H., Escribano J., Fariñas I. Pigment epithelium-derived factor is a niche signal for neural stem cell renewal. Nat. Neurosci. 2006;9:331–339. doi: 10.1038/nn1657. [DOI] [PubMed] [Google Scholar]
- 52.Tombran-Tink J., Mazuruk K., Rodriguez I.R., Chung D., Linker T., Englander E., Chader G.J. Organization, evolutionary conservation, expression and unusual Alu density of the human gene for pigment epithelium-derived factor, a unique neurotrophic serpin. Mol. Vis. 1996;2:11. [PubMed] [Google Scholar]
- 53.Kozaki K., Miyaishi O., Koiwai O., Yasui Y., Kashiwai A., Nishikawa Y., Shimizu S., Saga S. Isolation, purification, and characterization of a collagen-associated serpin, caspin, produced by murine colon adenocarcinoma cells. J. Biol. Chem. 1998;273:15125–15130. doi: 10.1074/jbc.273.24.15125. [DOI] [PubMed] [Google Scholar]
- 54.Meyer C., Notari L., Becerra S.P. Mapping the type I collagen-binding site on pigment epithelium-derived factor. Implications for its antiangiogenic activity. J. Biol. Chem. 2002;277:45400–45407. doi: 10.1074/jbc.M208339200. [DOI] [PubMed] [Google Scholar]
- 55.Doll J.A., Stellmach V.M., Bouck N.P., Bergh A.R., Lee C., Abramson L.P., Cornwell M.L., Pins M.R., Borensztajn J., Crawford S.E. Pigment epithelium-derived factor regulates the vasculature and mass of the prostate and pancreas. Nat. Med. 2003;9:774–780. doi: 10.1038/nm870. [DOI] [PubMed] [Google Scholar]
- 56.Quan G.M., Ojaimi J., Li Y., Kartsogiannis V., Zhou H., Choong P.F. Localization of pigment epithelium-derived factor in growing mouse bone. Calcif. Tissue Int. 2005;76:146–153. doi: 10.1007/s00223-004-0068-2. [DOI] [PubMed] [Google Scholar]
- 57.Tombran-Tink J., Barnstable C.J. Osteoblasts and osteoclasts express PEDF, VEGF-A isoforms, and VEGF receptors: Possible mediators of angiogenesis and matrix remodeling in the bone. Biochem. Biophys. Res. Commun. 2004;316:573–579. doi: 10.1016/j.bbrc.2004.02.076. [DOI] [PubMed] [Google Scholar]
- 58.Gerber H.P., Vu T.H., Ryan A.M., Kowalski J., Werb Z., Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 59.Akiyama T., Dass C.R., Shinoda Y., Kawano H., Tanaka S., Choong P.F. PEDF regulates osteoclasts via osteoprotegerin and RANKL. Biochem. Biophys. Res. Commun. 2010;391:789–794. doi: 10.1016/j.bbrc.2009.11.139. [DOI] [PubMed] [Google Scholar]
- 60.Cintas H.L., Siegel K.L., Furst G.P., Gerber L.H. Brief assessment of motor function: Reliability and concurrent validity of the Gross Motor Scale. Am. J. Phys. Med. Rehabil. 2003;82:33–41. doi: 10.1097/00002060-200301000-00006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.