

Abstract
Aneuploidy is associated with poor prognosis in solid tumours. Spontaneous chromosome mis-segregation events in aneuploid cells promote Chromosomal Instability (CIN) that may contribute to the acquisition of multi-drug resistance in vitro and heighten risk for tumour relapse in animal models. Identification of distinct therapeutic agents that target tumour karyotypic complexity has important clinical implications. In order to identify distinct therapeutic approaches to specifically limit the growth of CIN tumours we focussed on a panel of colorectal cancer (CRC) cell lines, previously classified as either chromosomally-unstable (CIN+) or diploid/near-diploid (CIN−), and treated them individually with a library of kinase inhibitors targeting components of signal transduction, cell cycle and trans-membrane receptor signalling pathways. CIN+ cell lines displayed significant intrinsic multi-drug resistance compared to CIN− cancer cell lines and this appeared to be independent of somatic mutation status and proliferation rate. Confirming the association of CIN rather than ploidy status with multi-drug resistance, tetraploid isogenic cells that had arisen from diploid cell lines displayed lower drug sensitivity than their diploid parental cells only with increasing chromosomal heterogeneity, and isogenic cell line models of CIN+ displayed multi-drug resistance relative to their CIN− parental cancer cell line derivatives. In a meta-analysis of CRC outcome following cytotoxic treatment, CIN+ predicted worse progression-free or disease-free survival relative to patients with CIN− disease. Our results suggest that stratifying tumour responses according to CIN status should be considered within the context of clinical trials to minimize the confounding effects of tumour CIN status on drug sensitivity.
Keywords: Chromosomal Instability, Drug Resistance, Colorectal Cancer, Tumour Heterogeneity, SNP Array
Introduction
Colorectal cancer (CRC) is associated with at least two distinct patterns of genomic instability (1). The more common form of genomic instability in CRC is chromosomal instability (CIN), resulting in ongoing numerical and structural chromosomal aberrations in cancer cells, leading to intra-tumour heterogeneity (2, 3). The less common pattern is microsatellite instability (MIN) caused by a deficiency in the mismatch repair apparatus. Due to the accumulation of replication errors, MIN results in length variation of microsatellite sequences in DNA. The majority of MIN+ CRC cell lines are near-diploid (4, 5) and chromosomally stable (CIN−). In contrast, CIN+ CRC cell lines are aneuploid and display a higher frequency of chromosomal missegregation errors during each mitosis relative to diploid cells (2). In human CRC, CIN+ is widely inferred through the measurement of tumour DNA ploidy (6); normal diploid cells are defined with a DNA index of 1.0 (7) and thus an increase in DNA index infers polyploidy or aneuploidy. Approximately 25% of human CRC are both CIN− and MIN− (6).
Consistent molecular mechanisms responsible for the CIN+ phenotype, and hence means to target this pattern of genome instability in colorectal and other solid tumours remain poorly defined. Putative mechanisms that may contribute to CIN include weakening of the spindle assembly checkpoint (SAC) (8, 9), defective sister chromatid cohesion (10), merotelic sister chromatid attachments (11), defective cytokinesis (12), centrosome amplification (13) and chromosome breakage-fusion-bridge cycles (14).
Increasing evidence suggests that CIN is associated with poor prognosis in solid tumours (6, 15, 16). It has been suggested that adverse outcome associated with CIN may be related to increased tumour cell heterogeneity driving the ability of tumours to adapt to environmental stresses (17-19). Consistent with a hypothesis whereby CIN may enhance tumour adaptation, transient initiation of CIN, following the brief induction of MAD2 expression in activated-KRAS initiated mouse lung tumour models, is associated with a high frequency of tumour recurrence following withdrawal of the KRAS oncogenic stimulus (20). Pre-clinical studies have demonstrated that CIN is associated with the rapid acquisition of multi-drug resistance in cell line systems (21) and intrinsic taxane resistance in vitro and in vivo (22). Recently, we and others have proposed the existence of a CIN− survival phenotype that allows CIN+ tumour cells to tolerate the impact of excessive chromosome gains and losses (22-24) that may in turn impact upon altered drug sensitivity.
Determining how CIN might impact upon prognosis and how this pattern of genomic instability might be specifically targeted remains an important research area (23, 25). Evidence in lower eukaryotes has demonstrated that aneuploid S. cerevisiae are dependent on increased glucose utilisation and are more sensitive to both hsp90 and proteosome inhibitors (26). Polyploid S. cerevisiae are dependent upon increased expression of genes involved in sister chromatid cohesion and mitotic spindle function (27). Roschke and colleagues have demonstrated the existence of anticancer compounds that may specifically target karyotypically complex cancer cells (25). These observations indicate that karyotypic instability may be specifically targeted in eukaryotic organisms and suggest that CIN might be an exploitable and targetable phenotype in cancer.
In order to identify distinct therapeutic approaches to limit the growth of CIN+ tumours relative to diploid cells, we focussed on a panel of CRC cell lines that had previously been classified as CIN+ or CIN− and used kinase inhibitor and cytotoxic libraries to identify agents that might be preferentially lethal towards CIN+ cells. Both isogenic and non-isogenic CIN+ cell lines displayed intrinsic multi-drug resistance in vitro relative to CIN− cell lines. Importantly, consistent with the proposal that CIN+ is associated with intrinsic multi-drug resistance, in a meta-analysis of patient outcome in CRC, CIN+ was associated with significantly worse clinical outcome relative to diploid cancers in both early and late stage disease following cytotoxic therapy.
Materials and Methods
Cell lines and FISH Analysis
27 CRC cell lines (Table 1, Supplementary Table 1) previously characterised for numerical/structural CIN, MIN status (2, 28-30) and subject to Affymetrix SNP 6.0 Array analysis where available (20 out of 27 cell lines) (Wellcome Trust Sanger Institute) were used.
Table 1.
Cell lines used in this study
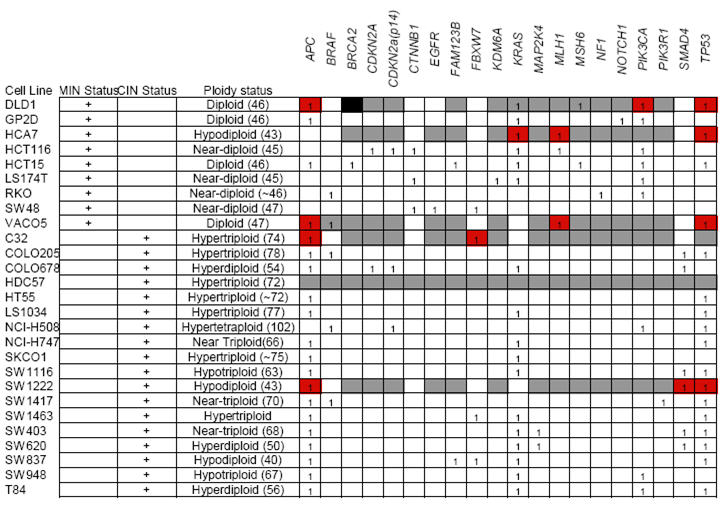
Black indicates presence of mutation
White indicates absence of mutation
Grey indicates mutation status not known
We used publicly available somatic mutation data from the Sanger Institute Cancer Cell Line Project (CLP) and COSMIC database (31). 15 CIN+ and 6 CIN− cell lines used in our analysis were present within the CLP database and a total of 20 out of the 61 genes resequenced in the project were found to have somatic mutations in at least 1 of those 21 cell lines. Additional information regarding the somatic mutation status of APC, CTNNB1, KRAS, MLH1, PIK3CA and TP53 were obtained from both published (32-35) and internal laboratory data. Isogenic HCT116 MAD2+/− cell lines (9) and HCT116 PTTG1−/− cell lines (10) were donated courtesy of Drs Benezra and Vogelstein respectively. To generate tetraploid HCT116 cells, naturally occurring tetraploid cells were isolated from the parental cell line and single cell sorted using flow cytometry. Clonal FISH was performed with Centromere Enumeration Probes (CEP) against Centromere 2 and 15.
Calbiochem Kinase Inhibitor Library and 5-FU Screen
Calbiochem Kinase Inhibitor Libraries I and II (EMD Biosciences) containing 160 inhibitors were used. Comprehensive data for these inhibitors including references documenting target inhibition or downstream signalling cascade inactivation can be found on the manufacturer’s website (36).
Cells were plated into 96-well tissue culture microplates at an initial seeding density of 4000 cells/well. After 24 hours, cells were treated with the inhibitors at a final concentration of 10μM/well. This concentration was selected following drug titration test analysis to give an optimal range of mean relative surviving cells following inhibitor treatment across all cell lines. After 72 hours of treatment, cell viability was assayed using the Celltiter-Blue Cell Viability Assay (Promega) according to the manufacturer’s instructions. The fluorescent readout for each inhibitor treated well was normalised to vehicle control wells. For the HCT116 MAD2 isogenic cell lines, a final inhibitor concentration of 1μM/well was used to allow for a larger range of number of surviving cells across all inhibitors.
5-FU was used at both 1μM and 10μM for a treatment length of 72 hours. The 27 CRC cell lines were plated and assayed using the same conditions and methods as above.
Biolog Anti-Cancer Agent Microplate Screen
Biolog Anti-Cancer Agent Microplates M11-M14 consisting of 92 anti-cancer agents at 4 increasing concentrations between 0.1 μM and 25μM per agent were used. Cells were plated at an initial seeding density of 5000 cells/well. After 72 hours, the number of surviving cells per well was assayed using Biolog Redox Dye Mix MA according to manufacturer’s instructions and normalised to the negative control wells.
Cell Proliferation Assay
We measured the proliferation rate of all 9 CIN− and 18 CIN+ CRC cell lines using the IncuCyte Long-term Cell Imaging System. Cell lines were plated in 96-well plates at an initial plating density of 4000 cells/well and phase contrast images were obtained 2-hourly over 70 hours allowing measurements of cell monolayer confluence. Outliers were removed manually and growth curves were fitted by splines with the R package grofit (37) with smoothing parameter smooth.gc = 0.7.
Meta-Analysis of Clinical Studies
Survival data were summarised using a log hazard ratio for comparison between CIN+ and CIN− groups. Data from individual studies were extracted using the methods described by (38) and pooled to generate the summary statistic and confidence intervals using a fixed-effects model with inverse variance weighting. All meta-analyses were performed using Stata 10.1 (Stata Corp, College Station, TX).
Statistical Methods
All tests were performed as two-sided unless otherwise mentioned. To remove outliers, drugs resulting in relative number of cells > 1.4 were eliminated from analysis. A Kolmogorov-Smirnov test was performed to test for overall differences in the distribution of relative cell numbers following inhibitor treatment between CIN+ and CIN− cell lines. Inhibitors that showed a fraction of surviving cells > 0.8 in > 75 % of the cell lines were excluded from the analysis. For comparisons of 2 cell lines, drugs resulting in relative number of cells >0.8 in both cell lines were removed from analysis. A Wilcoxon signed-rank test was used to test for differences between CIN+ and CIN− cells (for each concentration of drug in Biolog microplates). For the Biolog microplates, each concentration of drug was used in duplicate; therefore the replicates which showed the least difference in cell number between the 2 cell lines was used for further analysis.
The maximal slope of the growth curve (mu) for each cell line was used to test if the difference in drug sensitivity between CIN+ and CIN− colorectal cancer cell lines were not solely due to different proliferation rates. CIN+ and CIN− cell lines with mu < 1 were tested with a one-sided Wilcoxon-Mann-Whitney test. Next, we corrected for the influence of the proliferation rates of each cell line. We estimated a linear regression model for all cell lines with mu < 1, where we used mu as independent and the mean fraction of surviving cells over all inhibitors as dependent variable. The residuals of this analysis were tested for significant differences between CIN+ and CIN− with a one-sided Wilcoxon-Mann-Whitney-Test (39). An analysis of covariance model with interactions was applied, where the mean fraction of surviving cells over all inhibitors was used as dependent variable, mu as linear independent and CIN status as factor variable. The interaction term was utilised to test for significant differences in slopes between CIN+ and CIN− cell lines.
To test for significant differences in sensitivity to Thymidylate synthase inhibitors comparing HCT116 diploid parental and PTTG1−/− or MAD2+/− cells, we corrected for the influence of different concentrations by estimating a linear regression model for near linear correlations between concentration and sensitivity or a one-way ANOVA otherwise. In each case the concentration was used as the independent variable and the resulting residuals were tested for differences between CIN+ and CIN− cells with a Wilcoxon-Mann-Whitney test.
All statistical analyses were performed in R and can be found in the supplementary Sweave document.
Results
Classification of CIN+ Cell Lines and Relationship with Ploidy Status and Structural Chromosomal Complexity
We selected 9 CIN− and 18 CIN+ CRC cell lines (Table 1). CIN status for these cell lines had been previously described in terms of numerical and structural chromosomal aberrations (2, 28-30).
In order to confirm the utility of published approaches for defining the CIN status of the cell lines, where SNP Array data was available, we estimated the ploidy status of the cell lines using weighted mean integer copy numbers derived from the PICNIC (Predicting Integral Copy Numbers In Cancer) algorithm (40) (Figure 1A). Ploidy estimates classified cells as CIN+ if they surpassed a threshold of ploidy >2.2 (corresponding to an estimated DNA index of 1.1). Additionally, modal chromosomal number as determined previously using high quality metaphase spreads (28, 29) correlated well with ploidy estimates derived from weighted mean PICNIC copy number analysis using the SNP Array data (Pearson’s CC=0.94, Pearson’s correlation test p<0.0001) (Figure 1B). These data support the utility of SNP Array data for estimating cell line ploidy status and are consistent with ploidy estimates by traditional measures, confirming the CIN status of cell lines used in this analysis.
Figure 1.

A) Barplot of the ploidy status of 20 CRC cell lines determined using mean copy number PICNIC analysis of SNP Array data. Red line indicates ploidy value threshold of 2.2. CIN+ cell lines in black text and CIN− cell lines in red text throughout.
B) Correlation of modal chromosomal numbers and weighted mean copy numbers of the cell lines as determined by PICNIC. Pearson’s CC=0.94, p<0.0001.
C) Correlation of Structural Chromosomal Complexity Score with ploidy as determined by weighted mean copy number PICNIC analysis of SNP Array data. Pearson’s CC=0.746, p=0.0002.
Next we addressed the relationship between CIN status and structural chromosomal complexity using a summary Structural Chromosomal Complexity Score (SCCS) derived from the SNP Array datasets. This SCCS was determined by summarising (i) the number of breakpoints, (ii) loss of heterozygosity (LOH) events as predicted using PICNIC (40) and (iii) the Genome Integrity Index (GII) (41), into a single value for each cell line (Figure 1C). There was a highly significant correlation between ploidy status and the SCCS (Pearson’s CC =0.746, p=0.0002). Taken together, these analyses confirm that cells classified as CIN+ have significantly greater ploidy and structural chromosomal complexity compared to CIN− cells.
CIN+ Status is Associated with Intrinsic Multi-Drug Resistance
Next, we aimed to determine whether a specific kinase inhibitor could be identified to selectively target CIN+ cell lines. We used a small molecule library (Calbiochem Kinase Inhibitor Library I and II) that included 160 inhibitors to treat the 18 CIN+ and 9 CIN− cell lines. Preliminary drug titration experiments revealed that the majority of the CIN+ cell lines were resistant to concentrations up to 1μM (Supplementary Figure 1) and therefore 10μM was selected as the optimal drug concentration for cell growth inhibition across the majority of cell lines in order to attempt to identify drugs that were specifically active in CIN+ cells. No specific inhibitor or inhibitor family was found to be preferentially active in CIN+ cell lines compared to CIN− cell lines. In contrast, CIN+ cancer cell lines were significantly more resistant to the inhibitors (Kolmogorov-Smirnov test, p<0.0001) (Figure 2A-C). Following correction for multiple testing hypotheses, 45 inhibitors were identified which demonstrated significantly greater activity in CIN− cell lines compared to CIN+ cell lines (Supplementary Table 2).
Figure 2.

18 CIN+ and 9 CIN− CRC cell lines were treated with 160 kinase inhibitors at 10μM for 72 hours.
A) A higher fraction of cells survive inhibitor treatment in CIN+ cell lines compared to CIN− cell lines. (p<0.0001).
B) Boxplot of median cell number following inhibitor treatment across all inhibitors for all cell lines. The length of the whiskers was limited by maximal=1.5 times the IQR in this boxplot and throughout.
C) Heatmap showing the relative numbers of surviving cells following inhibitor treatment across the cell lines (Inhibitors that have minimal impact on cell growth defined as a surviving cell fraction of >0.8 in >75% of the cell lines tested have been excluded).
To address whether the variation in drug sensitivity between the CIN+ and CIN− cell lines was attributable to differences in proliferation rate, we calculated the maximum growth rate (maximal slope (mu) of growth curve) for each cell line. We observed that CIN+ and CIN− cell lines show significantly different slopes in regression lines (t-test, p = 0.007) with CIN− cell lines demonstrating a higher proliferation rate compared to the CIN+ cell lines (Wilcoxon-Mann-Whitney test, p = 0.003). These data are consistent with previous studies suggesting that aneuploidy (42) (43) or chromosomal segregation defects (44) (11) have a negative impact on cellular proliferation rate. We observed a significant correlation between increased sensitivity to the inhibitors at higher proliferation rates for CIN+ cell lines (Pearson’s CC = 0.61, p = 0.007). In contrast, no such correlation was observed between proliferation rate and drug sensitivity in CIN− cell lines (Pearson’s CC = 0.24, p = 0.55) (Supplementary Figure 2A). Neither ploidy index nor the SCCS showed a significant correlation with proliferation rate (data not shown).
We next investigated whether CIN+ and CIN− cell lines with similar proliferation rates displayed differential drug sensitivity (cell lines with mu < 1, 22 out of 27 cell lines). CIN+ cell lines remained multi-drug resistant compared to CIN− cell lines within this group (one-sided Wilcoxon-Mann-Whitney test p = 0.013). Next, we used a more conservative approach and corrected for the influence of proliferation rate of each individual cell line within this group. CIN+ cell lines remained significantly more drug resistant compared to CIN− cell lines (one-sided Wilcoxon-Mann-Whitney test, p = 0.049) (Supplementary Figure 2B). These data suggest that at similar growth rates, CIN+ cell lines remain more drug resistant compared to CIN− cell lines, indicating that proliferation rate is unlikely to be the main determinant of drug sensitivity.
Somatic Mutation Status and Drug Sensitivity
Next we addressed whether distinct tumour cell line somatic mutations might be the underlying determinant of drug resistance rather than genomic instability status. We investigated whether the somatic mutation status of 20 genes (Table 1) was associated with either altered sensitivity to inhibitors grouped according to target kinase family (Aurora kinase, AKT, CDK, EGFR, FLT-3, GSK-3, JAK3, JNK, MEK, PDGFR, PI3K and SYK) or to all inhibitors combined. We addressed whether there was an association of drug sensitivity with somatic mutation status in 13 genes for which cell line group sizes had sufficient statistical power to compare drug sensitivity in wild-type compared to mutated cell lines.
PIK3CA mutation was the only somatic mutation significantly associated with altered sensitivity to inhibitors grouped according to target kinase families (PIK3CA mutation associated with increased sensitivity to inhibitors targeting AKT, Aurora kinase, EGFR, PDGFR and PI3K, Wilcoxon-Mann-Whitney-Test, corrected p = 0.003, p = 0.038, p = 0.016, p = 0.038 and p = 0.004 respectively) (Supplementary Figure 3B). We found no evidence for a specific association with either PIK3CA exon 9 or exon 20 mutation status and drug sensitivity (data not shown). Notably, PIK3CA mutations were more likely to occur in CIN− than CIN+ cell lines (p=0.0066, Fisher’s exact test).
No single somatic mutation was associated with altered sensitivity to all inhibitors. Next, data for somatic mutation status and CIN status were pooled. When corrected for multiple testing with Benjamini-Hochberg under the assumption that the tests are either positively correlated or independent (45), CIN+ status was the only parameter significantly associated with multi-drug resistance (corrected p = 0.01) (Supplementary Figure 3B). Taken together, these data suggest that the association of PIK3CA mutation with CIN− status may confound the interpretation of the association of PIK3CA mutation status with sensitivity to specific inhibitors and may simply reflect the intrinsic drug sensitivity of CIN− cells.
Isogenic CIN+ CRC Cell Lines Display Intrinsic Multi-Drug Resistance
In order to support a direct role for the contribution of CIN to the multi-drug resistant phenotype, we assessed whether drug sensitivity was altered in isogenic CRC models of CIN. The HCT116 MAD2+/− cell line has one allele of the SAC gene, MAD2, deleted by homologous recombination, resulting in numerical CIN relative to its diploid parental cell line (9). We treated the isogenic HCT116 MAD2+/− cell and its parental diploid cell line with the kinase inhibitors and assessed the surviving cell line fraction after treatment. Consistent with the non-isogenic cell line data presented previously, the MAD2+/− cell line was found to be more resistant overall to the inhibitors tested compared to the parental diploid cell line (one-sided Wilcoxon signed-rank test, p=0.001) (Figure 3A, B). No single inhibitor appeared to specifically target the HCT116 MAD2 +/− cell line.
Figure 3.

A) Boxplot showing that following treatment with kinase inhibitors, there appeared to be a higher surviving fraction of cells in the HCT116 MAD2+/− cell line compared to its parental diploid cell line (p<0.001) following treatment with each equivalent inhibitor.
B) Heatmap showing the relative numbers of surviving cells following the inhibitor treatments compared to vehicle control across the HCT116 MAD2+/− and parental diploid cell lines (Inhibitors that show a surviving cell fraction of >0.8 in both cell lines have been excluded).
C) Biolog M11-M14 drug microplates were used at 4 increasing concentrations per drug (0.1μM to 25μM) to treat HCT116 MAD2+/− and PTTG1 −/− and their parental diploid cell lines for 72 hours. The boxplot shows difference in relative surviving cell number across all drugs at each of the four concentrations, comparing MAD2+/− and PTTG1 −/− cells to their specific isogenic parental cells. Significant p-values suggest higher resistance in MAD2+/− or PTTG1−/− cells compared to their parental diploid cells
D) Heatmap of surviving fraction of cells compared to negative control in HCT116 MAD2+/−, PTTG1 −/− and their parental diploid cell lines treated with Biolog drug microplates. Drugs resulting in a surviving cellular fraction of >0.8 compared to negative control in both isogenic cell lines were excluded.
In order to further test the association of CIN with intrinsic drug resistance to drugs other than kinase inhibitors, we challenged the HCT116 MAD2+/− and another CIN+ isogenic cell line, HCT116 PTTG1−/−, together with their isogenic parental diploid HCT116 cell lines with the Biolog cytotoxic library containing 92 anti-cancer cytotoxic agents. Consistent with the data from the kinase inhibitors, both the CIN+ MAD2+/− and PTTG1−/− cells were significantly more resistant (one-sided Wilcoxon signed-rank test, p<0.001, except p=0.035 at the lowest concentration of drug for PTTG1 −/−) to a diverse range of anti-cancer agents compared to the parental diploid cell lines (Figure 3C, D).
Importantly both the HCT116 MAD2+/− and parental diploid cell lines continue to display MIN+ (Supplementary Figure 4), indicating that MIN status is unlikely to sufficiently explain the altered drug sensitivity in the CIN+ isogenic models. These data suggest that CIN+ status, initiated by ongoing chromosome mis-segregation events driven by loss of two distinct proteins controlling mitotic fidelity, is the dominant phenotype associated with altered drug sensitivity.
CIN+ Not Tetraploidy is Associated with Drug Resistance
CIN+ CRC cell lines mis-segregate chromosomes at a high rate, in contrast to CIN− CRC cells that have a lower frequency of mitotic errors (1, 2). In addition, CIN− cells fail to tolerate the propagation of CIN when chromosome segregation errors are artificially induced by drug treatment (11), suggesting that sustaining CIN in a cell population may require a specialised survival phenotype. The majority of CIN+ CRC cell lines used in this study are triploid or tetraploid. We therefore considered whether altered ploidy status rather than ongoing CIN might be associated with enhanced drug resistance. Clonal tetraploid and diploid HCT116 cells were treated with the kinase inhibitors. There was no significant difference in the relative number of surviving cells following drug treatment between HCT116 Tetraploid Clone 4 (TC4) cell line and Diploid Clone 8 (DC8) cell line. However, Tetraploid Clone 9 (TC9) was significantly more resistant compared to the DC8 cell line (Wilcoxon signed-rank test, p<0.001) (Figure 4A). Further investigation by clonal FISH (Figure 4B) revealed that TC9 had a more heterogeneous karyotype compared to TC4 cell lines with a significantly higher proportion of cells that deviated from the mode of four copies of both Chromosomes 2 and 15 (Fisher’s exact test, p=0.05) (Figure 4C). This implies that karyotypic heterogeneity rather than increased ploidy might be responsible for increased drug resistance compared to karyotypically stable diploid cells. We cannot formally exclude the possibility that acquired mutations present in the drug resistant tetraploid clone that may have permitted the spontaneous tetraploid phenotype may primarily be responsible for increased drug resistance.
Figure 4.

A) Boxplot of relative surviving cell numbers comparing HCT116 Tetraploid Clone 4 (TC4) and Diploid Clone 8 (DC8) cell lines, and Tetraploid Clone 9 (TC9) with DC8 cell lines. The TC9 cell line was significantly more resistant compared to DC8 (p<0.001). The difference in drug sensitivity between TC4 and DC8 was not significant (p=0.078).
B) Representative FISH images for TC4 and TC9. Probes against Chromosome 15 in green.
C) Histogram showing distribution of number of markers per cell corresponding to Chromosome 2 (top two) and 15 (bottom two) in TC4 and TC9. TC9 had a statistically significant higher proportion of cells that deviated from having 4 copies of both Chromosome 2 and 15 (p=0.05) compared to TC4.
Taken together with the isogenic cell line datasets presented here, where CIN is artificially induced through loss of one allele of MAD2 or both copies of PTTG1, these results support the contribution of CIN rather than increased ploidy status or MIN in conferring altered drug sensitivity.
Relationship Between CIN status and Benefit From Cytotoxic Therapy in Clinical Datasets
Published clinical data support the view that CIN+ CRC is associated with a worse prognosis compared to CIN− tumours (6) and data presented here suggest that CIN+ cell lines display intrinsic multi-drug resistance. Conceivably the poorer prognosis of CIN+ disease may relate in part to intrinsic Thymidylate synthase inhibitor drug resistance, cytotoxics commonly used in the adjuvant treatment of CRC. Consistent with this hypothesis, both isogenic CIN+ cells are significantly more resistant to the majority of Thymidylate synthase inhibitors tested, including 5-FU, and non-isogenic CIN+ cells are more resistant to 5-FU compared to CIN− cell lines, at physiological concentrations (Supplementary Figure 6A, B).
Next, we asked whether CIN+ status might be associated with poorer outcome following adjuvant therapy with 5-FU-based regimens. A meta-analysis of studies examining the relationship between CIN and prognosis in loco-regional CRC, revealed that CIN+ disease (defined as aneuploidy/polyploidy determined using flow cytometry) confers a worse overall survival (30 studies) (Supplementary Figure 5A) and progression-free survival (15 studies) (Supplementary Figure 5B) compared to patients with diploid CRC. Similarly, if only patients who received chemotherapy are included (2 studies) (46) (47), CIN+ tumours are associated with a worse overall survival (Figure 5A).
Figure 5.

A) Impact of CIN+ on disease free survival, overall survival, and those receiving adjuvant chemotherapy in loco-regional CRC. Overall CIN+ appears to confer a worse prognosis compared to diploid.
B) Benefit derived from adjuvant 5-FU in patients with (near) diploid (top) and CIN+ (middle) CRC. Patients with diploid CRC appear to benefit more from chemotherapy compared to patients with CIN+ tumours. Combined analysis of CIN+ and diploid patients shows similar magnitude of benefit as would be expected from literature (bottom) (49).
Two studies explore the predictive value of CIN (46, 48) in patients with loco-regional CRC who received either adjuvant chemotherapy or no chemotherapy following surgery (Figure 5B). Patients with diploid CRC appear to benefit from 5-FU based therapy (N=262, HR=0.61; 95% CI 0.40-0.94, p=0.024; I2=0%, p=0.467) compared with untreated diploid controls, whereas there was no significant difference between treated and untreated CIN+ CRC (N=303, HR=0.81; 95% CI 0.57-1.16, p=0.250; I2=0%, p= 0.932). The combined analysis of all patients suggests a benefit following 5-FU treatment comparable to that reported in the literature for genetically unselected patients (49). Whilst these studies are limited, they are consistent with the view that patients with CIN+ CRC derive less benefit from 5-FU based adjuvant cytotoxic chemotherapy than patients with diploid CRC. Prospective evaluation of the association of CIN+ disease with the efficacy of combination regimens (eg 5-FU/oxaliplatin) in stage 3 and 5-FU/oxliplatin and irinotecan in stage 4 disease might be considered to assess whether clinical outcome following these more recent regimens has a similar association with CIN status.
Discussion
In this analysis we have provided evidence that CIN+ CRC cell lines display intrinsic multi-drug resistance compared to CIN− cell lines. No specific kinase inhibitor was identified that displayed greater activity in CIN+ cell lines. We cannot exclude the potential for off-target effects at the concentrations of kinase inhibitors used in this analysis, however the same conditions were applied to the CIN− cells and therefore off-target phenomena are unlikely to change the conclusions of this work. Furthermore, in the isogenic systems we observed significant drug resistance in the CIN+ cell lines relative to their isogenic parental CIN− pairs at all concentrations of cytotoxics tested (ranging from 0.1μM to 25μM). Intriguingly, the isogenic CIN+ and tetraploid cell line systems suggest that the primary association is between multi-drug resistance and CIN+ rather than tetraploidy. It has been previously demonstrated that aneuploid cell lines can acquire multi-drug resistance at an accelerated rate (50) that may be driven by cancer cell heterogeneity resulting from multiple chromosomal reassortments in aneuploid cells. The short time course of our experiments in comparison to this study suggests that multi-drug resistance is likely to be an intrinsic property of CIN+ cells rather than a process that is acquired in our cell systems over multiple generations. What might contribute to this intrinsic multidrug resistance phenotype in CIN+ cells? We speculate that either basal population heterogeneity in CIN+ cell lines is sufficiently diverse to confer a cell viability advantage following drug exposure or there is a specific CIN+ survival phenotype that initiates a tolerance of ongoing chromosomal rearrangements that is also associated with multi-drug resistance.
There is increasing evidence in support of a CIN+ survival phenotype and putative molecular coordinators of this property. Cell death after mitotic arrest may result from transcriptional inhibition due to condensed chromatin, precipitating the degradation of short-lived mRNA encoding pro-survival proteins (51). CIN+ cells may overexpress these pro-survival genes compared to diploid cells (22) that may drive the resistance of CIN+ cells to a mitotic arrest triggered by taxanes. Jeganathan and colleagues have demonstrated that tolerance of chromosome mis-segregation events can be conferred by a hypomorphic BUB1 allele in mouse embryonic fibroblasts (24). Recently, Thompson and Compton have demonstrated that chromosome mis-segregration in diploid human cells triggers an increase in nuclear p53 and that p53 null cells are able to tolerate chromosome mis-segregation events enabling the propagation of aneuploid genomes (52). A higher proportion of the CIN+ cell lines used in our study have mutant p53 in comparison to the CIN− cell lines. However when we pooled data for somatic mutation status and CIN status, CIN+ status was the only parameter significantly associated with resistance to these inhibitors.
Therefore, evidence exists for the coordination of apoptotic/cell death pathways following chromosome mis-segregation events. Conceivably, common molecular pathways regulating cell death following a chromosome mis-segregation event may become disrupted in CIN+ cells, simultaneously triggering tolerance of chromosome reassortments and, as an indirect consequence, resistance to drug exposure.
The observations that CIN+ cancer cell lines appear to be less sensitive to a range of anti-cancer agents compared to diploid cells and that poorer patient outcome follows cytotoxic treatment of CIN+ tumours compared to diploid counterparts, strongly suggest the need to consider tumour stratification according to CIN status in the design of clinical trials testing novel anti-cancer agents in CRC. This is particularly relevant to the advanced CRC setting where the incidence of CIN+ is greater than in early stage disease. Stratifying drug response according to CIN status may limit the risk of early drug attrition and heighten the chance of identifying responder populations in patients with diploid tumours. Importantly, these data indicate that specifically targeting cancer cells with CIN+ status using currently available kinase inhibitors appears challenging. An improved understanding of the mechanisms associated with the generation and survival of CIN+ CRC will be important to drive the development of new therapeutic approaches in order to improve patient outcome in this high risk disease subtype.
Supplementary Material
Acknowledgements
We thank Robert Benezra for the donation of the isogenic HCT116 MAD2+/− cell lines and Bert Vogelstein for the donation of the isogenic HCT116 PTTG1−/− cell lines.
CS is a senior Medical Research Council clinical research fellow and is funded by both Cancer Research UK (CRUK) and the Medical Research Council. AL is funded by CRUK. IT is supported by the Oxford Biomedical Research Centre and CRUK.
References
- 1.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–9. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 2.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–7. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 3.Roschke AV, Tonon G, Gehlhaus KS, et al. Karyotypic complexity of the NCI-60 drug-screening panel. Cancer Res. 2003;63:8634–47. [PubMed] [Google Scholar]
- 4.Lothe RA, Peltomaki P, Meling GI, et al. Genomic instability in colorectal cancer: relationship to clinicopathological variables and family history. Cancer Res. 1993;53:5849–52. [PubMed] [Google Scholar]
- 5.Thibodeau SN, French AJ, Cunningham JM, et al. Microsatellite instability in colorectal cancer: different mutator phenotypes and the principal involvement of hMLH1. Cancer Res. 1998;58:1713–8. [PubMed] [Google Scholar]
- 6.Walther A, Houlston R, Tomlinson I. Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut. 2008 doi: 10.1136/gut.2007.135004. [DOI] [PubMed] [Google Scholar]
- 7.Hiddemann W, Schumann J, Andreef M, et al. Convention on nomenclature for DNA cytometry. Cancer Genetics and Cytogenetics. 1984;13:181–3. doi: 10.1016/0165-4608(84)90059-1. [DOI] [PubMed] [Google Scholar]
- 8.Cahill DP, Lengauer C, Yu J, et al. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–3. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 9.Michel LS, Liberal V, Chatterjee A, et al. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 2001;409:355–9. doi: 10.1038/35053094. [DOI] [PubMed] [Google Scholar]
- 10.Jallepalli PV, Waizenegger IC, Bunz F, et al. Securin is required for chromosomal stability in human cells. Cell. 2001;105:445–57. doi: 10.1016/s0092-8674(01)00340-3. [DOI] [PubMed] [Google Scholar]
- 11.Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol. 2008;180:665–72. doi: 10.1083/jcb.200712029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukasawa K, Woude G Vande. Synergy between the Mos/mitogen-activated protein kinase pathway and loss of p53 function in transformation and chromosome instability. Mol Cell Biol. 1997;17:506–18. doi: 10.1128/mcb.17.1.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pihan GA, Purohit A, Wallace J, Malhotra R, Liotta L, Doxsey SJ. Centrosome Defects Can Account for Cellular and Genetic Changes That Characterize Prostate Cancer Progression. Cancer Res. 2001;61:2212–9. [PubMed] [Google Scholar]
- 14.Gisselsson D, Pettersson L, Hoglund M, et al. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5357–62. doi: 10.1073/pnas.090013497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043–8. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- 16.Sheffer M, Bacolod MD, Zuk O, et al. Association of survival and disease progression with chromosomal instability: A genomic exploration of colorectal cancer. Proceedings of the National Academy of Sciences. 2009;106:7131–6. doi: 10.1073/pnas.0902232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and darwinian selection in tumours. Trends in Cell Biology. 1999;9:M57–M60. [PubMed] [Google Scholar]
- 18.Nicholson JM, Duesberg P. On the karyotypic origin and evolution of cancer cells. Cancer Genetics and Cytogenetics. 2009;194:96–110. doi: 10.1016/j.cancergencyto.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Chandhok NS, Pellman D. A little CIN may cost a lot: revisiting aneuploidy and cancer. Current Opinion in Genetics & Development. 2009;19:74–81. doi: 10.1016/j.gde.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 20.Sotillo R, Schvartzman J-M, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010;464:436–40. doi: 10.1038/nature08803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duesberg P, Stindl R, Hehlmann R. Explaining the high mutation rates of cancer cells to drug and multidrug resistance by chromosome reassortments that are catalyzed by aneuploidy. Proc Natl Acad Sci U S A. 2000;97:14295–300. doi: 10.1073/pnas.97.26.14295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swanton C, Nicke B, Schuett M, et al. Chromosomal instability determines taxane response. Proceedings of the National Academy of Sciences. 2009;106:8671–6. doi: 10.1073/pnas.0811835106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McClelland SE, Burrell RA, Swanton C. Chromosomal instability: a composite phenotype that influences sensitivity to chemotherapy. Cell Cycle. 2009;8:3262–6. doi: 10.4161/cc.8.20.9690. [DOI] [PubMed] [Google Scholar]
- 24.Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. The Journal of Cell Biology. 2007;179:255–67. doi: 10.1083/jcb.200706015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roschke AV, Kirsch IR. Targeting cancer cells by exploiting karyotypic complexity and chromosomal instability. Cell Cycle. 2005;4:679–82. doi: 10.4161/cc.4.5.1687. [DOI] [PubMed] [Google Scholar]
- 26.Torres EM, Sokolsky T, Tucker CM, et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–24. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- 27.Storchova Z, Breneman A, Cande J, et al. Genome-wide genetic analysis of polyploidy in yeast. Nature. 2006;443:541–7. doi: 10.1038/nature05178. [DOI] [PubMed] [Google Scholar]
- 28.Woodford-Richens KL, Rowan AJ, Gorman P, et al. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9719–23. doi: 10.1073/pnas.171321498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaasenbeek M, Howarth K, Rowan AJ, et al. Combined Array-Comparative Genomic Hybridization and Single-Nucleotide Polymorphism-Loss of Heterozygosity Analysis Reveals Complex Changes and Multiple Forms of Chromosomal Instability in Colorectal Cancers. Cancer Res. 2006;66:3471–9. doi: 10.1158/0008-5472.CAN-05-3285. [DOI] [PubMed] [Google Scholar]
- 30.Abdel-Rahman WM, Katsura K, Rens W, et al. Spectral karyotyping suggests additional subsets of colorectal cancers characterized by pattern of chromosome rearrangement. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:2538–43. doi: 10.1073/pnas.041603298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cancer Genome Project. [Internet] Wellcome Trust Sanger Institute; Cambridge, UK: [updated 2010 Dec 20]. [cited 2010 Dec 23]. 2010. Available from: http://www.sanger.ac.uk/genetics/CGP. [Google Scholar]
- 32.Liu Y, Bodmer WF. Analysis of P53 mutations and their expression in 56 colorectal cancer cell lines. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:976–81. doi: 10.1073/pnas.0510146103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rowan AJ, Lamlum H, Ilyas M, et al. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:3352–7. doi: 10.1073/pnas.97.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ilyas M, Tomlinson IPM, Rowan A, Pignatelli M, Bodmer WF. Beta-Catenin mutations in cell lines established from human colorectal cancers. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:10330–4. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samuels Y, Diaz LA, Jr, Schmidt-Kittler O, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561–73. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 36.Inhibitor Libraries and Pathway Panels. [Internet] Merck KGaA; Darmstadt, Germany: [cited 2010 Dec 23]. 2010. Available from: http://www.emdchemicals.com/life-science-research/inhibitor-libraries-and-pathway-panels/c_bKSb.s1O9nQAAAEitTV5hDHD#docs. [Google Scholar]
- 37.Kahm GH Matthias, Hella Lichtenberg-Fraté, Jost Ludwig, Maik Kschischo. grofit: Fitting Biological Growth Curves with R. Journal of Statistical Software. 2010;33 [Google Scholar]
- 38.Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Stat Med. 1998;17:2815–34. doi: 10.1002/(sici)1097-0258(19981230)17:24<2815::aid-sim110>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 39.Anderson MJ. Permutation tests for univariate or multivariate analysis of variance and regression. Canadian Journal of Fisheries and Aquatic Sciences. 2001;58:626–39. [Google Scholar]
- 40.Greenman CD, Bignell G, Butler A, et al. PICNIC: an algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostatistics. 2010;11:164–75. doi: 10.1093/biostatistics/kxp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chin S, Teschendorff A, Marioni J, et al. High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer. Genome Biology. 2007;8:R215. doi: 10.1186/gb-2007-8-10-r215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams BR, Prabhu VR, Hunter KE, et al. Aneuploidy Affects Proliferation and Spontaneous Immortalization in Mammalian Cells. Science. 2008;322:703–9. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Segal DJ, McCoy EE. Studies on Down’s syndrome in tissue culture. I. Growth rates protein contents of fibroblast cultures. Journal of Cellular Physiology. 1974;83:85–90. doi: 10.1002/jcp.1040830112. [DOI] [PubMed] [Google Scholar]
- 44.Baker DJ, Jeganathan KB, Cameron JD, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–9. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- 45.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 1995;57:289–300. [Google Scholar]
- 46.Barratt PL, Seymour MT, Stenning SP, et al. DNA markers predicting benefit from adjuvant fluorouracil in patients with colon cancer: a molecular study. Lancet. 2002;360:1381–91. doi: 10.1016/s0140-6736(02)11402-4. [DOI] [PubMed] [Google Scholar]
- 47.Sinicrope FA, Rego RL, Halling KC, et al. Prognostic impact of microsatellite instability and DNA ploidy in human colon carcinoma patients. Gastroenterology. 2006;131:729–37. doi: 10.1053/j.gastro.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 48.Ahnen DJ, Feigl P, Quan G, et al. Ki-ras mutation and p53 overexpression predict the clinical behavior of colorectal cancer: a Southwest Oncology Group study. Cancer Res. 1998;58:1149–58. [PubMed] [Google Scholar]
- 49.Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol. 2004;22:1797–806. doi: 10.1200/JCO.2004.09.059. [DOI] [PubMed] [Google Scholar]
- 50.Duesberg P, Stindl R, Hehlmann R. Origin of multidrug resistance in cells with and without multidrug resistance genes: Chromosome reassortments catalyzed by aneuploidy. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11283–8. doi: 10.1073/pnas.201398998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blagosklonny MV. Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle. 2007;6:70–4. doi: 10.4161/cc.6.1.3682. [DOI] [PubMed] [Google Scholar]
- 52.Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. The Journal of Cell Biology. 2010;188:369–81. doi: 10.1083/jcb.200905057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.