

Abstract
EGF activates NF-κB and constitutively activated NF-κB contributes to EGFR mutation-associated tumorigenesis, but it remains unclear precisely how EGFR signaling leads to NF-κB activation. Here we report that CARMA3, a Caspase Recruitment Domain (CARD)-containing scaffold molecule, is required for EGF-induced NF-κB activation. CARMA3 deficiency impaired the activation of the IKK complex following EGF stimulation, resulting in a defect of EGF-induced IκBα phosphorylation and NF-κB activation. We found that CARMA3 and Bcl10 contributed to several characteristics of EGFR-associated malignancy, including proliferation, survival, migration, and invasion. Most importantly, CARMA3 contributed to tumor growth in vivo. Our findings elucidate a crucial link between EGFR-proximal signaling components and the downstream IKK complex, and they suggest a new therapeutic target for treatment of EGFR-driven cancers.
Keywords: EGF, NF-κB, cancer cell growth, CARMA3, Bcl10
Introduction
Nuclear Factor kappa B (NF-κB) is a family of transcription factors that regulates immunological and inflammatory responses, as well as cell survival (1). They are often constitutively activated in cancers (2). NF-κB controls the expression of pro-survival and anti-apoptotic genes, pro-inflammatory cytokines, and cyclins (3). Therefore, the constitutively activated NF-κB contributes to cancer cell survival, transformation and progression (2, 3), and up-regulates the expression of various proteins involving in the epithelial-to-mesenchymal transition (EMT) and metastasis of cancer cells (4). Thus, the end result of the constitutively activated NF-κB in cancer cells is their resistance to apoptotic stimuli and their increased proliferation and migration, which are all hallmarks of cancer (5). However, different cancers activate NF-κB through different mechanisms. Therefore, revealing various signaling pathways that lead to activation of NF-κB will provide the therapeutic targets for treatment of different cancers.
Multiple receptors, such as the Tumor Necrosis Factor alpha (TNFα) receptor (TNFR), Toll-like receptor (TLR), antigen receptor, and G-protein-coupled receptors (GPCRs), can activate this pathway leading to activation of NF-κB (1, 6, 7). Several growth factors, including Epidermal Growth Factor (EGF) (8, 9), Insulin-like Growth Factor (IGF) (10–12), Platelet-Derived Growth Factor (PDGF)(6, 13), and Fibroblast Growth Factor (bFGF)(14–16), can also induce weak, but notable NF-κB activation through their receptors that belong to a family of receptor tyrosine kinases (RTKs). Different human cancer cell lines have been shown to exhibit elevated levels of activated NF-κB, which are due to the over-expression or mutations of receptors for EGF, Heregulin (HRG), or Insulin-like Growth Factor (IGF). NF-κB activation is one of downstream events in EGF receptor (EGFR or erbB1/HER1) signaling (17), particularly in ER-negative breast cancer (9, 18). Stimulation of EGFR-expressing breast cancer cell lines with EGF induces NF-κB activation, which could be blocked by the inhibitor of IKK, an upstream enzyme complex of the NF-κB signal transduction cascade. Inhibition of NF-κB activation suppresses the growth of murine breast tumors with active NF-κB and inhibition of IKK incapacitates tumor formation (19). Although many reports implicate that Akt and IKK are involved in EGFR-induced NF-κB activation, the molecular mechanism by which EGFR signaling pathway leads to NF-κB activation remains largely unknown. Therefore, it is necessary to elucidate how EGFR link to NF-κB pathway, which will provide novel drug targets to inhibit cancer progression.
Caspase Recruitment Domain and Membrane-Associated Guanylate Kinase-like Domain Protein (CARMA) family of proteins are scaffold proteins. It contains three members, CARMA1 (also known as CARD11), CARMA2 (CARD14), and CARMA3 (CARD10) (20–23). These three proteins share similar structural motifs, with an N-terminal CARD domain, followed by a coiled-coil (CC) domain, a PDZ domain, a SH3 domain, and a C-terminal guanylate kinase-like (GUK) domain (21–23). However, they have distinct expression patterns with CARMA1 expressed in hematopoietic cells, CARMA2 in the placenta and several mucosal tissues (20), and CARMA3 in all non-hematopoietic cells (21–24). Early studies by our lab and others have demonstrated that CARMA1 plays an essential role in mediating antigen receptor-induced NF-κB activation (25–27). Recent studies demonstrated that CARMA3, a scaffold protein, is required for GPCR- and PKC-induced NF-κB activation (28, 29) and its potential role in cancer progression (30). In addition, several studies indicate that the downstream signaling components of CARMA3, namely B Cell Lymphoma protein 10 (BCL10) and Mucosa-associated lymphoid tissue lymphoma translocation gene 1 (MALT1), are also required for GPCR and PKC-induced NF-κB activation (29, 31, 32). Since PKC participates in NF-κB-activation induced by other receptors such as EGFR (18, 33), we hypothesized that CARMA3 might play an important role in EGFR-induced NF-κB activation. Here we show that CARMA3 is required for EGFR-induced NF-κB activation. We also found that CARMA3 deficiency affected cancer cell proliferation, survival, migration, and invasion. Importantly, suppressing CARMA3 expression significantly inhibited cancer cell growth in vivo. Together, our studies reveal a key signal transduction cascade that mediates EGFR-induced NF-κB activation, and the functional significance of this pathway in cancer cell growth.
Materials and Methods
Antibodies and Reagents
Phospho-specific antibodies for ERK1/2 (9101), IκBα (9246), and IKKα/β (2681), as well as total EGFR (2232) were purchased from Cell Signaling Technology. Antibodies against phospho-EGFR, IκBα (371, clone C21), IKKα/β (7607, clone H-470), and Actin (8432, clone C-2) were purchased from Santa Cruz Biotech. DNA-oligo probes for NF-κB (E3291), OCT1 (E3241), and AP1 (E3201) were purchased from Promega. AG1478, AG825, Genistein, and mouse EGF was from Sigma. TNFα was from Endogen. Lentivirus-encoded shRNA were purchased from Sigma.
MDA468 cells were purchased from ATCC in 2008, and aliquots of these cells (less than 5 passages from the purchasing vial) were kept in liquid nitrogen before usage. A431 cells were tested for their authenticity in the Characterized Cell Line Core Facility in MDA Cancer Center in August of 2009.
Electrophoretic mobility shift assay (EMSA)
To conduct the EMSA experiments, 1~2×106 cells were starved overnight, stimulated for appropriate times with various agents, and nuclear extracts were prepared. Nuclear extracts (5–10 μg) were incubated with 1×105 cpm of 32P-labelled probes at room temperature for 15 minutes. The samples were separated on a native Tris-Borate-EDTA polyacrylamide gel, which was dried at 80°C for 1–2 hours, and then exposed to X-ray film.
Lentivirus infection for shRNA knockdown
Lentiviruses were generated by transfecting HEK293T with packaging vectors encoding VSV-G using the calcium precipitation method. One or two days later, the supernatant was collected from the cells, spun at 3000 × g for 10 minutes to pellet any cell debris, and applied directly to target cells. Cells were placed under the selection with media containing Puromycin, and resultant pools were used for experiments.
MTT Assay
To conduct the MTT assay, cells were seeded in wells of a 96-well plate in 200 ml of complete or serum-free media. At the indicated time, 20 ml of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide (MTT) was dissolved at 5 mg/ml in sterile PBS, was added to the cells. 2 hours later, the media was removed, the metabolized MTT was re-suspended in DMSO, and the absorbance was measured at 570 nm. The experiment was performed in triplicates.
Colony formation assay
The soft agar colony formation assays were performed as following. First, 2.4% agarose in complete 1 × DMEM (diluted from complete 2xDMEM at pH 7.2) was melted in 37° water bath. Then the agarose was mixed with complete media to a final concentration of 0.6% agarose, and layered 0.5ml on the bottom of each well in 24-well plates. For A431 cells, 1,250 cells were mixed with stock agarose and complete media to a final concentration of 0.3% agarose in 0.5ml, and 10 ng/ml of EGF were added to this mixture. Layer 0.5ml of this mixture on top of the solidified 0.6% lower layer. The cell cultures were maintained in incubator for two weeks to allow colony formation. Every 4–6 days after preparing agarose, 100 ul of half-complete media with 10 ng/ml of EGF were added to keep the cells growth. Two weeks later, cells were fixed with 4% paraformadehyde in PBS, and stained with 0.5 ml of 0.005% Crystal Violet for more than 1 hour. Put the top layer on the microscope slides and dry till it became membrane. Colonies of diameter >45um were counted manually using a microscope. For MDA468 cells, 1×104 cells were mixed with 0.3% agarose, and layered down on the top of the lower gel. The experiments were repeated for three times.
Cell cycle distribution
Cells were harvested by centrifugation and washed with PBS. The cells were fixed with ice cold 70% ethanol at 4°C for 18 hours. The fixed cell suspensions were washed with PBS, and then treated with 500 ml 50 μg/ml Propidium iodide (PI), 2.5μl 0.1 mg/ml RNase A, and 0.05% Triton X-100 for 40 minutes at 37°C in the dark. Cell cycle profile was analyzed by a flow cytometry (Becton Dickinson FACS Canto II).
Anoikis and apoptosis assay
Anoikis assay was mainly based on previously described procedures (34). Briefly, cells were seeded in 25 mm2 low cell-binding flasks (coated with 2-Hydroxyethyl methacrylate, Poly-HEMA, 10g/L in 100% ethanol) at a density of 1 × 106 cells per flask in serum-free media containing EGF, and incubate vertically at 37°C, 5% CO2 for 24 hours. Cells were collected by centrifuge, fixated and staining with Propidium iodide (PI). The number of total and viable cells was determined using a flow cytometer (Becton Dickinson FACS Canto II).
For apoptosis assay, cells were collected by trypsinization and washed with cold-PBS, cells (1×105 and 1×106 cells) were re-suspended in 100 μl of Annexin V incubation solution, which includes 10μl 10× binding buffer, 10μl Propidium iodide (PI), 1μl Annexin V-FITC and water. These cells were incubated in the dark for 15 min at room temperature. The cells were analyzed within one hour and the percent of apoptotic (Annexin V-positive) cells is determined by the flow cytometry.
Cell migration and invasion assay
Cell migration ability was analyzed in modified BD Falcon Cell Culture Insert System (35). The lower chambers (24 well plates) were placed with 500 μl serum-free migration buffer containing EGF, and cells were added to the insert, the upper migration chamber. Plates were incubated for 16–18h at 37°C in a 5% CO2 supplemented atmosphere. Following incubation, cells that did not migrate were removed from the top of the insert with sterile cotton swabs. Cells that migrated to the lower side of the filter disk were then fixed, stained and quantified. All experiments were performed in triplicates. Cellular invasion ability was assayed to determining the ability of cells to invade a synthetic basement membrane (Matrigel; BD Biosciences) based on a procedure described previously (36). Polycarbonate membranes (8-μm pore size) of the insert of BD Falcon Cell Culture Insert System were coated with Matrigel at 50 μl/cm2 of growth surface. Cell suspension (1.25×104) was added to the insert, and the lower chamber was filled with 500 μl serum-free medium with EGF. Then, the tumor invasion system was incubated at 37°C for and allowed to invade through the Matrigel barrier for 24 hours. Following incubation, membranes were fixed with 4% paraformadehyde in PBS and stained (Giemsa; Sigma-Aldrich). Non-invading cells were removed using a clean cotton swab, whereas invading cells on the underside of the membrane were counted using an inverted microscope. All experiments were performed in triplicates.
Mouse xenograft tumor model
6–8 weeks age of BALB/c nude (Nu/Nu) mice were purchased from Harlan laboratories Inc. A431 cells (1 × 106) or MDA468 cells (3 × 106) were suspended in a total volume of 100 μl of a 1:1 mixture of appropriate medium vs Matrigel (BD Biosciences). The cell suspension was injected subcutaneously in male of nude mice, or at the fat pad of female of nude mice, respectively. One week after inoculating, tumors volumes were measured twice a week and calculated by the formula π/6 [length (mm) × width (mm)]2. All the animal procedures were conducted under the institution’s guidelines.
Results
CARMA3 contributes to cancer cell growth
Previous studies demonstrate that the CARMA3-Bcl10-MALT1 (CBM) complex plays an essential role for GPCR-induced NF-κB activation (28, 29, 31, 32), and CARMA3 is also implicated downstream of Ras signaling pathway in cancer cells (30). To determine whether CARMA3 has a role in cancer cell growth, we obtained two lentiviral constructs encoding CARMA3 shRNA (sh47 and sh48), which could significantly suppress CARMA3 mRNA expression (Supplemental Fig. 1). We first infected MDA468 cells with the lentivirus encoded CARMA3 shRNAs or Green-Fluorescent-Protein (GFP) shRNA, and then examined the growth rate of these cells using MTT assay. With CARMA3 shRNA knockdown, MDA468 cells displayed reduced MTT-converting activity comparing to the negative and GFP shRNA control (Fig. 1A). Similar to MDA468 cells, we found that knockdown of CARMA3 also reduced the growth rate of A431 cells (Fig. 1B) and several cancer cell lines (Supplemental Fig. 2A). To further confirm the role of the CBM complex in cell growth, we also examined the growth rate of mouse embryonic fibroblast (MEF) from wild type and CARMA3- or Bcl10-deficient (KO) mice using the MTT assay. We found that both of CARMA3 KO and Bcl10 KO MEF cells grew significantly slower than wild type cells (Supplemental Fig. 2B). Together, these data indicate that the CARMA3-Bcl10-MALT1 (CBM) complex plays an important role for cancer cell growth.
Figure 1. CARMA3 deficiency affects cell cycle and cancer cell growth.
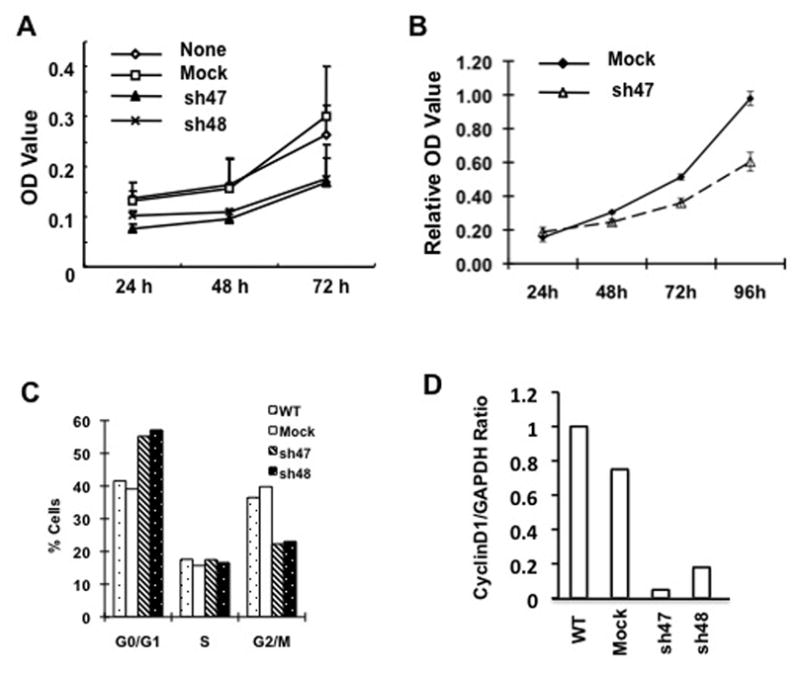
(A) MDA-468 cells with stably expressing CARMA3 shRNA (sh47 and sh48) or negative control (GFP) were seeded in 96-well plates. Cell proliferation was examined at 24, 48, and 72 hours, respectively, by the MTT assay. The absorbance at 570 nm at different time points was determined. The experiment was repeated for three times, and the result from one of these experiments is presented. (B) MTT assay was similarly performed to examine the growth rate of A431 cells with CARMA3 shRNA (sh47) or mock control. (C) Above MDA cells were grown in DMEM complete media for overnight. The cells were harvested, fixed, and subjected to Propidium Iodide (PI) staining for cell cycle analysis by FACS. Data from different cell cycle phase was presented as percentage of cells (**P<0.01, *p>0.05). (D) Real-time PCR were performed to detect the expression level of Cyclin D1 using total RNA were prepared from MDA468 cells (WT), or MDA468 cells transduced with control shRNA for GFP (Mock) or CARMA3 shRNA (sh47 and sh48). The expression level of GAPDH was used as internal controls.
To further determine the effect of CARMA3 on cell proliferation, we analyzed the cell cycle profile of MDA468 breast cancer cells with or without CARMA3 knockdown, and found that the percentage of G0/G1 phase was increased in cells with CARMA3 knockdown, whereas the percentage of G2/M phase was decreased in these cells comparing to non-transduced (WT) or GFP shRNA (Mock) control cells (Fig. 1C), indicating that suppression of CARMA3 expression induces a G0/G1 arrest. We also checked the expression of cyclin D1 by quantitative PCR, and found that the expression of cyclin D1 mRNA was significantly lower in MDA468 cells with CARMA3 knockdown than wild type and mock control (Fig. 1D). Together, these results suggest that inhibiting CARMA3 expression suppresses cell growth.
CARMA3 is required for EGF-induced NF-κB activation
Among above investigated cancer cell lines, MDA231, MDA468, MDA453, A431, H1975, and PC3 cells have higher expression levels of EGFR, and also displayed a more significant reduction of growth rate with CARMA3 knockdown (Supplemental Fig. 2A). Therefore, we hypothesized that the CBM complex may involve in EGF-induced signal transduction. Since it has been shown that EGF can induce NF-κB activation (9), we decided to determine whether CARMA3 is required for EGF-induced NF-κB activation in cancer cells. To address this question, we prepared mouse embryonic fibroblast (MEF) cells from wild type (Carma3+/+) and CARMA3-deficinent (Carma3−/−)mice and stimulated them with EGF. We found that EGF could effectively induce NF-κB activation in a dose-dependent manner in wild type cells, but this activation was completely abolished in CARMA3-deficient cells (Fig. 2A). In contrast, TNFα could effectively activate NF-κB in both wild type and CARMA3-deficient cells (Fig. 2A). Since EGF induces both EGFR homodimer and EGFR/HER2 heterodimer formation, we sought to determine which receptor was responsible for EGF-induced NF-κB activation. To do this, we pretreated MEF cells with the non-specific tyrosine kinase inhibitor Genistein, the EGFR-specific inhibitor AG1478, the HER2-specific inhibitor AG825, or the latter two drugs together. We observed that Genistein completely abolished EGF-induced nuclear localization NF-κB (Supplemental Fig. 3). For either AG1478 or AG825 alone, they could only partially reduce nuclear localization of NF-κB. However, combination of the two drugs led to an almost complete inhibition of EGF-induced nuclear localization of NF-κB (Supplemental Fig. 3), indicating that both EGFR and HER2 are required for EGF-induced NF-κB activation. Together, our data indicate that CARMA3 is selectively involved in EGF-, but not TNFα-induced NF-κB activation.
Figure 2. CARMA3 is required for EGF-induced NF-κB activation.
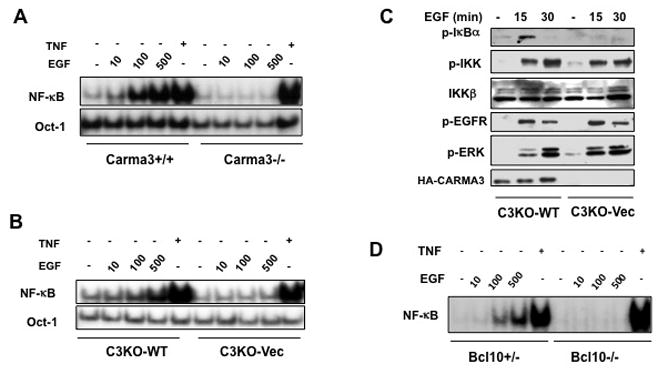
(A) MEF cells from Carma3+/+ or Carma3−/− mice were stimulated with indicated doses of EGF (μM) or TNFα (10 ng/ml). (B) CARMA3-knockout cells were reconstituted with wild type CARMA3 (C3KO-WT) or vector control (C3KO-Vec) were stimulated with indicated doses of EGF or TNFα (10 ng/ml). Nuclear extracts were prepared and then subjected to EMSA using 32P-labeled NF-κB or Oct-1 probe. (D) C3KO-WT or C3KO-Vec cells were stimulated with EGF (100 μM) for indicated time and then cell lysates were subjected to immunoblotting analysis using indicated antibodies. (C) C3KO-WT or C3KO-Vec cells were stimulated with EGF (100 μM) or TNFα (10 ng/ml) for indicated time. Cell lysates were subjected to immunoblotting analysis using indicated antibodies. (D) MEF cells from Bcl10+/− or Bcl10−/− mice were stimulated with indicated concentration of EGF (μM) or TNFα (10 ng/ml) for one hour. Nuclear extracts were prepared from these cells, and then subjected to EMSA using 32P-labeled NF-κB probe.
To further verify that the observed defect in Carma3−/− cells was due to CARMA3 deficiency, CARMA3-deficient cells that were reconstituted with and without CARMA3 (C3KO-WT and C3KO-Vec) were stimulated with EGF. Consistent with above results, we found that expression of CARMA3 in CARMA3-deficient cells restored the EGF-induced NF-κB activation (Fig. 2B). CARMA3 deficiency resulted in a defect of EGF-induced IκBα phosphorylation (Fig. 2C), indicating that CARMA3 is required for EGF-induced IKK activation. Similar to our previous finding that CARMA3 is not required for GPCR-induced IKK phosphorylation (28), we also interestingly found that EGF-induced IKK phosphorylation was not defective in CARMA3-deficient cells (Fig. 2C). In contrast to the defect of IKK and NF-κB activation, EGF-induced phosphorylation of EGFR and ERK was intact in these cells (Fig. 2C). Together, these results indicate that CARMA3 plays a specific role in EGFR signaling pathway leading to NF-κB activation.
Since Bcl10 binds to CARMA3 and is also required for GPCR-induced NF-κB activation (29, 31, 32) and formed a complex with CARMA3, we decided to determine whether Bcl10 is also involved in EGF-induced NF-κB activation. To address this question, MEF cells prepared from Bcl10-heterozygous (Bcl10+/−) or Bcl10-deficient (Bcl10−/−) mouse embryos were stimulated with increasing doses of EGF. We found that the Bcl10-deficient cells were also defective in EGF-induced, but not TNFα-induced NF-κB activation (Fig. 2D). This result suggests that the CBM complex is required for EGF-induced NF-κB activation.
To further demonstrate the role of the CBM complex in EGF-induced NF-κB activation in cancer cells, A431 cells and MDA468 cells that were transduced with CARMA3 shRNA (shC3-47 and shC3-48)) and Bcl10 shRNA (shBcl10), were stimulated with EGF. We found that knockdown of CARMA3, which decreased CARMA3 expression in A431 and MDA468 cells (Supplemental Fig. 4), impaired EGF-induced NF-κB activation (Fig. 3A–B). Similarly, knockdown of Bcl10 in A431 cells also impaired EGF-induced NF-κB activation (Fig. 3A). In contrast, TNFα-induced NF-κB activation was not affected in these cells (Fig. 3A). Although EGF-induced NF-κB activation was defected in CARMA3 or Bcl10 knockdown cells, EGF-induced ERK activation was not affected in these cells (Fig. 3C–D). Together, these results indicate that CARMA3 and Bcl10 play a specific role in EGFR signaling pathway leading to NF-κB activation.
Figure 3. CARMA3 and Bcl10 mediate EGF-induced NF-κB activation in cancer cells.
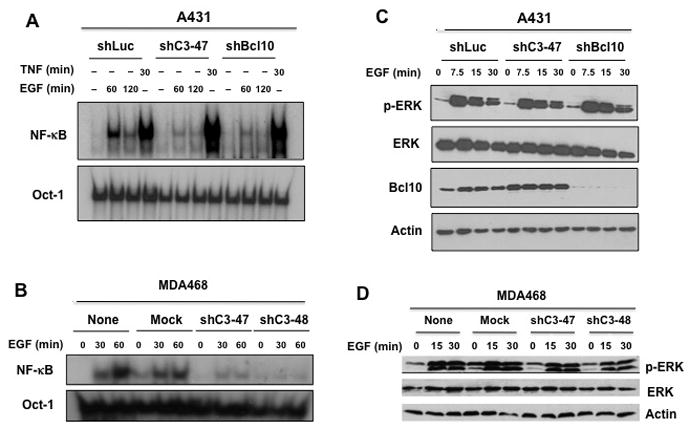
(A) A431 cells transduced with shRNA for luciferase (shLuc), for CARMA3 (shC3), and for Bcl10 (shBcl10) were stimulated with or without 10 ng/ml of EGF or 10 ng/ml of TNFα for indicated times. (B) MDA468 cells (None) or MDA468 cells transduced with control shRNA for GFP (Mock) or with CARMA3 shRNA (shC3-47 and shC3-48) were stimulated with EGF (10 ng/ml) for 0, 30, and 60 minutes. Nuclear extracts were prepared from cells in (A) or (B), and then subjected to EMSA using 32P-labeled NF-κB or Oct-1 probe. (C) A431 cells in (A) were stimulated with EGF (10 ng/ml) for 0, 7.5, 15, and 30 minutes. Total cell lysates were prepared from these cells and then subjected to immunoblotting analysis with indicated antibodies. (D) MDA468 cells in (B) were stimulated with 10 ng/ml of EGF for 0, 15, or 30 minutes. Total cell lysates were prepared from these cells and then subjected to immunoblotting analysis with indicated antibodies.
Knockdown of CARMA3 inhibits the characteristics of cancer cells
To determine whether CARMA3 plays an important role in tumorigenesis, we compared the colony formation ability of A431 cells transduced with control shRNA (shLuc), CARMA3 shRNA (shC3), or Bcl10 shRNA (shBcl10) in soft agar cultures. Cells with control shRNA (shLuc) grew well in soft agar with well developed colonies, whereas the colony number of CARMA3 shRNA- or Bcl10 shRNA-transduced cells was significantly reduced (Fig. 4A). Similarly, the colony formation ability of MDA468 cells with CARMA3 knockdown was also significantly impaired (Supplemental Fig. 5).
Figure 4. CARMA3 deficiency affects the characteristics of cancer cells.
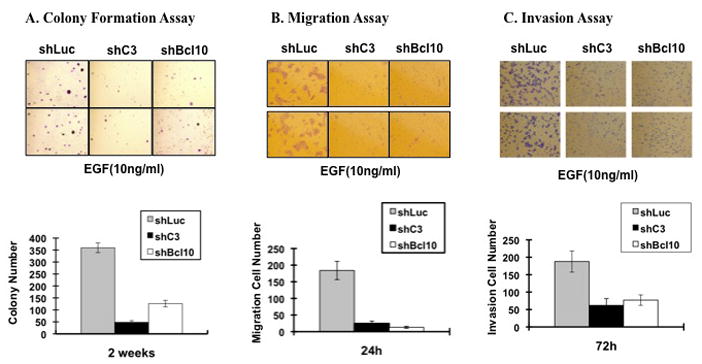
(A) A431 cells transduced with shRNA for control (shLuc), CARMA3 (shC3), or Bcl10 (shBcl10) were subjected to colony formation assay in the present of 10 ng/ml of EGF for two weeks. Two fields per plate were photographed and presented (Upper panels). The bar graph indicates the mean and differences among the groups in colony number, and the standard deviation was calculated from the average of colony numbers in 3 different plates (Lower panels). (B) Above cells were seeded in the upper chamber of Transwell Boyden Chamber, and then 10 ng/ml of EGF were added in the lower chamber to allow them migrating for 24 hours. After staining, random 5 fields were photographed, and 2 of them were presented (Upper panels). The bar graph indicates the mean and difference cell numbers among the photographs, and the standard derivation was calculated from the average of cells in the 5 fields (Lower panels). (C) Above cells were seeded in the upper chamber of Transwell Boyden Chamber with matrix-gel, and then 10 ng/ml of EGF were added in the lower chamber to allow them migrating for 72 hours. After staining, random 5 fields were photographed, and 2 of them were presented (Upper panels). The bar graph indicates the mean and difference cell numbers among the photographs, and the standard derivation was calculated from the average of cells in the 5 fields (Lower panels). All of these experiments were repeated for three times and similar results were obtained.
To further examine the contribution of CARMA3 and Bcl10 to cancer cell malignancy, we examined whether CARMA3 and Bcl10 contributes to migratory and invasive phenotypes of cancer cells, which are characteristics of malignant tumors. Using the Boyden-chamber assay, we found that the migration ability of CARMA3- and Bcl10-knockdown A431 cancer cells was significantly lower than the shLuc control cells (Fig. 4B). Furthermore, we examined invasion ability of these cells in the Matrigel-coated Transwell system, and found that the invasive ability of CARMA3-knockdown cells was also reduced compared with the mock control cells (Fig. 4C). Similarly, the knockdown of CARMA3 in MDA468 cells also reduced the migration and invasion ability of these cells (Supplemental Fig. 6). Together, these results demonstrate that CARMA3 plays an important role in controlling cancer cell migration and invasion.
CARMA3-mediated signaling plays a critical role for cancer cell survival
Another common feature of carcinoma development and growth is the ability of transformed cells to survive under the anchorage-independent “anoikis” condition. To determine if CARMA3 contributes to the resistance of anoikis, we cultured MDA468 cancer cell in 2-hydroxyethyl methacrylate-coated flasks that provide cancer cells an anchorage-independent environment. We found that CARMA3 knockdown MDA468 cells were very sensitive to anoikis and resulted in 97.1% (shC3-47) and 97.5% (shC3-48) of apoptotic cells, respectively, after 24 hours cultured in the anchorage-independent condition (Fig. 5A). In contrast, non-transduced and mock control cells were relatively resistant to anoikis with 60.9% and 50% of apoptotic cells in this culture condition (Fig. 5A). Since NF-κB activation plays a critical role for cell survival, we next determine whether CARMA3-mediated signaling contributes to the survival of cancer cells by examining the apoptosis rate of MDA468 cells in the condition with the treatment of chemotherapy drug, 5-Fluorouracil (5-FU). We found that the apoptosis rate of MDA468 cells with CARMA3 knockdown was significantly increased in the presence of 5-FU (Fig. 5B). Therefore, these results indicate that CARMA3 may contribute to cancer cell survival.
Figure 5. CARMA3 is required for resistance for 5-FU-induced cell death.
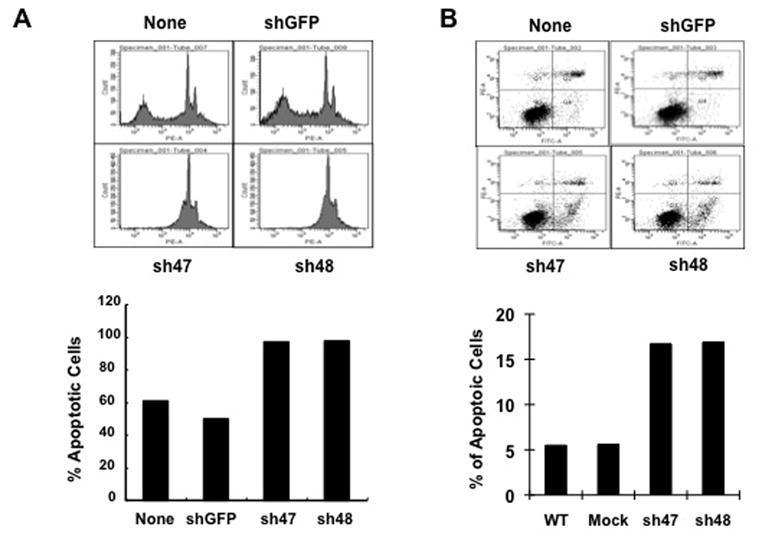
MDA468 cells, without transducing shRNA (None) or with transducing GFP shRNA (mock) or CARMA3 shRNA (sh47 and sh48), were seeded at equal density, grown to sub-confluence, starved, and incubated with EGF (100 μM) for overnight. These cells were then trypsinized, and then cultured in single cell suspensions in 2-hydroxyethyl methacrylate-coated flasks for 24 hours. Cells were harvested, fixed, stained with Annexin V staining, and subjected to FACS analysis (Upper panels). The percentage of Annexin V-positive cells was plotted into the bar graph (Lower panel). (B) Above cells were starved in serum-free media and stimulated with 20 ug/ml of 5-FU and 100 ng/ml of EGF for 48 hours, and then stained with PE-conjugated PI and FITC-labeled Annexin V. The resulted cells were subjected to FACS for apoptosis analysis. The early apoptosis cells (Annexin V-positive Q4 cells) were plotted into the bar graph. These experiments were repeated for three times, and similar results were obtained.
Suppression of CARMA3 expression reduces tumor growth in vivo
To further investigate the role of CARMA3 in tumor growth and progression in vivo, we inoculated A431 cells, with or without CARMA3 shRNA knockdown (shC3 or mock), subcutaneously in nude mice to induce xenograft tumors. After tumors established a week after inoculation, tumor size was measured twice a week and calculated by the formula: π/6[Length (mm)] × [Width (mm)]2. In this xenograft model, A431 cells with CARMA3 shRNA knockdown showed a significant reduction of tumors growth than that of mock control (Fig. 6A). The average tumor weight of mock and CARMA3-knockdown A431 cells was 0.44g ± 0.11 and 0.17g ± 0.12 (P<0.001), respectively, at the day 18 after inoculation (Fig. 6B, and Supplemental Fig. 7). In a similar set of experiment, we also inoculated MDA468 cells with or without CARMA3 knockdown (shC3 or mock) on each side of fat pads of nude mice to induce xenograft tumors. We found that MDA468 cells with CARMA3 knockdown also showed a similar reduction of tumor growth (Supplemental Fig. 8), although the size difference between mock (0.30g ± 0.14) and CARMA3-knockdown (0.19g ± 0.09) in MDA468 cell-induced tumors at the end of 30 days after inoculation of these cells (Supplemental Fig. 8) was less than those in A431 cell-induced tumors (Fig. 6B). Together, the similar effect of CARMA3 depletion on the in vivo tumor growth of both A431 and MDA468 cancer cell lines indicates that CARMA3 plays a critical role in tumor growth and progression.
Figure 6. Suppressing CARMA3 expression inhibits tumor xenografts in nude mice.

(A) A431 cells with control GFP shRNA (mock) or CARMA3 shRNA (shC3) were subcutaneously on each side of male nude mice to develop xenograft tumors. Tumor volume (mm3) was determined twice weekly and plotted against time (days), expressed as mean ± SE (P<0.01). (B) Tumor weight (g) was measured at the end of this study, expressed as mean ± SE (P<0.01).
Discussion
Many literatures have reported that EGFR family members can induce NF-κB activation, and suggest a role of NF-κB in EGFR-induced tumorigenesis. However, the molecular mechanism by which EGFR signaling cascade links to the NF-κB activation remains elusive and controversial. In this study, we provide the genetic evidence showing that CARMA3 and Bcl10, which are essential components for G protein-coupled receptor (GPCR)-induced NF-κB, are also required for EGF-induced NF-κB activation. Similar to the role of GPCR signaling pathway, we have found that CARMA3 deficiency causes a loss in EGF-induced IκBα phosphorylation and NF-κB activation. Furthermore, we have shown that CARMA3 contributes to cancer cell proliferation and survival, and regulates various characteristics of cancer cells, including colony-forming ability, survival, migration, and invasion. Finally, we show that suppressing CARMA3 expression significantly reduced tumor growth in vivo. Based on our current findings, we propose that CARMA3 is functioned downstream of PKC, and forms a complex with Bcl10, which leads to the K63-linked ubiquitination of the IKK complex as proposed for GPCR signaling pathway (28). At the same time, another kinase, such as MEKK3 (37), functions downstream of PKC and phosphorylates the IKK complex. The phosphorylation and ubiquitination of the IKK complex is required for EGFR-induced NF-κB activation (Fig. 7). Together, our studies reveal a genetic link between EGFR-proximal signaling components to the downstream NF-κB activation complex, and demonstrate that CARMA3-associated signaling plays a critical role in cancer progression.
Figure 7. Working model for EGFR-induced NF-κB activation.
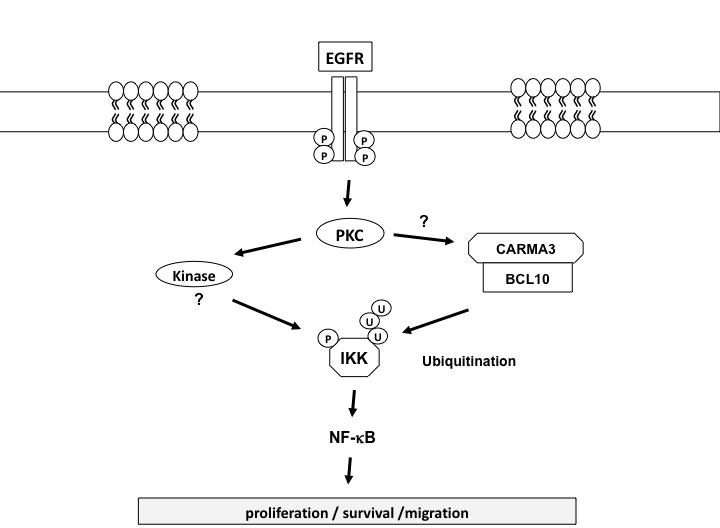
The impact of CARMA3-mediated NF-κB activation on tumor growth in vivo is likely due to several NF-κB-associated mechanisms. First, the cell cycle progression in CARMA3-knockdown cells is slower and cell metabolism rate is decreased. Second, cell apoptosis rate is increased in CARMA3 knockdown cells. These functional outcomes of CARMA3 deficiency likely contribute to tumor growth in vivo directly. The impact of CARMA3-mediated NF-κB activation on tumor growth can also be due to an indirect mechanism. In this case, our preliminary studies have found that suppression of CARMA3 expression blocks EGF-induced expression of several cytokines such as IL-6 (Jiang and Lin, unpublished results). Since IL-6 can activate Stat3 that has been shown to contribute to EGFR-associated cancer cell growth (38, 39). Therefore, CARMA3-mediated cytokine production may also contribute to cancer cell growth in vivo. In addition to cancer cell growth, our studies also indicate that CARMA3 deficiency affects cancer cell migration and invasion. This effect is likely due to CARMA3 deficiency may affect the expression of some genes that are involved in cell migration and invasion. Therefore, it will be interesting to address whether CARMA3 deficiency affect cancer cell metastasis in the future studies.
Our results also indicate the functional similarity among the CARMA family members. Previous studies have been determined that CARMA1, the hematopoietic homolog of CARMA3, functions downstream of the T cell receptor (TCR) and B cell receptor (BCR) to activate NF-κB. Although TCR and BCR do not contain kinase activity, they recruit several tyrosine kinases, Lck, ZAP70, and Syk. Like EGFR signaling pathways, tyrosine kinases in TCR and BCR signaling pathways lead to activation of PKC family members that activate NF-κB through phosphorylating CARMA1. Similar to these signaling pathways, PKC inhibitors also completely abolish EGFR-induced NF-κB activation (data not shown). Therefore, we predict that a specific isoform(s) of PKC may phosphorylate CARMA3 in the EGFR signaling pathway, leading to activation of NF-κB. Identification of such a PKC isoform and determination of phosphorylation sites in CARMA3 may provide the detailed mechanism by which EGF signaling leads to activation of the CARMA3-associated signaling cascade.
In TCR signaling pathway, it has been shown that CARMA1 is recruited into the TCR complex by a hematopoietic tissue-specific adaptor protein, ADAP (40). To determine whether CARMA3 is directly or indirectly recruited into the EGFR complex, we have immunoprecipitated EGFR complex from A431 cells following EGF stimulation. However, we were unable to detect CARMA3 in this immunoprecipitated complex (data not shown), suggesting that CARMA3 may be also recruited to EGFR complex through an adaptor molecule(s) in epithelial cells. Therefore, it will require a more systemic approach to identify the molecule that links CARMA3 to the EGFR complex in the future studies.
It has been shown that CARMA1 plays a critical role for a subset of Diffused Large B Cell Lymphoma (DLBCL), and suppression the expression is highly sensitive for the survival of these lymphoma cells (41). In addition, mutations of CARMA1 have been found in a significant part of DLBCL patients (42, 43). It is possible that similar mutations in CARMA3 or overexpression of CARMA3 may result in a constitutively activated NF-κB, and contribute to tumor progression of cancer in epithelial cells. Therefore, it will be interested to examine whether there are any mutation(s) or overexpression of CARMA3 in non-hematopoietic malignant cells in the future studies.
Mutation or aberrant expression of receptor tyrosine kinases (RTK) has been appreciated as a causative mechanism of tumorigenesis in multiple types of cancer including breast, lung, and brain cancers for a long time. Despite this, inhibitors of these receptors have yet to reach their full clinical potential. Furthermore, targeting common downstream signaling pathways of RTKs will be an alternative therapeutic approach for different types of cancers. Therefore, identification of common signaling pathways downstream of these receptors would provide the molecular insight for designing the therapeutic agents for various cancers. It has been shown that multiple RTKs, such as PDGFR, FGFR, and IGFR, can induce NF-κB activation. Since these RTKs share similar downstream signaling cascades with EGFR, we predict that these RTKs may also utilize the CBM complex to activate NF-κB. However, due to technical difficulty, we have not been able to shown these receptors can activate NF-κB in the wild type MEF cells (data not shown). Therefore, the activation of NF-κB by these receptors may be tissue-specific events. It will need to identify and use a specific cell type to determine whether these RTKs also utilize the CARMA3-mediated pathway to activate NF-κB.
Additionally, various hematological malignancies depend upon RTK signaling. For example, some of multiple myeloma depends on FGFR3, and Acute Myeloid Leukemia (AML) depends heavily on FLT3. Both FGFR3 and FLT3 have been implicated to activate NF-κB. Therefore, it will be important to determine whether NF-κB activation contributes to tumorigenesis of multiple myeloma and AML. Since CARMA1, but not CARMA3, is expressed in cells from the myeloid lineage, it is possible that CARMA1 may play a role in mediating NF-κB activation downstream of RTKs in hematopoietic cells. Therefore, it will be important to determine whether CARMA1 plays an essential role in RTK signaling pathway in hematopietic cells. Such studies will likely provide novel therapeutic targets for treatment of various cancers.
Supplementary Material
Acknowledgments
This work was partially supported by NIH grants, RO1GM079451 and RO1GM065899, and an institutional pilot grant from MD Anderson Cancer Center to X. Lin.
References
- 1.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–62. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 2.Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest. 2001;107:241–6. doi: 10.1172/JCI11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 4.Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 6.Olashaw NE, Kowalik TF, Huang ES, Pledger WJ. Induction of NF-kappa B-like activity by platelet-derived growth factor in mouse fibroblasts. Mol Biol Cell. 1992;3:1131–9. doi: 10.1091/mbc.3.10.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye RD. Regulation of nuclear factor kappaB activation by G-protein-coupled receptors. J Leukoc Biol. 2001;70:839–48. [PubMed] [Google Scholar]
- 8.Sun L, Carpenter G. Epidermal growth factor activation of NF-kappaB is mediated through IkappaBalpha degradation and intracellular free calcium. Oncogene. 1998;16:2095–102. doi: 10.1038/sj.onc.1201731. [DOI] [PubMed] [Google Scholar]
- 9.Biswas DK, Cruz AP, Gansberger E, Pardee AB. Epidermal growth factor-induced nuclear factor kappa B activation: A major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc Natl Acad Sci U S A. 2000;97:8542–7. doi: 10.1073/pnas.97.15.8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaliman P, Canicio J, Testar X, Palacin M, Zorzano A. Insulin-like growth factor-II, phosphatidylinositol 3-kinase, nuclear factor-kappaB and inducible nitric-oxide synthase define a common myogenic signaling pathway. J Biol Chem. 1999;274:17437–44. doi: 10.1074/jbc.274.25.17437. [DOI] [PubMed] [Google Scholar]
- 11.Pons S, Torres-Aleman I. Insulin-like growth factor-I stimulates dephosphorylation of ikappa B through the serine phosphatase calcineurin (protein phosphatase 2B) J Biol Chem. 2000;275:38620–5. doi: 10.1074/jbc.M004531200. [DOI] [PubMed] [Google Scholar]
- 12.Kim HJ, Litzenburger BC, Cui X, et al. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol Cell Biol. 2007;27:3165–75. doi: 10.1128/MCB.01315-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 14.Byrd VM, Ballard DW, Miller GG, Thomas JW. Fibroblast growth factor-1 (FGF-1) enhances IL-2 production and nuclear translocation of NF-kappaB in FGF receptor-bearing Jurkat T cells. J Immunol. 1999;162:5853–9. [PubMed] [Google Scholar]
- 15.Bushdid PB, Chen CL, Brantley DM, et al. NF-kappaB mediates FGF signal regulation of msx-1 expression. Dev Biol. 2001;237:107–15. doi: 10.1006/dbio.2001.0356. [DOI] [PubMed] [Google Scholar]
- 16.Vandermoere F, El Yazidi-Belkoura I, Adriaenssens E, Lemoine J, Hondermarck H. The antiapoptotic effect of fibroblast growth factor-2 is mediated through nuclear factor-kappaB activation induced via interaction between Akt and IkappaB kinase-beta in breast cancer cells. Oncogene. 2005;24:5482–91. doi: 10.1038/sj.onc.1208713. [DOI] [PubMed] [Google Scholar]
- 17.Biswas DK, Iglehart JD. Linkage between EGFR family receptors and nuclear factor kappaB (NF-kappaB) signaling in breast cancer. J Cell Physiol. 2006;209:645–52. doi: 10.1002/jcp.20785. [DOI] [PubMed] [Google Scholar]
- 18.Biswas DK, Dai SC, Cruz A, Weiser B, Graner E, Pardee AB. The nuclear factor kappa B (NF-kappa B): a potential therapeutic target for estrogen receptor negative breast cancers. Proc Natl Acad Sci U S A. 2001;98:10386–91. doi: 10.1073/pnas.151257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biswas DK, Martin KJ, McAlister C, et al. Apoptosis caused by chemotherapeutic inhibition of nuclear factor-kappaB activation. Cancer Res. 2003;63:290–5. [PubMed] [Google Scholar]
- 20.Blonska M, Lin X. NF-kB signaling pathways regulated by CARMA family of scaffold proteins. Cell Research. 2011 doi: 10.1038/cr.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaide O, Martinon F, Micheau O, Bonnet D, Thome M, Tschopp J. Carma1, a CARD-containing binding partner of Bcl10, induces Bcl10 phosphorylation and NF-kappaB activation. FEBS Lett. 2001;496:121–7. doi: 10.1016/s0014-5793(01)02414-0. [DOI] [PubMed] [Google Scholar]
- 22.McAllister-Lucas LM, Inohara N, Lucas PC, et al. Bimp1, a MAGUK family member linking protein kinase C activation to Bcl10-mediated NF-kappaB induction. J Biol Chem. 2001;276:30589–97. doi: 10.1074/jbc.M103824200. [DOI] [PubMed] [Google Scholar]
- 23.Wang L, Guo Y, Huang WJ, et al. Card10 is a novel caspase recruitment domain/membrane-associated guanylate kinase family member that interacts with BCL10 and activates NF-kappa B. J Biol Chem. 2001;276:21405–9. doi: 10.1074/jbc.M102488200. [DOI] [PubMed] [Google Scholar]
- 24.Bertin J, Wang L, Guo Y, et al. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J Biol Chem. 2001;276:11877–82. doi: 10.1074/jbc.M010512200. [DOI] [PubMed] [Google Scholar]
- 25.Wang D, You Y, Case SM, et al. A requirement for CARMA1 in TCR-induced NF-kappa B activation. Nat Immunol. 2002;3:830–5. doi: 10.1038/ni824. [DOI] [PubMed] [Google Scholar]
- 26.Gaide O, Favier B, Legler DF, et al. CARMA1 is a critical lipid raft-associated regulator of TCR-induced NF-kappa B activation. Nat Immunol. 2002;3:836–43. doi: 10.1038/ni830. [DOI] [PubMed] [Google Scholar]
- 27.Pomerantz JL, Denny EM, Baltimore D. CARD11 mediates factor-specific activation of NF-kappaB by the T cell receptor complex. EMBO J. 2002;21:5184–94. doi: 10.1093/emboj/cdf505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grabiner BC, Blonska M, Lin PC, et al. CARMA3 deficiency abrogates G protein-coupled receptor-induced NF-{kappa}B activation. Genes Dev. 2007;21:984–96. doi: 10.1101/gad.1502507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McAllister-Lucas LM, Ruland J, Siu K, et al. CARMA3/Bcl10/MALT1-dependent NF-kappaB activation mediates angiotensin II-responsive inflammatory signaling in nonimmune cells. Proc Natl Acad Sci U S A. 2007;104:139–44. doi: 10.1073/pnas.0601947103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahanivong C, Chen HM, Yee SW, Pan ZK, Dong Z, Huang S. Protein kinase C alpha-CARMA3 signaling axis links Ras to NF-kappa B for lysophosphatidic acid-induced urokinase plasminogen activator expression in ovarian cancer cells. Oncogene. 2008;27:1273–80. doi: 10.1038/sj.onc.1210746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang D, You Y, Lin PC, et al. Bcl10 plays a critical role in NF-kappaB activation induced by G protein-coupled receptors. Proc Natl Acad Sci U S A. 2007;104:145–50. doi: 10.1073/pnas.0601894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klemm S, Zimmermann S, Peschel C, Mak TW, Ruland J. Bcl10 and Malt1 control lysophosphatidic acid-induced NF-kappaB activation and cytokine production. Proc Natl Acad Sci U S A. 2007;104:134–8. doi: 10.1073/pnas.0608388103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banan A, Zhang LJ, Farhadi A, Fields JZ, Shaikh M, Keshavarzian A. PKC-beta1 isoform activation is required for EGF-induced NF-kappaB inactivation and IkappaBalpha stabilization and protection of F-actin assembly and barrier function in enterocyte monolayers. Am J Physiol Cell Physiol. 2004;286:C723–38. doi: 10.1152/ajpcell.00329.2003. [DOI] [PubMed] [Google Scholar]
- 34.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–26. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang Z, Wu T, Lou H, et al. Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer Res. 2004;64:4302–8. doi: 10.1158/0008-5472.CAN-03-3958. [DOI] [PubMed] [Google Scholar]
- 36.Albini A, Iwamoto Y, Kleinman HK, et al. A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res. 1987;47:3239–45. [PubMed] [Google Scholar]
- 37.Sun W, Li H, Yu Y, et al. MEKK3 is required for lysophosphatidic acid-induced NF-kappaB activation. Cell Signal. 2009;21:1488–94. doi: 10.1016/j.cellsig.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia R, Bowman TL, Niu G, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20:2499–513. doi: 10.1038/sj.onc.1204349. [DOI] [PubMed] [Google Scholar]
- 39.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Medeiros RB, Burbach BJ, Mueller KL, et al. Regulation of NF-kappaB activation in T cells via association of the adapter proteins ADAP and CARMA1. Science. 2007;316:754–8. doi: 10.1126/science.1137895. [DOI] [PubMed] [Google Scholar]
- 41.Ngo VN, Davis RE, Lamy L, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–10. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- 42.Lenz G, Davis RE, Ngo VN, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319:1676–9. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- 43.Compagno M, Lim WK, Grunn A, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–21. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.