

Abstract
Two cocrystal X-ray structures of the exceptionally potent α-ketoheterocycle inhibitor 1 (Ki = 290 pM) bound to a humanized variant of rat fatty acid amide hydrolase (FAAH) are disclosed, representing noncovalently and covalently bound states of the same inhibitor with the enzyme. Key to securing the structure of the noncovalently bound state of the inhibitor was the inclusion of fluoride ion in the crystallization conditions that is proposed to bind the oxyanion hole precluding inhibitor covalent adduct formation with stabilization of the tetrahedral hemiketal. This permitted the opportunity to detect important noncovalent interactions stabilizing the binding of the inhibitor within the FAAH active site independent of the covalent reaction. Remarkably, noncovalently bound 1 in the presence of fluoride appears to capture the active site in the same “in action” state with the three catalytic residues Ser241–Ser217–Lys142 occupying essentially identical positions observed in the covalently bound structure of 1, suggesting that this technique of introducing fluoride may have important applications in structural studies beyond inhibiting substrate or inhibitor oxyanion hole binding. Key insights to emerge from the studies include the observations that noncovalently bound 1 binds in its ketone (not gem diol) form, that the terminal phenyl group in the acyl side chain of the inhibitor serves as the key anchoring interaction overriding the intricate polar interactions in the cytosolic port, and that the role of the central activating heterocycle is dominated by its intrinsic electron-withdrawing properties. These two structures are also briefly compared with five X-ray structures of α-ketoheterocycle-based inhibitors bound to FAAH recently disclosed.
Introduction
Fatty acid amide hydrolase (FAAH)1,2 hydrolyzes endogenous lipid primary amides3 and ethanolamides4–7 including anandamide8–10 and oleamide,11–14 serving the role of regulating signaling fatty acid amides at their sites of action (Figure 1A).5,15 Preventing the degradation of such endogenous signaling molecules provides an attractive approach for the development of next-generation therapeutics that may avoid the side effects associated with direct agonists of cell surface receptors including cannabinoid receptor agonists.15 Since FAAH inhibition only potentiates an activated signaling pathway, increasing the endogenous levels of released lipid signaling molecules at their sites of action, it provides a temporal and spatial pharmacological control that is not available to a classical receptor agonist.
Figure 1.

(A) Endogenous substrates of FAAH. (B) FAAH inhibitors 1–6.
FAAH is the only well-characterized mammalian member of the amidase signature class of serine hydrolases that possesses an unusual Ser–Ser–Lys catalytic triad (Ser241–Ser217–Lys142 in FAAH). Its catalytic mechanism involves the formation of a tetrahedral intermediate derived from nucleophilic attack of the catalytic Ser241 residue on the carbonyl group of the substrate. The tetrahedral intermediate collapses to release the amine and provides the enzyme-bound acyl intermediate. Lys142 acts as a general base-general acid, mediating the deprotonation of Ser241 and the subsequent protonation of the leaving group that are shuttled through Ser217.16 The reaction terminates with hydrolysis of the acyl intermediate and release of the free fatty acid. FAAH hydrolyzes both ester and amide substrates16 with primary amides being hydrolyzed 2-fold faster than ethanolamides,6 and it preferentially hydrolyzes arachidonoyl or oleoyl substrates (arachidonoyl > oleoyl, 3-fold).6,7 In addition to its atypical catalytic core, FAAH contains a series of channels and cavities that are important. These include the membrane access channel (MAC) connecting the active site to an opening located at the membrane anchoring face of the enzyme, the cytosolic port that allows exit of hydrophilic products from the active site, and the acyl chain-binding pocket (ABP) that interacts with the substrate acyl chain during the catalytic reaction.17,18
Two major classes of inhibitors have been disclosed that provide opportunities for therapeutic development.19 One class is the aryl carbamates and ureas20–31 that irreversibly acylate the FAAH active site Ser241.30 A second class is the α-ketoheterocycle-based inhibitors that bind through reversible hemiketal formation with the active site serine. Following early efforts that defined FAAH as a serine hydrolase capable of inhibition by substrate-inspired compounds bearing electrophilic carbonyls,32,33 we described the systematic exploration of an extensive series of α-ketoheterocycle-based inhibitors.34–44 In these efforts, efficacious FAAH inhibitors were developed and used to validate FAAH as a new therapeutic target for the treatment of pain and inflammatory disorders.44
In recent disclosures, we reported the X-ray crystal structures of five α-ketoheterocycle inhibitors, 2 (OL-135) and 3–6, bound to FAAH (Figure 1B).18 These structures confirmed covalent attachment through nucleophilic attack of Ser241 on the inhibitor electrophilic carbonyl, captured the catalytic residues in a unique “in action” state, revealed an unusual Ser217 OH–π H-bond to the activating heterocycle distinct from active site interactions observed in work with serine proteases, and identified a potential anion binding site in the cytosolic port. In addition to defining key active site interactions stabilizing inhibitor binding, they also defined a distinguishing acyl chain-binding pocket/membrane access channel flexibility, and revealed a prominent role for cytosolic port-bound solvent (H2O) in stabilizing inhibitor binding.
Herein, we report two X-ray crystal structures of one of the most potent and selective α-ketoheterocycle inhibitors of FAAH disclosed to date, the α-ketooxadiazole 137 (Ki = 290 pM, Figure 1B), representing a covalently and a noncovalently bound state of the same inhibitor with the enzyme. Key to securing the structure of the noncovalently bound state of the inhibitor and constituting a general technique to utilize with other enzymes, fluoride ion included in the crystallization conditions is proposed to bind the oxyanion hole precluding covalent adduct formation. Complementing the recently disclosed studies of the inhibitors 2–6 and reinforcing the key features of the inhibitor binding observed in their cocrystal structures,18 the comparison of the two states of the bound inhibitor 1 further revealed that such inhibitors noncovalently bind the active site of FAAH in their ketone versus gem diol form, established that the terminal phenyl group in the acyl tail of the inhibitor serves as the key anchoring interaction overriding those found in the cytosolic port, confirmed that the role of the central activating heterocycle is dominated by its intrinsic electron-withdrawing properties, and clarified subtle interactions responsible for the exceptionally potent activity of the oxadiazole-based inhibitors important for future design. Just as significantly, noncovalently bound 1 crystallized in the presence of fluoride ion appears to capture the active site in the same “in action” state with the three catalytic residues Ser241–Ser217–Lys142 occupying essentially identical positions observed in the covalently bound structure of 1, suggesting that this technique of introducing fluoride may have important applications in structural studies beyond inhibiting substrate or inhibitor oxyanion hole binding.
Results and Discussion
Cocrystal Structures
The covalently and noncovalently bound structures of FAAH with 1 were solved at a resolution of 2.20 Å and 1.78 Å, respectively. The relatively high resolution of these structures, especially that of the noncovalently bound state of the inhibitor, resulted in an unambiguous assignment of the inhibitor in the active sites and led to Rfree values of 19.0% and 17.6%, respectively. The structure refinement statistics are provided in Table 1. The overall structures of FAAH are nearly identical to the previously determined structures with FAAH bound to 2–6 (root mean squared deviations based on Cα atoms is less than 0.3 Å) and the small differences are restricted to the subtle active site distinctions discussed below. Unbiased electron density maps defined the orientation of the inhibitor in the active site and confirmed that the first, which was conducted with crystallization in the absence of fluoride, is covalently bound to the catalytic Ser241 through reaction with the electrophilic carbonyl whereas the second, which was conducted with crystallization in the presence of NaF (100 mM), is noncovalently bound in the active site in its ketone versus gem diol state and fluoride ion is proposed to occupy the oxyanion hole.
Table 1.
| FAAH–1 noncovalent interaction | FAAH–1 covalent interaction | |
|---|---|---|
| X-ray source | SSRL BL11-1 | APS-GM/CA-CAT |
| PDB ID | 3PPM | 3PR0 |
| Crystal data | ||
| Space group | P3321 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 103.69, 103.69, 254.71 | 102.24, 105.47, 149.15 |
| α=β, γ (°) | 90.0, 120.0 | 90.0, 90.0 |
| Data collection | ||
| Processing software | HKL2000 | XDS |
| Wavelength (Å) | 0.97945 | 1.03320 |
| Resolution (Å) | 40.0-1.78(1.81-1.78) | 30.0-2.20(2.26-2.20) |
| Rmerge (%) | 5.8(45.4) | 8.8(67.1) |
| Wilson B factor | 22.4 | 36.4 |
| I/σI | 20.0(2.0) | 11.8(2.4) |
| Completeness (%) | 99.6(99.0) | 99.9(99.9) |
| No. of unique reflections | 151855 | 82372 |
| Redundancy | 3.9(2.8) | 5.2(5.0) |
| Refinement | ||
| Resolution (Å) | 1.78(1.80-1.78) | 2.20(2.23-2.20) |
| Rwork/Rfree (%) | 15.1(27.6)/17.6(29.3) | 15.0(21.7)/19.0(25.2) |
| No. atoms | 9708 | 9150 |
| Protein | 8558 | 8506 |
| Ligand/ion | 96 | 65 |
| Water | 1054 | 579 |
| Average B overall (Å2) | 25.5 | 40.4 |
| R.m.s.d. bond length (Å) | 0.007 | 0.007 |
| R.m.s.d. bond angle (°) | 1.081 | 1.063 |
| Ramachandran Plot | ||
| Preferred (%) | 97.3 | 97.1 |
| Allowed (%) | 2.7 | 2.9 |
| Outliers (%) | 0.0 | 0.0 |
Values in parenthesis are referring to highest resolution shell data. Rmerge is defined as: ΣhΣi(|Ii(h) − <I(h)>|)/ΣhΣiIi(h), where Ii(h) is the ith integrated intensity of a given reflection and <I(h)> is the weighted mean of all measurements of I(h).
Inhibition of FAAH by Fluoride Ion
FAAH inhibition assays, as used to measure the activity of the α-ketoheterocycle inhibitors, were conducted in the presence of fluoride and chloride anions (NaF and NaCl). Fluoride anion (Ki = 45 mM) was found to competitively and reversibly inhibit the enzymatic hydrolysis of the endogenous substrate oleamide whereas chloride anion showed no inhibition up to 1000 mM, indicating that fluoride, but not chloride, can inhibit catalysis at relatively low concentrations presumably by competitively binding in the oxyanion hole. The crystallization conditions used to secure noncovalently bound 1 contained 100 mM of NaF.
Covalently Bound 1
The phenhexyl chain of covalently bound 1 was found to overlay precisely with the phenhexyl chains of 2–4 benefiting from key interactions with the residues lining the hydrophobic channel (Figure 2). The π-system of the bound phenyl group is engaged in an aromatic CH–π interaction with an aryl ring hydrogen of Phe381, mimicking the stabilizing interactions that support unsaturated fatty acid side chain binding. Phe192 is oriented to provide a second weak CH–π interaction with the terminal phenyl group of 1. The mobile residues Phe432, Met495, and Met436 adopt the conformation that leads to a broadened and open membrane access channel with closure of the acyl chain-binding pocket.17,18 Phe432 makes a key aryl CH–π contact with the inhibitor phenyl ring while the sulfur lone pair electrons of the two methionines are oriented towards the bound phenyl hydrogens engaging in two aromatic CH–π interactions. These latter three residues, Phe381 and perhaps Phe192 provide key anchoring interactions for binding inhibitors related to 1–3. The comparison of the covalent complex of 1 with the complexes with 2–4 reveals that the bound disposition of the phenhexyl chain is identical, indicates that its positioning is independent of the choice of central activating heterocycle or its attached substituents, and suggests that the placement of phenyl group in this special location serves as a key anchoring interaction for such inhibitors.
Figure 2.
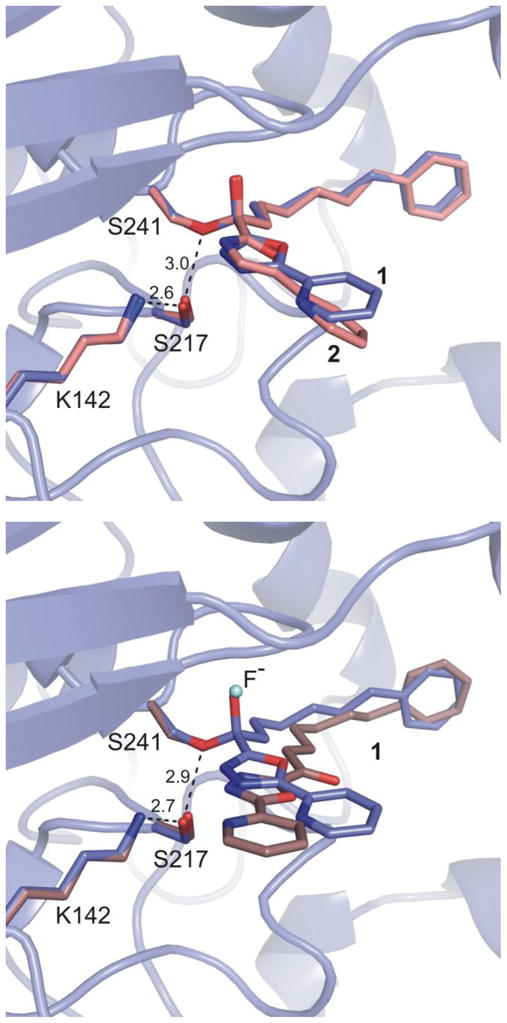
Top: Overlay of inhibitor 1 in the two bound states. Bottom: Overlay of covalently bound 1 and 2 (OL-135). Covalently bound inhibitor 1 is shown in light blue, noncovalently bound inhibitor 1 in brown, and 2 in pink. Interatomic distances of the noncovalent structure (top) and covalent structure (bottom) are shown with dashes and measured in Å.
The electron density at the active site established that 1forms a covalent complex with FAAH like 2–6, resulting from Ser241 attack on the electrophilic carbonyl (Figure 3). The resulting hemiketal alkoxide binds in the oxyanion hole defined by the four main-chain amide N-H groups of Ile238, Gly239, Gly240, and Ser241 at the center of the oxyanion hole, and with the shortened distances of 2.8, 2.8, 3.25, and 2.74 Å, respectively, indicative of oxyanion (–O−) versus protonated hemiketal (–OH) binding. Its axis is perpendicular to the plane of the four amino acids, the γ-oxygen of Ser241 and the bound carbon of the inhibitor is pulled towards the oxyanion hole, and the relevant atoms of 1 and the preceding five inhibitors (2–6) are virtually superimposable, Figure 4.
Figure 3.
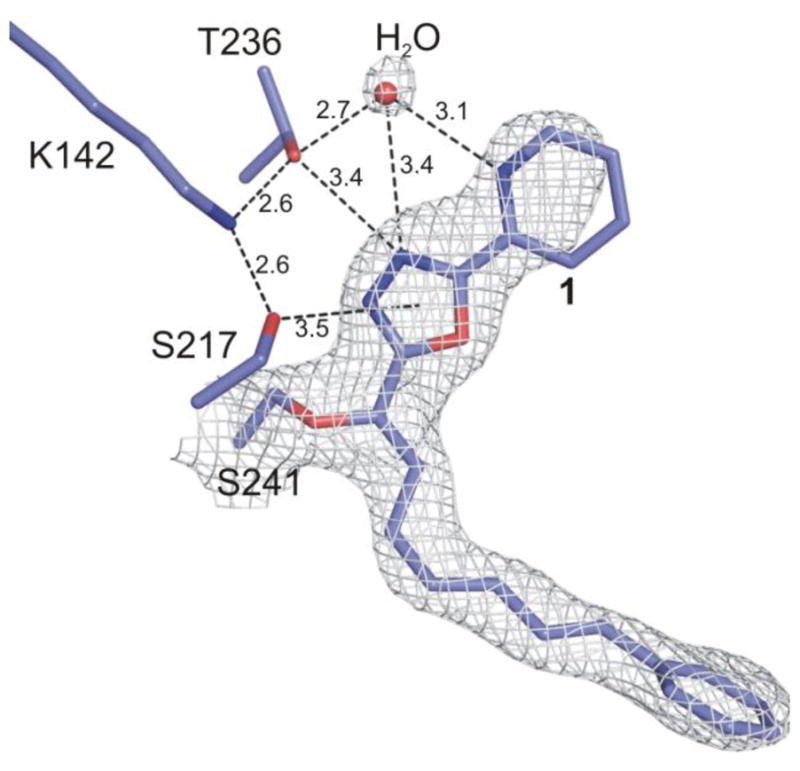
Density for covalently bound 1 at 1.0σ contour is shown with white meshes
Figure 4.
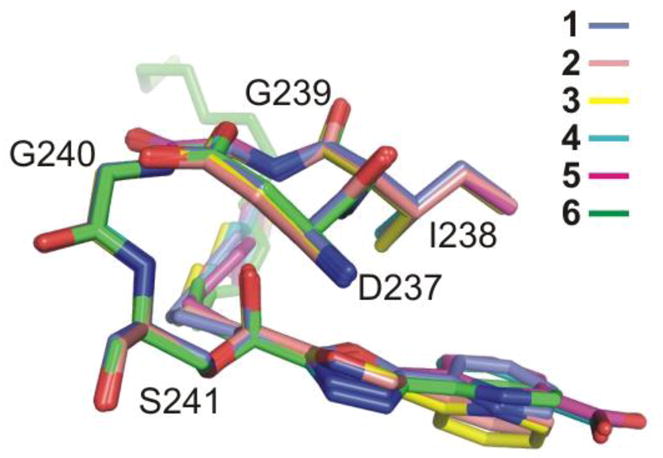
Superposition of the oxyanion holes for covalently bound 1 and the five prior structures of FAAH covalently bound to oxa(dia)zole inhibitors 2–6.
The oxadiazole-based inhibitors are uniquely potent relative to other activating α-ketoheterocycles, being >10-fold more potent than the corresponding oxazoles. Consequently, the most interesting insights emerged from examining the interaction of 1 with the cytosolic port of the enzyme. For the covalently bound complex of 1, the pyridine and oxadiazole are essentially coplanar (≤ 2° dihedral angle) and the pyridyl ring is oriented in the same direction observed with 2–6 and its nitrogen lies syn to the oxadiazole N4 and anti to the oxadiazole O. A distinction between 1 and the bound oxazoles 2–5 is the coplanar arrangement of the oxadiazole and pyridine (< 2° vs 15–20°), reflecting the removal of the destabilizing torsional strain (N4 vs C4H) inherent in the oxazole coplanar arrangement that has been estimated to reduce binding 1–2 kcal/mol.45 This likely contributes to the enhanced oxadiazole versus oxazole binding. The pyridyl nitrogen atom lies very close to its position found in 2 and 3 and identical to its location found with 6, and it is engaged in the same H-bond with the ordered cytosolic port water (3.0 Å vs 2.8–3.1 Å in 2–3). The second nitrogen of the oxadiazole that is not found in the oxazole inhibitors (N4 vs C4) may be H-bonded to this same cytosolic port water (3.5 Å), contributing to the subtle reorientation of the biaryl axis of 1 versus 2 and 3. The net result is that the activating heterocycle and attached pyridine substituent are rotated closer to the catalytic triad including Lys142 as well as Thr236 than observed with 2 or 3. Although the geometry is not optimally aligned, the Thr236 OH is now in plane and closer to the oxadiazole N4 nitrogen potentially providing another, albeit weakly stabilizing (3.4 Å), H-bond. This intricate H-bond network of the cytosolic port water and Thr236 OH with the pyridyl substituent and activating oxadiazole N4 also likely contributes to the >10-fold increase in inhibitor potency observed with the 1,3,4-oxadiazoles37,42 and its closely related isomers. This does require a slight reorientation of the oxadiazole plane relative to that observed with oxazole and this appears to partially disrupt the more ideal Ser217 OH–π H-bond to the activating heterocycle observed with 2 and 3 (2.7–3.0 Å). However, the less optimal geometry for this Ser217 OH–π H-bond (3.5 Å) is compensated for by the dual hydrogen bond interaction of the oxadiazole with the key cytosolic port bound water.
Noncovalently Bound 1
Unbiased electron density maps defined the orientation of the inhibitor in the active site and confirmed that the second structure represents 1 noncovalently bound in the active site in its ketone versus gem diol state with fluoride ion proposed to occupy the oxyanion hole (Figure 5). The binding of the phenhexyl chain of noncovalently bound 1 extends into the same cavity up to and terminating at the proximal portion of the channel leading to the membrane (Figure 2). The terminal phenyl is bound at precisely the same location and in a nearly identical orientation as with its covalently bound complex (Figure 2). The terminal phenyl group is now rotated ca. 18° in the plane of the ring, it is tilted only slightly (ca. 10°), and its centroid is displaced by only ≤ 0.5 Å (Figure 2). These minor changes in the orientation of the bound terminal phenyl group do not alter the nature or the extent of the key interactions with the enzyme. In fact, the protein conformation in this region with noncovalently bound 1 is practically identical to that found with covalently bound 1, including the adoption of the closed acyl chain-binding pocket. The intervening linking hexyl chain also adopts a nearly fully extended conformation with one intervening gauche turn that occurs at the same chain site, the terminal three methylenes of the linking chain of covalently and noncovalently bound 1 occupy similar sites, but the initial three methylenes and the carbonyl are now displaced away from Ser241 and the oxyanion hole. The comparison of the covalent and noncovalent complexes in this region firmly establishes that the terminal phenyl group serves as the key anchoring interaction for such inhibitors.
Figure 5.
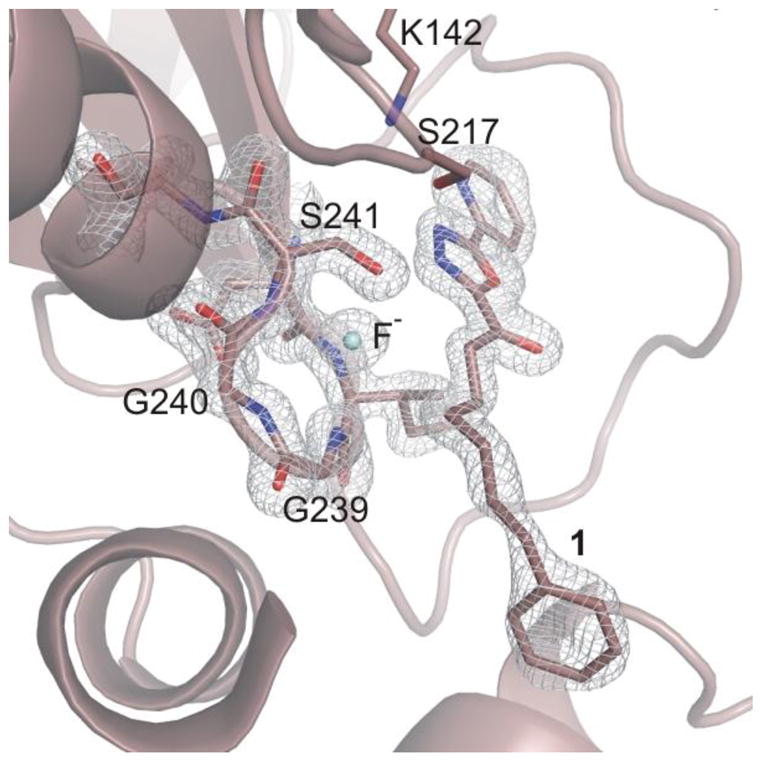
FAAH active site with noncovalently bound 1 and the proposed bound fluoride. The 2Fo-Fc electron density maps for noncovalently bound 1 at 1.5σ contour is shown with white meshes.
As described earlier, fluoride anion is proposed to occupy the oxyanion hole when the crystallization of 1 with FAAH was conducted in the presence of sodium fluoride (100 mM). Under these conditions, a strong positive density (> 6σ in the 2Fo-Fc maps) was found for a noncovalently bound, single atom in the oxyanion hole. Because fluorine does not have a strong anomalous signal, the unambiguous crystallographic assignment cannot be definitively made. Modeling of either an oxygen atom (water molecule or hydroxide ion), a chloride ion, and a fluoride ion were examined during refinement. Interpretation of the electron density at the oxyanion hole considered not only the properties of the atoms, i.e. atomic radii and atomic scattering factors,46 but especially their occupancy and thermal motion. Examination of difference maps and B-factors (~30 A2 vs 14–17 A2 for the surrounding backbone atoms) led to exclusion of the presence of a chloride ion. The temperature factor variance analysis coupled to the strong bonding environment supports fluoride ion coordination at the OAH, with a B-factor of ca. 17 A2 (vs 14–17 A2 for the interacting atoms of the surrounding protein backbone) compared to 13 A2 of a modeled water. Finally, the formation of a hydroxide ion that may coordinate to the OAH is rather unlikely, especially at the acidic pH of the crystallization buffer. Collectively, the experimental enzymatic inhibition of FAAH by fluoride and not chloride ion and the specific crystallization conditions utilized (presence of fluoride and pH) combined with the interpretation of the electron density maps and the B-factors of the atoms at the catalytic core, the electron density found at the OAH is assigned to a fluoride ion in tight coordination to the four amide groups in the oxyanion hole, preventing the covalent binding of inhibitor 1.
The fluoride anion interacts with the oxyanion hole backbone amide N-H’s of Ile238 Ser241 (2.6, 2.6, 3.1 and 2.7 Å, respectively) (Figure 6). The inhibitor 1 binds in the ketone state (Figure 5), not as a gem diol, the carbonyl is nearly coplanar with the activating heterocycle (ca. 15°) and syn to the oxadiazole oxygen (not N3), and the carbonyl carbon is displaced away from the oxyanion hole and is located 2.9–3.0 Å away from its position observed with covalently bound 1. Beautifully, it is still positioned for nucleophilic attack by Ser241, requiring the carbonyl rotate ca. 110–120° to enter the oxyanion hole passing through a conformation that places Ser241 at an angle stereoelectronically required for nucleophilic addition (105 ±5°). One of the most provocative observations to emerge from the studies is the ketone versus gem diol binding of 1 at the active site of FAAH indicating that such inhibitors, even with the most powerful electron-withdrawing heterocycles, are not prone to gem diol or nonselective hemiketal formation with proteins. Rather, the results would seem to suggest that they require oxyanion hole stabilization of the adduct for observation.
Figure 6.
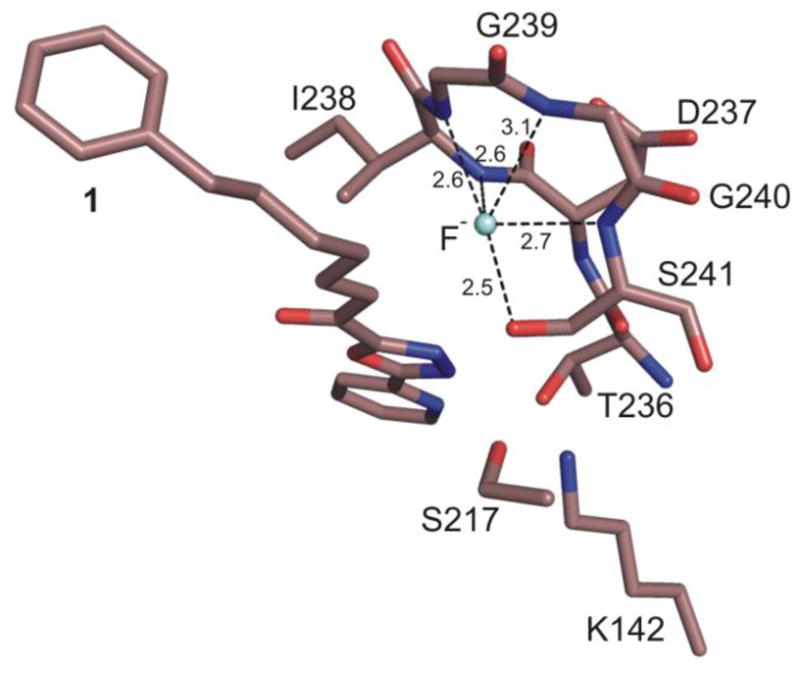
Fluoride binding in the oxyanion hole of the noncovalent FAAH–1 structure.
The covalent structures of the α-ketoheterocycles examined to date reveal that the FAAH catalytic triad is trapped in an interrupted “in action” state with Lys142 H-bonded to Ser217 that in turn was engaged in an unusual OH–π H-bond to the activating heterocycle.18 An ordered cytosolic port bound water was found to mediate an indirect and flexible H-bond interaction between Thr236 and the pyridyl nitrogen of the heterocycle substituent locking it into one of two possible orientations and providing a second anchoring interaction for such inhibitors. In turn, Thr236 was H-bonded to the protonated Lys142, an integral residue in the Ser241–Ser217–Lys142 catalytic triad. Remarkably, noncovalently bound 1 captures the active site in the same state with the three catalytic residues occupying essentially identical positions as observed in the inhibitor covalently-bound structures (Figure 2). Thus, the fluoride–oxyanion hole binding also captures the enzyme in an “in action” state by virtue of its apparent H-bonding to Ser241 (2.5 Å) and suggests that this technique of introducing fluoride ion may have additional important applications in structural studies for examining apo or Michaelis complex states of enzymes.
With noncovalently bound 1, the central heterocycle and the attached pyridine are again essentially coplanar (dihedral angle is 3–6°) and they are displaced relative to covalently bound 1, but their relative orientations are identical with the pyridyl nitrogen syn to the oxadiazole N4. The planes of the two oxadiazoles are 10–15° apart, but the position of the ring centroids differ by only 0.9 Å. The most significant change is in the locations of the pyridine ring. Because of its displacement, the pyridyl nitrogen is now directly H-bonded to Thr236 (2.7 Å) and occupies the site of the cytosolic port water molecule that mediates the key anchoring H-bond between Thr236 and the pyridyl nitrogen in the structure of covalently bound 1 (Figure 7). Notably, the location of the active site Thr236 between the two structures is essentially unaltered, and it is the inhibitor that moves in its location. Both Ser217 and Ser241 in the active site remain in intimate H-bond contact (2.9 Å, ca. 103° CβS217-OS217-OS241angle), but they now play a role in solvating the oxadiazole. The Ser241 OH is now in the same plane of the central heterocycle and at a distance (less than 3.2 Å) and angle (93°) indicative of a stabilizing H-bond interaction with the oxadiazole N3. Additionally, Ser217 OH lies at a distance (2.9 Å) that might suggest a solvating interaction with the oxadiazole N3, but it lies below the plane of the oxadiazole and out of alignment for a conventional H-bond. Similarly, the oxadiazole N4 is within interaction distances of Thr236 (3.1 Å) and perhaps Lys142 (3.4 Å), with angles that approach 125–130°.
Figure 7.

The cocrystal structure of covalently bound FAAH–1 (in blue) and noncovalently bound FAAH–1 (in light brown) display the differences in cytosolic port water H-bonds with the oxadiazole core.
Again and because of the comparison with covalently bound 1, this highlights one of the additional most interesting interactions observed at the catalytic core that is mediated by Ser217. Rather than lying in the plane of the activating heterocycle and aligned to H-bond to one of its heteroatoms, this residue is located below and oriented toward the center of the heterocycle π-system at a distance of 3.5 Å for covalently bound 1 and 3.7 Å for noncovalently bound 1. The corresponding lack of a stabilizing H-bond with the basic nitrogen of the heterocycle in the covalent complex with 1 (oxadiazole N3) is in contrast to its key role with serine proteases first defined by Edwards.47 Like the cases subsequently explored,48 an activating heterocycle H-bonds through nitrogen to a catalytic residue (typically His) preferentially stabilizing the bound tetrahedral complex. In contrast, the FAAH Ser217 engages in a SerOH–π H-bond with the activating heterocycle. Thus, the role of the activating heterocycle is intrinsically different and this accounts for the pronounced substituent effects observed in our work.
Conclusions
Herein we disclose the use of fluoride ion in protein cocrystallization studies to preferentially bind an oxyanion hole, trapping a serine hydrolase in an “in action” state and inhibiting reactive substrate or inhibitor covalent binding at the active site. This was utilized to provide a cocrystal structure of an exceptionally potent and selective49 inhibitor of FAAH, α-ketooxadiazole 1 (Ki = 290 pM), in its noncovalently bound state (in presence of fluoride) for comparison with its covalently bound state (in absence of fluoride). The comparisons revealed that such inhibitors noncovalently bind FAAH preferentially in their ketone state and not as a gem diol, and that the hydrophobic interactions of terminal acyl chain phenyl region of such inhibitors dominate their noncovalent interactions at the enzyme active site in preference to the intricate polar interactions observed in the cytosolic port. The studies reinforce the observations that the primary role of the central activating heterocycle is dominated by its intrinsic electron-withdrawing properties, distinguishing it from the role it has been shown to play in protease inhibition, and provide a clear picture of the major and subtle origins of the remarkable potency of oxadiazole versus oxazole based inhibitors in this series.50 Lastly, these studies provide insights into the participation of an anion or a water molecule at the enzymatic core that may play a role in binding the oxyanion hole in the resting state of the enzyme and that may play a role in serine activation or preorganization of the active site catalytic residues.
Experimental
Inhibitor 1
The inhibitor was prepared in studies disclosed previously.37
FAAH Inhibition by Fluoride Ion
14C-labeled oleamide was prepared from 14C-labeled oleic acid as described.13 Truncated rat FAAH (rFAAH) was expressed in E. coli and purified as described51 and the inhibition assays were performed as detailed.13 The initial rates of hydrolysis (≤10–20% reaction) were monitored by following the breakdown of 14C-oleamide and using enzyme concentrations of 1 nM. In brief, the enzyme reaction was initiated by mixing 1 nM of rFAAH with 20 μM of 14C-labeled oleamide in 100 μL of reaction buffer (125 mM TrisCl, 1 mM EDTA, 0.2% glycerol, 0.02% Triton X-100, 0.4 mM Hepes, pH 9.0) at room temperature in the presence of three different concentrations (10, 100 and 1000 mM) of additives (NaF or NaCl). The enzyme reaction was terminated by transferring 20 μL of the reaction mixture to 500 μL of 0.1 N HCl at three different time points. The 14C-labeled oleamide (substrate) and oleic acid (product) were extracted with EtOAc and analyzed by TLC as detailed.13 The Ki’s were calculated using a Dixon plot as described.32
FAAH Expression, Purification, and Crystallization
The procedures used here were described previously.17 In brief, the N-terminal transmembrane-deleted (ΔTM) form (amino acids 30–579) of the humanized/rat (h/r) FAAH17b gene was expressed in the E. coli strain BL21 A.I. (Invitrogen) and purified using three chromatography steps including metal affinity, cation exchange, and size exclusion chromatography. The protein sample was concentrated to 30 mg/mL in a buffer containing 10 mM Hepes (pH 7.0), 500 mM NaCl, 0.08% n-undecyl-β-D-maltoside (Anatrace), and 2 mM dithiothreitol. Protein concentrations were determined using the reducing-agent compatible-BCA protein assay kit (Pierce Biotechnology). The protein sample was added to 6% dimethyl formamide (DMF) and inhibitor 1 at a concentration of 0.5 mM (from a 100 mM stock dissolved in DMF). The additives xylitol (Sigma) and benzyldimethyl(2-dodecyloxyethyl)ammonium chloride (Aldrich) were added to the protein sample up to a concentration of 12% and 1%, respectively. The covalent and noncovalent FAAH–1 structures were obtained by mixing (1:1) the protein solution to two different crystallization buffers with and without the presence of sodium fluoride (100 mM). The structures with the covalently and noncovalently bound inhibitor were obtained from crystals grown with a buffer containing 30% PEG400, 100 mM Hepes (pH 7.5), 100 mM NaCl; and 30% PEG400, 100 mM Mes pH 5.5, 100 mM KCl, 100 mM NaF, and 8% polypropylene glycol-P400, respectively. Crystals were grown by sitting drop vapor diffusion at 14 °C in 96-well plates (Innovaplate SD-2; Innovadyne Technologies) and frozen by plunging into liquid nitrogen directly after harvesting. The data for the cocrystal structure of FAAH with covalently bound 1 was collected at 100 K from a single crystal at the GM/CA-CAT beamline of the Advanced Photon Source (APS, Argonne, IL) using a 10-μm beam collimator. The data for the noncovalently bound inhibitor structure was collected at the Stanford Synchrotron Radiation Laboratory (SSRL, Menlo Park, CA) on beamline 11-1 at 100 K. For data reduction, we used the XDS (covalently bound structure) and HKL2000 (noncovalently bound structure) programs. Structures were solved and refined by using programs contained in the CCP4 package. The software suite Phenix was used to refine individual atomic displacement parameters. Results from data processing and structure refinement are provided in Table 1. The crystal lattices were found in the P212121 (covalently bound structure) and P3221 (noncovalently bound structure) space groups, containing a FAAH dimer in the asymmetric unit. The structures were determined at a resolution of 2.20 Å (covalent, PDB code: 3PR0), and 1.78 Å (noncovalent, PDB code: 3PPM), and solved by molecular replacement using the coordinates of the FAAH–2 structure (PDB code: 2WJ1) as a search model. Chemical parameters for the inhibitors were calculated by the Dundee PRODRG Web server.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (DA015648, DLB; DA017259, RCS and BFC) and the Skaggs Institute for Chemical Biology. JG was a Skaggs Fellow. We are especially grateful to Gye Won Han for helping with the PDB quality check and deposition. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Footnotes
Supporting Information Available: Complete refs 17a, 22c, 25c, and 31e. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 2.Giang DK, Cravatt BF. Proc Natl Acad Sci USA. 1997;94:2238–2242. doi: 10.1073/pnas.94.6.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ezzili C, Otrubova K, Boger DL. Bioorg Med Chem Lett. 2010;20:5959–5968. doi: 10.1016/j.bmcl.2010.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patricelli MP, Cravatt BF. Vit Hormones. 2001;62:95–131. doi: 10.1016/s0083-6729(01)62002-8. [DOI] [PubMed] [Google Scholar]
- 5.Egertova M, Cravatt BF, Elphick MR. Neuroscience. 2003;119:481–496. doi: 10.1016/s0306-4522(03)00145-3. [DOI] [PubMed] [Google Scholar]
- 6.Boger DL, Fecik RA, Patterson JE, Miyauchi H, Patricelli MP, Cravatt BF. Bioorg Med Chem Lett. 2000;10:2613–2616. doi: 10.1016/s0960-894x(00)00528-x. [DOI] [PubMed] [Google Scholar]
- 7.Patricelli MP, Cravatt BF. Biochemistry. 2001;40:6107–6115. doi: 10.1021/bi002578r. [DOI] [PubMed] [Google Scholar]
- 8.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 9.Martin BR, Mechoulam R, Razdan RK. Life Sci. 1999;65:573–595. doi: 10.1016/s0024-3205(99)00281-7. [DOI] [PubMed] [Google Scholar]
- 10.Schmid HHO, Schmid PC, Natarajan V. Prog Lipid Res. 1990;29:1–43. doi: 10.1016/0163-7827(90)90004-5. [DOI] [PubMed] [Google Scholar]
- 11.Boger DL, Henriksen SJ, Cravatt BF. Curr Pharm Des. 1998;4:303–314. [PubMed] [Google Scholar]
- 12.Cravatt BF, Lerner RA, Boger DL. J Am Chem Soc. 1996;118:580–590. [Google Scholar]
- 13.Cravatt BF, Prospero–Garcia O, Suizdak G, Gilula NB, Henriksen SJ, Boger DL, Lerner RA. Science. 1995;268:1506–1509. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]
- 14.Lerner RA, Siuzdak G, Prospero–Garcia O, Henriksen SJ, Boger DL, Cravatt BF. Proc Natl Acad Sci USA. 1994;91:9505–9508. doi: 10.1073/pnas.91.20.9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Cravatt BF, Lichtman AH. Curr Opin Chem Biol. 2003;7:469–475. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]; (b) Ahn K, McKinney MK, Cravatt BF. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ahn K, Johnson DS, Cravatt BF. Exp Opin Drug Discov. 2009;4:763–784. doi: 10.1517/17460440903018857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) McKinney MK, Cravatt BF. Ann Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]; (b) McKinney MK, Cravatt BF. J Biol Chem. 2003;278:37393–37399. doi: 10.1074/jbc.M303922200. [DOI] [PubMed] [Google Scholar]; (c) Patricelli MP, Cravatt BF. J Biol Chem. 2000;275:19177–19184. doi: 10.1074/jbc.M001607200. [DOI] [PubMed] [Google Scholar]; (d) Patricelli MP, Lovato MA, Cravatt BF. Biochemistry. 1999;38:9804–9812. doi: 10.1021/bi990637z. [DOI] [PubMed] [Google Scholar]; (e) Patricelli MP, Cravatt BF. Biochemistry. 1999;38:14125–14130. doi: 10.1021/bi991876p. [DOI] [PubMed] [Google Scholar]
- 17.(a) Ahn K, et al. Chem Biol. 2009;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mileni M, Johnson DS, Wang Z, Everdeen DS, Liimatta M, Pabst B, Bhattacharya K, Nugent RA, Kamtekar S, Cravatt BF, Ahn K, Stevens RC. Proc Natl Acad Sci USA. 2008;105:12820–12824. doi: 10.1073/pnas.0806121105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 18.(a) Mileni M, Garfunkle J, DeMartino JK, Cravatt BF, Boger DL, Stevens RC. J Am Chem Soc. 2009;131:10497–10506. doi: 10.1021/ja902694n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mileni M, Garfunkle J, Ezzili C, Kimball FS, Cravatt BF, Stevens RC, Boger DL. J Med Chem. 2010;53:230–240. doi: 10.1021/jm9012196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Seierstad M, Breitenbucher JG. J Med Chem. 2008;51:7327–7343. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]; (b) Deng HF. Expert Opin Drug Discovery. 2010;5:961–993. doi: 10.1517/17460441.2010.513378. [DOI] [PubMed] [Google Scholar]
- 20.(a) Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]; (b) Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Br J Pharmacol. 2006;147:281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. J Med Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]; (b) Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D. J Med Chem. 2003;46:2352–2360. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]; (c) Tarzia G, Duranti A, Gatti G, Piersanti G, Tontini A, Rivara S, Lodola A, Plazzi PV, Mor M, Kathuria S, Piomelli D. ChemMedChem. 2006;1:130–139. doi: 10.1002/cmdc.200500017. [DOI] [PubMed] [Google Scholar]; (d) Mor M, Lodola A, Rivara S, Vaconido F, Duranti A, Tontini A, Sanchini S, Oiersanti G, Clapper JR, King AR, Tarzia G, Piomelli D. J Med Chem. 2008;51:3487–3498. doi: 10.1021/jm701631z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Normanbhoy T, Alexander JP, Cravatt BF. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]; (b) Johnson DS, Ahn K, Kesten S, Lazerwith SE, Song Y, Morris M, Fay L, Gregory T, Stiff C, Dunbar JB, Jr, Liimatta M, Beidler D, Smith S, Nomanbhoy TK, Cravatt BF. Bioorg Med Chem Lett. 2009;19:2865–2869. doi: 10.1016/j.bmcl.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Johnson DS, et al. ACS Med Chem Lett. 2011;2 doi: 10.1021/ml100190t. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Abouab–Dellah A, Burnier P, Hoornaert C, Jeunesse J, Puech F. 2004/099176. (Sanofi); WO. ; (b) Abouab–Dellah A, Almario GA, Froissant J, Hoornaert C. 2005/077898. Sanofi; WO. ; (c) Abouab–Dellah A, Almario GA, Hoornaert C, Li AT. 2005/070910. Sanofi; WO. ; (d) Abouab–Dellah A, Almario GA, Hoornaert C, Li AT. 2007/027141. Sanofi; WO.
- 24.(a) Sit SY, Xie K, Deng H. 2003/06589. Bristol–Myers Squibb; WO. ; (b) Sit SY, Xie K. 2002/087569. Bristol–Myers Squibb; WO. ; (c) Sit SY, Conway C, Bertekap R, Xie K, Bourin C, Burris K, Deng H. Bioorg Med Chem Lett. 2007;17:3287–3291. doi: 10.1016/j.bmcl.2007.04.009. [DOI] [PubMed] [Google Scholar]; (d) Sit SY, Conway CM, Xie K, Berktekap R, Bourin C, Burris KD. Bioorg Med Chem Lett. 2010;20:1272–1277. doi: 10.1016/j.bmcl.2009.11.080. [DOI] [PubMed] [Google Scholar]
- 25.(a) Apodaca R, Breitenbucher JG, Pattabiraman K, Seierstad M, Xiao W. 2006/0173184. J&J; US. ; (b) Apodaca R, Breitenbucher JG, Pattabiraman K, Seierstad M, Xiao W. 2007/004741. J&J; US. ; (c) Keith JM, et al. Bioorg Med Chem Lett. 2008;18:4838–4843. doi: 10.1016/j.bmcl.2008.07.081. [DOI] [PubMed] [Google Scholar]; (d) Karbarz MJ, Luo L, Chang L, Tham CS, Palmer JA, Wilson SJ, Wennerholm ML, Brown SM, Scott BP, Apodaca RL, Keith JM, Wu J, Breitenbucher JG, Chaplan SR, Webb M. Anesth Analg. 2009;108:316–329. doi: 10.1213/ane.0b013e31818c7cbd. [DOI] [PubMed] [Google Scholar]
- 26.(a) Matsumoto T, Kori M, Miyazaki J, Kiyota Y. 2006054652. Takeda; WO. ; (b) Matsumoto T, Kori M, Kouno M. 2007020888. Takeda; WO.
- 27.Ishii T, Sugane T, Maeda J, Narazaki F, Kakefuda A, Sato K, Takahashi T, Kanayama T, Saitoh C, Suzuki J, Kanai C. 2006/088075. Astellas; WO.
- 28.(a) Moore SA, Nomikos GG, Dickason-Chesterfield AK, Sohober DA, Schaus JM, Ying BP, Xu YC, Phebus L, Simmons RM, Li D, Iyengar S, Felder CC. Proc Natl Acad Sci USA. 2005;102:17852–17857. doi: 10.1073/pnas.0507470102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Alexander JP, Cravatt BF. J Am Chem Soc. 2006;128:9699–9704. doi: 10.1021/ja062999h. [DOI] [PubMed] [Google Scholar]
- 29.Minkkila A, Myllymaki MJ, Saario SM, Castillo-Melendez JA, Koskinen AMP, Fowler CJ, Leppanen J, Nevalainen T. Eur J Med Chem. 2009;44:2294–3008. doi: 10.1016/j.ejmech.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 30.Alexander JP, Cravatt BF. Chem Biol. 2005;12:1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.For additional FAAH inhibitors, see: Onnis V, Congiu C, Bjorklund E, Hempel F, Soderstrom E, Fowler CJ. J Med Chem. 2010;53:2286–2298. doi: 10.1021/jm901891p.Vincent F, Nguyen MT, Emerling DE, Kelly MG, Duncton MAJ. Bioorg Med Chem Lett. 2009;19:6793–6796. doi: 10.1016/j.bmcl.2009.09.086.Wang JL, Bowen SJ, Schweitzer BA, Madsen HM, McDonald J, Pelc MJ, Tenbrink RE, Beidler D, Thorarensen A. Bioorg Med Chem Lett. 2009;19:5970–5974. doi: 10.1016/j.bmcl.2009.07.101.Feledziak M, Michaux C, Urbach A, Labar G, Muccioli GG, Lambert DM, Marchand-Brynaert J. J Med Chem. 2009;52:7054–7068. doi: 10.1021/jm9008532.Hart T, et al. Bioorg Med Chem Lett. 2009;19:4241–4244. doi: 10.1016/j.bmcl.2009.05.097.Wang X, Sarris K, Kage K, Zhang D, Brown SP, Kolasa T, Surowy C, El Kouhen OF, Muchmore SW, Briono JD, Stewart AO. J Med Chem. 2009;52:170–180. doi: 10.1021/jm801042a.Minkkila A, Saario SM, Kasnanen H, Leppanen J, Poso A, Nevalainen T. J Med Chem. 2008;51:7057–7060. doi: 10.1021/jm801051t.Urbach A, Muccioli GG, Stern E, Lambert DM, Marchand-Brynaert J. Bioorg Med Chem Lett. 2008;18:4163–4167. doi: 10.1016/j.bmcl.2008.05.081.Myllymaki MJ, Saario SM, Kataja AO, Castillo-Melendez JA, Nevalainen T, Juvonen RO, Jarvinen T, Koskinen AMP. J Med Chem. 2007;50:4236–4242. doi: 10.1021/jm070501w.Muccioli GG, Fazio N, Scriba GKE, Poppitz W, Cannata F, Poupaert JH, Wouters J, Lambert DM. J Med Chem. 2006;49:417–425. doi: 10.1021/jm050977k.Saario SM, Poso A, Juvonen RO, Jarvinen T, Salo-Ahen OMH. J Med Chem. 2006;49:4650–4656. doi: 10.1021/jm060394q.Du W, Hardouin C, Cheng H, Hwang I, Boger DL. Bioorg Med Chem Lett. 2005;15:103–106. doi: 10.1016/j.bmcl.2004.10.025.Patricelli MP, Patterson JE, Boger DL, Cravatt BF. Bioorg Med Chem Lett. 1998;8:613–616. doi: 10.1016/s0960-894x(98)00073-0.
- 32.Patterson JE, Ollmann IR, Cravatt BF, Boger DL, Wong CH, Lerner RA. J Am Chem Soc. 1996;118:5938–5945. [Google Scholar]
- 33.Boger DL, Sato H, Lerner AE, Austin BJ, Patterson JE, Patricelli MP, Cravatt BF. Bioorg Med Chem Lett. 1999;9:265–270. doi: 10.1016/s0960-894x(98)00734-3. [DOI] [PubMed] [Google Scholar]
- 34.Boger DL, Sato H, Lerner AE, Hedrick MP, Fecik RA, Miyauchi H, Wilkie GD, Austin BJ, Patricelli MP, Cravatt BF. Proc Natl Acad Sci USA. 2000;97:5044–5049. doi: 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boger DL, Miyauchi H, Hedrick MP. Bioorg Med Chem Lett. 2001;11:1517–1520. doi: 10.1016/s0960-894x(01)00211-6. [DOI] [PubMed] [Google Scholar]
- 36.Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Leung D, Acevedo O, Guimaráes CRW, Jorgensen WL, Cravatt BF. J Med Chem. 2005;48:1849–1856. doi: 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leung D, Du W, Hardouin C, Cheng H, Hwang I, Cravatt BF, Boger DL. Bioorg Med Chem Lett. 2005;15:1423–1428. doi: 10.1016/j.bmcl.2004.12.085. [DOI] [PubMed] [Google Scholar]
- 38.Romero FA, Hwang I, Boger DL. J Am Chem Soc. 2006;128:14004–14005. doi: 10.1021/ja064522b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romero FA, Du W, Hwang I, Rayl TJ, Kimball FS, Leung D, Hoover HS, Apodaca RL, Breitenbucher BJ, Cravatt BF, Boger DL. J Med Chem. 2007;50:1058–1068. doi: 10.1021/jm0611509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardouin C, Kelso MJ, Romero FA, Rayl TJ, Leung D, Hwang I, Cravatt BF, Boger DL. J Med Chem. 2007;50:3359–3368. doi: 10.1021/jm061414r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kimball FS, Romero FA, Ezzili C, Garfunkle J, Rayl TJ, Hochstatter DG, Hwang I, Boger DL. J Med Chem. 2008;51:937–947. doi: 10.1021/jm701210y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garfunkle J, Ezzili C, Rayl TJ, Hochstatter DG, Hwang I, Boger DL. J Med Chem. 2008;51:4392–4403. doi: 10.1021/jm800136b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeMartino JK, Garfunkle J, Hochstatter DG, Cravatt BF, Boger DL. Bioorg Med Chem Lett. 2008;18:5842–5846. doi: 10.1016/j.bmcl.2008.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.(a) Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BF. J Pharmacol Exp Ther. 2004;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]; (b) Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, Breitenbucher JG, Chaplan SR, Webb M. Br J Pharmacol. 2006;148:102–113. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Palmer JA, Higuera ES, Chang L, Chaplan SR. Neuroscience. 2008;154:1554–1561. doi: 10.1016/j.neuroscience.2008.04.047. [DOI] [PubMed] [Google Scholar]; (d) Timmons A, Seirestad M, Apodaca R, Epperson M, Pippel D, Brown S, Chang L, Scoot B, Webb M, Chaplan SR, Breitenbucher JG. Bioorg Med Chem Lett. 2008;18:2109–2113. doi: 10.1016/j.bmcl.2008.01.091. [DOI] [PubMed] [Google Scholar]; (e) Schlosburg JE, Boger DL, Cravatt BF, Lichtman AH. J Pharmacol Exp Ther. 2009;329:314–323. doi: 10.1124/jpet.108.150136. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kinsey SG, Long JZ, O’Neal ST, Abdulla RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. J Pharmacol Exp Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guimaráes CRW, Boger DL, Jorgensen WL. J Am Chem Soc. 2005;127:17377–17384. doi: 10.1021/ja055438j.See also: Tubert-Brohman I, Acevedo O, Jorgensen WL. J Am Chem Soc. 2006;128:16904–16913. doi: 10.1021/ja065863s.
- 46.Qin J, Chai G, Brewer JM, Lovelace LL, Lebioda L. Biochemistry. 2006;45:793–800. doi: 10.1021/bi051558s.For additional observations of fluoride ion in protein crystal structures, see: Pei XY, Erixon KM, Luisi BF, Leeper FJ. Biochemistry. 2010;49:1727–1736. doi: 10.1021/bi901864j.Edwards SL, Poulos TL, Kraut J. J Biol Chem. 1984;259:12984–12988.For a comprehensive review of fluoride enzyme inhibition or activation, see: Adamek E, Pawlowska-Goral K, Bober K. Rocz Pomor Akad Med Szczecinie. 2005;51:69–85.
- 47.(a) Edwards PD, Meyer EFJ, Vijayalakshmi J, Tuthill PA, Andisik DA, Gomes B, Strimpler A. J Am Chem Soc. 1992;114:1854–1863. [Google Scholar]; (b) Edwards PD, Bernstein PR. Med Res Rev. 1994;14:127–194. doi: 10.1002/med.2610140202. [DOI] [PubMed] [Google Scholar]; (c) Edwards PD, Zottola MA, Davis M, Williams J, Tuthill PA. J Med Chem. 1995;38:3972–3982. doi: 10.1021/jm00020a011. [DOI] [PubMed] [Google Scholar]; (d) Edwards PD, Andisik DW, Bryant CA, Ewing B, Gomes B, Lewis JJ, Rakiewicz D, Steelman G, Strimpler A, Trainor DA, Tuthill PA, Mauger RC, Veale CA, Wildonger RA, Williams JC, Wolanin DJ, Zottola M. J Med Chem. 1997;40:1876–1885. doi: 10.1021/jm960819g. [DOI] [PubMed] [Google Scholar]
- 48.Maryanoff BE, Costanzo MJ. Bioorg Med Chem. 2008;16:1562–1595. doi: 10.1016/j.bmc.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 49.Leung D, Hardouin C, Boger DL, Cravatt BF. Nat Biotechnol. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 50.Abbreviations: FAAH, fatty acid amide hydrolase; MAC, membrane access channel; ABP, acyl chain-binding pocket; OAH, oxyanion hole.
- 51.Patricelli MP, Lashuel HA, Giang DK, Kelly JW, Cravatt BF. Biochemistry. 1998;37:15177–15187. doi: 10.1021/bi981733n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.