


Abstract
The visual detection of specific double-stranded DNA sequences possesses great potential for the development of diagnostics. Zinc finger domains provide a powerful scaffold for creating custom DNA-binding proteins that recognize specific DNA sequences. We previously demonstrated sequence-enabled reassembly of TEM-1 β-lactamase (SEER–LAC), a system consisting of two inactive fragments of β-lactamase each linked to engineered zinc finger proteins (ZFPs). Here the SEER–LAC system was applied to develop ZFP arrays that function as simple devices to identify bacterial double-stranded DNA sequences. The ZFP arrays provided a quantitative assay with a detection limit of 50 fmol of target DNA. The method could distinguish target DNA from non-target DNA within 5 min. The ZFP arrays provided sufficient sensitivity and high specificity to recognize specific DNA sequences. These results suggest that ZFP arrays have the potential to be developed into a simple and rapid point-of-care (POC) diagnostic for the multiplexed detection of pathogens.
INTRODUCTION
Sensitive and specific detection of DNA is of great interest and demand for an increasing number of biomedical studies and applications, including clinical diagnostics. The development of simple, robust and rapid technologies for the detection of pathogens is particularly crucial, considering the high rate of mortality associated with bloodborne infection (1). The availability of portable devices for the multiplexed detection of pathogens in blood would also be highly significant for disaster management, as well as treatment of patients in resource-limited settings. For pathogen detection, lab-on-chip technologies are well suited for point-of-care (POC) diagnostics. Incorporating molecular diagnostics on a microfluidic technology would facilitate the development of POC device.
Polymerase chain reaction (PCR)-based methods have been established as quick and sensitive methods for the detection of pathogens, in contrast to laborious culture methods (2,3). However, diagnostic situations often require multiplexed pathogen detection (4,5). Multiplex PCR can identify several genes in one reaction. On the other hand, DNA microarrays offer the capability to detect a much larger number of pathogens simultaneously. To take advantage of this capability, the combination of PCR with microarray detection has been studied (6–10). Most of the detection methods are based on DNA hybridization with single-stranded DNA probes (6,11–13). However, DNA hybridization is time consuming, and duplex formation can be affected by the formation of secondary structures in the probes (6,12,13). In contrast, sequence specific DNA-binding proteins read the sequence information directly from double-stranded DNA, avoiding the need for denaturation and subsequent renaturation with probes under controlled conditions.
A common type of DNA-binding domain is the Cys2-His2 zinc finger, which contains about 30 amino acids (14). The Cys2-His2 zinc finger domain provides a versatile scaffold to construct customized DNA-binding proteins that specifically recognize virtually any desired DNA sequence (14,15). The amino acids of the zinc finger form a ββα fold, which is stabilized by hydrophobic interactions and a zinc ion coordinated by the two conserved cysteine and histidine residues. Each finger typically recognizes three to four nucleotides of DNA. Zinc finger domains can be linked into tandem arrays that allow mutifinger proteins to recognize extended DNA sequences. To modify the binding specificity of naturally occurring zinc finger domains, combinatorial mutagenesis and selection methods have been used to generate zinc fingers that could recognize three base pairs of DNA (16–19). Using these predefined modules, six zinc finger domains have the theoretical capacity to bind 18 base pairs of contiguous DNA, which would be sufficient to describe a unique site within all known genomes (16,19,20). This modular assembly approach enables the rapid construction of multifinger domains to bind almost any desired DNA sequence (21). Many engineered zinc finger proteins (ZFPs) show high specificity, with binding affinities in 0.5–50 nM range (18,20). Engineered zinc finger domains have been fused to effectors such as transcriptional regulatory domains and nucleases for gene regulation and genome modification (22,23).
Previously, we demonstrated a new methodology for the direct detection of specific double-stranded DNA sequences called SEER (sequence-enabled reassembly) (24,25). SEER consists of split-protein domains that can reassemble into an active complex only in the presence of a cognate DNA sequence (24). To construct SEER–β-lactamase(LAC), Escherichia coli TEM-1 β-lactamase was dissected into two halves (26), with each fragment linked to a ZFP (24). The two split fragments of β-lactamase (LacA and LacB) were reassembled when brought into close proximity upon ZFP binding to target DNA sequences. The activity of the reassembled β-lactamase could be easily measured by the hydrolysis of the substrate nitrocefin that changes from yellow to red when the β-lactam ring is hydrolyzed (26). The nitrocefin assay enabled the visual detection of a signal generated by the specific binding of the ZFPs to the target DNA. The SEER–LAC system allowed the sensitive detection of a single-base substitution in 18 base pairs of sequence, with a detection limit of 20 nM target DNA (24).
In this study, we employed a SEER–LAC system to develop ZFP arrays that could detect a double-stranded bacterial DNA sequence. Engineered ZFPs were deposited on a hydrogel-coated surface, creating a simple device to identify target DNA sequences from E. coli. The enzymatic signal amplification of the SEER–LAC system allowed for the rapid and sensitive visual detection of the target DNA. The method could distinguish target DNA from non-target DNA within 5 min, and with high specificity. These results suggest that ZFP arrays can be potentially developed for a simple and portable POC diagnostic for the multiplexed detection of pathogens.
MATERIALS AND METHODS
Construction, expression and purification of rrsA SEER ZFPs
All ZFPs were constructed by the modular assembly method using the Barbas set of modules in a modified Sp1C framework (27). The DNA coding regions for each ZFP were commercially synthesized by Bio Basic. ZFP(A)s (rrsA27, rrsA125 and rrsA1175) were subcloned between the XmaI and HindIII sites of pMAL-c2X LacA-Zif268 (24), replacing the C-terminal Zif268 ZFP. ZFP(B)s (rrsA62, rrsA160 and rrsA1192) were subcloned between the SmaI and AgeI sites of pMAL-c2X PBSII-LacB (24), replacing the N-terminal PBSII ZFP. The vector enables bacterial expression of the proteins as fusions with an N-terminal maltose binding protein (MBP) as a purification tag. Proteins were expressed in E. coli BL21 (Invitrogen) upon induction with 0.3 mM isopropyl β-d-1-thiogalactopyranoside at an OD600 of 0.6–0.8 for 5 h at 37°C. Cells were pelleted and resuspended in Zinc Buffer A (ZBA: 100 mM Tris base, 90 mM KCl, 1 mM MgCl2 and 100 µM ZnCl2 at pH 7.5) including 5 mM dithiothreitol (DTT) and 50 µg/µl RNase A. After sonication, proteins in cell lysates were applied to an amylose resin column pre-equilibrated with ZBA/5 mM DTT, washed with 2 M NaCl/ZBA and ZBA/1 mM tris(2-carboxyethyl)phosphine (TCEP), and eluted in ZBA/10 mM Maltose/1 mM TCEP. Concentration and purity were assessed by Coomassi-stained polyacrylamide gel electrophoresis with sodium dodecyl sulfate (SDS–PAGE) using bovine serum albumen (BSA) standards. Purified protein was stored in a ZBA/1 mM TCEP solution at −20°C until use.
Coating glass slides with poly(ethylene glycol) hydrogel
Prior to covering the surface with hydrogel, glass slides were silanized according to the procedure reported by us earlier (28). The silanization introduced on the surface acrylate groups for covalent linking to the hydrogel. PEG hydrogel films were made using a precursor solution of poly(ethylene glycol) (PEG)-diacrylate (DA) (MW575) with 2% w/v photoinitiator, DMPA. This solution was spun onto silanized glass slides at 1000 rpm for 5 s using a spin-coater (Machine World Inc., Redding, CA, USA). The glass slides coated with a layer of liquid PEG-DA were then exposed to 365 nm, 10-mW/cm2 UV light (Cannon PLA 501, San Jose, CA, USA), cross-linking prepolymer and resulting in a hydrogel coating. Gel-coated glass slides were lyophilized for 24 h in order to ensure rapid absorption of ZFP molecules to the surface.
ZFP array and DNA binding
DNA target oligonucleotides were prepared by heating to 95°C for 10 min, then slowly cooling to room temperature to form hairpins containing a four-thymidine loop. The sequences of hairpin DNA target oligonucleotides are provided in the Supplementary Data. Escherichia coli genomic DNA (E. coli strain B) was purchased from Sigma. The ZFP immobilization/enzymatic reaction area was confined by placing a silicone gasket with a diameter of 6 mm and a well depth of 1 mm (Grace Bio-Labs, Bend OR) on the PEG hydrogel surface before arraying the ZFPs. 5.3 µl of purified MBP-LacA-ZFP(A) at a concentration of 1.6 µM was pipetted onto the PEG hydrogel and incubated for 40 min. About 10 µl of hairpin target DNA solution was added on the ZFP array and incubated for 20 min to allow DNA binding to the ZFP. The slide was washed with ZBA/50 mM KCl and ZBA/0.05% Tween-20, followed by air-drying. About 10 µl of MBP-ZFP(B)-LacB was added on the ZFP array and incubated for 20 min to allow the ZFP to bind the DNA that was complexed with MBP-LacA-ZFP(A). The slide was washed with ZBA/50 mM KCl and ZBA/0.05% Tween-20, followed by air-drying.
Nitrocefin assay
After placing the slide onto a 96-well plate and aligning the arrays with the wells, 20 µl of 1 mM nitrocefin (Calbiochem, San Diego, CA, USA) was added to the ZFP array. Absorbance at 486 nm was monitored with a Cambrex ELX 808 (Bio-TEK Instruments, Winooski, VT, USA). All experiments were repeated in duplicate, and the standard error was calculated from duplicate samples. The slopes shown in Table 2 were defined as the change in signal intensity over incubation time from the linear portion of each plot. Normalized slope values ± standard errors were calculated by dividing each slope by the slope of the substrate-only plot.
Table 2.
| ZFP |
|||
|---|---|---|---|
| rrsA27/rrsA62 | rrsA125/rrsA160 | rrsA1175/rrsA1192 | |
| Sensitivity | |||
| 2.5 µM | 115.33 ± 3.67a | 24.00 ± 7.00 | 29.38 ± 1.88 |
| 0.25 µM | 84.32 ± 6.98 | 15.50 ± 3.50 | 20.75 ± 1.50 |
| 5 nM | 43.82 ± 1.52 | 4.92 ± 0.42 | 9.13 ± 0.38 |
| 0.5 nM | 37.82 ± 0.48 | ND | ND |
| No DNA | 30.52 ± 2.82 | 3.00 ± 2.00 | 7.50 ± 0.25 |
| Specificity | |||
| Target site 1 | 86.83 ± 11.17 | 7.17 ± 0.17 | 17.33 ± 1.00 |
| Target site 2 | 36.33 ± 3.33 | 24.17 ± 4.17 | 15.50 ± 0.50 |
| Target site 3 | 26.50 ± 1.83 | 5.33 ± 0.67 | 42.17 ± 0.50 |
| Zif268/PBSII | 18.67 ± 1.33 | 4.33 ± 0.33 | 13.50 ± 1.50 |
| Genomic DNA | |||
| No | 284.5 ± 1.50 | 42.25 ± 0.25 | 63.00 ± 19.00 |
| Equal mass | 183.5 ± 15.50 | 34.00 ± 2.00 | 31.67 ± 0.00 |
| 2-fold | 165.0 ± 5.00 | 31.50 ± 1.00 | 30.00 ± 3.33 |
| 4-fold | 163.0 ± 10.00 | 27.25 ± 3.25 | 25.00 ± 0.33 |
| 8-fold | 140.0 ± 9.00 | 23.50 ± 0.50 | 23.17 ± 2.17 |
aNormalized slope values ±SE.
ND, not done.
Electromobility shift assay
Complementary pairs of 5′-biotin labeled forward and 5′-poly T reverse oligonucleotides were annealed to obtain double-stranded target DNAs. Electromobility shift assay (EMSA) was performed with the Light Shift Chemiluminescent EMSA Kit (Pierce, Rockford, IL, USA) according to the manufacturer’s protocol. The following parameters were used: binding reactions were performed for 1 h at room temperature (22°C) in ZBA containing 150 mM KCl, 5 mM DTT, 10% glycerol, 0.1 mg/ml BSA, 0.05% NP-40, 25–55 pM target DNA and purified ZFPs with a concentration of 0.025–1000 nM. Gel electrophoresis was performed on a 10% native polyacrylamide gel in 0.5 X TBE buffer. After blotting on a nylon membrane, the DNA was cross-linked by a UV cross-linker (Stratagene) for 4 min.
RESULTS
ZFP design and array
The design goal was to construct a simple two-probe complementation assay to detect specific DNA sequences (Figure 1). One sequence-specific ZFP probe [ZFP(A)] would be immobilized on the array surface. Double-stranded DNA fragments and a second ZFP probe [ZFP(B)] would then be applied to the surface sequentially. After a brief incubation, a wash step would remove unbound or weakly-bound molecules. Detection of the binding event would take advantage of the SEER–LAC system (24). Each probe would carry an inactive fragment of the enzyme TEM-1 β-lactamase, LacA and LacB. LacA-ZFP(A) and ZFP(B)-LacB would be designed to bind to contiguous sites on their target DNA, thus juxtaposing the enzyme fragments upon binding. Enzyme reassembly would only occur when both target sequences were present in the correct spacing and orientation. Enzyme activity would be measured in a final step, using the colorimetric substrate nitrocefin. Nitrocefin is converted from yellow to red (486 nm) upon hydrolysis, producing a signal that is both quantitative and visual. This system therefore provides an isothermic enzymatic amplification of a visual signal that indicates the presence of a specific, user-defined DNA sequence.
Figure 1.

Schematic representation of ZFP array with the SEER–LAC system for detection of double-stranded bacterial DNA sequences. The colored dots represent the digital image of nitrocefin assay, showing the color change from yellow to red due to hydrolysis of the substrate during the incubation.
As an initial proof of concept, ZFPs were designed to detect a non-pathogenic strain of E. coli. The 16S rDNA sequence (rrsA) is only found in bacteria, and the detection of this sequence could confirm the presence or absence of pathogenic and non-pathogenic bacteria in sterile bodily tissues such as blood (29). ZFPs were designed so that each pair [ZFP(A) and ZFP(B)] targeted three different locations in rrsA (Supplementary Figure S1). Two pairs of six-finger ZFPs (rrsA27/rrsA62 and rrsA125/rrsA160) and one pair of three-finger ZFPs (rrsA1175/rrsA1192) were constructed (Table 1). LacA was appended at the N-terminus of the ZFP(A)s and LacB at the C-terminus of the ZFP(B)s.
Table 1.
Sequences of zinc fingers and their intended DNA target sequences
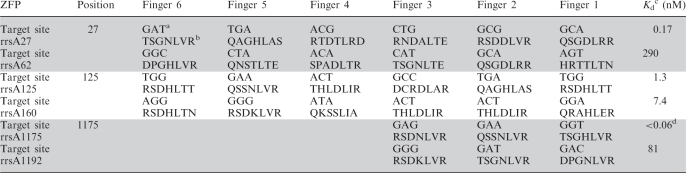 |
aThe most C-terminal zinc finger binds the 5′-end of the target site, therefore Finger 6 is shown to bind the first 5′-GAT-3′ of an 18-bp target site starting at position 27 of the rrsA gene.
bAmino acids at positions −1, 1, 2, 3, 4, 5 and 6 of the α-helix of each finger are shown.
cThe Kd-value of ZFP LacA-Zif268 was determined to be 7.7 nM under the conditions used in this assay. All values reported represent the results of at least two independent experiments and have a standard error of ±50%.
dThe Kd-value of ZFP LacA-rrsA1175 could not be accurately determined under the conditions of this assay because the value was lower than the concentration of the DNA probe (55 pM).
We initially fabricated the ZFP microarrays using a pin-spotting method. Immobilization of ZFPs on the array was demonstrated by the fluorescent detection of an antibody to the ZFP (Supplementary Figure S2A). The ability of the immobilized ZFP to retain its specific DNA binding function was shown using fluorescently-labeled target oligonucleotides (Supplementary Figure S2B). Using the spotted array format, our initial experiments applying SEER–LAC to detect the E. coli DNA sequences produced the anticipated, visually-detectable color signal in proportion to the concentration of target DNA (Supplementary Figure S3). However, we found a dramatic improvement in sensitivity could be achieved by depositing LacA-ZFP(A) within a confined area of a silicone gasket on the PEG gel slide, rather than printing with a microarrayer. Such protein arrays were bigger and denser than conventional pin-spotting microarrays, and allowed for more rapid signal kinetics (Figures 2–4). The improved format was used for all subsequent experiments.
Figure 2.

Determination of DNA binding sensitivity of engineered ZFPs with the SEER–LAC system. The SEER–LAC signal was monitored using ZFP LacA-rrsA27 and rrsA62-LacB (A), ZFP LacA-rrsA125 and rrsA160-LacB (B) and ZFP LacA-rrsA1175 and rrsA1192-LacB (C) on the array at the different target DNA concentrations. Inset (A) represents digital image of nitrocefin assay after 30 min incubation.
Figure 4.

Detection of the SEER–LAC signal by ZFP LacA-rrsA26 and rrsA62-LacB (A), ZFP LacA-rrsA125 and rrsA160-LacB (B) and ZFP LacA-rrsA1175 and rrsA1192-LacB (C) on the array in the presence of E. coli genomic DNA. Escherichia coli genomic DNA was added to the concentration equal in moles of base pairs to 2.5 µM concentration of the target oligonucleotide (red), 2-fold (light green), 4-fold (light purple) and 8-fold (light blue) higher than the concentration of the target oligonucleotide. Target DNA only (blue), substrate only (yellow).
Escherichia coli target sequence limit of detection
To test the binding of the engineered ZFPs to their cognate target DNAs and determine the minimum detectable level of DNA by the SEER–LAC system on the slide, target DNA oligonucleotides were applied at various concentrations (5 nM, 0.25 µM and 2.5 µM). β-lactamase activity was assayed using nitrocefin. The ZFP array using the SEER–LAC detection system produced a DNA-dependent response that was linear and quantitative over a range of 5 nM–2.5 µM of target DNA (Figure 2 and Table 2). The ZFP pair rrsA27 and rrsA62 was sensitive enough to detect 5 nM of DNA that is equal to ∼1.4 ng or 50 fmol of oligonucleotide target. A small increase in the signal for no DNA was observed during the incubation. For the other two pairs of ZFPs, the signal for 5 nM of target DNA was similar to the signal for no DNA, indicating a limit of detection >5 nM.
Specificity of ZFP recognition
To examine the DNA binding specificity of the ZFPs, each pair was incubated with its cognate target oligonucleotide and those of the two others. Also included was a target with sites for Zif268 and PBSII as an irrelevant sequence that was not found in the E. coli genome. All the engineered ZFP pairs were able to distinguish their own targets from non-targets (Figure 3). The slope of the signal change over time for the ZFP pair rrsA125 and rrsA160 was higher for the cognate target site 2 than for target site 1, target site 3 and the Zif268/PBSII site by 3.4-, 4.5- and 5.6-fold, respectively (Table 2). The corresponding values for the ZFP pair rrsA27 and rrsA62 were higher for the cognate site 1 than for target site 2, target site 3 and the Zif268/PBSII site by 2.4-, 3.3- and 4.7-fold, respectively. Based on the slopes, the ZFP pair rrsA125 and rrsA160 had the highest specificity among the three rrsA ZFPs.
Figure 3.

Determination of DNA binding specificity of engineered ZFPs with the SEER–LAC system. (A) ZFP LacA-rrsA27 and rrsA62-LacB (Target site 1), (B) ZFP LacA-rrsA125 and rrsA160-LacB (Target site2) and (C) ZFP LacA-rrsA1175 and rrsA1192-LacB (Target site 3) on the array.
Target site detection in the presence of genomic DNA
In order to identify pathogenic DNA in a real-world context, a device would need to detect the target sequence in the presence of the rest of the chromosomal DNA. Since any 36-bp site that is not the intended 36-bp target site can be considered non-target, the E. coli chromosome is equivalent to approximately 106 copies of non-target sites. All ZFPs have a low-affinity for genomic (i.e. non-specific) DNA, with a large variability in the ratio of specific versus non-specific binding affinity among ZFPs (30). The abundance of low-affinity genomic DNA might therefore act as a competitor of specific binding. To investigate if the presence of complex genomic DNA would interfere with the assay system, E. coli genomic DNA was added to the target DNA oligonucleotides. The initial amount of added E. coli genomic DNA was equal in mass to 2.5 µM of target DNA (0.715 µg for the six-finger protein pairs and 0.423 µg for the three-finger protein pair). The genomic DNA was then increased to 2-, 4- and 8-fold higher concentrations in subsequent samples. Although all ZFP pairs could still detect the target DNA in the presence of excess non-target sites, the signal decreased in the presence of an equal mass of E. coli genomic DNA and further decreased in the presence of an increased amount (Figure 4 and Table 2).
DNA binding affinity of engineered ZFPs
Engineered ZFPs can display a fairly wide range of affinities and specificities for their intended target sites (21,31–34). To examine if the observed sensitivity and specificity behaviors of the ZFP pairs correlated with the affinities of the engineered ZFPs, quantitative equilibrium constants were measured by EMSA (Table 1 and Supplementary Figure S4). Because Kd-values are highly dependant on the reaction conditions, ZFP LacA-Zif268 was measured as a control and determined to 7.7 nM, consistent with previously published values for Zif268 (16,18). Affinities were observed in a range from <0.06 to 290 nM, consistent with values reported for other three and six-finger ZFPs (16,31,32). Surprisingly, the affinities for ZFP pairs rrsA27 and rrsA62 as well as rrsA1175 and rrsA1192 differed by >1000-fold, while those of rrsA125 and rrsA160 were similar and at the midpoint of the observed range on a log scale. While these values may not allow an easy correlation with the detection characteristics of the ZFP pairs, the data might suggest that alternative strategies can be used to specify the correct target DNA.
DISCUSSION
In this study, we designed ZFP arrays that could visually detect double-stranded bacterial DNA sequences. Such arrays could be used in the development of a pathogen detection device. The SEER–LAC system was applied to engineered ZFPs deposited on PEG gel slides allowed for the rapid visual detection of the desired target DNA sequence. The SEER–LAC signal was DNA concentration dependent and linear. The limit of detection sensitivity was 5 nM of target DNA, equivalent to 50 fmol of target sites. Target DNA could be distinguished from non-targets within 5 min. The ZFP arrays were, therefore, able to detect specific DNA sequences with high sensitivity and specificity.
Several parameters impact the sensitivity of the assay. Immobilization of the ZFPs on the hydrogel as performed here might have limited the ability of some ZFP molecules to fully interrogate the DNA, which could be addressed in future studies by using an orientation-dependent immobilization method. Immobilization might also have reduced the capture of some analytes, since DNAs in the 3D sample solution were required to diffuse to the 2D slide surface. Indeed, we observed that applying ZFPs broadly to the surface instead of pin-spotting produced a stronger detection signal per unit time. In principle, having the ZFPs in solution may have captured more analytes. However, immobilization on the slide provided several advantages over our previous solution-based detection methods (24,25,35). It allowed us to remove low-affinity interactions by washing that could not be performed in a solution assay. It also enabled an eventual multiplexed array format that could be used to detect multiple sequences for a large number of potential pathogens. We therefore believe that immobilization provides a superior platform for the subsequent development of a POC detection device.
Another parameter affecting the sensitivity and specificity of the assay is the affinity and specificity of the ZFPs. Our results here and reported previously also demonstrate that the utility of this assay will depend heavily on the properties of the engineered ZFPs. We previously reported an example in which a single base substitution at one ZFP binding site could reduce the SEER–LAC detection signal to background levels, while a similar substitution at the other ZFP binding site reduced the signal only 30% (24). In the current study, all the rrsA ZFP pairs provided a detection signal for the target DNA that was 3–6-fold stronger than for an irrelevant non-target DNA (Table 2). The rssA27/rssA62 pair displayed the highest sensitivity and the rssA125/rrsA160 pair displayed slightly better specificity, but the reasons are not obvious from the affinity data (Table 1). Six zinc fingers did not provide markedly better sensitivity or specificity than three zinc fingers in this study. Some pairs differed in affinity by >1000-fold. These results suggest that the fortuitous combination of high- and low-affinity proteins was able to produce a similar net effect as having two moderate-affinity proteins. Interestingly, the highest affinity member of each pair happened to be the ZFP immobilized on the array (rrsA27, rrsA125 and rrsA1175). Conceivably, the higher affinity ZFP may have retained a DNA on the slide surface for a relatively long period, during which the lower affinity (but high initial concentration) ZFP could sample the bound molecules. It might be interesting to examine a situation in which these roles were reversed. It is also not clear if the combination of two very high-affinity ZFPs would result in improved assay sensitivity, and if this would come at the expense of assay specificity. Our results warrant more studies of these important parameters.
We also evaluated the capability of our system to recognize specific DNA sequences in the presence of a complex genome. The system was still able to detect the target DNA over background in the presence of an equal or greater mass of non-target DNA, though the detection signal was reduced. Further improvements in system performance will likely be required to apply this system in an actual pathogen detection scenario, in which detection of the target DNA would need to occur in the presence of an equimolar concentration of the millions of non-target sites on the chromosome. It is possible that other ZFP pairs, or the further evolution of these pairs (34,36–39), might present proteins with more optimal affinity and specificity characteristics for this assay. For example, having both proteins possess subnanomolar affinity for their target sites might allow for more stringent washing and thus reduce non-specific interactions with non-target chromosomal DNA. Genome-wide in vivo binding assays have shown that natural mammalian ZFPs display distinct, although somewhat degenerate, binding site preferences in the context of billions of non-target sites (40,41). These observations suggest that a more extensive optimization of engineered ZFPs and in vitro binding conditions could produce performance characteristic sufficient to detect specific pathogen DNAs in the presence of millions of non-target sites. The recently described TAL effector DNA binding domains might also have characteristics favorable for this purpose (42,43). Furthermore, the detection method used in this study was the SEER–LAC system that relies on enzyme reassembly to detect target DNAs. An alternative ‘sandwich’ type assay, in which a full-length enzyme such as alkaline phosphatase or β-lactamse is linked to the second probe, could also be investigated.
POC applications are becoming increasingly important in health care (1). POC devices require an unambiguous and rapid readout, and should be suitable for use by an untrained person (1). Our ZFP array could address several of these requirements. In principle, engineered zinc finger or TAL effector probes could be designed to detect any desired pathogen-specific DNA sequences. Multiple probes could be arrayed for the multiplexed detection of pathogens. The array provides the simple readout of a color change within 5 min. In addition, the method does not require DNA labeling, hybridization or specialized detection instrumentation (6,7,11,44,45). This combination of features suggests that ZFP arrays have a great potential to be developed into POC devices.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (GM077403 to D.J.S.); National Institutes of Health, University of California, Davis-Lawrence Livermore National Laboratories Point-of-Care Technologies Center (pilot grant EB007959). Funding for open access charge: National Institutes of Health (GM077403).
Conflict of interest statement. None declared.
REFERENCES
- 1.Kaittanis C, Santra S, Perez JM. Emerging nanotechnology-based strategies for the identification of microbial pathogenesis. Adv. Drug Deliv. Rev. 2010;62:408–423. doi: 10.1016/j.addr.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johansson A, Ibrahim A, Goransson I, Eriksson U, Gurycova D, Clarridge JE, III, Sjostedt A. Evaluation of PCR-based methods for discrimination of Francisella species and subspecies and development of a specific PCR that distinguishes the two major subspecies of Francisella tularensis. J. Clin. Microbiol. 2000;38:4180–4185. doi: 10.1128/jcm.38.11.4180-4185.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mothershed EA, Whitney AM. Nucleic acid-based methods for the detection of bacterial pathogens: present and future considerations for the clinical laboratory. Clin. Chim. Acta. 2006;363:206–220. doi: 10.1016/j.cccn.2005.05.050. [DOI] [PubMed] [Google Scholar]
- 4.Sekine K, Revzin A, Tompkins RG, Toner M. Panning of multiple subsets of leukocytes on antibody-decorated poly(ethylene) glycol-coated glass slides. J. Immunol. Methods. 2006;313:96–109. doi: 10.1016/j.jim.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 5.Zhu H, Stybayeva G, Macal M, Ramanculov E, George MD, Dandekar S, Revzin A. A microdevice for multiplexed detection of T-cell-secreted cytokines. Lab Chip. 2008;8:2197–2205. doi: 10.1039/b810244a. [DOI] [PubMed] [Google Scholar]
- 6.Cleven BE, Palka-Santini M, Gielen J, Meembor S, Kronke M, Krut O. Identification and characterization of bacterial pathogens causing bloodstream infections by DNA microarray. J. Clin. Microbiol. 2006;44:2389–2397. doi: 10.1128/JCM.02291-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palka-Santini M, Cleven BE, Eichinger L, Kronke M, Krut O. Large scale multiplex PCR improves pathogen detection by DNA microarrays. BMC Microbiol. 2009;9:1. doi: 10.1186/1471-2180-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang C, Wu B, Amer S, Luo J, Zhang H, Guo Y, Dong G, Zhao B, He H. Phylogenetic analysis and molecular characteristics of seven variant Chinese field isolates of PRRSV. BMC Microbiol. 2010;10:146. doi: 10.1186/1471-2180-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang D, Coscoy L, Zylberberg M, Avila PC, Boushey HA, Ganem D, DeRisi JL. Microarray-based detection and genotyping of viral pathogens. Proc. Natl Acad. Sci. USA. 2002;99:15687–15692. doi: 10.1073/pnas.242579699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warsen AE, Krug MJ, LaFrentz S, Stanek DR, Loge FJ, Call DR. Simultaneous discrimination between 15 fish pathogens by using 16S ribosomal DNA PCR and DNA microarrays. Appl. Environ. Microbiol. 2004;70:4216–4221. doi: 10.1128/AEM.70.7.4216-4221.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang T, Yoo SM, Yoon I, Lee SY, Kim B. Patterned multiplex pathogen DNA detection by Au particle-on-wire SERS sensor. Nano Lett. 2010;10:1189–1193. doi: 10.1021/nl1000086. [DOI] [PubMed] [Google Scholar]
- 12.Peplies J, Lau SC, Pernthaler J, Amann R, Glockner FO. Application and validation of DNA microarrays for the 16S rRNA-based analysis of marine bacterioplankton. Environ. Microbiol. 2004;6:638–645. doi: 10.1111/j.1462-2920.2004.00588.x. [DOI] [PubMed] [Google Scholar]
- 13.Wu L, Thompson DK, Li G, Hurt RA, Tiedje JM, Zhou J. Development and evaluation of functional gene arrays for detection of selected genes in the environment. Appl. Environ. Microbiol. 2001;67:5780–5790. doi: 10.1128/AEM.67.12.5780-5790.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct. 2000;29:183–212. doi: 10.1146/annurev.biophys.29.1.183. [DOI] [PubMed] [Google Scholar]
- 15.Beerli RR, Barbas CF., III Engineering polydactyl zinc-finger transcription factors. Nat. Biotechnol. 2002;20:135–141. doi: 10.1038/nbt0202-135. [DOI] [PubMed] [Google Scholar]
- 16.Dreier B, Beerli RR, Segal DJ, Flippin JD, Barbas CF., III Development of zinc finger domains for recognition of the 5'-ANN-3' family of DNA sequences and their use in the construction of artificial transcription factors. J. Biol. Chem. 2001;276:29466–29478. doi: 10.1074/jbc.M102604200. [DOI] [PubMed] [Google Scholar]
- 17.Dreier B, Segal DJ, Barbas CF., III Insights into the molecular recognition of the 5'-GNN-3' family of DNA sequences by zinc finger domains. J. Mol. Biol. 2000;303:489–502. doi: 10.1006/jmbi.2000.4133. [DOI] [PubMed] [Google Scholar]
- 18.Segal DJ, Dreier B, Beerli RR, Barbas CF., III Toward controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 5'-GNN-3' DNA target sequences. Proc. Natl Acad. Sci. USA. 1999;96:2758–2763. doi: 10.1073/pnas.96.6.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Q, Segal DJ, Ghiara JB, Barbas CF., III Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc. Natl Acad. Sci. USA. 1997;94:5525–5530. doi: 10.1073/pnas.94.11.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beerli RR, Segal DJ, Dreier B, Barbas CF., III Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc. Natl Acad. Sci. USA. 1998;95:14628–14633. doi: 10.1073/pnas.95.25.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Segal DJ, Beerli RR, Blancafort P, Dreier B, Effertz K, Huber A, Koksch B, Lund CV, Magnenat L, Valente D, et al. Evaluation of a modular strategy for the construction of novel polydactyl zinc finger DNA-binding proteins. Biochemistry. 2003;42:2137–2148. doi: 10.1021/bi026806o. [DOI] [PubMed] [Google Scholar]
- 22.Camenisch TD, Brilliant MH, Segal DJ. Critical parameters for genome editing using zinc finger nucleases. Mini Rev. Med. Chem. 2008;8:669–676. doi: 10.2174/138955708784567458. [DOI] [PubMed] [Google Scholar]
- 23.Blancafort P, Beltran AS. Rational design, selection and specificity of Artificial Transcription Factors (ATFs): the influence of chromatin in target gene regulation. Comb. Chem. High Throughput Screen. 2008;11:146–158. doi: 10.2174/138620708783744453. [DOI] [PubMed] [Google Scholar]
- 24.Ooi AT, Stains CI, Ghosh I, Segal DJ. Sequence-enabled reassembly of beta-lactamase (SEER-LAC): a sensitive method for the detection of double-stranded DNA. Biochemistry. 2006;45:3620–3625. doi: 10.1021/bi0517032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stains CI, Porter JR, Ooi AT, Segal DJ, Ghosh I. DNA sequence-enabled reassembly of the green fluorescent protein. J. Am. Chem. Soc. 2005;127:10782–10783. doi: 10.1021/ja051969w. [DOI] [PubMed] [Google Scholar]
- 26.Galarneau A, Primeau M, Trudeau LE, Michnick SW. Beta-Lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat. Biotechnol. 2002;20:619–622. doi: 10.1038/nbt0602-619. [DOI] [PubMed] [Google Scholar]
- 27.Bhakta MS, Segal DJ. The generation of zinc finger proteins by modular assembly. Methods Mol. Biol. 2010;649:3–30. doi: 10.1007/978-1-60761-753-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Revzin A, Tompkins RG, Toner M. Surface engineering with poly(ethylene glycol) photolithography to create high-density cell arrays on glass. Langmuir. 2003;19:9855–9862. [Google Scholar]
- 29.Wilson KH, Blitchington RB, Greene RC. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J. Clin. Microbiol. 1990;28:1942–1946. doi: 10.1128/jcm.28.9.1942-1946.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hurt JA, Thibodeau SA, Hirsh AS, Pabo CO, Joung JK. Highly specific zinc finger proteins obtained by directed domain shuffling and cell-based selection. Proc. Natl Acad. Sci. USA. 2003;100:12271–12276. doi: 10.1073/pnas.2135381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Q, Xia Z, Zhong X, Case CC. Validated zinc finger protein designs for all 16 GNN DNA triplet targets. J. Biol. Chem. 2002;277:3850–3856. doi: 10.1074/jbc.M110669200. [DOI] [PubMed] [Google Scholar]
- 32.Sander JD, Zaback P, Joung JK, Voytas DF, Dobbs D. An affinity-based scoring scheme for predicting DNA-binding activities of modularly assembled zinc-finger proteins. Nucleic Acids Res. 2009;37:506–515. doi: 10.1093/nar/gkn962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolfe SA, Greisman HA, Ramm EI, Pabo CO. Analysis of zinc fingers optimized via phage display: evaluating the utility of a recognition code. J. Mol. Biol. 1999;285:1917–1934. doi: 10.1006/jmbi.1998.2421. [DOI] [PubMed] [Google Scholar]
- 34.Cornu TI, Thibodeau-Beganny S, Guhl E, Alwin S, Eichtinger M, Joung JK, Cathomen T. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol. Ther. 2008;16:352–358. doi: 10.1038/sj.mt.6300357. [DOI] [PubMed] [Google Scholar]
- 35.Stains CI, Furman JL, Segal DJ, Ghosh I. Site-specific Detection of DNA methylation utilizing mCpG-SEER. J. Am. Chem. Soc. 2006;128:9761–9765. doi: 10.1021/ja060681j. [DOI] [PubMed] [Google Scholar]
- 36.Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol. Cell. 2008;31:294–301. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo J, Gaj T, Barbas CF., III Directed evolution of an enhanced and highly efficient FokI cleavage domain for zinc finger nucleases. J. Mol. Biol. 2010;400:96–107. doi: 10.1016/j.jmb.2010.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gersbach CA, Gaj T, Gordley RM, Barbas CF., III Directed evolution of recombinase specificity by split gene reassembly. Nucleic Acids Res. 2010;38:4198–4206. doi: 10.1093/nar/gkq125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tschulena U, Peterson KR, Gonzalez B, Fedosyuk H, Barbas CF., III Positive selection of DNA-protein interactions in mammalian cells through phenotypic coupling with retrovirus production. Nat. Struct. Mol. Biol. 2009;16:1195–1199. doi: 10.1038/nsmb.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 41.Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 43.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 44.Lagally ET, Scherer JR, Blazej RG, Toriello NM, Diep BA, Ramchandani M, Sensabaugh GF, Riley LW, Mathies RA. Integrated portable genetic analysis microsystem for pathogen/infectious disease detection. Anal. Chem. 2004;76:3162–3170. doi: 10.1021/ac035310p. [DOI] [PubMed] [Google Scholar]
- 45.Park TJ, Yoo SM, Keum KC, Lee SY. Microarray of DNA-protein complexes on poly-3-hydroxybutyrate surface for pathogen detection. Anal. Bioanal. Chem. 2009;393:1639–1647. doi: 10.1007/s00216-008-2574-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.