

Abstract
The importance of white matter (WM) injury to stroke pathology has been underestimated in experimental animal models and this may have contributed to the failure to translate potential therapeutics into the stroke clinic. Histone deacetylase (HDAC) inhibitors are neuroprotective and also promote neurogenesis. These properties make them ideal candidates for stroke therapy. In a pure WM tract (isolated mouse optic nerve), we show that pan- and class I-specific HDAC inhibitors, administered before or after a period of oxygen and glucose deprivation (OGD), promote functional recovery of axons and preserve WM cellular architecture. This protection correlates with the upregulation of an astrocyte glutamate transporter, delayed and reduced glutamate accumulation during OGD, preservation of axonal mitochondria and oligodendrocytes, and maintenance of ATP levels. Interestingly, the expression of HDACs 1, 2, and 3 is localized to astrocytes, suggesting that changes in glial cell gene transcription and/or protein acetylation may confer protection to axons. Our findings suggest that a therapeutic opportunity exists for the use of HDAC inhibitors, targeting mitochondrial energy regulation and excitotoxicity in ischemic WM injury.
Introduction
Acetylation is a key posttranslational modification of proteins and responsible for regulating critical intracellular pathways. Protein acetylation is the transfer of an acetyl moiety from acetyl-coenzyme A to the ε-amino group of a lysine residue. The acetylation of lysine is reversible and controlled by the opposing actions of histone acetyltransferase (HAT) and histone deacetylase (HDAC). Although histones represent the most thoroughly studied substrates for these enzymes (Clayton et al., 2006), the activity of diverse nonhistone proteins are also modified by HATs and HDACs, including transcription factors such as p53 and signal transduction mediators (Haberland et al., 2009). Predictably, HDACs regulate fundamental cellular events including the cell cycle, differentiation, and apoptosis.
Inhibitors of HDACs promote the demise of tumor cells and are of significant interest in the treatment of various cancers (Minucci and Pelicci, 2006). The same drugs promote the differentiation of neural progenitor cells (Balasubramaniyan et al., 2006; Siebzehnrubl et al., 2007) and sustain mature neurons in the face of a variety of insults in vitro (Langley et al., 2008; Uo et al., 2009) and in vivo (for review, see Langley et al., 2009), thereby generating considerable interest in their potential for use in acute and neurodegenerative conditions (Chuang et al., 2009; Haberland et al., 2009; Thomas, 2009).
A number of drugs with HDAC inhibitory activity have been shown to protect neurons against ischemic damage induced by transient or permanent arterial ligation (for review, see (Gibson and Murphy, 2010). It is now clear that white matter (WM) is injured in most strokes and contributes importantly to clinical deficits. The current concept of “neuroprotection” places little attention on the involvement or on the treatment of WM during stroke, which may explain why neuroprotective treatments have thus far failed in clinical trials. We investigated the effects of the pan-HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) and the class I HDAC inhibitor N-(2-aminophenyl)-4-[N-(pyridine-3-yl-methoxy-carbonyl)aminomethyl]benzamide (MS-275) in an in vitro ischemic WM injury model, the isolated mouse optic nerve (MON). The optic nerve offers several advantages to study the mechanisms of WM injury, including the capacity to quantitatively assess axon function using electrophysiology and WM cellular components, and cytoskeleton using immunohistochemistry and confocal imaging (Tekkök et al., 2007; Baltan et al., 2008). An in vivo ischemia model that selectively targets WM in adults is unavailable; however, the optic nerve preparation offers intact, three-dimensional interaction between glial cells and myelinated axons, but without neuronal cell bodies.
We found that administration of HDAC inhibitors, before or after a period of oxygen and glucose deprivation (OGD), promoted functional recovery of axons and preserved WM cellular architecture. This protection correlated with the upregulation of an astrocyte glutamate transporter, delayed and reduced glutamate accumulation during OGD, preservation of axonal mitochondria and oligodendrocytes, and maintenance of ATP levels. Because significant protection was also observed when HDAC inhibitor was added after OGD and the accumulation of glutamate, HDAC inhibition must have at least two distinct sites of action during the sequential course of ischemic WM injury (Baltan, 2009).
Materials and Methods
Materials.
SAHA and MS-275 were obtained from Selleck Chemicals. The sources for other chemicals were described previously (Baltan et al., 2008; Uo et al., 2009). The mitoCFP mice (Misgeld et al., 2007) were originally purchased from The Jackson Laboratory and bred at the University of Washington. The Institutional Animal Care and Use Committee approved all experimental procedures.
Preparation of optic nerves, recording techniques, and OGD.
MONs were obtained from adult male Swiss Webster mice and from mice expressing mitochondrial-targeted cyan fluorescent protein (CFP) on a C57BL/6 background (Thy-1 CFP) (Misgeld et al., 2007). Optic nerves were gently freed from their dural sheaths, placed in a perfusion chamber superfused with artificial CSF (ACSF), and continuously aerated by a humidified gas mixture of 95%O2/5% CO2. All experiments were performed at 37°C. Suction electrodes back-filled with ACSF were used for stimulation (Isostim 520; WPI) and for recording the compound action potential (CAP). The recording electrode was connected to an Axoclamp 2A amplifier and the signal was amplified 50 times, filtered at 30 kHz, and acquired at 20–30 kHz. Stimulus pulse (30 μs duration) strength was adjusted to evoke the maximum CAP possible, and then increased another 25% for supramaximal stimulation. The MONs were allowed to equilibrate for at least 15 min in the chamber in normal ACSF. During experiments, the supramaximal CAP was elicited every 30 s. The OGD stress was induced by switching to glucose-free ACSF (replaced with equimolar sucrose to maintain osmolarity) and a gas mixture containing 95% N2/5% CO2. The OGD was applied for 60 min, ACSF and O2 were restored, and CAPs were recorded for up to 5 h.
Glutamate assay.
Glutamate release from MON into the superfusate, under control and OGD conditions with or without HDAC inhibitor (1 μm MS-275), was measured using HPLC, similar to previous reports (Tekkök et al., 2007; Baltan et al., 2008). Samples of extracellular perfusion fluid were collected continuously such that every vial contained 2 min of superfusate, and glutamate content was measured in every other vial (i.e., every 4 min). Samples were centrifuged at 16,000 × g for 3 min and supernatants were transferred for HPLC analysis. Amino acids were precolumn derivatized with o-phthaldialdehyde (Sigma-Aldrich), separated, and measured using standard techniques. Glutamate measurements, normalized to baseline glutamate release (i.e., the average for 20 min under control conditions immediately before OGD or MS-275 treatment), were made from MONs treated identically with those studied electrophysiologically, and the results were plotted against time. Total glutamate accumulation was quantified as the area under the time course plots. The rate and release pattern of glutamate was monitored for 60 min before, during 60 min OGD, and was continued for at least 60 min after the end of OGD. The MS-275 was applied starting 30 min before OGD, during OGD (60 min), and 30 min after the end of OGD. Glutamate levels were corrected for protein content of MONs before comparison. Technical support and analysis of experiments were performed by a person blinded to the experimental conditions.
Immunohistochemistry.
This was performed in perfusion-fixed (4% paraformaldehyde in PBS) MON or coronal brain slices. Cryoprotection was achieved in 30% sucrose for 16–18 h. Ten- to 30-μm-thick sections from each MON were blocked and permeabilized in 5–40% normal goat/donkey (50% by volume) serum (Sigma-Aldrich) and 0.3% Triton X-100 (Sigma-Aldrich) for 60 min at room temperature. Fifty 200-μm-thick sections from each brain were blocked and permeabilized in 5% normal donkey serum (Sigma-Aldrich) and 1% Triton X-100 for 60 min at room temperature. All primary antibodies (Table 1) were prepared in their respective solutions. Brain sections were incubated in primary antibodies at 4°C for 2 d with constant shaking, whereas MONs were incubated in primary antibodies at 4°C overnight without shaking. After a thorough wash in PBS, the tissue was exposed to a secondary antibody, prepared in 2% normal goat serum for overnight incubation. Donkey anti-rabbit Cy5, anti-mouse Cy5, anti-mouse Cy3, anti-chicken Cy3, and anti-guinea pig (Jackson ImmunoResearch) were used at 1:100. Sections were double or triple labeled.
Table 1.
Antibodies used in immunohistochemistry
| Antibody 1 | Species | Dilution | Source | Catalog # |
|---|---|---|---|---|
| HDAC1 | Anti-rabbit | 1:250 | Sigma-Aldrich | H3284 |
| HDAC2 | Anti-rabbit | 1:250 | Sigma-Aldrich | H3159 |
| HDAC2 | Anti-mouse | 1:250 | Sigma-Aldrich | H2663 |
| HDAC3 | Anti-rabbit | 1:250 | Sigma-Aldrich | H3034 |
| HDAC4 | Anti-rabbit | 1:250 | Sigma-Aldrich | H9411 |
| HDAC6 | Anti-rabbit | 1:250 | Sigma-Aldrich | H2287 |
| GFP | Anti-chicken | 1:100 | Abcam | AB13970 |
| Hsp 60 (H-300) | Anti-rabbit | 1:100 | Santa Cruz | sc-13966 |
| NF-200 | Anti-rabbit | 1:400 | Millipore Bioscience Research Reagents | Ab1989 |
| Anti-mouse | 1:400 | Sigma-Aldrich | N0142 | |
| GFAP | Anti-mouse | 1:1000 | Millipore Bioscience Research Reagents | MAB360 |
| Anti-rabbit (prediluted 6×) | 1:15 | ImmunoStar/DiaSorin | 22522 | |
| Anti-mouse | 1:1000 | Sigma-Aldrich | C9205 | |
| APC | Anti-mouse | 1:100 | Calbiochem | OP80 |
| GLT-1 | Anti-rabbit | 1:500 | J. Rothstein (Baltimore, MD) | |
| Anti-guinea pig | 1:500 | Millipore (Millipore Bioscience Research Reagents) | AB1783 | |
| GLAST | Anti-guinea pig | 1:2000 | Millipore (Millipore Bioscience Research Reagents) | AB1782 |
| Sytox (Green) | 1:25,000 | Millipore (Invitrogen) | 57020 |
Confocal microscopy and pixel intensity measurements.
The expression of GFAP, Sytox, GLT1, NF-200, or CFP was imaged using an Olympus FV1000 upright laser-scanning confocal microscope. Sections were scanned with an argon and He–Ne laser for Cy3/543, for eCFP/458, for Cy5/633, and for Cy2/488 fluorescence. Two to three adjacent sections from each MON were imaged for a total of three or eight areas of interest per MON. A total of 30 optical sections of 1 μm thickness at 1024 × 1024 pixel size were collected in the z-axis from a single microscopic field using the 60× (PlanApo, oil-immersion; numerical aperture, 1.42; Olympus) or 40× (UPlanFL, oil-immersion; numerical aperture, 1.30; Olympus) objective lens under fixed gain, laser power, pinhole, and PMT settings. To compare and quantify immunohistochemical staining, all sections were processed concurrently. Images were acquired with Olympus FluoView imaging software in sequential mode using multiple channels simultaneously. Z-stacks were projected into a single plane image before analysis and assessment of pixel intensity or colocalization. Care was taken to analyze CFP pixel intensity in freshly sectioned tissue for all groups.
ATP measurement.
ATP extraction was performed as previously described (Khan, 2003) and modified for optic nerve. In brief, MON was cut into two to three small pieces using a small needle and placed in 75 μl of 10% perchloric acid (HClO4). The sample was sonicated five times for 1 s and centrifuged at 4500 rpm for 10 min at 4°C. The supernatant was collected, neutralized with 30 μl of 2.5 m potassium hydroxide (KOH), and centrifuged at 14,000 rpm for 10 min at 4°C. The precipitate was removed and the supernatant was kept on ice for ATP measurement. Total cellular ATP was measured using a bioluminescence assay kit (Roche; catalog #11 699 709 001). Samples were diluted in buffer provided by the manufacturer and mixed with luciferase reagent. The absorbance was measured at 560 nm. The blank values were subtracted from the raw data and ATP concentrations calculated from a log-log plot of the standard curve and normalized to protein concentration. The values are expressed as micromoles of ATP per milligram of protein. The MONs that had undergone OGD with or without drug treatment and reperfusion (60 min baseline plus 30 min drug plus 60 min OGD plus 60 min reperfusion; 3.5 h in total) were frozen as a pair attached to the optic chiasm in liquid nitrogen. Protein content was quantified in one nerve and ATP level measured in the other.
Data analysis.
Optic nerve function was monitored quantitatively as the area under the supramaximal CAP. The CAP area is proportional to the total number of excited axons and represents a convenient and reliable means of monitoring optic nerve axon function (Cummins et al., 1979; Stys et al., 1991). Irreversible injury was measured by determining residual CAP area, normalized to control CAP area, 5 h after the conclusion of OGD. Original data were normalized by setting the mean of initial baseline values (measured over 15 min) to a value of 1.0. Results from several nerves were pooled, averaged, and plotted against time. All data are presented as mean ± SEM. In time course plots, SEM bars are only shown every 3 min for simplicity. Statistical significance was determined by unpaired two-tailed Student's t test or ANOVA. The ANOVA tests were followed by Bonferroni's post hoc test (Prism 4; GraphPad). The n values indicate number of optic nerves.
For morphological evaluation, representative microscopic fields in the MONs were imaged by 60× objective lens with constant exposure times. Scoring for cell counts and nuclear morphology was performed with Olympus FluoView, using a predetermined grid (5 × 5). Of 25 fields, immunoreactive cells in every other field were counted only if their nuclei were visible with Sytox staining in the specific section. Cell numbers were expressed as the percentage of immunoreactive cells to the total number of nuclei present. In each experimental condition, the total number of nuclei also was compared to ensure that differences observed between the groups were not attributable to a change in the number of cells. Sytox-stained pyknotic nuclei were counted in the same fields as immunoreactive cells.
Results
HDAC inhibition preserves WM function during OGD
The isolated optic nerve, a purely myelinated CNS tract (Foster et al., 1982), has been successfully used to study ischemic injury (Tekkök et al., 2007; Baltan et al., 2008). The effects of OGD on axon recovery (Fig. 1) were determined in Thy-1 mitoCFP mice (Misgeld et al., 2007), which allowed fluorescent identification of mitochondrial shape, size, and location. The functional integrity of optic nerve axons was monitored by quantifying the area under the CAPs evoked by a supramaximal stimulus. After a 30 min baseline recording, SAHA was introduced for 30 min and the superfusion conditions were maintained during 60 min of OGD and the initial 30 min of reperfusion time. The MON was then reperfused in ACSF for 5 h in the absence of HDAC inhibitors. The onset of OGD caused a transient initial increase in the evoked CAP area before axon excitability rapidly diminished (Fig. 1a). After OGD, the CAP area recovered to 12.7 ± 2% (n = 6) of the maximum recorded CAP area. Whereas the lower dose of SAHA (0.1 μm) improved the CAP area recovery on reperfusion to 22.8 ± 1.1% (n = 4; p < 0.05, one-way ANOVA), a higher dose (1 μm) prevented the complete loss of CAP area during OGD (26.5 ± 6.5%) and improved CAP area recovery to 47.1 ± 2.8% (n = 6; p < 0.001 compared with 0.1 μm SAHA and p < 0.001 compared with OGD, one-way ANOVA).
Figure 1.

Pan- and class 1 HDAC inhibitors promote functional recovery of MON from different mouse strains after OGD. a, Pretreatment with SAHA improved CAP area in a dose-dependent manner in response to OGD (60 min). The percentage CAP area recovery after 60 min of OGD was slightly but significantly improved by 0.1 μm SAHA (22.8 ± 1.1%) compared with OGD (12.7 ± 2.0%). SAHA at 1 μm concentration was more beneficial to axon function (47.1 ± 2.8%). b, SAHA (1 μm) and MS-275 (1 μm) improved percentage CAP area recovery after OGD in MON from both Swiss Webster (SW) and C57BL/6 Thy-1 CFP mice. The CAP area recovered slightly better in MONs from SW when pretreated with MS-275. ***p < 0.0001, two-way ANOVA; #p < 0.0003, unpaired Student's t test. Error bars indicate SEM.
The zinc-containing HDACs are categorized as class I (HDAC 1–3, 8) or class II (HDAC 4–7, 9, 10), based on sequence similarity (Carey and La Thangue, 2006). A specific inhibitor of class I HDACs (Hess-Stumpp et al., 2007), MS-275, proved to be even more effective than SAHA (1 μm) in promoting CAP area recovery (Figs. 1b, 2). After MS-275 (1 μm) pretreatment, CAP area recovery significantly improved (69.0 ± 1.6%, n = 4; compared with 12.7 ± 2.0% after OGD alone, n = 6; p < 0.0001, two-way ANOVA) in C57BL/6 mice. To ensure that the protective effect of HDAC inhibition against OGD was not strain-specific, similar experiments were done using MONs from Swiss Webster mice. The protection conferred by treatment with SAHA and MS-275 was also observed in this second strain (Fig. 1b). The CAP area recovery after 60 min OGD (20.3 ± 3.6%, n = 12) improved to 49.6 ± 5.6% with SAHA (n = 6; p < 0.0001, two-way ANOVA) and to 80.7 ± 1.2% with MS-275 (n = 6; p < 0.0001, two-way ANOVA), demonstrating that the effect of HDAC inhibition is a general phenomenon and is not determined by strain differences (p = 0.6105, two-way ANOVA). Neither SAHA nor MS-275 had any baseline effects on axon function in MONs (Figs. 1a, 2a). This is in contrast to another HDAC inhibitor, Na valproate, which depresses action potentials because of a prominent blockade of voltage-dependent Na+ channels, as well as its interference with K+ and Ca2+ channels (Johannessen, 2000).
Figure 2.
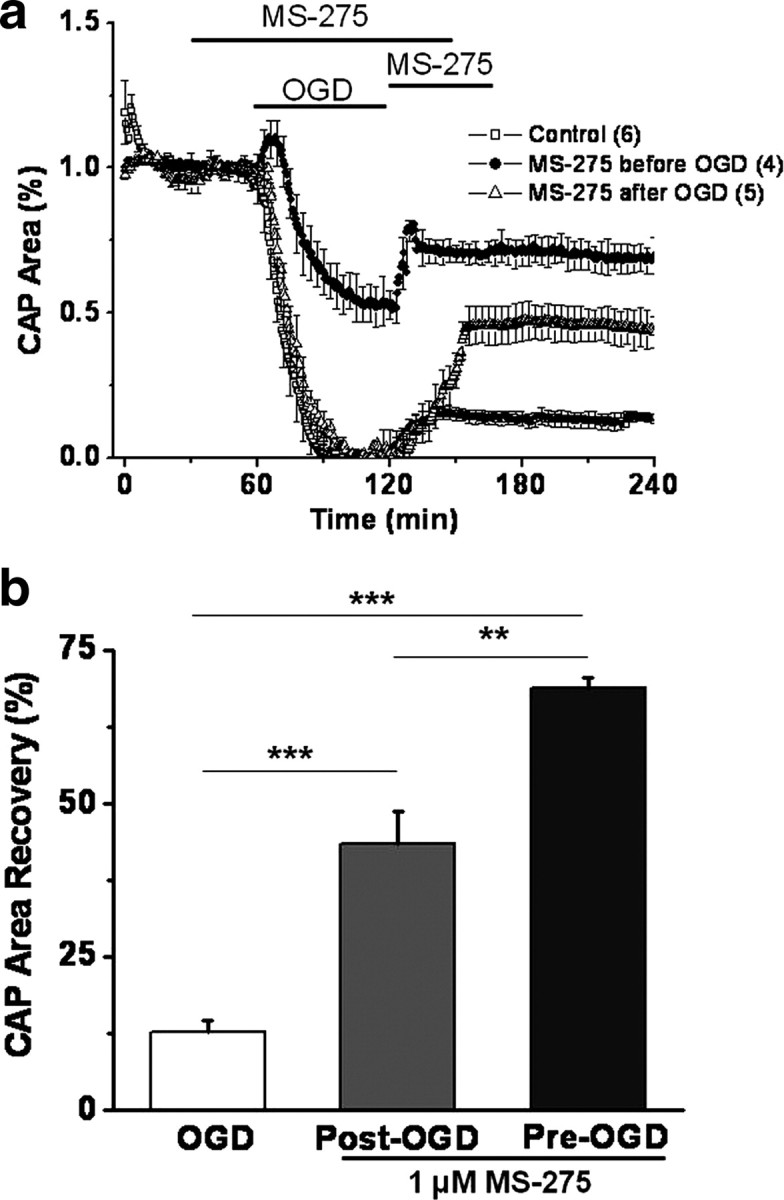
Pretreatment or posttreatment with class I HDAC inhibitor promotes functional recovery of MON against OGD. a, Pretreatment with MS-275 (1 μm; closed circles) improved CAP area in response to OGD (60 min; open squares). Posttreatment with MS-275 (open triangles) promoted a consistent and sustained CAP area recovery. MS-275 application started right at the end of OGD and continued for 45 min before switching to regular ACSF. b, Class 1 HDAC inhibition preserved axon function independent of onset of application, although pretreatment with MS-275 was more beneficial to axon function. The CAP area recovery after 60 min of OGD was robust when MONs were pretreated with MS-275 (69.0 ± 1.6%), whereas the CAP area recovered to 43.3 ± 5.4% when MONs were posttreated with MS-275. **p < 0.01, ***p < 0.001, one-way ANOVA. Error bars indicate SEM.
To determine whether MS-275 application was effective postinjury, drug was applied starting right after the end of OGD for 45 min before switching to regular ACSF (Fig. 2a). Whereas OGD, predictably, suppressed the CAP area completely, MS-275 (1 μm) application slowly but steadily promoted CAP area recovery to 43.4 ± 5.4% (Fig. 2) (n = 5; p < 0.001, one-way ANOVA compared with OGD). This sustained recovery of the CAP area was accompanied by a corresponding preservation of CFP (+) mitochondrial pixel intensity (see below) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (25.9 ± 0.7%, n = 13 for OGD; 40.2 ± 7.4%, n = 17 for post-OGD MS-275 application; p < 0.05). Although pretreatment of MONs with MS-275 before OGD was more effective (Fig. 2; supplemental figure, available at www.jneurosci.org as supplemental material), the consistent protection conferred by postinjury MS-275 application suggests a window of opportunity to attenuate ischemic WM injury.
Class 1 HDACs are selectively expressed in axonal and glial cell compartments
We evaluated the expression and localization of HDACs 1, 2, and 3 in MONs, using immunohistochemistry and isoform-specific antibodies in conjunction with confocal imaging, to support a biological basis for HDAC inhibitor action in the MON preparation (Fig. 3). The expression of HDAC 1 colocalized with GFAP (+) astrocyte nuclei and some processes, and with NF-200 (+) axons (Fig. 3, top panels, merged images). Some oligodendrocyte nuclei were strongly immunoreactive for HDAC 1 (data not shown). The expression of HDAC 2 was principally restricted to astrocyte nuclei and cell bodies, and along the entire length of their processes and end feet. It also colocalized with some oligodendrocyte nuclei. The HDAC 3 enzyme was also observed in astrocytes, but the distribution pattern was punctate in appearance and principally limited to cell processes. Immunolabeling was also evident on distal microglial processes (data not shown). The extensive expression of these HDACs in glial cells, in addition to axons, implicates them as cellular targets of SAHA and MS-275. Labeling characteristics and the specificity of the antibodies were further verified in coronal mouse brain slices containing corpus callosum, together with cortex and hippocampus (data not shown).
Figure 3.
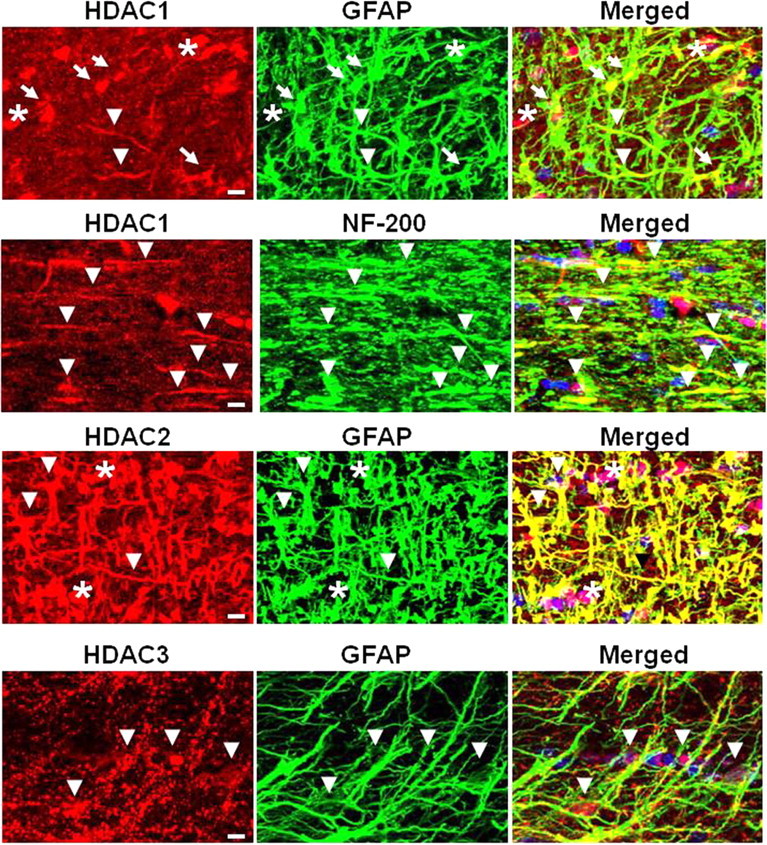
Expression of HDACs 1, 2, and 3 in MON. To determine the localization and pattern of HDAC protein expression, MON was labeled for each HDAC together with GFAP to identify astrocytes or NF-200 for axonal neurofilaments and Sytox for nuclei. The arrows point to astrocyte nuclei, the arrowheads show astrocyte processes or axons, and the asterisks indicate nuclei of oligodendrocytes. Scale bar, 10 μm.
HDAC inhibition preserves WM structural integrity during OGD
The protective effect of HDAC inhibition revealed by electrophysiological analysis was confirmed in a series of imaging studies (Fig. 4). We evaluated the nuclear morphology and density of oligodendrocytes, and the axon cytoskeleton, in MONs under control conditions and OGD with and without HDAC inhibitors, since these critical elements of optic nerve show widespread damage after ischemic insult (Tekkök and Goldberg, 2001; Tekkök et al., 2007). There was no significant change in oligodendrocyte nuclear morphology and density, or in axon cytoskeleton over 12 h in vitro in the absence of injury (data not shown). Uninjured oligodendrocyte nuclei labeled with Sytox were observed as dimly fluorescent, oval cell bodies with clear cytoplasm containing three to five nuclear inclusions (Fig. 4a, control). Exposing MONs to OGD resulted in a loss of adenomatus polyposis coli [APC (+)] oligodendrocytes and a gain in the appearance of pyknotic nuclei with brighter Sytox fluorescence in MONs imaged 5 h after 60 min of OGD (Fig. 4a, OGD, white arrows). The period of OGD rapidly reduced the counts of APC (+) cells to 6.8 ± 2.2% (Fig. 4b) (n = 6; p < 0.001, one-way ANOVA) compared with 67.4 ± 2.6% under control conditions. In parallel, pyknotic nuclei increased from 3.3 ± 0.7 to 73.8 ± 5.2% (Fig. 4b) (n = 6; p < 0.001, one-way ANOVA). Comparably, the intensity of neurofilament labeling and the linear structure of individual axons under control conditions (Fig. 4a, NF-200 panel) was completely disrupted and replaced by bright spots of axonal head and retraction ball formation (asterisks) after OGD. Only a few individual linear axons with dim staining were observed in this group of MONs (Fig. 4a, OGD). Consistent with this, the NF-200 pixel intensity decreased to 23.9 ± 5.1% of control (Fig. 3) (n = 6; p < 0.001, one-way ANOVA). Treatment of MONs with SAHA (1 μm) or MS-275 (1 μm) preserved oligodendrocyte numbers (Fig. 4a,b) (64.6 ± 2.1 and 63.5 ± 1.9%; n = 6; p < 0.001, one-way ANOVA), ameliorated axonal neurofilament loss (Fig. 4a,c), and reversed APC (+) cell loss, and kept pyknotic nuclei counts to the level observed under control conditions.
Figure 4.
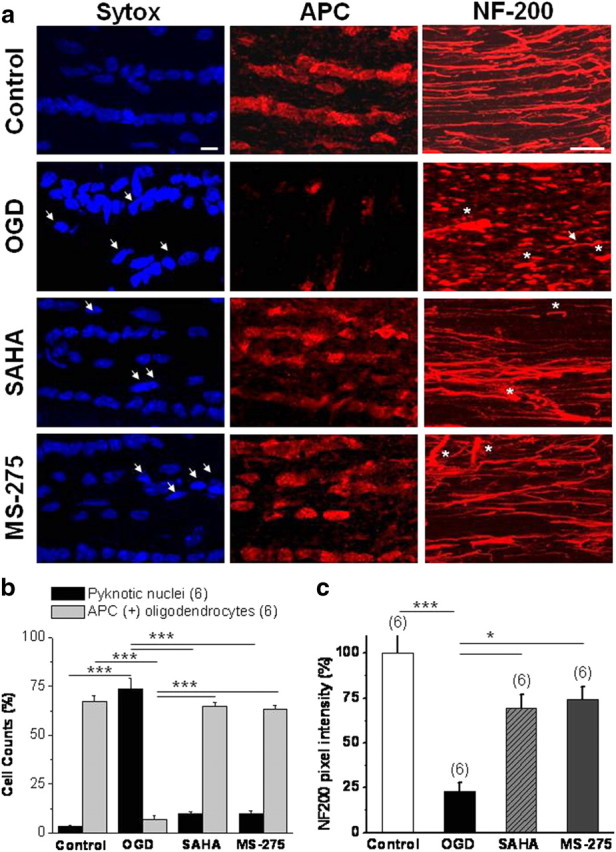
HDAC inhibitors prevent oligodendrocyte death in MON and preserve axon integrity in response to OGD. a, Numerous APC (+) oligodendrocytes (middle top panel) with ellipsoid nuclei labeled with Sytox (left top panel) were observed in 8-week-old Thy-1 mitoCFP control MONs. NF-200 (+) neurofilaments extended along the MON as linear individual fibers (right top panel). A period of OGD (60 min) caused a significant loss of APC (+) oligodendrocytes, a gain in the appearance of pyknotic nuclei (dense, brighter nuclei, white arrows, OGD panel), and loss of NF-200 (+) axon structures, which were replaced with axonal head and bulb formation (white asterisks). Pretreatment with SAHA (1 μm) or MS-275 (1 μm) effectively preserved APC (+) oligodendrocytes, together with numerous linear individual NF-200 (+) axons. Note fewer pyknotic nuclei (white arrows, SAHA and MS-275 panels) after OGD in MONs treated with SAHA or MS-275. b, c, More than 90% of oligodendrocytes [of total APC (+) cells] (b) and 75% of NF-200 immunolabeling (c) were lost after OGD. APC (+) cell loss was accompanied by a steep increase in pyknotic cell counts. SAHA or MS-275 treatment restored expression of APC (+) cells and reduced the number of pyknotic cells. *p < 0.05, ***p < 0.0001. Scale bars, 10 μm. Error bars indicate SEM.
HDAC inhibitors upregulate GLT-1 expression leading to delayed and reduced glutamate accumulation during OGD
The GLT-1 transporter, involved in clearing extracellular glutamate and therefore essential for neuroprotection (Rothstein et al., 2005), is almost exclusively located on astrocytes in the MON from young mice (Baltan et al., 2008). In 8-week-old MONs from Thy-1 mitoCFP mice, GLT-1 (Fig. 5a, red) and GFAP (Fig. 5a, green) expression showed substantial colocalization, confirming that GLT-1 was located principally on astrocytes (Fig. 5a, merged). Note the GLT-1 immunoreactivity outlining GFAP (+) astrocytes processes (white arrows). The period of OGD differentially modulated GLT-1 and GFAP expression. Pixel intensity for GLT-1 was significantly suppressed (Fig. 5a, white columns) (64.5 ± 3%; n = 12; p < 0.001, one-way ANOVA), whereas GFAP pixel intensity was upregulated threefold in conjunction with the appearance of reactive astrocytes (Fig. 5b, gray columns) (256.6 ± 11.6%; n = 12; p < 0.001, one-way ANOVA). The protective doses of SAHA and MS-275 significantly elevated GLT-1 levels (134.8 ± 4.0%, n = 15, p < 0.001, one-way ANOVA for SAHA; 140.9 ± 2.3%, n = 15, p < 0.001, one-way ANOVA for MS-275), whereas both SAHA (145.4 ± 5.1%; n = 15; p < 0.001, one-way ANOVA) and MS-275 reduced OGD-induced changes in GFAP expression (79.4 ± 4.4%; n = 15; p < 0.001, one-way ANOVA) and astrocyte morphology (Fig. 5a,b). Note that the effects of OGD or HDAC inhibition on GLT-1 or GFAP pixel intensity did not reflect a change in the number of astrocytes. Because astrocytes are natural anaerobes and hydrolyze glycogen to maintain enough ATP to function during ischemia (Goldberg and Choi, 1993), astrocyte counts remained stable under all conditions tested (black columns). For instance, under control conditions, GFAP (+) astrocytes accounted for 28.8 ± 1.5% of all glial cells. After 60 min of OGD, astrocyte counts were 26.4 ± 2.3% and pretreatment of MONs with SAHA or MS-275 before OGD maintained astrocyte counts at 27.2 ± 3.4 and 26.9 ± 2.2%, respectively. The HDAC inhibitor did not cause a similar increase in glutamate–aspartate transporter (GLAST) immunoreactivity, another Na+-dependent glutamate transporter that is known to colocalize with GFAP (+) astrocytes (data not shown).
Figure 5.
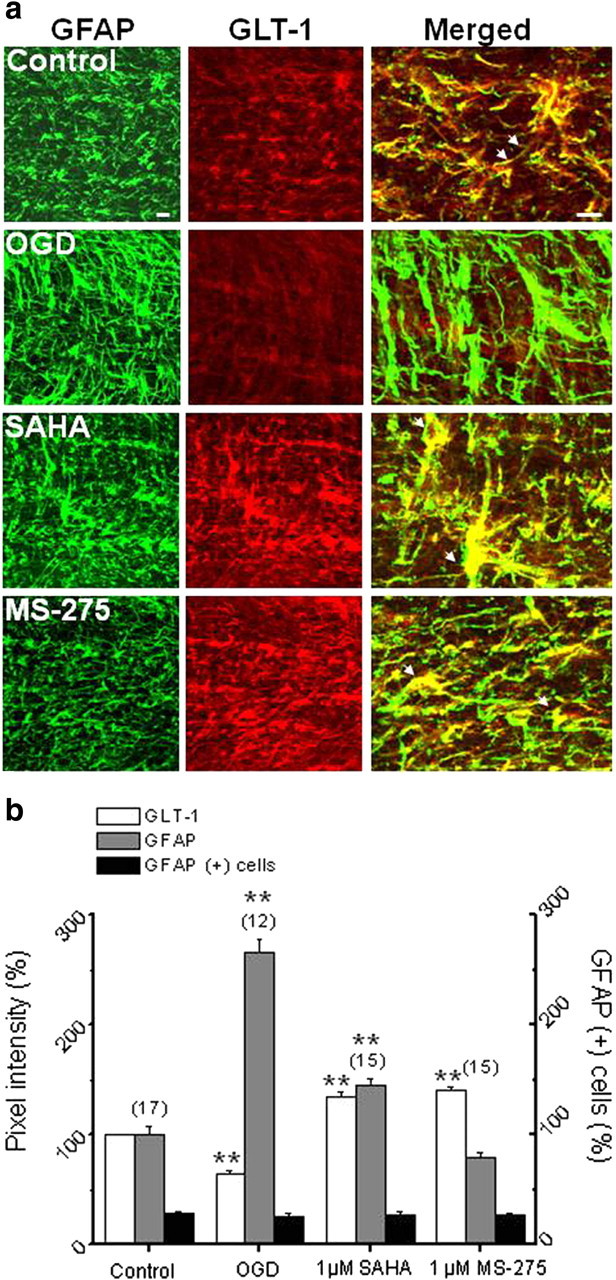
HDAC inhibitors upregulate GLT-1 expression. a, The colocalization of GLT-1 and GFAP labeling under control conditions indicated that GLT-1 is predominantly expressed by astrocytes (merged, higher magnification) in MON. The period of OGD caused a significant increase in GFAP expression (265.6 ± 11.7%) but depressed GLT-1 immunolabeling (64.5 ± 3.0%). Pretreatment of MON with SAHA (1 μm) or MS-275 (1 μm) significantly attenuated this increase in GFAP expression and upregulated GLT-1 pixel intensity (arrows). Scale bar, 10 μm. b, Quantitative changes in GFAP (gray columns) and GLT-1 (white columns) expression under control, OGD, SAHA, or MS-275 conditions were not attributable to changes in the number of astrocytes (black columns). **p < 0.001, one-way ANOVA. Error bars indicate SEM.
A role for GLT-1 upregulation in the induction of tolerance to ischemia in gray matter is well established for both in vitro and in vivo conditions (Romera et al., 2004; Kawahara et al., 2005; Kosugi et al., 2005; Zhang et al., 2007). The increase in GLT-1 expression with HDAC inhibition could increase the capacity of astrocytes to remove glutamate and delay extracellular glutamate accumulation, thus playing an important role in attenuating excitotoxicity and ischemic injury to WM. We directly tested this hypothesis by quantifying the glutamate release from MONs at 37°C under control and OGD conditions, with or without MS-275 (1 μm). Using a pair of MONs, baseline glutamate release levels were quantified for 60 min before OGD by sampling the perfusion solution (Tekkök et al., 2007; Baltan et al., 2008). For comparison, all glutamate release data were normalized to the average control release levels, which were in the range of 2–5 nm (data not shown). Glutamate is only detected in these experiments after it has escaped from the extracellular space of WM, which is diluted by the bath solution (1000- to 10,000-fold), and the data were plotted for every 4 min value. Initially during OGD, glutamate release remained stable at the baseline level for at least 30 min for all MONs (Fig. 6, inset, black squares). As previously reported (Tekkök et al., 2007; Baltan et al., 2008), glutamate release steadily increased starting at 30 min of OGD (Fig. 6, inset, black squares) and reached maximal levels that were sixfold higher compared with basal levels at the end of OGD. On return to control ACSF, glutamate levels declined rapidly toward baseline within 15 min. Glutamate release was delayed when MONs were treated with MS-275, and the amount of glutamate release was lower at every time point measured (Fig. 6, inset, red circles) (*p < 0.05, **p < 0.001). To compare glutamate accumulation, the areas under the glutamate release time course plots were quantified and normalized, and the data with MS-275 were expressed as a percentage of glutamate released by OGD alone (Fig. 6). Inhibition with MS-275 reduced glutamate accumulation to 46.0 ± 7.4% (n = 6) of OGD levels (100.0 ± 3.2%; n = 4; p < 0.0005, Student's t test).
Figure 6.
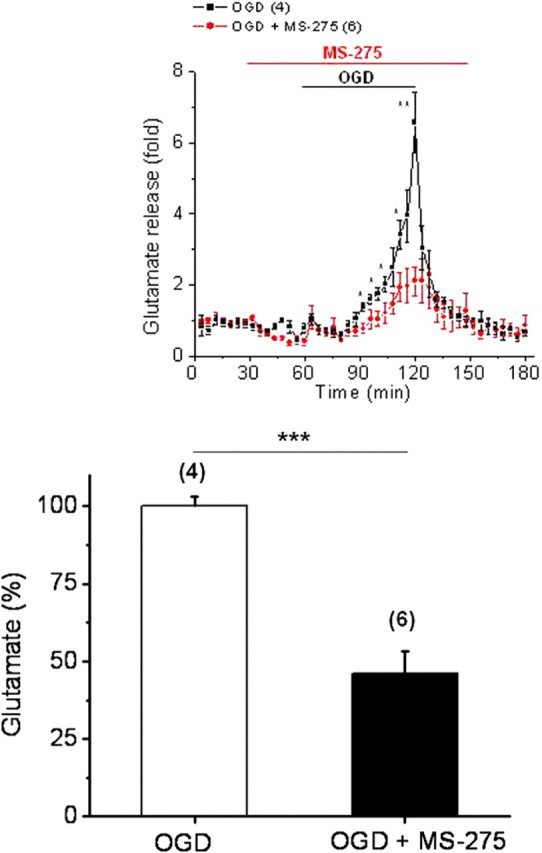
Class 1 HDAC inhibition delays and reduces glutamate accumulation during OGD. Glutamate accumulation was quantified by the area under the glutamate release time course plots and normalized data with MS-275 were expressed as a percentage of OGD. HDAC inhibition with MS-275 (1 μm) during OGD reduced glutamate accumulation to 46.0 ± 7.4% (n = 6) of OGD levels (100.0 ± 3.2%; n = 4; p < 0.0005, Student's t test). Inset, Baseline glutamate release remained stable under control conditions. Glutamate release steadily increased starting at 30 min after the onset of OGD and reached a level sixfold greater than baseline (black squares; n = 4). Under identical conditions, glutamate release was significantly lower and delayed at every time point during OGD with MS-275. *p < 0.05, **p < 0.001, ***p < 0.0005. Student's t test. Error bars indicate SEM.
HDAC inhibition preserves mitochondrial complement and ATP levels in MON
Ischemia initiates an ATP crisis that facilitates glutamate release in WM (Tekkök et al., 2007). Excitotoxicity and elevated Ca2+ induce marked changes in mitochondrial morphology, arrest their motion (Rintoul et al., 2003; Barsoum et al., 2006; Chang and Reynolds, 2006), and generate reactive oxygen species in neurons (for review, see Nicholls et al., 2007). Because HDAC inhibition delayed glutamate accumulation (Fig. 6), and reduced excitotoxic injury, we reasoned that their effects on mitochondrial density and ATP levels should be evident. Axonal mitochondria in MONs can be selectively imaged in the mitoCFP mouse in which a neuron-specific Thy-1 promoter drives expression of mitochondrial-targeted CFP (Misgeld et al., 2007) (Fig. 7a). Mitochondria in the untreated MON displayed a short, tubular morphology (Fig. 7a, control, inset). After OGD, there was a dramatic reduction in CFP fluorescence (Fig. 7a) (19.5 ± 0.5%; n = 6; p < 0.001, one-way ANOVA). Remaining CFP (+) mitochondria presented a punctate morphology (Fig. 7a, OGD, inset) in agreement with ischemia-induced fission (Barsoum et al., 2006). Pretreatment of MONs with SAHA, and pretreatment or posttreatment with MS-275, preserved CFP pixel intensity (Fig. 7b) (SAHA, 70.2 ± 0.7%, n = 7, p < 0.001; MS-275, 69.1 ± 0.8%, n = 6, p < 0.001, one-way ANOVA) (supplemental figure, available at www.jneurosci.org as supplemental material) (posttreatment with MS-275, 73.2 ± 1.6%, n = 17, p < 0.001) and mitochondrial morphology (Fig. 7a; supplemental figure, available at www.jneurosci.org as supplemental material). We confirmed that the ischemia-mediated loss of cyan signal is attributable to the actual loss of CFP protein by labeling with GFP antibody, and by monitoring mitochondrial damage with Hsp60 immunohistochemistry in gray and white matter (data not shown).
Figure 7.
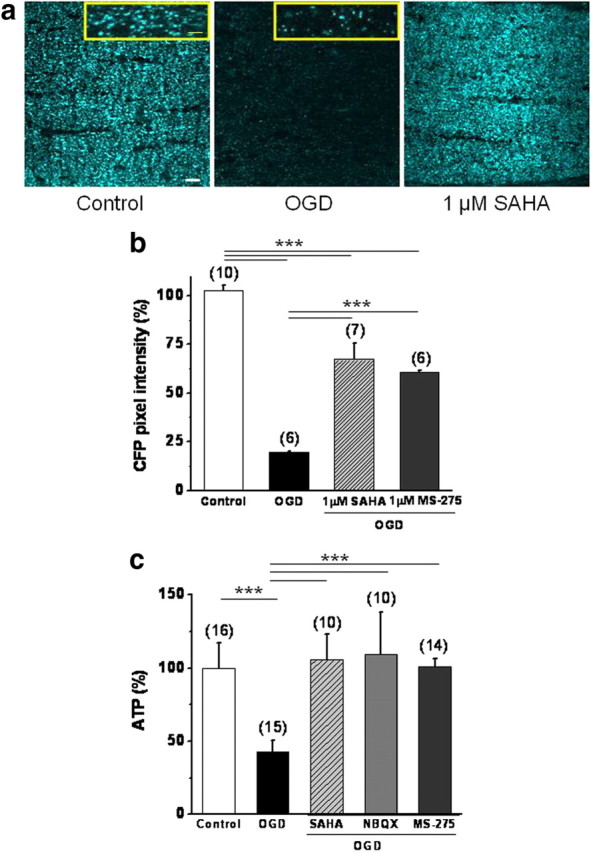
HDAC inhibitors preserve CFP (+) axonal mitochondria and ATP levels in response to OGD. a, OGD drastically reduced CFP fluorescence in MONs from mitoCFP (+) mice and pretreatment with SAHA (1 μm) protected against this loss. Note the change in mitochondrial morphology from small tubular to tiny punctuated form with OGD (insets). Scale bar, 2 μm. b, SAHA and MS-275 were equally effective in preserving CFP pixel intensity. c, Consistent with the preservation of CFP pixel intensity, SAHA and MS-275 pretreatment conserved ATP levels in MONs. NBQX (30 μm) was included as a positive control (Tekkök et al., 2007). ***p < 0.0001, one-way ANOVA. Scale bar, 10 μm. n value is in parentheses. Error bars indicate SEM.
These results suggest that the maintenance of functional integrity within mitochondria can reduce excitotoxic WM injury, presumably via the preservation of the local energy homeostasis of the axon. Consistent with a loss of axonal mitochondria as suggested by the reduction in CFP fluorescence, we observed a significant reduction in MON ATP levels after OGD (60 min, followed by 60 min reperfusion) (Fig. 7c) (42.8 ± 0.8%; n = 15; p < 0.001, one-way ANOVA). Preservation of CFP (+) mitochondria (Fig. 7) and axon function (Figs. 1, 2) by SAHA (1 μm) and MS-275 (1 μm) treatment correlated with the maintenance of ATP levels (Fig. 7c) (SAHA, 106.2 ± 17.2%, n = 10, p < 0.001; MS-275, 100.9 ± 5.6%, n = 14, p < 0.001). Consistent with this finding, blockade of AMPA/kainate receptors with NBQX (30 μm), which promotes axon function recovery after OGD (Tekkök et al., 2007), also effectively preserved ATP levels (109.4 ± 28.9%; n = 10; p < 0.001, one-way ANOVA) and CFP pixel intensity (data not shown), confirming that suppression of excitotoxicity prevents mitochondrial dysfunction and ATP depletion.
Discussion
We directly tested, and confirmed, that inhibition of HDAC activity, specifically inhibition of class I HDACs, before or after an ischemic episode promotes functional recovery of MON axons and preserves WM structure. Key correlates of these effects included the upregulation of GLT-1 transporter expression on astrocytes, a delayed and diminished glutamate accumulation, inhibition of astrocyte hypertrophy, maintenance of mitochondrial integrity in axons, and preservation of MON ATP levels. Our findings suggest that a therapeutic opportunity exists for the use of HDAC inhibitors in targeting mitochondrial energy regulation and excitotoxicity in ischemic WM injury.
One novel finding in our study was that HDAC inhibition preserved axon structure and function in WM. This effect was apparent in two different strains of mice, suggesting that this is a general response to HDAC inhibition. Consistent with these findings, glial cells and axons were found to express class I HDACs, specifically HDAC 1, 2, and 3. The consistent expression pattern and cellular localization of HDAC 1, 2 and 3 in WM components was intriguing and suggests a universal role for HDACs in WM. By comparison, HDAC 4 and 6 (class II HDACs) expression levels were very low in both neuronal and glial elements (data not shown), despite recent evidence for HDAC 6 involvement in CNS injury (Langley et al., 2009).
A provocative aspect of our study was the demonstrated efficacy of the class I HDAC inhibitor, MS-275, when administered after an ischemic episode. Since protection was observed when this drug was added after the maximal accumulation of glutamate had occurred, this finding demonstrates that MS-275 may have multiple distinct sites of action; one site related to glutamate accumulation (GLT-1 expression) and the other involving postexcitotoxic mechanisms. Ischemic injury in young WM is mediated by integration of multiple pathways; axons are injured directly by ionic deregulation leading to toxic accumulation of Na+ and Ca2+ (Stys et al., 1990; Fern et al., 1995; Wolf et al., 2001; Underhill and Goldberg, 2007), whereas oligodendrocytes and their myelin are subject to excitotoxicity because of reversal of Na+-dependent glutamate transporters, mainly GLT-1 (Tekkök et al., 2007). Finally, the oxidative pathway attacks WM constituents because of formation of free radicals mediated by glutamate competing with cysteine at the glutamate–cysteine pump (Oka et al., 1993), or by AMPA/kainate receptor activation. The ionic pathway triggers the injury process, which subsequently reverses the glutamate transporter(s); however, it is the accumulation of glutamate that dictates the threshold for irreversible injury (Tekkök et al., 2007; Baltan, 2009).
We propose that the consistent protection via HDAC inhibition, whether before or after injury, may be a result of the drug simultaneously targeting excitotoxic and oxidative pathways, during the spatiotemporal progression of WM injury. Several lines of evidence support this proposal. First, a central finding of this study was the increase in expression of astrocyte GLT-1 when HDAC inhibition was achieved before injury. The onset of OGD, alone, reduced GLT-1 expression on astrocytes, consistent with previous reports for ischemia (Torp et al., 1995), epilepsy (Rothstein et al., 1996; Tanaka et al., 1997), Alzheimer's disease (Li et al., 1997; Dabir et al., 2006), Huntington's disease (Lipton and Rosenberg, 1994), and amyotrophic lateral sclerosis (Rothstein and Kuncl, 1995; Lin et al., 1998). Second, GLT-1 upregulation by HDAC inhibitors delayed and attenuated extracellular glutamate accumulation caused by OGD in WM, similar to previous reports in astrocyte cultures (Romera et al., 2004; Kawahara et al., 2005; Kosugi et al., 2005). The cause of glutamate release during OGD is a loss of the transmembrane Na+ gradient, subsequently reversing Na+-dependent glutamate transport because of ATP depletion and failure of Na+-K+ ATPase. By delaying and reducing glutamate accumulation, HDAC inhibition suppressed subsequent injury pathways, preserving mitochondrial function and ATP levels, which in turn maintains the function of GLT-1 providing an energy efficient cycle to minimize glutamate accumulation. As a result, excitotoxic oligodendrocyte death and subsequent axonal cytostructural disruption were drastically reduced. It is therefore of great interest that HDAC inhibition offers a pharmacological strategy to upregulate GLT-1, a target gene critically involved in various disease conditions including stroke, so as to effectively ameliorate excitotoxicity. Pan-HDAC inhibitors, such as SAHA, have been shown to enhance glutamate transport, increase expression of GLT-1 and GLAST in astrocytes (Wu et al., 2008; Allritz et al., 2009), and also GLT-1 expression in the striatum (Morland et al., 2004). Therefore, HDAC inhibition before injury interrupts excitotoxicity and subsequent downstream injury pathways. Consequently, the increased extent of axon function recovery with preinjury application compared with postinjury reflects early intervention of HDAC inhibitors at the excitotoxicity step in the sequential course of WM injury. Additional confirmation was provided when NBQX comparably preserved ATP levels by blockade of excitotoxicity mediated by AMPA/kainate receptor activation. It is possible that HDAC inhibition may affect downstream components of excitotoxicity or oxidative injury, or spare mitochondria to reduce glutamate release and so exert protective effects.
Another significant finding in our study was the demonstration that the axon-protective action associated with HDAC inhibition correlated with preservation of mitochondrial structure/function in axons and ATP levels in MONs. This effect was evident whether HDAC inhibitors were applied before or after the injury. Axon functional recovery was twofold higher with preinjury HDAC inhibitor application than with postinjury. Axon function directly correlates with tissue energy reserves, since Na+-K+ ATPase activity is intimately dependent on ATP levels. As a result, OGD caused a significant reduction in ATP levels and CFP (+) mitochondria, which were profoundly restored after HDAC inhibition. However, the extent of ATP and CFP (+) mitochondria preservation depended on the time of application of the HDAC inhibitor as preinjury treatment resulted in sustained CFP levels. Importantly, HDAC application postinjury consistently provided significant protection of axon function that correlated with preservation of CFP (+) mitochondria, suggesting intervention in the oxidative pathway, which is a later step downstream of glutamate accumulation in the sequential course of ischemic WM injury.
The present study provides the first evidence that inhibition of class I HDACs confers long-lasting benefits to CNS WM function and structure after acute ischemic injury. Our results suggest that preinjury HDAC inhibition acts on the excitotoxic pathway, interrupting subsequent mitochondrial dysfunction and ATP depletion, whereas postinjury HDAC inhibition acts on the oxidative pathway and/or downstream components of excitotoxicity. Accordingly, the extent of protection is higher with preinjury application compared with postinjury. Attenuating inflammation, alleviating endoplasmic reticulum stress, or direct activation of mGluR receptors (Chiechio et al., 2009) are additional possible sites of HDAC inhibitor action, and these remain to be explored in WM.
Our findings concerning the effectiveness of HDAC inhibitors have implications for treating various forms of brain injury. The WM is injured in most strokes, contributing to the disability associated with clinical deficits. HDAC inhibition may provide a suitable option to protect WM in the clinical manifestations of stroke to facilitate successful translation of experimental stroke research to clinical trials.
Footnotes
This work was supported by the American Heart Association (S.B., R.S.M.), National Institutes of Health (NIH)–National Institute on Aging Grant AG033720 (S.B.), and NIH–National Institute of Neurological Disorders and Stroke Grant NS065319 (S.P.M., R.S.M.) and Grants NS03553 and NS056031 (R.S.M.). We thank Dr. Yoshito Kinoshita for generating transgenic mice.
References
- Allritz C, Bette S, Figiel M, Engele J. Endothelin-1 reverses the histone deacetylase inhibitor-induced increase in glial glutamate transporter transcription without affecting histone acetylation levels. Neurochem Int. 2009;55:22–27. doi: 10.1016/j.neuint.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Balasubramaniyan V, Boddeke E, Bakels R, Küst B, Kooistra S, Veneman A, Copray S. Effects of histone deacetylation inhibition on neuronal differentiation of embryonic mouse neural stem cells. Neuroscience. 2006;143:939–951. doi: 10.1016/j.neuroscience.2006.08.082. [DOI] [PubMed] [Google Scholar]
- Baltan S. Ischemic injury to white matter: an age-dependent process. Neuroscientist. 2009;15:126–133. doi: 10.1177/1073858408324788. [DOI] [PubMed] [Google Scholar]
- Baltan S, Besancon EF, Mbow B, Ye Z, Hamner MA, Ransom BR. White matter vulnerability to ischemic injury increases with age because of enhanced excitotoxicity. J Neurosci. 2008;28:1479–1489. doi: 10.1523/JNEUROSCI.5137-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Gräber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey N, La Thangue NB. Histone deacetylase inhibitors: gathering pace. Curr Opin Pharmacol. 2006;6:369–375. doi: 10.1016/j.coph.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Chang DT, Reynolds IJ. Differences in mitochondrial movement and morphology in young and mature primary cortical neurons in culture. Neuroscience. 2006;141:727–736. doi: 10.1016/j.neuroscience.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Chiechio S, Zammataro M, Morales ME, Busceti CL, Drago F, Gereau RW, 4th, Copani A, Nicoletti F. Epigenetic modulation of mGlu2 receptors by histone deacetylase inhibitors in the treatment of inflammatory pain. Mol Pharmacol. 2009;75:1014–1020. doi: 10.1124/mol.108.054346. [DOI] [PubMed] [Google Scholar]
- Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton AL, Hazzalin CA, Mahadevan LC. Enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell. 2006;23:289–296. doi: 10.1016/j.molcel.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Cummins KL, Perkel DH, Dorfman LJ. Nerve fiber conduction-velocity distributions. I. Estimation based on the single-fiber and compound action potentials. Electroencephalogr Clin Neurophysiol. 1979;46:634–646. doi: 10.1016/0013-4694(79)90101-9. [DOI] [PubMed] [Google Scholar]
- Dabir DV, Robinson MB, Swanson E, Zhang B, Trojanowski JQ, Lee VM, Forman MS. Impaired glutamate transport in a mouse model of tau pathology in astrocytes. J Neurosci. 2006;26:644–654. doi: 10.1523/JNEUROSCI.3861-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fern R, Ransom BR, Waxman SG. Voltage-gated calcium channels in CNS white matter: role in anoxic injury. J Neurophysiol. 1995;74:369–377. doi: 10.1152/jn.1995.74.1.369. [DOI] [PubMed] [Google Scholar]
- Foster RE, Connors BW, Waxman SG. Rat optic nerve: electrophysiological, pharmacological and anatomical studies during development. Dev Brain Res. 1982;3:371–386. doi: 10.1016/0165-3806(82)90005-0. [DOI] [PubMed] [Google Scholar]
- Gibson CL, Murphy SP. Benefits of histone deacetylase inhibitors for acute brain injury; a systematic review of animal studies. J Neurochem. 2010;115:806–813. doi: 10.1111/j.1471-4159.2010.06993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess-Stumpp H, Bracker TU, Henderson D, Politz O. MS-275, a potent orally available inhibitor of histone deacetylases—the development of an anticancer agent. Int J Biochem Cell Biol. 2007;39:1388–1405. doi: 10.1016/j.biocel.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Johannessen CU. Mechanisms of action of valproate: a commentatory. Neurochem Int. 2000;37:103–110. doi: 10.1016/s0197-0186(00)00013-9. [DOI] [PubMed] [Google Scholar]
- Kawahara K, Kosugi T, Tanaka M, Nakajima T, Yamada T. Reversed operation of glutamate transporter GLT-1 is crucial to the development of preconditioning-induced ischemic tolerance of neurons in neuron/astrocyte co-cultures. Glia. 2005;49:349–359. doi: 10.1002/glia.20114. [DOI] [PubMed] [Google Scholar]
- Khan HA. Bioluminometric assay of ATP in mouse brain: determinant factors for enhanced test sensitivity. J Biosci. 2003;28:379–382. doi: 10.1007/BF02705114. [DOI] [PubMed] [Google Scholar]
- Kosugi T, Kawahara K, Yamada T, Nakajima T, Tanaka M. Functional significance of the preconditioning-induced down-regulation of glutamate transporter GLT-1 in neuron/astrocyte co-cultures. Neurochem Res. 2005;30:1109–1116. doi: 10.1007/s11064-005-8219-z. [DOI] [PubMed] [Google Scholar]
- Langley B, D'Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, Yang L, Ko B, Fisher M, Cho S, Beal MF, Ratan RR. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21waf1/cip1 in cell cycle-independent neuroprotection. J Neurosci. 2008;28:163–176. doi: 10.1523/JNEUROSCI.3200-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, Brochier C, Rivieccio MA. Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke. 2009;40:2899–2905. doi: 10.1161/STROKEAHA.108.540229. [DOI] [PubMed] [Google Scholar]
- Li S, Mallory M, Alford M, Tanaka S, Masliah E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol. 1997;56:901–911. doi: 10.1097/00005072-199708000-00008. [DOI] [PubMed] [Google Scholar]
- Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, Rothstein JD. Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron. 1998;20:589–602. doi: 10.1016/s0896-6273(00)80997-6. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW. Imaging axonal transport of mitochondria in vivo. Nat Methods. 2007;4:559–561. doi: 10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- Morland C, Boldingh KA, Iversen EG, Hassel B. Valproate is neuroprotective against malonate toxicity in rat striatum: an association with augmentation of high-affinity glutamate uptake. J Cereb Blood Flow Metab. 2004;24:1226–1234. doi: 10.1097/01.WCB.0000138666.25305.A7. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Johnson-Cadwell L, Vesce S, Jekabsons M, Yadava N. Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J Neurosci Res. 2007;85:3206–3212. doi: 10.1002/jnr.21290. [DOI] [PubMed] [Google Scholar]
- Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci. 1993;13:1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci. 2003;23:7881–7888. doi: 10.1523/JNEUROSCI.23-21-07881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romera C, Hurtado O, Botella SH, Lizasoain I, Cárdenas A, Fernández-Tomé P, Leza JC, Lorenzo P, Moro MA. In vitro ischemic tolerance involves upregulation of glutamate transport partly mediated by the TACE/ADAM17-tumor necrosis factor-α pathway. J Neurosci. 2004;24:1350–1357. doi: 10.1523/JNEUROSCI.1596-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Kuncl RW. Neuroprotective strategies in a model of chronic glutamate-mediated motor neuron toxicity. J Neurochem. 1995;65:643–651. doi: 10.1046/j.1471-4159.1995.65020643.x. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Siebzehnrubl FA, Buslei R, Eyupoglu IY, Seufert S, Hahnen E, Blumcke I. Histone deacetylase inhibitors increase neuronal differentiation in adult forebrain precursor cells. Exp Brain Res. 2007;176:672–678. doi: 10.1007/s00221-006-0831-x. [DOI] [PubMed] [Google Scholar]
- Stys PK, Ransom BR, Waxman SG, Davis PK. Role of extracellular calcium in anoxic injury of mammalian central white matter. Proc Natl Acad Sci U S A. 1990;87:4212–4216. doi: 10.1073/pnas.87.11.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Na+-Ca2+ exchanger mediates Ca2+ influx during anoxia in mammalian central nervous system white matter. Ann Neurol. 1991;30:375–380. doi: 10.1002/ana.410300309. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tekkök SB, Goldberg MP. AMPA/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci. 2001;21:4237–4248. doi: 10.1523/JNEUROSCI.21-12-04237.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekkök SB, Ye Z, Ransom BR. Excitotoxic mechanisms of ischemic injury in myelinated white matter. J Cereb Blood Flow Metab. 2007;27:1540–1552. doi: 10.1038/sj.jcbfm.9600455. [DOI] [PubMed] [Google Scholar]
- Thomas EA. Focal nature of neurological disorders necessitates isotype-selective histone deacetylase (HDAC) inhibitors. Mol Neurobiol. 2009;40:33–45. doi: 10.1007/s12035-009-8067-y. [DOI] [PubMed] [Google Scholar]
- Torp R, Lekieffre D, Levy LM, Haug FM, Danbolt NC, Meldrum BS, Ottersen OP. Reduced postischemic expression of a glial glutamate transporter, GLT1, in the rat hippocampus. Exp Brain Res. 1995;103:51–58. doi: 10.1007/BF00241964. [DOI] [PubMed] [Google Scholar]
- Underhill SM, Goldberg MP. Hypoxic injury of isolated axons is independent of ionotropic glutamate receptors. Neurobiol Dis. 2007;25:284–290. doi: 10.1016/j.nbd.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uo T, Veenstra TD, Morrison RS. HDAC inhibitors prevent p53-dependent and -independent Bax-mediated neuronal apoptosis through two distinct mechanisms. J Neurosci. 2009;29:2824–2832. doi: 10.1523/JNEUROSCI.6186-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21:1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY, Niu FN, Huang R, Xu Y. Enhancement of glutamate uptake in 1-methyl-4-phenylpyridinium-treated astrocytes by trichostatin A. Neuroreport. 2008;19:1209–1212. doi: 10.1097/WNR.0b013e328308b355. [DOI] [PubMed] [Google Scholar]
- Zhang M, Li WB, Geng JX, Li QJ, Sun XC, Xian XH, Qi J, Li SQ. The upregulation of glial glutamate transporter-1 participates in the induction of brain ischemic tolerance in rats. J Cereb Blood Flow Metab. 2007;27:1352–1368. doi: 10.1038/sj.jcbfm.9600441. [DOI] [PubMed] [Google Scholar]