

Abstract
Biomolecular interactions between proteins and synthetic surfaces impact diverse biomedical fields. Simple, quantitative, label-free technologies for the analysis of protein adsorption and binding of biomolecules are thus needed. Here, we report the use of a novel type of substrate, poly-p-xylylenes coating prepared by chemical vapor deposition (CVD) polymerization, for surface plasmon resonance enhanced ellipsometry (SPREE) studies and assess the reactive coatings as spatially-resolved biomolecular sensing arrays. Prior to use in binding studies, reactive coatings were fully characterized by Fourier transform infrared spectroscopy (FTIR), electrochemical impedance spectroscopy (EIS), and ellipsometry. As a result, chemical structure, thickness, and homogenous coverage of the substrate surface were confirmed for a series of CVD-coated samples. Subsequent SPREE imaging and fluorescence microscopy indicated that the synthetic substrates supported detectable binding of a cascade of biomolecules. Moreover, analysis revealed a useful thickness range for CVD films in the assessment of protein and/or antigen-antibody binding via SPREE imaging. With a variety of functionalized end groups available for biomolecule immobilization and ease of patterning, CVD thin films are useful substrates for spatially-resolved, quantitative binding arrays.
Keywords: Imaging surface plasmon resonance enhanced ellipsometry, chemical vapor deposition, reactive coatings, biomolecular sensing, bioconjugation
INTRODUCTION
Specific interactions between proteins and surface-bound ligands are important in controlling most biological events that may occur at a synthetic materials surface including protein adsorption, cell adhesion, and cell proliferation.1, 2 Thus, quantitative analysis of protein adsorption or binding of biomacromolecules to surface-immobilized recognition sites has been an area of intense research and a plethora of different methods have been investigated.3 Among the most widely used methods are fluoriometry,4 enzyme-linked immunosorbent assay (ELISA),5 radiometry,6 or photoluminescence analysis.7 Although widely used in current biology, these methods require cumbersome assay optimization (ELISA) or are associated with the need for diverse labels prior to detection, such as fluorescent, radioactive, or photoluminescent groups.8 Such labels require additional chemical or biological reactions and separation steps. In addition, chemical modification changes the properties of the target molecules.9 Thus, label-free analytical tools, such as quartz crystal microbalance (QCM), MEMS-based sensors, or surface plasmon resonance (SPR) have increasingly attracted attention in the biological community.10, 11 In spite of the undoubted success of these methods in recent years, the spatially defined quantitative analysis of biomolecule/surface interactions remains highly elusive.12 In principle, ellipsometry, and more specifically imaging ellipsometry in surface plasmon resonance conditions, is well positioned to overcome these limitations.13 Surface plasmon resonance enhanced ellipsometry (SPREE) imaging14 has enhanced sensitivity, as compared to conventional surface plasmon resonance methods, because it provides phase information – in addition to intensity.15 Outputs in this configuration are the ellipsometric parameters delta (Δ) and psi (Ψ), where psi is analogous to the reflectivity intensity provided by conventional SPR.15 Phase information, provided by the parameter Δ in this work, has been found to be more sensitive, (10−7-10−8 refractive index units versus 10−5-10−6 refractive index units for conventional SPR),16, 17 to biomolecular interactions than reflective intensity alone, because phase changes abruptly in response to variations in the bulk refractive index of the medium and thus is associated with higher signal-to-noise ratio.18 Imaging ellipsometry can deliver spatially-resolved, quantitative data, is sufficiently sensitive for many biological questions (adsorption thickness detection in the picometer range), and can be easily used in aqueous environments. So far, progress with imaging ellipsometry for biological questions has been hampered by the availability of flexible binding substrates for protein immobilization. Self-assembled monolayers of thiols on gold have been pursued in the past, but have limited stability and shelf-life.19 Dextran matrices have also been utilized but require chemical modification in situ, typically N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) chemistry.20 Star-PEGs21 and dendrimers22 are other surface modification strategies that have been employed to study protein and DNA interactions for biomedical and bio-analytical applications. Recently, self-assembled gold fusion proteins were employed as recognition elements for antibody detection.23
Vapor-based reactive polymer coatings have the potential to function as versatile, yet chemically well-defined binding substrates, when deposited on Au-coated substrates. These reactive coatings are made by chemical vapor deposition polymerization of functionalized [2.2]paracyclophanes, and are known as poly-p-xylylenes. Functionalized poly-p-xylylenes containing aldehydes,24 amines,25 anhydrides,26 or active esters,27-29 have been used to immobilize a wide range of biomolecules.29-31 In addition, CVD based reactive coatings can be micro- and nanostructured with a number of well established patterning methods, including microcontact printing,32 vapor-assisted micropatterning,33 supramolecular nanostamping,34 and photolithography.35 In this study, we demonstrate based on a representative model coating, poly(4-pentafluoropropionyl-p-xylylene-co-p-xylylene) (PPX-COC2F5), that reactive coatings can be deposited as sufficiently thin, yet pinhole-free coatings, that support label-free, spatially-controlled and quantitative studies of protein binding cascades for immobilization studies via imaging surface plasmon resonance enhanced ellipsometry. As such, CVD coatings are novel substrates for studying biomolecular-surface interactions, when quantitative and spatial information is desired.
EXPERIMENTAL PART
Chemical Vapor Deposition Polymerization
Polymer coatings were prepared using a custom-designed chemical vapor deposition (CVD) system comprised of three working sections; a sublimation zone, pyrolysis zone, and a deposition zone.36 In this instance, the starting material was a [2.2]paracyclophane functionalized with trifluoropropinyl groups (COC2F5)37. The sublimation and pyrolysis temperatures were 120°C and 660°C for all samples. In order to generate different polymer thicknesses, the amount of starting material was varied and ranged from 3-13 mg with more material being utilized for thicker films. Gold-coated SPREE slides from Nanofilm (Germany) served as the substrate for polymer deposition. These slides were placed on a rotating sample holder to ensure homogenous surface coverage during deposition and the holder was maintained at 13°C. Fourier transform infrared (FTIR) spectroscopy was utilized to confirm polymer structure and ellipsometry was used to determine film thicknesses. Analysis of the polymer structure was undertaken with a Thermo Nicolet 6700 spectrometer (Waltham, MA, USA) with an 85° grazing angle and 128 scans for each sample at a resolution of 4cm−1. All thicknesses were determined with a Nanofilm EP3 imaging ellipsometer at a 532 nm wavelength with an angle of incidence of 60° and polarizer range of 15°. Both ellipsometric parameters, delta and psi, were utilized in a thickness model and the refractive index of the polymer was assumed to be 1.41 as determined by optical modeling.
Electrochemical Impedance Spectroscopy
The presence and quality of the polymer films was confirmed via electrochemical impedance spectroscopy (EIS).38 Impedance samples consisted of a conducting section and a CVD-coated surface area. The base substrate, prior to CVD deposition was silicon coated with gold. Electrochemical analysis was carried out using a three electrode electrochemical cell where the CVD sample served as the working electrode, a saturated calomel electrode [SCE] functioned as the reference electrode, and the platinum mesh was operated as the counter electrode. Phosphate buffered saline (PBS) acted as the electrolyte solution. Impedance measurements were taken with a Gamry PC14/300 potentiostat and utilized EIS300 software (Warminster, PA, USA). The applied potential had an ac amplitude of 10 mV r.m.s. and a frequency range from 1 to 100,000 Hz, with a dc bias of 0 mV with respect to the SCE. Amplitude and phase angle of the current response were recorded at 10 points per decade in frequency.
Atomic Force Microscopy
The surface roughness of the thin films was assessed by atomic force microscopy (AFM). AFM was conducted in tapping mode in air at room temperature. A Nanoscope IIIa from Digital Instruments/Veeco (Plainview, NY, USA) using an EV scanner (15 μm × 15 μm maximum scan size) was used in the analysis. NSC15 cantilevers (MikroMasch, San Jose, CA, USA) with spring constants ranging from 20 N/m to 75 N/m and resonance frequencies of 265-400 kHz served as the AFM tips. Each image depicted herein represents a 5 μm × 5 μm scan size at a scan rate of 1 Hz.
Surface Modification of CVD Coatings
Surface modification of the CVD-coated Au slide prior to SPREE analysis consisted of several surface modification steps, as outlined in Figure 1. Briefly, a PDMS stamp with a 400 by 400 micron square array was utilized to microcontact print a CVD-coated Au slide.26 In order to increase the hydrophilicity of the stamp, the surface was oxidized via UV ozone treatment for 25 minutes (UV-Ozone Cleaner; Model no. 342, Jelight Company Inc., Irvine, CA, USA). The inking solution for the PDMS stamp consisted of 2 mg of biotin-LC-hydrazide (Pierce, Rockford, IL, USA) dissolved in 1 ml of absolute ethanol. The stamp was inked and dried in a stream of air before being placed in contact with the CVD-coated Au slides. Contact was maintained for 3-5 minutes and the stamp removed. To limit non-specific adsorption, the patterned surface was further modified with poly(ethyleneglycol) (PEG) hydrazide (MW=10,000 g/mol from Laysan Bio, Arab, AL, USA) for 12-16 hours and subsequently rinsed several times with deionized water.
Figure 1.

Schematic diagram of sample preparation and analysis of biomolecular binding. (A) CVD film deposited to a desired thickness on Au-coated SPREE slide. (B) Immobilization of biotin containing ligand on the CVD surface and PEG hydrazide incubation to limit nonspecific adsorption.
Monitoring of Surface Reactions by Imaging Surface Plasmon Resonance Enhanced Ellipsometry
An imaging null-ellipsometer (EP3 Nanofilm, Germany) equipped with a fluid cell, utilizing the Kretschmann configuration,39 was employed in the analysis of protein and antibody interactions with the CVD-coated Au surfaces. Filtered and degassed phosphate buffered saline served as the sampling medium for all experimental steps. The experimental set-up consisted of a laser beam, flow cell, 60° SF-10 prism, two syringe pumps (one to pump the buffer solution and the other to pump solutions of biomolecules). The syringe pumps were attached to a y-connector with two needles in order to prevent mixing of the buffer and the biomolecular solution before transport to the flow cell. Ellipsometric measurements were taken at 10 second intervals with a 10× magnification objective at a wavelength of 532 nm and an angle of incidence of 60°.The field of view was 0.5 mm2. Prior to and following biomolecular exposure, the surface was rinsed with PBS. The initial PBS rinse established a baseline signal for the surface and the subsequent washing removed biomolecules that were non-specifically adsorbed. Flow rates were constant at 50 μl/min for all buffer and analyte solutions. Biomolecules of interest were streptavidin labeled with a TRITC fluorophore (Pierce, Rockford, IL, USA), a biotinylated fibrinogen antibody (abcam, Cambridge, MA, USA), and fibrinogen labeled with a FITC fluorophore (Invitrogen, San Diego, CA, USA). Analyte concentrations were 40 μg/ml, 20 μg/ml, and 333 μg/ml for streptavidin, biotinylated antibody, and fibrinogen, respectively. Each analyte was exposed to the surface for 20 minutes. Ellipsometric signal changes for each step of the cascade were determined by subtracting the reference signal (area coated with PEG) from the analyte signal. Though many of the techniques used to study biomolecular adsorption do not measure protein density directly, they have means of correlating sensor outputs with changes in mass. 40 SPREE sensing is similar in that surface density of the adsorbed protein can be determined, if optical parameters of the protein and buffer as well as the adsorbed thickness are known. This relation is given by the de Feijter equation41:
| (1) |
where σ is the surface density, dp is the thickness of the protein layer, np is the refractive index of the protein, nb is the refractive index of the buffer and dn/dc is the refractive index increment of the molecules. In this work, np was 1.45,42 nb was 1.33,43 and dn/dc is 0.183 cm3/g.44 The change in delta can be correlated with thickness changes. An optical model of the sensing cascade was utilized to determine the expected change in delta per nanometer of adsorbed molecule by taking the slope of the fit in the linear regime. The inverse of this value was then multiplied by the change in delta observed in the SPREE sensogram to determine the layer thickness. Thus the surface density of the biomolecules in this instance was determined using optical modeling in conjunction with equation (1). In addition to SPREE imaging, fluorescence microscopy was employed to provide further confirmation of the biomolecular immobilization steps. Samples were rinsed three times with PBS and three times with a solution of PBS containing Tween 20 (0.02% w/v) and bovine serum albumin (0.1 % w/v) prior to fluorescence imaging.
RESULTS AND DISCUSSION
CVD Polymerization and Chemical/Electrical Characterization
Ultra-thin films of poly(4-pentafluoropropionyl-p-xylylene-co-p-xylylene) were conformally deposited onto gold-coated SPREE substrates by CVD polymerization. The chemical structure of the films was confirmed by grazing angle FTIR spectroscopy and was in agreement with previously reported polymers.23 Deposition of the reactive coating resulted in characteristic vibrational bands at 1712 cm−1, which is indicative of the carbonyl stretch. The bands at 1066, 1232, and 1352 cm−1 can be assigned to the C-F stretches (Figure 2a). The FTIR spectra also revealed systematic changes in the intensity of characteristic vibrational bands with increasing amounts of starting material, which ranged from 3 to 13 mg in this study. We attributed the increase in signal intensity to an increased film thickness. This finding provided initial evidence that the amount of starting material can be utilized to control the thickness of the resulting polymer films. Further evidence was obtained by ellipsometry. Based on a Cauchy model assuming a refractive index of 1.41 for the polymer film (as determined from optical fitting) and a refractive index of 0.7 for the gold layer (provided by the manufacturer), the film thickness of coatings deposited under identical conditions with 3, 4.5, 6.5, 10, and 13 mg of starting material were determined to be 3, 10, 20, 40, and 60 nm, respectively.
Figure 2.
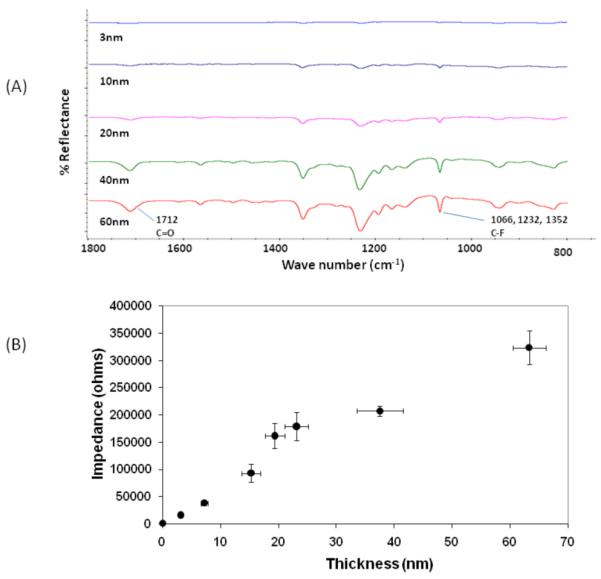
The presence of thin CVD films was confirmed by means of FTIR and electrochemical impedance. (A) FTIR spectroscopy of CVD films at various thicknesses. Characteristic carbonyl and C-F stretches are apparent. (B) Impedance output of the CVD film as a function of film thickness (n=3 per thickness).
Because the CVD films were utilized as reactive interfaces for a cascade of biomolecular immobilization reactions, it is highly desirable that the thin films are pinhole-free. The reason is that the presence of pinholes in the films will be an indication of incomplete coverage and will result in an undefined substrate for binding studies. To confirm complete coverage of the substrate, polymer films with a range of different thickness were studied by electrochemical impedance spectroscopy (EIS). This level of coverage is indicated by the electrochemical permeability of an electrode which should be significantly different from that of the base substrate after a polymer has been deposited. The impedance technique is limited to the assessment of 95% surface coverage.45 As shown in Figure 2b, the impedance increases from 1.5 kohm to 17 kohm after deposition of a 3 nm thick CVD film. At a surface coverage of at least 95%, pinholes are not likely to adversely impact SPREE sensing. As the thickness of the CVD films is further increased, the electrochemical impedance increases accordingly. Further analysis revealed a linear correlation between film thickness and impedance. Physically, this is expected as the film is a dielectric between two conductors (the metal substrate and the electrolyte) and the impedance of the system is directly proportional to the distance between the two conductors, i.e., the thickness of the polymer film. Additional support for films having few, if any pinholes, was provided by imaging ellipsometry which has a 1 μm lateral resolution. This technique did not reveal any pinholes and imaging indicated that substrate patterning was consistent across the substrate.
Surface Roughness
For ultra-thin films, morphology and roughness of the surface may change with surface thickness. Additionally, the roughness of the sensor interface may also play a role in ellipsometric sensing on functionalized CVD coatings. In fact, surface roughness has been shown to be an important factor in protein adsorption.46, 47 Because an important aspect of this work is to investigate the role of film thickness on the usefulness of CVD films for biomolecular sensing, it was necessary to ascertain that surface roughness did not significantly vary among polymer coatings of different thicknesses. Figure 3 shows AFM images for samples coated with 3, 10, and 60 nm thick CVD films. In spite of the significant thickness differences of these samples, only slight morphological differences were observed. The 3 nm coating had sharper growth cones, while plateau size increased with increasing amount of deposited polymer. However, the root mean square roughness (rms) values, which were 2.0, 1.8, and 1.9 nm for 3, 10, and 60 nm films, did not vary significantly. Based on the AFM study, we concluded that differences in surface morphology and roughness can be neglected and should not affect SPREE sensing.
Figure 3.
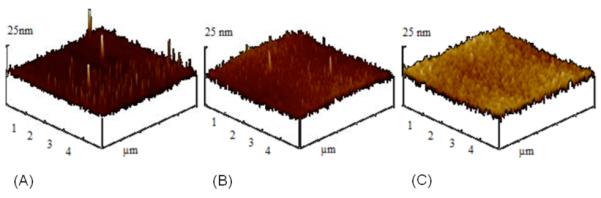
Surface roughness analysis using atomic force microscopy in tapping mode. (A) 3 nm thick film with surface roughness Rms = 2 nm (B) 10nm film with surface roughness Rms = 1.8 nm (C) 60 nm film with surface roughness Rms = 1.9 nm in 3D representations.
Microfluidic SPREE imaging
Surface plasmon resonance spectroscopy is a real time, in situ optical technique to study biomolecule interactions on model substrates.48 Typical substrates include gold, SAMs, or thiol-modified dextrans on gold, and silver.49, 50 Here, we tested the feasibility of using vapor-deposited polymer coatings as biomolecular immobilization layer. Samples for SPREE imaging studies were prepared as shown in Figure 1. Syringe pumps were utilized to deliver the buffer and analyte solutions to the sensor surface at a steady flow rate of 50 μl/min and the experimental set-up is depicted in Figure 4a. Analyte exposure of the reactive CVD coatings consisted of a cascade of biomolecules, starting with streptavidin labeled with a rhodamine fluorophore. Streptavidin is a 53 kDa protein that was selected because of its high-affinity interaction with the vitamin biotin.51 The second component in the cascade was a biotinylated antibody that specifically recognized the blood plasma protein fibrinogen. Here, the biotin served as the recognition element for the protein streptavidin previously bound to the sensor surface. The antibody acted as a recognition element for the final target of the binding cascade, which was fibrinogen labeled with fluorescein fluorophores. Fibrinogen is a large, 340 kDa, protein that plays an important role in blood clotting and blood surface interactions.52 In fact, several studies have demonstrated relationships between fibrinogen concentration and disease states, such as esophageal,53 lung, and colon cancer,54 making it a relevant model molecule for ellipsometric sensing. To reduce non-specific binding of proteins, we modified the unreacted background regions with poly(ethylene glycol). The second inset in Figure 4a displays each component of the system architecture from the base substrate to the final protein layer. In each experiment, patterned areas were compared to a non-reactive background. As seen in Figure 4b, the patterned area was distinguishable from the background based on thickness differences. Bound protein was reported as the difference in adsorbed protein between binding areas and the protein-resistant background. In this study, CVD thicknesses of 3, 10, 20, 40, and 60 nm were assessed. A representative surface response for each step in the cascade for both reference and analyte areas is presented in SPREE sensograms for a 10 nm film in Figure 5. The change in the ellipsometric signal after each analyte exposure indicates that the surface is reactive and that the analytes of interest interact with the CVD array. Analyte adsorption is evident on the order of minutes and adsorption is also independently confirmed by fluorescence microscopy. The density of the adsorbed biomolecules is determined and was found to be approximately 122 ng/cm2, 46 ng/cm2 and 11 ng/cm2 for streptavidin TRITC, biotinylated fibrinogen antibody, and fibrinogen FITC, respectively on films 20 nm or less. In the corresponding microscopy images provided in Figure 4c, the patterned array is readily apparent for both the first step in the cascade, binding of TRITC-streptavidin to surface-immobilized biotin, and the final step in the cascade, FITC-fibrinogen binding to antibody molecules presented at the surface. Evidence of a height dependence on technique sensitivity is highlighted in Figure 6, which compares the ellipsometric response for each step in the cascade as a function of sensor film thickness. From the graphs, it is apparent that there is an upper bound to the useful range of CVD thickness for SPREE imaging sensing. The thinnest films (3 nm and 10 nm thickness range) have a significantly larger response (p< 0.05) than the thicker films (≥20 nm) for each step in the cascade. A substantial difference is also observed between the thicker films for the first step of the cascade and the 40 nm and 60 nm films as compared to the 20 nm films in the second step of the cascade. The similarity of the ellipsometric response to fibrinogen for the thicker films (≥20 nm) likely results from reduced detection sensitivity as binding occurs further from the sensor surface as a consequence of previous immobilization steps.55 Even at the end of a 3-step binding cascade the sensor was able to detect concentrations of fibrinogen at a level of 333 μg/ml, which is about an order of magnitude lower than the average blood plasma levels of 2-4 mg/ml. 56
Figure 4.
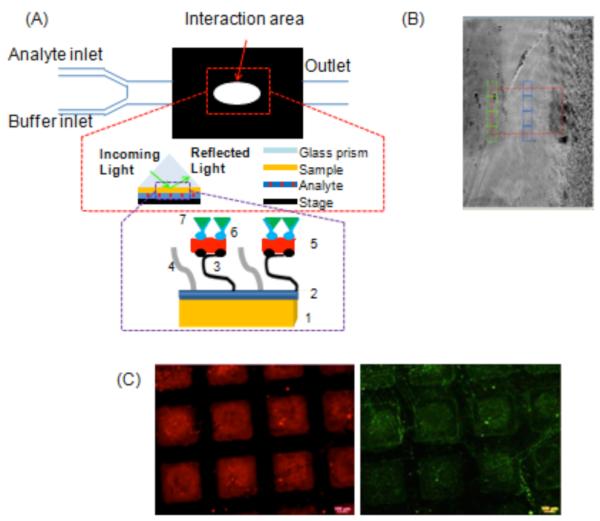
SPREE analysis set-up and fluorescence microscopy. (A) SPREE imaging was utilized to analyze biomolecular interactions with the CVD surface. Experimental set-up consisted of an inlet for buffer and analyte flow and the fluids were delivered by syringe pumps to the interaction area. The second inset is a schematic of the sample architecture with each component represented numerically as follows: 1. Au coated SPREE slide 2. PPXCOC2F5 3. Biotin hydrazide long chain 4. 10k PEG hydrazide 5. Streptavidin TRITC 6. Biotinylated Fibrinogen Antibody 7. Fibrinogen FITC (B) Prior to analysis, portions of the sample patterned with biotin hydrazide and unpatterned areas reacted with PEG hydrazide were selected for data collection (C) Secondary confirmation of protein binding via fluorescence microscopy. The left image is the rhodamine channel which indicates the binding of streptavidin TRITC and the right image is the fluorescein channel which indicates binding of fibrinogen FITC.
Figure 5.

The patterned CVD surface was exposed to a cascade of biomolecules. Because the surface is patterned, an internal reference (unpatterned area) is utilized. Biomolecular immobilization is indicated by a change in ellipsometric delta signal. Thin lines on the graphs indicate the reference signal while the thick lines indicate the signal from the patterned area. Representative SPREE sensograms for a 10nm CVD film in response to sequential analyte exposure of (A) streptavidin TRITC, followed by (B) fibrinogen antibody, and then (C) fibrinogen FITC.
Figure 6.

The change in ellipsometric delta is the signal difference between the patterned and unpatterned areas and is provided for each step of the biomolecular cascade for various film thicknesses. (A) Plot of streptavidin TRITC ellipsometric delta as a function of CVD film thickness (B) Plot of fibrinogen antibody ellipsometric delta as a function of CVD film thickness (C) Plot of fibrinogen FITC ellipsometric delta as a function of CVD film thickness. Error bars indicate the standard deviation of the mean of 3 samples. Symbols indicate a significant difference (p<0.05) between CVD thicknesses with * representing comparison to 3 and 10 nm films, & representing comparisons to 20 nm films, and # representing comparisons to 40 nm films.
Platforms for SPREE imaging studies can be utilized to obtain a wide range of data for surface-ligand interactions. These include but are not limited to determining concentration of molecules, kinetics of binding, relative binding patterns, and the specificity of binding.55 Like dextran matrices and thiols, CVD substrates are well suited for this purpose. However, CVD films have better stability and shelf-life as compared to thiols as indicated by the long term maintenance of functional groups at the film surface (see supplementary materials Figure 1). In addition to long-term stability, the advantage of the CVD platform is that it is spatially-resolved and has range of functionalities which can be incorporated onto the same surface via numerous modification techniques. This is advantageous for high throughput screening and assay optimization. In this work, fibrinogen at a concentration of 333 μg/ml was detected after a cascade of biomolecules. However, the authors in no way purport this value to be the detection limit of the platform. The detection limit of the platform is influenced not only by the concentration of the biomolecule but also its size and binding affinity.57
CONCLUSIONS
Herein, we describe a new binding platform for ellipsometric binding studies, which uses vapor-based reactive polymer coatings as a biomolecular immobilization substrate. These novels substrates may be advantageous in that CVD coatings provide a broad range of surface chemistries. The antigen-antibody sensor showed the ability to sense a cascade of biomolecules. Furthermore, the impact of CVD film thickness on signal intensity was determined. From this analysis, a desirable range of CVD thicknesses for ellipsometric studies was established to be less than 20 nm. Surface response was fairly rapid occurring on the order of minutes indicating the potential for high throughput use. Thus it is feasible to utilize CVD reactive polymers as a platform for imaging surface plasmon resonance enhanced ellipsometry and the ease, with which these films can be modified make them attractive for a plethora of sensing applications.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Himabindu Nandivada and Srijanani Bhaskar for their help with the SPREE fluid delivery set-up and Dr. Peter Thiesen for his help with the optical modeling. AMR acknowledges support from a graduate fellowship from the National Institute of Health through the University of Michigan Microfluidics in Biomedical Sciences Training grant (T32 EB005582-06) and from the GEM fellowship program. J.L. gratefully acknowledges support from the National Science Foundation (NSF) in the form of a CAREER grant (DMR-0449462).
REFERENCES
- (1).Anderson JM, Rodriguez A, Chang DT. Semin. Immunol. 2008;20:86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wilson CJ, Clegg RE, Leavesley DI, Pearcy MJ. Tissue Eng. 2005;11:1–18. doi: 10.1089/ten.2005.11.1. [DOI] [PubMed] [Google Scholar]
- (3).Davis CB, Shamansky LM, Rosenwald S, Stuart JK, Kuhr WG, Brazill SA. Biosens. Bioelectron. 2003;18:1299–1307. doi: 10.1016/s0956-5663(03)00071-x. [DOI] [PubMed] [Google Scholar]
- (4).Uniewicz KA, Ori A, Xu R, Ahmed Y, Wilkinson MC, Fernig DG, Yates EA. Anal. Chem. 2010;82:3796–3802. doi: 10.1021/ac100188x. [DOI] [PubMed] [Google Scholar]
- (5).Yu DQ, Ghosh R. Langmuir. 2010;26:924–929. doi: 10.1021/la902395v. [DOI] [PubMed] [Google Scholar]
- (6).Yamaguchi N, Zhang L, Chae B-S, Palla CS, Furst EM, Kiick KL. J. Am. Chem. Soc. 2007;129:3040–3041. doi: 10.1021/ja0680358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yuan C-X, Tao X-T, Wang L, Yang J-X, Jiang M-H. J. Phys. Chem. C. 2009;113:6809–6814. [Google Scholar]
- (8).Ray S, Mehta G, Srivastava S. Proteomics. 2010;10:731–748. doi: 10.1002/pmic.200900458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bally M, Halter M, Voros J, Grandin HM. Surf. Interface Anal. 2006;38:1442–1458. [Google Scholar]
- (10).Hartwell SK, Grudpan K. Microchim. Acta. 2010;169:201–220. [Google Scholar]
- (11).James T, Mannoor MS, Ivanov DV. Sensors. 2008;8:6077–6107. doi: 10.3390/s8096077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zernla J, Lekka M, Wiltowska-Zuber J, Budkowski A, Rysz J, Raczkowska J. Langmuir. 2008;24:10253–10258. doi: 10.1021/la801313u. [DOI] [PubMed] [Google Scholar]
- (13).Arwin H, Poksinski M, Johansen K. Phys. Status Solidi A. 2008;205:817–820. [Google Scholar]
- (14).Fabre RM, Talham DR. Langmuir. 2009;25:12644–12652. doi: 10.1021/la901920y. [DOI] [PubMed] [Google Scholar]
- (15).Klenkar G, Brian B, Ederth T, Stengel G, Hook F, Piehler J, Liedberg B. Biointerphases. 2008;3:29–37. doi: 10.1116/1.2921867. [DOI] [PubMed] [Google Scholar]
- (16).Nelson SG, Johnston KS, Yee SS. Sens. Actuators, B. 1996;35:187–191. [Google Scholar]
- (17).Schasfoort RBM, Tudos AJ, editors. Handbook of Surface Plasmon Resonance. The Royal Society of Chemistry; Cambridge: 2008. [Google Scholar]
- (18).Nabok A, Tsargorodskaya A. Thin Solid Films. 2008;516:8993–9001. [Google Scholar]
- (19).Scarano S, Mascini M, Turner APF, Minunni M. Biosens. Bioelectron. 2009;25:957–966. doi: 10.1016/j.bios.2009.08.039. [DOI] [PubMed] [Google Scholar]
- (20).Lofas S, Malmqvist M, Ronnberg I, Stenberg E, Liedberg B, Lundstrom I. Sens. Actuators, B. 1991;5:79–84. [Google Scholar]
- (21).Gasteier P, Reska A, Schulte P, Salber J, Offenhausser A, Moeller M, Groll J. Macromol. Biosci. 2007;7:1010–1023. doi: 10.1002/mabi.200700064. [DOI] [PubMed] [Google Scholar]
- (22).Benters R, Niemeyer CM, Wohrle D. Chembiochem. 2001;2:686–694. doi: 10.1002/1439-7633(20010903)2:9<686::AID-CBIC686>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- (23).Park TJ, Hyun MS, Lee HJ, Lee SY, Ko S. Talanta. 2009;79:295–301. doi: 10.1016/j.talanta.2009.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nandivada H, Chen HY, Lahann J. Macromol. Rapid Commun. 2005;26:1794–1799. [Google Scholar]
- (25).Kashima Y, Munakata T, Matoba A. Opt. Rev. 1997;4:69–71. [Google Scholar]
- (26).Lahann J, Langer R. Macromolecules. 2002;35:4380–4386. [Google Scholar]
- (27).Lahann J, Balcells M, Lu H, Rodon T, Jensen KF, Langer R. Anal. Chem. 2003;75:2117–2122. doi: 10.1021/ac020557s. [DOI] [PubMed] [Google Scholar]
- (28).Jiang XW, Chen HY, Galvan G, Yoshida M, Lahann J. Adv. Funct. Mater. 2008;18:27–35. [Google Scholar]
- (29).Lahann J, Choi IS, Lee J, Jenson KF, Langer R. Angew. Chem., Int. Ed. 2001;40:3166–3169. doi: 10.1002/1521-3773(20010903)40:17<3166::AID-ANIE3166>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- (30).Lahann J, Hocker H, Langer R. Angew. Chem., Int. Ed. 2001;40:726–728. doi: 10.1002/1521-3773(20010216)40:4<726::aid-anie7260>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- (31).Himabindu N, Hsien-Yeh C, Lidija B, Joerg L. Angew. Chem., Int. Ed. 2006;45:3360–3363. [Google Scholar]
- (32).Lahann J, Balcells M, Rodon T, Lee J, Choi IS, Jensen KF, Langer R. Langmuir. 2002;18:3632–3638. [Google Scholar]
- (33).Chen HY, Lahann J. Adv. Mater. 2007;19:3801–3808. [Google Scholar]
- (34).Thévenet S, Chen HY, Lahann J, Stellacci F. Adv. Mater. 2007;19:4333–4337. [Google Scholar]
- (35).Chen HY, Lahann J. Anal. Chem. 2005;77:6909–6914. doi: 10.1021/ac050964e. [DOI] [PubMed] [Google Scholar]
- (36).Lahann J. Polym. Int. 2006;55:1361–1370. [Google Scholar]
- (37).Elkasabi Y, Yoshida M, Nandivada H, Chen HY, Lahann J. Macromol. Rapid Commun. 2008;29:855–870. [Google Scholar]
- (38).Ganesh V, Lakshminarayanan V. Langmuir. 2006;22:1561–1570. doi: 10.1021/la0519296. [DOI] [PubMed] [Google Scholar]
- (39).Raether H. Springer Tracts Mod. Phys. 1988;111:1–133. [Google Scholar]
- (40).Goda T, Miyahara Y. Anal. Chem. 2010;82:1803–1810. doi: 10.1021/ac902401y. [DOI] [PubMed] [Google Scholar]
- (41).Defeijter JA, Benjamins J, Veer FA. Biopolymers. 1978;17:1759–1772. [Google Scholar]
- (42).Beketov GV, Shirshov YM, Shynkarenko OV, Chegel VI. Sens. Actuators, B. 1998;48:432–438. [Google Scholar]
- (43).Topolancik J, Vollmer F. Biophys. J. 2007;92:2223–2229. doi: 10.1529/biophysj.106.098806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ball V, Ramsden JJ. Biopolymers. 1998;46:489–492. [Google Scholar]
- (45).Rehak M, Hall EAH. Analyst. 2004;129:1014–1025. doi: 10.1039/b411363b. [DOI] [PubMed] [Google Scholar]
- (46).Rechendorff K, Hovgaard MB, Foss M, Zhdanov VP, Besenbacher F. Langmuir. 2006;22:10885–10888. doi: 10.1021/la0621923. [DOI] [PubMed] [Google Scholar]
- (47).Lord MS, Foss M, Besenbacher F. Nano Today. 2010;5:66–78. [Google Scholar]
- (48).Homola J. Anal. Bioanal. Chem. 2003;377:528–539. doi: 10.1007/s00216-003-2101-0. [DOI] [PubMed] [Google Scholar]
- (49).Green RJ, Frazier RA, Shakesheff KM, Davies MC, Roberts CJ, Tendler SJB. Biomaterials. 2000;21:1823–1835. doi: 10.1016/s0142-9612(00)00077-6. [DOI] [PubMed] [Google Scholar]
- (50).Homola J. Chem. Rev. 2008;108:462–493. doi: 10.1021/cr068107d. [DOI] [PubMed] [Google Scholar]
- (51).Turková J. J. Chromatogr., B: Biomed. Sci. Appl. 1999;722:11–31. doi: 10.1016/s0378-4347(98)00434-4. [DOI] [PubMed] [Google Scholar]
- (52).Marchant RE, Barb MD, Shainoff JR, Eppell SJ, Wilson DL, Siedlecki CA. Thromb. Haemostasis. 1997;77:1048–1051. [PubMed] [Google Scholar]
- (53).Hiroya T, Shunji I, Yuko K, Atsushi S, Takashi O, Yoh I, Kiyoshi K, Masaki K, Sumio M. J. Gastroenterol. Hepatol. 2007;22:2222–2227. [Google Scholar]
- (54).Wojtukiewicz MZ, Zacharski LR, Moritz TE, Hur K, Edwards RL, Rickles FR. Blood Coagul. Fibrinolysis. 1992;3:429–437. [PubMed] [Google Scholar]
- (55).Liedberg B, Lundström I, Stenberg E. Sens. Actuators, B. 1993;11:63–72. [Google Scholar]
- (56).Wolberg AS. Blood Rev. 2007;21:131–142. doi: 10.1016/j.blre.2006.11.001. [DOI] [PubMed] [Google Scholar]
- (57).Fan XD, White IM, Shopoua SI, Zhu HY, Suter JD, Sun YZ. Anal. Chim. Acta. 2008;620:8–26. doi: 10.1016/j.aca.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.