

Abstract
NMDA receptors are ionotropic glutamate receptors that mediate excitatory synaptic transmission and have been implicated in several neurological diseases. We have evaluated the mechanism of action of a class of novel subunit-selective NMDA receptor antagonists, typified by (E)-4-(6-methoxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)-benzoic acid (QNZ46). We found that QNZ46 inhibits NMDA receptor function in a noncompetitive and voltage-independent manner by an unconventional mechanism that requires binding of glutamate to the GluN2 subunit, but not glycine binding to the GluN1 subunit. This dependency of antagonist association on glutamate binding to GluN2 renders these compounds nominally use-dependent, since inhibition will rely on synaptic release of glutamate. Evaluation of the structural determinants responsible for the subunit-selectivity of QNZ46 revealed that these compounds act at a new site that has not previously been described. Residues residing in the part of the agonist binding domain immediately adjacent to the transmembrane helices appear to control selectivity of QNZ46 for GluN2C- and GluN2D-containing receptors. These residues are well-positioned to sense glutamate binding to GluN2 and thus to mediate glutamate-dependent actions. This new class of noncompetitive antagonists could provide an opportunity for the development of pharmacological tools and therapeutic agents that target NMDA receptors at a new site and modulate function by a novel mechanism.
Introduction
NMDA receptors are members of the larger family of ionotropic glutamate receptors that also comprises the AMPA and kainate receptors. NMDA receptors are expressed throughout the mammalian CNS, where they play essential roles in a wide range of normal processes such as neural development, perception of pain, and synaptic plasticity (Traynelis et al., 2010). However, NMDA receptors are also implicated in a plethora of neuropathological conditions, including Alzheimer's and Parkinson's diseases, depression, schizophrenia, epilepsy, ischemia, hypoxia, and traumatic brain injury, and have therefore received considerable interest as targets for therapeutic intervention (Kalia et al., 2008; Traynelis et al., 2010). To this end, it could be beneficial to modify neuronal function using small-molecule modulators that reduce or tune down NMDA receptor function in some neuropathological conditions.
NMDA receptors are assembled from two GluN1 and two GluN2 subunits (Ulbrich and Isacoff, 2007), and are activated by simultaneous binding of glycine and glutamate to the GluN1 and GluN2 subunits, respectively (Kleckner and Dingledine, 1988). The different GluN2 subunits (GluN2A–D) show distinct temporal and spatial expression in the brain (Ishii et al., 1993; Watanabe et al., 1993; Monyer et al., 1994), and endow the NMDA receptors with markedly different biophysical and pharmacological properties (Monyer et al., 1992; Vicini et al., 1998; Yuan et al., 2009; Traynelis et al., 2010). Despite the involvement of NMDA receptors in brain functions and neurological disorders, there remains a surprising lack of ligands that act on NMDA receptors with sufficient subunit-selectivity to allow evaluation of the roles of individual GluN2 subunits in normal brain function and disease.
We have recently described a new class of subunit-selective noncompetitive antagonists (Mosley et al., 2010). Here, we describe the novel mechanism and structural determinants for NMDA receptor inhibition by this new class of ligands. The data presented here suggest that voltage-independent, subunit-selective, noncompetitive NMDA receptor antagonists exist that inhibit function in a glutamate-dependent manner by acting at a previously unrecognized modulatory site.
Materials and Methods
DNA constructs and ligands.
Wild-type cDNAs for rat GluN1-1a (GenBank accession numbers U11418 and U08261; hereafter called GluN1), rat GluN2A (D13211), rat GluN2B (U11419), rat GluN2C (M91563), and rat GluN2D (L31611) were provided by Drs. S. Heinemann (Salk Institute, San Diego, CA), S. Nakanishi (Kyoto University, Kyoto, Japan), and P. Seeburg (University of Heidelberg, Heidelberg, Germany). The GluN2D amino-terminal domain deletion construct (2D-ΔATD) and the chimeras with amino-terminal domain interchanged between GluN2A and GluN2D [2A-(2D ATD) and 2D-(2A ATD)] were generated as previously described (Yuan et al., 2009). All other chimeras were generated using standard molecular biology methods. Detailed information about the GluN2A-GluN2D chimeric junctions is summarized in supplemental Table 1 (available at www.jneurosci.org as supplemental material). Site-directed mutagenesis was performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's protocol and verified by DNA sequencing. The amino acids are numbered according to the full-length protein, including the signal peptide. For expression in Xenopus oocytes, DNA constructs were linearized by restriction enzymes to produce cRNAs using the mMessage mMachine kit (Ambion). (E)-4-(6-methoxy-2-(3-nitrostyryl)-4-oxoquinazolin-3(4H)-yl)-benzoic acid (QNZ46) was synthesized and analyzed for purity as previously described (Mosley et al., 2010). The maximum solubility of QNZ46 in extracellular oocyte recording solution was 70 μm (see Mosley et al., 2010). All other ligands were purchased from Sigma-Aldrich.
Two-electrode voltage-clamp recordings from Xenopus oocytes.
Injection of cRNA and two-electrode voltage-clamp recordings from Xenopus laevis oocytes were performed as previously described (Traynelis et al., 1998). Following cRNA injection, the oocytes were stored at 15°C in Barth's solution containing the following (in mm): 88 NaCl, 2.4 NaHCO3, 1 KCl, 0.33 Ca(NO3)2, 0.41 CaCl2, 0.82 MgSO4, 5 Tris-HCl, pH 7.4 with NaOH. The ratio of injected GluN1 to GluN2 cRNA was 1:2. Two-electrode voltage-clamp recordings were performed 3–4 d postinjection at room temperature (23°C). The extracellular oocyte recording solution contained the following (in mm): 90 NaCl, 1 KCl, 10 HEPES, 0.5 BaCl2, 0.01 EDTA, pH 7.4 with NaOH. Solutions were applied by gravity, and solution exchange was controlled through an 8-modular valve positioner (Digital MVP Valve, Hamilton). Voltage and current electrodes were filled with 0.3 and 3.0 m KCl, respectively, and current responses were recorded at a holding potential of −40 mV unless otherwise stated. Data acquisition and voltage control were accomplished with a two-electrode voltage-clamp amplifier (OC725, Warner Instrument). Glutamate (100 μm) and glycine (30 μm) were used in all oocyte experiments unless otherwise stated. DMSO concentration was kept constant in experiments where ≥10 μm QNZ46 was used and never exceeded 0.25%.
Whole-cell voltage-clamp electrophysiology.
HEK293 cells (CRL 1573, ATCC) were plated on coated (0.1 mg/ml poly-d-lysine) glass coverslips (Warner Instruments) ∼48 h before the experiments. The culture medium was DMEM with GlutaMax-I (Invitrogen) supplemented with 10% dialyzed fetal bovine serum, 10 units/ml penicillin, and 10 μg/ml streptomycin. Cells were transiently cotransfected using the calcium phosphate precipitation method with plasmid cDNAs encoding GluN1 and GluN2 subunits as well as green fluorescent protein. Immediately following transfection, 200 μm d,l-2-amino-5-phosphonovalerate and 200 μm 7-chlorokynurenic acid were added to the culture medium. The cells were used for experiments ∼24 h following transfection.
Whole-cell voltage-clamp recordings were performed at −60 mV unless otherwise stated using an Axopatch 200B amplifier (Molecular Devices) at room temperature (23°C). Recording electrodes (3–4 MΩ) were made from thin wall glass micropipettes (TW150F-4, World Precision Instruments) pulled using a vertical puller (PP-830, Narishige). The electrodes were filled with internal solution containing the following (in mm): 110 d-gluconate, 110 CsOH, 30 CsCl, 5 HEPES, 4 NaCl, 0.5 CaCl2, 2 MgCl2, 5 BAPTA, 2 NaATP, and 0.3 NaGTP, pH 7.35 with CsOH. The extracellular recording solution was composed of the following (in mm): 150 NaCl, 10 HEPES, 3 KCl, 0.5 CaCl2, and 0.01 EDTA, pH 7.4 with NaOH.
Rapid solution exchange was achieved on lifted cells with a two-barrel theta-glass pipette controlled by a piezoelectric translator (Burleigh Instruments). Junction currents were used to estimate speed of open tip solution exchange after recordings and typically had 10–90% rise times of 0.4–0.8 ms. The solution exchange around lifted whole cells voltage-clamped at 0 mV was also determined by rapidly changing the regular extracellular solution to a solution in which 150 mm KCl replaced the 150 mm NaCl. The 10–90% rise time of the change in holding current upon this solution exchange was 4.5 ± 1.3 ms (N = 7) (K. M. Vance and S. F. Traynelis, unpublished observations), which is 1.0- to 2.8-fold faster than receptor activation rise times and 16- to 79-fold faster than the time constants for inhibition by 10 μm QNZ46.
Homology modeling.
The published crystal structure of the glutamate-bound isolated agonist binding domain of GluN2A (Protein Data Bank ID 2A5S) (Furukawa et al., 2005) was used as a template to generate a homology model of the isolated agonist binding domain of GluN2D with bound glutamate. Sequence alignment and molecular modeling were performed using Modeler 9v7 (Eswar et al., 2008) with the EasyModeller 2 user interface (Kuntal et al., 2010) and default parameters. All structural figures were generated using PyMOL 1.3 (Schrödinger).
Data analysis.
Concentration-response data were analyzed using GraphPad Prism 5.0. Data for individual oocytes were fitted to the Hill equation using variable slope (unless otherwise stated) and a fixed minimum of zero. Fitted EC50 or IC50 values and Hill coefficients (nH) from individual oocytes were used to calculate the mean and SEM. For graphic presentation, data points from individual oocytes were normalized to the maximum current response in the same recording and averaged. The averaged data points (i.e., composite data) were then fitted to the Hill equation and plotted together with the resulting curve.
Current responses from whole-cell voltage-clamp recordings using HEK293 cells were analyzed using ChanneLab (Synaptosoft). The deactivation time courses of the current responses were fitted by two exponential components using the following:
where Itotal is the total current response, Ifast and Islow are the fast and slow current components, and τfast and τslow are the deactivation time constants for the fast and slow components, respectively. The time courses for the onset and recovery from QNZ46 inhibition of current responses were best fitted by one exponential component. In experiments for which solution was rapidly switched from QNZ46 with or without glycine or glutamate into glutamate plus glycine, the instantaneous current was determined by extrapolating the time course of the slow increase of the current to full-amplitude steady-state current (as QNZ46 unbinds) to the time point of the initial rise in current response for receptor activation (i.e., the first data point that rises off the baseline).
Unpaired t test (two-tailed) or ANOVA (one-way ANOVA with Tukey–Kramer post-test) was used for statistical comparisons as indicated (p < 0.05 was considered significant). All data are presented as mean ± SEM.
Results
QNZ46 is a noncompetitive antagonist of NMDA receptors
QNZ46 is a member of a recently described class of recombinant NMDA receptor antagonists that contain the quinazolin-4-one backbone (compound 46, see Mosley et al., 2010), which is shared with the previously reported noncompetitive AMPA receptor antagonist CP-465,022 (Welch et al., 2001; Lazzaro et al., 2002; Menniti et al., 2003) (Fig. 1a). QNZ46 inhibits GluN2C/D-containing receptors more potently than GluN2A/B receptors (Mosley et al., 2010) (Table 1; see also supplemental Fig. S1, available at www.jneurosci.org as supplemental material), but the mechanism and structural determinants of action are unknown. To determine the mechanism of action, we recorded whole-cell current responses under voltage-clamp from recombinant GluN1/GluN2D receptors expressed in HEK293 cells using rapid solution exchange (Fig. 1b). Both the onset and offset of QNZ46 inhibition of steady-state responses to saturating glutamate (1000 μm) and glycine (100 μm) can be adequately described using monoexponential fits with the time constants for inhibition (τinhibition) ranging from 91 to 356 ms, depending on the QNZ46 concentration (20–1 μm, respectively) (Table 2). The time constant for recovery from inhibition (τoff) was independent of QNZ46 concentration and was between 455 and 492 ms (Fig. 1c; supplemental Table 2, available at www.jneurosci.org as supplemental material). Evaluation of the association and dissociation rate of QNZ46 binding as a function of concentration allowed us to estimate a KD for antagonist binding to GluN1/GluN2D of 4.9 μm (N = 4) (Fig. 1c), which is in agreement with IC50 values of 3.2 μm (N = 4) and 3.9 μm (N = 12) for inhibition of GluN1/GluN2D receptors expressed in HEK293 and Xenopus oocytes, respectively (Table 1; supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
Figure 1.
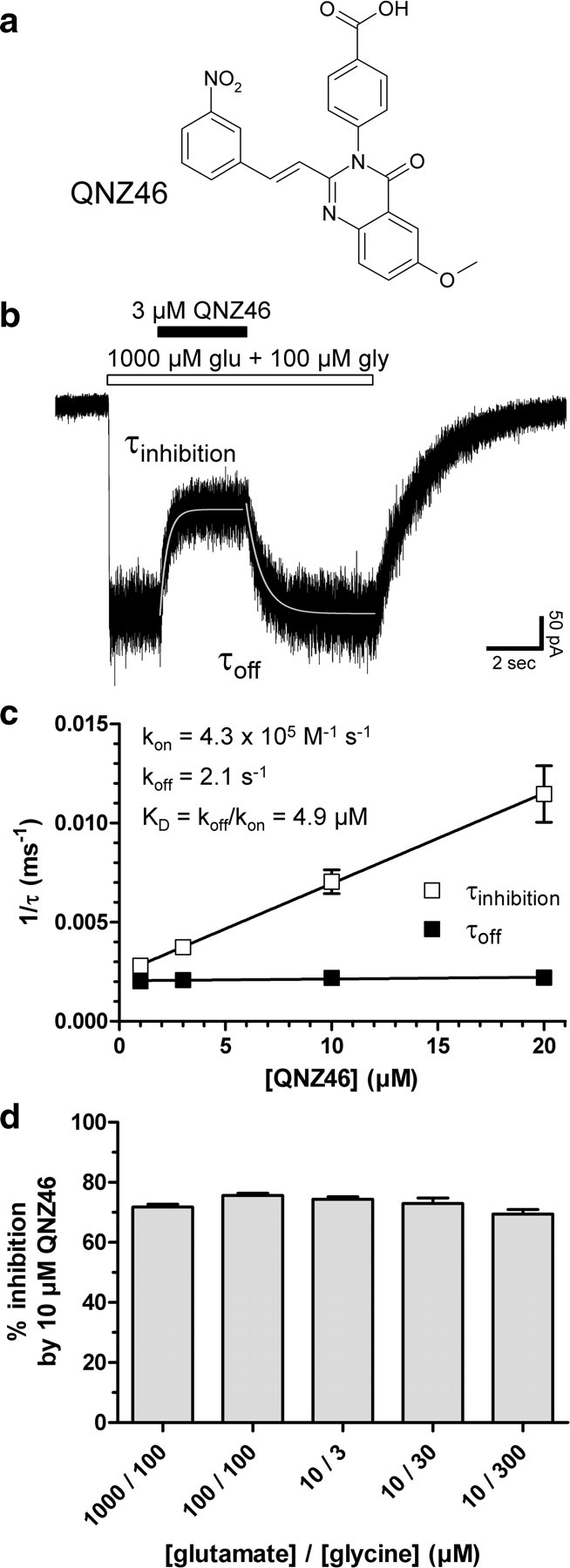
QNZ46 is a noncompetitive NMDA receptor antagonist. a, Chemical structure of QNZ46. QNZ46 represents a new class of NMDA receptor antagonists and is ∼50-fold selective for GluN2C- and GluN2D-containing receptors over receptors containing GluN2A or GluN2B (Mosley et al., 2010) (see Table 1; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). b, Representative whole-cell current response recorded under voltage-clamp from recombinant GluN1/GluN2D receptors expressed in a HEK293 cell using rapid solution exchange. The onset and offset of QNZ46 inhibition of steady-state responses to saturating glutamate and glycine can be described using monoexponential fits (see Table 2). c, τinhibition and τoff were used to determine the association and dissociation rates of QNZ46 binding as a function of concentration, allowing estimation of KD for antagonist binding to GluN1/GluN2D (N = 4). d, Inhibition of GluN1/GluN2D by QNZ46 was independent of agonist concentration and could not be surmounted by increasing concentrations of glutamate or glycine (values from 3 to 11 cells were not significantly different; p > 0.05; one-way ANOVA with Tukey–Kramer post-test). This is consistent with a noncompetitive mechanism of action (see also supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Table 1.
IC50 values for QNZ46 at wild-type NMDA receptor subtypes
| NMDA receptor subtype | IC50 (μm) | nH | N | Relative IC50 (IC50 2X/IC50 2D) |
|---|---|---|---|---|
| GluN1/GluN2A | 182 ± 24 | 1.0a | 7 | 47 |
| GluN1/GluN2B | 193 ± 45 | 1.0a | 8 | 49 |
| GluN1/GluN2C | 7.1 ± 0.4 | 1.0 | 7 | 1.8 |
| GluN1/GluN2D | 3.9 ± 0.2 | 1.1 | 12 | 1.0 |
QNZ46 IC50 ± SEM was determined using two-electrode voltage-clamp recordings at GluN2A–D subunits coexpressed with GluN1 in Xenopus oocytes and was used to calculate the fold shift in IC50 at the indicated receptor (IC50 2X) relative to the IC50 at GluN1/GluN2D (IC50 2D). The receptors were activated by 100 μm glutamate plus 30 μm glycine. N is the number of oocytes used to generate the data.
aThe parameter was fixed during fitting to the Hill equation (see Materials and Methods).
Table 2.
Kinetic parameters for QNZ46 inhibition of GluN1/GluN2D NMDA receptors
| [QNZ46] (μm) | N | QNZ46 applied after activation |
QNZ46 present before, during, and after activation |
|||
|---|---|---|---|---|---|---|
| τinhibition (ms) | Inhibition (%) | N | τinhibition (ms) | Inhibition (%) | ||
| 1 | 4 | 356 ± 6 | 23 ± 1 | 4 | 344 ± 6 | 28 ± 3 |
| 3 | 4 | 268 ± 11 | 50 ± 2 | 4 | 244 ± 5 | 46 ± 3 |
| 10 | 4 | 145 ± 13 | 75 ± 1 | 7 | 116 ± 3 | 73 ± 2 |
| 20 | 4 | 91 ± 11 | 88 ± 1 | 7 | 74 ± 2 | 85 ± 2 |
Kinetic parameters for inhibition of GluN1/GluN2D receptors by QNZ46 determined using whole-cell voltage-clamp recordings from HEK293 cells. Receptors were activated by 1000 μm glutamate and 100 μm glycine. QNZ46 was applied to the cell after receptor activation [i.e. after application of glutamate and glycine, as shown in Fig. 1b] or was present before, during, and after receptor activation [i.e. glutamate was applied in the continuous presence of QNZ46 plus glycine, as shown in Fig. 3b]. N is the number of cells; Inhibition is the percentage inhibition of steady-state responses relative to control in the absence of QNZ46. All values are mean ± SEM.
Inhibition of GluN1/GluN2D by QNZ46 was independent of agonist concentration, since it could not be surmounted by increasing concentrations of glutamate or glycine, consistent with a noncompetitive mechanism of action (Fig. 1d; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). QNZ46 did not reduce the potency of glutamate or glycine, confirming that it did not inhibit receptors through competition with agonist binding. The EC50 of glutamate at GluN1/GluN2D is 0.44 μm (N = 17) in the absence of QNZ46 and 0.22 μm (N = 8) in the presence of 10 μm QNZ46, and the glycine potency at GluN1/GluN2D is 0.17 μm (N = 8) in the absence of QNZ46 and 0.13 μm (N = 8) in the presence of 10 μm QNZ46 (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The potency of QNZ46 did not change for any NMDA receptor subtype when activated by the lower affinity, partial glutamate site agonist NMDA (supplemental Table 3, available at www.jneurosci.org as supplemental material) compared with when activated by glutamate (Table 1), consistent with the idea that QNZ46 does not interact with the agonist binding site.
Although QNZ46 is negatively charged at physiological pH, we evaluated the voltage-dependence of inhibition to determine whether it might be acting in a manner that is sensitive to the transmembrane electric field. QNZ46 shows a similar degree of inhibition for inward and outward currents, and the time course of QNZ46 association and dissociation is independent of membrane potential (Fig. 2). In addition, the IC50 values as well as the KD calculated from kinetic analysis at −60 and −30 mV were virtually identical (Fig. 1; supplemental Fig. S2, available at www.jneurosci.org as supplemental material). These data strongly suggest that QNZ46 does not act within the transmembrane ion channel pore as a use-dependent channel blocker.
Figure 2.

Inhibition by QNZ46 is voltage-independent. a, Representative whole-cell current responses recorded under voltage-clamp at +30 and −30 mV from recombinant GluN1/GluN2D receptors. 10 μm QNZ46 and 1000 μm glutamate plus 100 μm glycine (glu + gly) were applied to the cell for the duration of the respective bars above the recorded traces. b, Inhibition by 10 μm QNZ46 is independent of membrane holding potential (values from 5 to 7 cells were not significantly different; p > 0.05; one-way ANOVA with Tukey–Kramer post-test). c, Time constants for inhibition (τinhibition) and recovery from inhibition (τoff) by 10 μm QNZ46 at GluN1/GluN2D are independent of membrane holding potential (values from 5 to 7 cells were not significantly different; p > 0.05; one-way ANOVA with Tukey–Kramer post-test). Error bars are SEM, shown when larger than symbol.
Inhibition by QNZ46 is use-dependent
We next examined the effect of QNZ46 on responses to glutamate application (1000 μm) in the continuous presence of glycine (100 μm) (Fig. 3). In the absence of QNZ46 (control), the response to a glutamate application has a 6.9 ± 0.3 ms rise time (10–90%) and is deactivated with a dual-exponential time course described by time constants of 1830 ± 100 ms (40%) and 5130 ± 120 ms (60%) (N = 7) (Fig. 3a, Table 3; supplemental Table 2, available at www.jneurosci.org as supplemental material). There was virtually no detectable desensitization of GluN1/GluN2D receptors activated by glutamate and glycine. However, the response time course was dramatically changed for cells recorded in the continuous presence of QNZ46, with QNZ46 present before, during, and after agonist application. Rapid application of glutamate in the presence of QNZ46 induces a rapid response that peaks and then relaxes to a steady-state level (Fig. 3b,c). Several observations argue that this relaxation to a steady-state response is not caused by QNZ46-induced receptor desensitization, but rather reflects the time course of QNZ46 association with its binding site and subsequent receptor inhibition. For example, the time course of relaxation to the steady-state response in the continuous presence of QNZ46 at different concentrations can be described by a monoexponential time course that is similar to the time course for inhibition of glutamate plus glycine-activated steady-state responses (compare Figs. 3c, 1b). That is, the time constants for the relaxation to steady-state responses (τinhibition) are similar to the time constants for inhibition of steady-state responses (Table 2). Moreover, the level of inhibition achieved with QNZ46 applied either before or after receptor activation is the same (Table 2). In addition, the time constant describing the relaxation is inversely related to antagonist concentration, as expected if it reflects association of QNZ46 with its binding site. The concentration-dependent association rate constant determined from linear regression of 1/τinhibition and concentration for QNZ46 (5.7 × 105 m−1 s−1) (Fig. 3c, Table 2) is similar to the association rate constant determined from inhibition of steady-state responses (4.3 × 105 m−1 s−1) (Fig. 1c, Table 2), consistent with the idea that the relaxation time course reflects QNZ46 binding, rather than a gradually developing set of conformational changes that yield varying degrees and rates of desensitization. The altered response waveform observed when GluN1/GluN2D is activated in the presence of QNZ46 suggests a use-dependent mechanism, in which QNZ46 association depends on either agonist binding or subsequent protein conformational changes leading to channel opening.
Figure 3.

Inhibition by QNZ46 is use-dependent. a, b, Representative whole-cell current responses from recombinant GluN1/GluN2D receptors. Glutamate (1000 μm; glu) was applied to the cell using rapid solution exchange in the continuous presence of 100 μm glycine alone (gly; control) or in continuous presence of 100 μm glycine plus 3 μm QNZ46 (+ QNZ46). Rapid application of glutamate in the continuous presence of QNZ46 induces a rapid response that peaks and then relaxes to a steady-state level. c, Overlay of the traces in a and b. d, Representative whole-cell current responses activated by NMDA from recombinant GluN1/GluN2D receptors. NMDA (200 μm) was applied to the cell using rapid solution exchange in the continuous presence of 100 μm glycine alone (gly; control) or in continuous presence of 100 μm glycine plus 10 μm QNZ46 (+ QNZ46). Similar to activation by glutamate, activation by the partial glutamate site agonist NMDA in the continuous presence of QNZ46 induces a rapid response that peaks and then relaxes to a steady-state level. e, Representative whole-cell current responses activated by glycine from recombinant GluN1/GluN2D receptors. Glycine (300 μm) was applied to the cell using rapid solution exchange in the continuous presence of 10 μm glutamate alone (glu; control) or in continuous presence of 10 μm glutamate plus 10 μm QNZ46 (+ QNZ46). Activation by glycine in the continuous presence of QNZ46 and glutamate leads to responses that show considerable inhibition of the instantaneous current, evidenced by the small peak current in the presence QNZ46 compared with control. f, Summary of peak current responses (Ipeak) in the continuous presence of QNZ46 relative to control peak current responses in the absence of QNZ46. The activating agonist is indicated; when glutamate and NMDA were activating agonists, the cells were bathed in glycine, and when glycine was activating agonist, the cells were bathed in glutamate (N = 5–8) (see also supplemental Table 4, available at www.jneurosci.org as supplemental material).
Table 3.
Time constants for deactivation of GluN1/GluN2D NMDA receptors in the continuous presence of QNZ46 plus glycine
| [QNZ46] (μm) | N | τfast (ms) | Deactivation |
|
|---|---|---|---|---|
| τslow (ms) | Slow (%) | |||
| 0 | 7 | 1830 ± 100 | 5130 ± 120 | 60 |
| 1 | 4 | 1210 ± 150 | 5640 ± 160 | 78 |
| 3 | 4 | 1340 ± 50 | 7100 ± 180 | 79 |
| 10 | 7 | 8150 ± 330 | 100 | |
| 20 | 7 | 10,130 ± 280 | 100 | |
Receptors were activated by 1000 μm glutamate in the continuous presence of 100 μm glycine plus increasing concentrations of QNZ46. That is, QNZ46 was present before, during, and after receptor activation, as shown in Fig. 3b (see also Fig. 5). The deactivation time courses in the absence of QNZ46 and in the presence of 1–3 μm QNZ46 were best described using dual-exponential fits, and therefore two time constants are listed (τfast and τslow); whereas, the deactivation time course in the presence of 10–20 μm QNZ46 was best described using a monoexponential fit, and therefore only one time constant is listed (τslow). N is the number of cells. All values are mean ± SEM.
Inhibition by QNZ46 depends on glutamate binding
To further explore the nature of the apparent use-dependence of QNZ46 inhibition, we performed experiments in which the receptors were activated by glycine (300 μm) in the continuous presence of glutamate (10 μm), or were activated by glutamate (1000 μm) or NMDA (200 μm) in the continuous presence of glycine (100 μm). Rapid application of glycine in the continuous presence of QNZ46 (10 μm) plus glutamate leads to responses that show considerable inhibition of the instantaneous current recorded in response to rapid agonist application (compare small peak current, Fig. 3e, control). We interpret this result to suggest that QNZ46 binds in the presence of glutamate alone before activation. This is in stark contrast to application of glutamate or NMDA following preincubation of the receptor in the continuous presence of QNZ46 plus glycine, which show peak responses (i.e., instantaneous activation) that are virtually identical to the control response (Fig. 3f; supplemental Table 4, available at www.jneurosci.org as supplemental material). These data suggest that QNZ46 inhibition may depend on agonist binding to the glutamate site on the GluN2 subunit rather than glycine binding or channel opening.
To directly test the idea that QNZ46 binding to GluN1/GluN2D receptors is enhanced following glutamate but not glycine binding, we performed experiments in which we compared the instantaneous current response following a rapid solution exchange out of QNZ46 alone into glutamate plus glycine (Fig. 4a) to that observed for a rapid solution exchange out of glycine plus QNZ46 into glutamate plus glycine (Fig. 4b) or out of glutamate plus QNZ46 into glutamate plus glycine (Fig. 4c). This experimental design allows us to determine whether QNZ46 binds to the receptor in the presence of either glutamate or glycine alone by assessing the degree of preinhibition of the instantaneous current before washout of bound QNZ46. Because QNZ46 binds far more slowly than receptor activation, the preinhibition of the instantaneous current reflects the fraction of receptors to which QNZ46 has already bound before activation of the receptor. When GluN1/GluN2D receptors are pretreated with QNZ46 alone, a rapid solution exchange into glutamate plus glycine produces an instantaneous current of nearly the same amplitude as control, followed by a slow relaxation to full-amplitude steady state of the QNZ46 that is receptor bound before glutamate plus glycine application unbinds (Fig. 4a). This suggests that 10 μm QNZ46 can bind to (and thus inhibit) only ∼11% of the receptors in the nominal absence of glutamate plus glycine (Fig. 4d; supplemental Table 4, available at www.jneurosci.org as supplemental material). Surprisingly, repeating this experiment with preapplication of a maximally effective concentration of glycine produces no significant difference in the degree of inhibition of the instantaneous current (12%) (Fig. 4b,d). This suggests that QNZ46 interacts with reduced affinity in the absence of both agonists, and that occupation of the glycine site does not enhance or alter QNZ46 binding rate constants. By contrast, QNZ46 produces ∼66% inhibition of the instantaneous current when a maximally effective concentration of glutamate is preapplied (Fig. 4c,d), suggesting that glutamate binding to the GluN2D subunit enhances QNZ46 binding.
Figure 4.

Inhibition by QNZ46 depends on glutamate binding. a–c, Representative whole-cell current responses from recombinant GluN1/GluN2D receptors. a, Responses from a cell that experienced a solution exchange from ligand-free extracellular solution (left) or QNZ46 (10 μm) alone (right) to glutamate (1000 μm) plus glycine (300 μm). b, Responses from a cell that experienced a solution exchange from glycine (100 μm) (left) or glycine plus QNZ46 (10 μm) (right) to glutamate (1000 μm) plus glycine (100 μm). c, Responses from a cell that experienced a solution exchange from glutamate (10 μm) (left) or glutamate plus QNZ46 (10 μm) (right) to glutamate (10 μm) plus glycine (300 μm). d, Preinhibition of GluN1/GluN2D pretreated with QNZ46 alone (no agonist; as in a) or glycine plus QNZ46 (glycine; as in b) were significantly different from preinhibition of receptors pretreated with glutamate plus QNZ46 (glutamate; as in c) or glutamate plus glycine plus QNZ46 (glutamate + glycine; as in Fig. 1b) (N = 4–7; p < 0.05; one-way ANOVA with Tukey–Kramer post-test; see supplemental Table 4, available at www.jneurosci.org as supplemental material). e, The time constants (τoff) for the slow relaxation to full amplitude steady-state following receptor washout of 10 μm QNZ46 are independent of whether glycine, glutamate, or no agonist was present before receptor activation (values from 4 to 7 cells were not significantly different; p > 0.05; one-way ANOVA with Tukey–Kramer post-test; see supplemental Table 4, available at www.jneurosci.org as supplemental material). f, Concentration-response data for preinhibition by QNZ46 represented as the instantaneous current normalized to the full amplitude steady-state responses (i.e., 100% corresponds to no preinhibition). The IC50 values for QNZ46 preinhibition were 3.2 μm (N = 4) when both glutamate and glycine were present during pretreatment (see also supplemental Fig. S2, available at www.jneurosci.org as supplemental material) and 6.4 μm (N = 5) when glutamate alone was present during pretreatment. In the presence of glycine alone or no agonists, the IC50 values were 85 μm (N = 5) or 86 μm (N = 5), respectively.
To further assess the agonist dependency of QNZ46 potency for inhibition of GluN1/GluN2D, we determined the relationship between the concentration of QNZ46 present during pretreatment before activation by glutamate and glycine and preinhibition of the instantaneous currents (Fig. 4f). The IC50 value of QNZ46 was 3.2 μm (N = 4) when both glutamate and glycine were present during pretreatment (see supplemental Fig. S2, available at www.jneurosci.org as supplemental material), and the IC50 value of QNZ46 was 6.4 μm (N = 5) when glutamate alone was present during pretreatment. However, in the presence of glycine alone or no agonists, the IC50 values of QNZ46 were 85 μm (N = 5) or 86 μm (N = 5), respectively (Fig. 4f). Consequently, glutamate binding to the GluN2D subunit greatly enhances QNZ46 binding, resulting in more than a 10-fold increase in QNZ46 potency. Moreover, the dependence of QNZ46 binding on glutamate binding accounts for the use-dependent features of QNZ46 and demonstrates that QNZ46 inhibition is not dependent on receptor activation per se and opening of the channel pore.
The time constants (τoff) for the slow relaxation to full amplitude following washout of QNZ46 coincident with receptor activation are independent of whether glycine, glutamate, or no agonist was present before receptor activation (Fig. 4e). Moreover, the time constant describing this time course was the same as τoff determined when QNZ46 was applied to receptors already activated by glutamate plus glycine. This result demonstrates that QNZ46 unbinds from the same binding site with the same affinity once the receptor becomes activated, independent of the timing of the QNZ46 application and the state of the receptor before QNZ46 binding. It is therefore unlikely that inhibition of the instantaneous current observed in Figure 4 reflects association of QNZ46 to a secondary low-affinity binding site in the absence of receptor activation.
In the absence of QNZ46, the receptor deactivation following removal of glutamate (in continuous presence of glycine) is controlled by the time it takes for the ion channel to close, the receptor to traverse the kinetics steps back to a state from which glutamate can unbind, and the time for glutamate to unbind. Analyses of the deactivation time course following removal of glutamate in the continuous presence of QNZ46 (e.g., the response shown in Fig. 3b) show that the time constants for deactivation significantly increase up to approximately 2-fold in the presence of QNZ46 (e.g., 10,130 ± 280 ms in the presence of 20 μm QNZ46; N = 7) (Fig. 5a,b, Table 3; see supplemental Table 2, available at www.jneurosci.org as supplemental material). The observation that QNZ46 prolongs glutamate deactivation suggests that QNZ46 binding to GluN2D prevents or slows one or more of the steps leading to glutamate unbinding from the receptor. One interpretation of this finding is that QNZ46 must unbind before glutamate can unbind, an idea that is consistent with a use-dependent mechanism of QNZ46 inhibition. QNZ46 also prolongs deactivation of responses to the partial agonist NMDA in the continuous presence of glycine (Fig. 5c; see also supplemental Table 4, available at www.jneurosci.org as supplemental material). In support of the results showing that QNZ46 inhibition depends on agonist binding to the glutamate site in GluN2 but not the glycine site in GluN1, we also found that QNZ46 had minimal effects on deactivation of responses to rapid glycine application in the continuous presence of glutamate (Fig. 5c; see also supplemental Table 4, available at www.jneurosci.org as supplemental material).
Figure 5.

QNZ46 prolongs receptor deactivation time. a, Overlay of whole-cell current recordings as shown in Figure 3b normalized to steady-state responses to glutamate plus glycine in the absence (control) or presence of 3 or 10 μm QNZ46. The deactivation time courses in the absence of QNZ46 and in the presence of 3 μm QNZ46 were best described using dual-exponential fits (2 exp), whereas the deactivation time course in the presence of 10 μm QNZ46 was best described using a monoexponential fit (1 exp). b, Analyses of the deactivation time course following removal of glutamate in the continuous presence of QNZ46 show that the time constants for deactivation (τweighted) significantly increase up to approximately 2-fold in the presence of QNZ46 (1–20 μm) (see supplemental Table 2, available at www.jneurosci.org as supplemental material). *Significantly different from τweighted in the absence of QNZ46 (white bar) (p < 0.05; one-way ANOVA with Tukey–Kramer post-test). Data are from four to seven cells. c, QNZ46 prolongs deactivation of responses to both glutamate and the partial agonist NMDA in the continuous presence of glycine. The QNZ46-mediated prolongation of deactivation glycine responses in the continuous presence of glutamate is less pronounced compared with the effects on glutamate and NMDA deactivation (see also supplemental Table 4, available at www.jneurosci.org as supplemental material). *Significantly different from control in the absence of QNZ46 (indicated by the dashed line; p < 0.05; two-tailed unpaired t test). Data are from five to seven cells.
We also examined whether the recovery from QNZ46 inhibition proceeds by the same time course when agonist is bound as when no agonist is bound. We found that agonist binding to the GluN2 subunit has no significant effect on the rate of recovery from QNZ46 inhibition (Fig. 6). The time constants (τoff) for the recovery from QNZ46 inhibition were 320 ± 20 (N = 6) in the continuous presence of NMDA (200 μm) and 290 ± 10 (N = 6) in the absence of agonist. This result suggests that the primary effect of agonist binding to the GluN2 subunit on QNZ46 potency occurs on the association rate of QNZ46 for its binding site.
Figure 6.
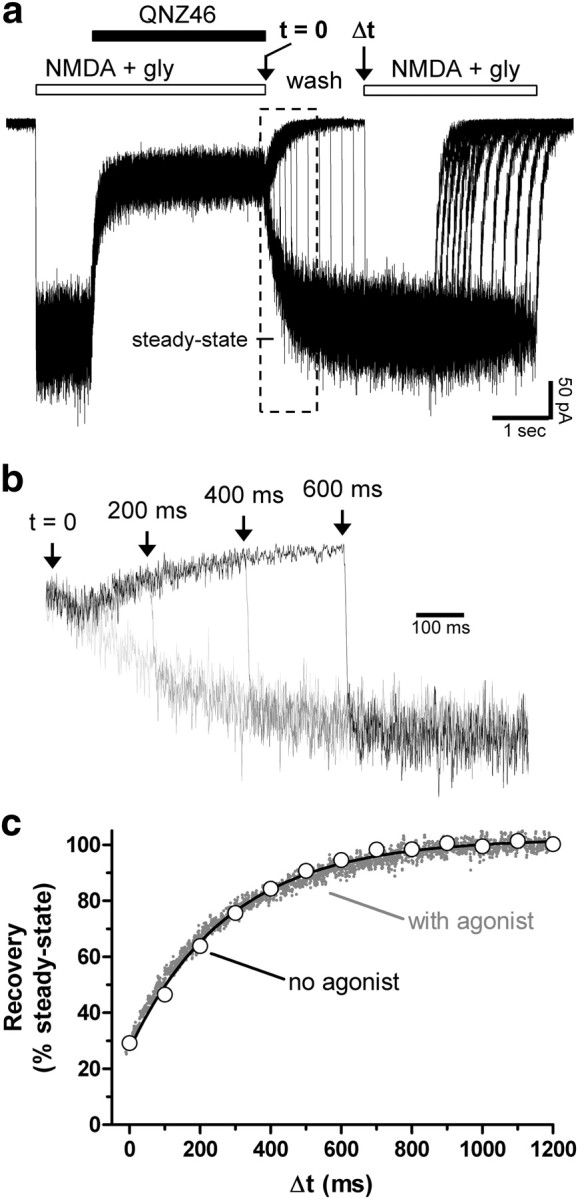
Recovery from QNZ46 inhibition is not use-dependent. a, Representative overlay of whole-cell current recordings from one HEK293 cell expressing recombinant GluN1/GluN2D receptors. The cell is initially stepped into NMDA (200 μm) plus glycine (300 μm), and the resulting response is then inhibited by QNZ46 (10 μm). Subsequent to this inhibition, the cell is stepped into wash (i.e., no agonists and no QNZ46) at time 0 (t = 0). The cell is then stepped back into NMDA plus glycine at different time intervals (Δt). QNZ46 unbinding and recovery from inhibition occur from the step into wash (t = 0) to the time point where the steady-state response is reached following the step back into NMDA plus glycine. NMDA was used as glutamate site agonist because the time constant for NMDA deactivation (τweighted 92 ms; see supplemental Table 4, available at www.jneurosci.org as supplemental material) is 41-fold lower (i.e.faster) than τ weighted for glutamate (3810 ms; see supplemental Table 2, available at www.jneurosci.org as supplemental material), and 3.5-fold lower than τoff for QNZ46 in the continuous presence of NMDA (320 ms). By contrast, τweighted for glutamate is 8.3-fold higher (i.e., slower) than τoff for QNZ46 in the continuous presence of glutamate (460 ms; see supplemental Table 2, available at www.jneurosci.org as supplemental material). b, Representative recordings showing recovery from inhibition on an expanded time scale (a, boxed area). c, Mean recovery from inhibition at different intervals in wash (no agonist) measured as the instantaneous current response relative to the steady-state response from the same recording are shown as white circles with the monoexponential fit (black line) (τoff = 290 ± 10; N = 6). The averaged time course for recovery from inhibition in the continuous presence of NMDA from the same cells is shown as gray dots (with agonist) (τoff = 320 ± 20; N = 6). The time constants for recovery from QNZ46 inhibition in the presence and absence of agonist are not significantly different (p > 0.05, two-tailed unpaired t test).
Structural determinants of QNZ46 subunit-selectivity
To identify portions of the receptor responsible for the actions of QNZ46, we exploited the subunit-selectivity of QNZ46 between receptors that contain GluN2A and GluN2D (Mosley et al., 2010). Concentration-response data suggest a ∼50-fold difference in QNZ46 IC50 values between receptors containing these GluN2 subunits (Table 1), allowing us to evaluate chimeric GluN2A-GluN2D receptors to identify the regions of GluN2D that are sufficient to enhance QNZ46 potency at GluN2A-containing receptors (Figs. 7–9). We initially evaluated the QNZ46 sensitivity of a series of five chimeric receptors that introduced the amino-terminal domain or segments of the agonist binding domain from GluN2D into GluN2A, as well as a deletion mutant of GluN2D without the amino-terminal domain (GluN2D-ΔATD) (Fig. 7c–e). These studies revealed that the S2 portion of the agonist binding domain contained the structural determinants that control the QNZ46 potency at GluN2D and GluN2A; swapping these regions transferred the effects of QNZ46 to GluN2A (Figs. 7e, 9a, Table 4). A series of 12 chimeric receptors that progressively dissect the S2 region identified three distinct regions of S2 that significantly influence QNZ46 selectivity (Figs. 7f, 8a).
Figure 7.

Structural determinants for subunit-selective QNZ46 inhibition are located in the S2 segment of the agonist binding domain. a, b, Representative two-electrode voltage-clamp recordings from GluN1/GluN2A (a) and GluN1/GluN2D (b) receptors expressed in Xenopus oocytes. The receptors were activated by glutamate (100 μm) and inhibited by QNZ46 (10 μm). Glycine (30 μm) was present in all solutions. c, NMDA receptors are comprised of four semiautonomous domains; the extracellular amino-terminal domain (ATD) and agonist binding domain, the transmembrane domain containing three transmembrane helices (M1, M2, and M4) and a membrane re-entrant loop (M2), and the intracellular C-terminal domain. The agonist binding domain is formed by two amino acid segments (S1 and S2). d, Linear representations of the polypeptide chains of GluN2A (gray) and GluN2D (blue), as well as chimeric GluN2A-GluN2D subunits (for chimeric junctions, see supplemental Table 1, available at www.jneurosci.org as supplemental material). e, Summary of the inhibition of the response to glutamate (100 μm) by 10 μm QNZ46 in the continuous presence of glycine (30 μm) (as shown in a, b) for five chimeric GluN2 subunits and a deletion mutant of GluN2D without the amino-terminal domain (all GluN2 subunits coexpressed with GluN1 in Xenopus oocytes). *Significantly different from GluN1/GluN2A (white bar) (p < 0.05; one-way ANOVA with Tukey–Kramer post-test). ns, Not significantly different from GluN1/GluN2D (black bar) (p > 0.05; one-way ANOVA with Tukey–Kramer post-test). Data are from 4 to 13 oocytes. f, The QNZ46-sensitivity of 2A-(2D S2) chimeras (coexpressed with GluN1) that progressively dissect the S2 region identified three distinct regions that significantly influence QNZ46 selectivity (see also Fig. 8a). *Significantly different from 2A-(2D S2) (S2; white bar) (p < 0.05; one-way ANOVA with Tukey–Kramer post-test). Data are from 4 to 12 oocytes.
Figure 8.

Individual residues that influence subunit-selective QNZ46 inhibition. a, Amino acid sequence alignment of the S2 segment from GluN2A-D. The regions marked above the sequences were converted to the GluN2A sequence in the 2A-(2D S2) chimera of the indicated letter. For example, Thr686, Ser688, and Arg693 were converted to Gln, Thr, and Lys, respectively, in the 2A-(2D S2a) chimera. −, the residue was mutated in GluN2D with no effect on QNZ46 sensitivity; +, QNZ46 sensitivity was significantly changed when the residue in GluN2D was mutated. b, Individual nonconserved residues in the three regions identified by the 2A-(2D S2b), 2A-(2D S2c), and 2A-(2D S2i) chimeras (see Fig. 7f), as well as surrounding residues were mutated in GluN2D to the corresponding residues in GluN2A. Furthermore, selected residues in GluN2D that are conserved in GluN2A and are located close to Gln699, Glu700, Gln701, Leu705, and Lys779 in a homology model of GluN2D (see Fig. 10) were mutated to alanine (or valine in case of Ala752). Nine mutations significantly changed the sensitivity to QNZ46. *Significantly different from GluN1/GluN2D (white bar) (p < 0.05; one-way ANOVA with Tukey–Kramer post-test). Data are from 4 to 20 oocytes.
Figure 9.
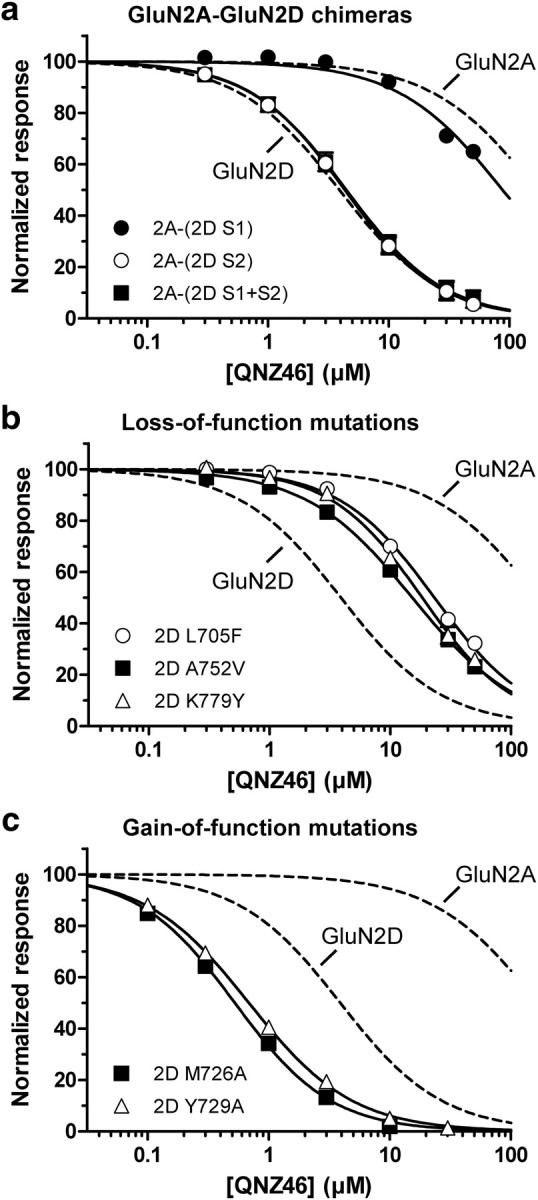
Individual residues that influence QNZ46 inhibition. a–c, Concentration-response data for QNZ46 inhibition of GluN2A-GluN2D chimeras (a), GluN2D loss-of-function mutants (b), and GluN2D gain-of-function mutants (coexpressed with GluN1) (c) (for IC50 values, see Table 4). For comparison, the concentration-response curves for QNZ46 at GluN1/GluN2A and GluN1/GluN2D are shown as dashed lines only (data points are included in supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Table 4.
IC50 values for QNZ46 at mutant NMDA receptors
| GluN2 subunit (GluN1/GluN2X) | IC50 (μm) | nH | N | Relative IC50 (IC50 2X/IC50 2D) |
|---|---|---|---|---|
| GluN2A mutant subunits | ||||
| 2A-(2D S1) | 91 ± 9 | 1.0a | 7 | 23 |
| 2A-(2D S2) | 4.6 ± 0.5 | 1.1 | 6 | 1.2 |
| 2A-(2D S1 + S2) | 4.7 ± 0.7 | 1.1 | 4 | 1.2 |
| HDYS/QEQY + F680L + Y754K | 17 ± 1 | 1.2 | 5 | 4.4 |
| GluN2D mutant subunits | ||||
| QEQY/HDYS | 10 ± 1 | 1.0 | 6 | 2.6 |
| Q701Y | 7.7 ± 0.7 | 1.0 | 6 | 2.0 |
| L705F | 23 ± 2 | 1.1 | 6 | 5.9 |
| K779Y | 19 ± 2 | 1.2 | 6 | 4.9 |
| QEQY/HDYS + L705F + K779Y | 15 ± 1 | 1.0 | 4 | 3.8 |
| Y723A | 7.6 ± 0.6 | 1.3 | 6 | 1.9 |
| M726A | 0.5 ± 0.02 | 1.1 | 9 | 0.1 |
| Y729A | 0.7 ± 0.04 | 1.0 | 10 | 0.2 |
| A752V | 16 ± 1 | 1.0 | 6 | 4.1 |
QNZ46 IC50 ± SEM was determined using two-electrode voltage-clamp recordings at GluN2 subunits coexpressed with GluN1 in Xenopus oocytes and was used to calculate the fold shift in IC50 at the indicated receptor (IC50 2X) relative to the IC50 at GluN2D (IC50 2D). IC50 values of wild-type GluN1/GluN2A and GluN1/GluN2D were 182 ± 24 μm and 3.9 ± 0.2 μm, respectively, and are also listed in Table 1. N is the number of oocytes used to generate the data.
aThe parameter was fixed during fitting to the Hill equation (see Materials and Methods). QEQY/HDYS and HDYS/QEQY refer to the quadruple-mutant GluN2D Q699H + E700D + Q701Y + Y702S and the quadruple mutant GluN2A H674Q + D675E + Y676Q + S677Y, respectively.
We subsequently examined individual residues in these three regions as well as surrounding residues, initially focusing only on those residues that differ between GluN2A and GluN2D. By mutating these residues in GluN2D to the corresponding residues in GluN2A, we identified five residues that impact inhibition by 10 μm QNZ46, which were GluN2D Gln699, Glu700, Gln701, Leu705, and Lys779 (Fig. 8b). GluN2D subunits with individual or all of Gln699, Glu700, Gln701, Leu705, and Lys779 mutated to the corresponding residues in GluN2A markedly reduced the potency of QNZ46 (up to 6-fold increase in QNZ46 IC50 values compared to wild-type GluN2D), consistent with the findings that these residues influence the subunit-selectivity of QNZ46 (Table 4). Mutating the same residues in GluN2A to the corresponding GluN2D residues resulted in a nearly complete transfer of QNZ46 sensitivity into GluN2A (Table 4).
Selected residues in GluN2D that are conserved in GluN2A and are located close to Gln699, Glu700, Gln701, Leu705, and Lys779 in a homology model of GluN2D (see Materials and Methods) were also mutated to alanine (or valine in case of Ala752). Functional evaluation of these mutants identified four additional residues that affect the sensitivity to 10 μm QNZ46 (Fig. 8b). Two of these GluN2D mutations (Y723A and A752V) reduced the potency of QNZ46 (Fig. 9b), whereas two mutations (M726A and Y729A) increased the potency of QNZ46 (Fig. 9c). None of the mutations that affected QNZ46 sensitivity in this study markedly changed the EC50 value for glutamate (supplemental Table 5, available at www.jneurosci.org as supplemental material).
Discussion
The most important finding of this study is that QNZ46, a member of a new class of noncompetitive antagonists, acts at a previously unrecognized site to inhibit NMDA receptors in a subunit-selective fashion by a novel mechanism. QNZ46 is a proof-of-concept molecule that we have used to explore the mechanism and site of action for a new class of noncompetitive antagonists (Mosley et al., 2010). Inhibition by QNZ46 clearly depends on the presence of glutamate, which represents a new mechanism for inhibition of glutamate receptors not previously reported. Moreover, because synaptic release of glutamate is the primary means by which NMDA receptors at synapses are activated, at the functional level, this form of inhibition will be use-dependent, since it will require synaptic activation to promote NMDA receptor inhibition. This may have important implications for how antagonists with this mechanism will act on circuit function in the CNS, and may provide opportunities to modulate circuit function in a more sophisticated manner than full inhibition of all receptors by nonselective antagonists.
In addition to acting by a novel and intriguing mechanism, we interpret our mutagenesis data to suggest that QNZ46 acts at a previously unrecognized site on the NMDA receptor. The QNZ46 binding site is not contained in the amino-terminal domain, and QNZ46 potency is unaffected by GluN2A-GluN2D chimeric receptors exchanging residues in the S1 portion of the agonist binding domain, which comprises the central part of the dimer interface between GluN1 and GluN2 agonist binding domains (Furukawa et al., 2005). Thus, the data suggest that QNZ46 does not bind at the dimer interface between GluN1 and GluN2 agonist binding domains. By contrast, the S2 portion of the agonist binding domain contained the structural determinants that control the selectivity of QNZ46 for GluN2D. Although the S2 region per se has been implicated in NMDA receptor modulation by carboxylated naphthalene and phenanthrene derivatives (Costa et al., 2010), as well as the use-dependent and voltage-independent actions of endogenous sulfated neurosteroids (Jang et al., 2004; Petrovic et al., 2005; Horak et al., 2006; Kussius et al., 2009), there is as yet no information implicating any specific residues, and thus it is not known whether any residues forming these binding sites overlap with residues that control QNZ46 potency.
The residues in the S2 segment of the agonist binding domain that affect QNZ46 sensitivity reside in different portions of the linear polypeptide chain. However, evaluation of a homology model of GluN2D based on the crystal structure of the glutamate-bound isolated GluN2A agonist binding domain (Furukawa et al., 2005) suggests that these residues are closely clustered in space given the tertiary receptor structure (Fig. 10). This raises the possibility that this region of the agonist binding domain clamshell proximal to the membrane of the receptor not only controls the subunit-selectivity, but may also contribute to a binding site for QNZ46.
Figure 10.
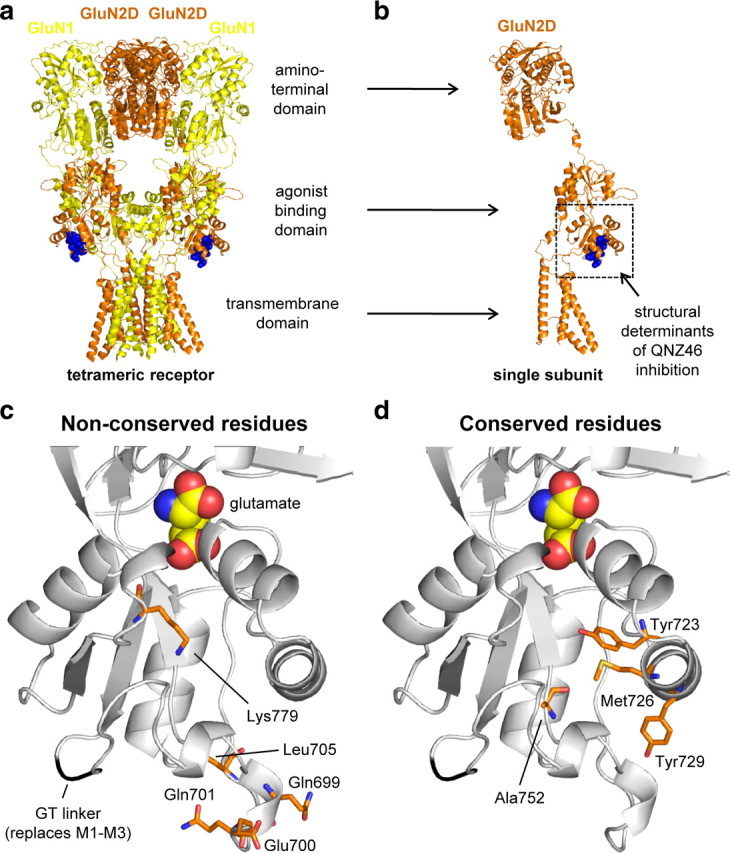
Mapping of residues that influence QNZ46 inhibition. a, Illustration of residues important for QNZ46 inhibition in a hypothetical tetrameric NMDA receptor structure using the crystal structure of the tetrameric membrane-spanning GluA2 as guide (Protein Data Bank ID 3KG2) (Sobolevsky et al., 2009). The GluN1 (yellow) and GluN2D (orange) subunits are colored according to the position they would occupy in a tetrameric NMDA receptor (Sobolevsky et al., 2009). Residues in GluA2 homologous to residues in GluN2D that influence QNZ46 activity are highlighted as blue spheres. b, Same as in a, but here only a single subunit is shown. c, Close-up of the lower portion of the agonist binding domain proximal to the membrane from a homology model of the isolated GluN2D agonist binding domain with bound glutamate (see Materials and Methods). Bound glutamate in the agonist binding pocket is shown as spheres. The main chain of the Gly-Thr (GT) linker that replaces the transmembrane helices M1–M3 is shown in black (Furukawa et al., 2005). The five residues identified in this study that influence QNZ46 inhibition and are nonconserved between GluN2A and GluN2D are highlighted as orange sticks. d, The four identified residues that influence QNZ46 inhibition and are conserved between GluN2A and GluN2D are highlighted as orange sticks.
However, two observations suggest that the residues identified in this study are unlikely to directly participate in the binding site for QNZ46. First, the effects on QNZ46 potency of the single mutations in GluN2D were relatively small (less than 6-fold), and none of the multiple point mutations in GluN2D could replicate the full 50-fold difference in QNZ46 potency between GluN2A and GluN2D (see Table 4). Second, the residues identified in this study are not conserved between GluN2A/B and GluN2C/D, as would be expected if they formed the binding site for QNZ46 that is selective for GluN2C/D- over GluN2A/B-containing NMDA receptors. Consequently, it is conceivable that the residues identified in this study influence steric or electrostatic interactions that indirectly shape the QNZ46 binding site, which is likely located close to the membrane-proximal part of the agonist binding domain and the peptide linkers, which connect the agonist binding domain to the transmembrane ion channel pore. This site of action for QNZ46 appears distinct from the residues implicated in the activity of the noncompetitive AMPA receptor antagonist CP-465,022 by evaluation of GluA3-GluK2 chimeras (Balannik et al., 2005).
Interaction with the lower S2 portion of the agonist binding domain in the receptor is ideally positioned to influence the long-range intraprotein conformational changes that are induced by glutamate binding to GluN2 and lead to opening of the receptor pore. Furthermore, structural determinants for QNZ46 sensitivity in the lower portion of the agonist binding domain are consistent with the results showing that QNZ46 binding is strongly dependent on glutamate binding to GluN2. In support of this idea, it was recently shown that NMDA receptor activation increases the accessibility to cysteine-modifying reagents for some GluN2 residues located in the peptide linkers, which connect the agonist binding domain to the transmembrane ion channel pore (Talukder et al., 2010). These GluN2 linker residues are located between the transmembrane ion channel pore and the lower portion of the agonist binding domain, and are therefore also in close proximity to the structural determinants for QNZ46 sensitivity identified in this study. The synthesis of our findings and studies using the substituted cysteine-scanning method (Sobolevsky et al., 2007; Talukder et al., 2010) suggests that in response to glutamate binding, subunit-specific conformational changes occur in the lower portion of the GluN2 agonist binding domain and the peptide linkers to the transmembrane ion channel pore. Furthermore, the mechanism of action for QNZ46 demonstrates that these subunit-specific conformational changes can be exploited for subunit-selective and noncompetitive inhibition of NMDA receptors.
Figure 11 combines functional and structural ideas presented here to illustrate a working hypothesis as to how we predict that inhibition by QNZ46 proceeds. The accessibility of the QNZ46 binding site is either enhanced or the binding site is placed in a higher affinity conformation subsequent to glutamate but not glycine binding. This is consistent with the modular arrangement of glutamate receptor subunit structure (Wo and Oswald, 1995; Sobolevsky et al., 2009), which should allow autonomous conformational changes within individual subunit domains (Rosenmund et al., 1998; Robert and Howe, 2003; Prieto and Wollmuth, 2010). Occupation of the QNZ46 binding site in GluN2D then inhibits opening of the gate by increasing the activation energy for rearrangement of transmembrane helices. This model is also compatible with a prolongation of the glutamate deactivation time course if QNZ46 occupancy of its site alters the manner by which the agonist binding domain clamshell can open and release glutamate (Fig. 5c). By contrast, QNZ46 occupancy should not alter the glycine deactivation time course, since QNZ46 binding has no detectable effects on the agonist binding domain of GluN1 and vice versa. QNZ46 binding is therefore not expected to markedly change glycine unbinding (Fig. 5c).
Figure 11.

Working hypothesis for noncompetitive and glutamate-dependent inhibition of NMDA receptors by QNZ46. Glutamate binding promotes cleft closure of the GluN2 agonist binding domain, and a conformational change within the GluN2 subunit either increases the accessibility of the QNZ46 binding site or places the binding site in a higher affinity conformation (indicated by red arrow). Occupation of the QNZ46 binding site then inhibits opening of the gate. The glutamate-dependent enhancement of QNZ46 binding accounts for the apparent use-dependence of QNZ46 inhibition, since the receptor can enter the open state before QNZ46 can bind. Glycine binding to the GluN1 subunit alone without occupation at the glutamate binding site in GluN2 does not promote rearrangement of GluN2 in a manner that enhances QNZ46 binding.
The observation that QNZ46 occupancy prolongs the time courses for glutamate and NMDA deactivation suggests that QNZ46 unbinds before glutamate or NMDA can unbind. The model in Figure 11 is consistent with this idea, as well as the result showing that QNZ46 unbinding is not use-dependent (Fig. 6), as expected if QNZ46 unbinding primarily occurs from receptors with agonist bound to the GluN2 subunit (i.e., glutamate or NMDA) as opposed to unbinding from both unliganded and agonist-bound receptors with two different rates.
In summary, these experiments reveal new features of NMDA receptor structure, function, and modulation that significantly advance our understanding of this class of receptors. The mechanism elucidated here provides an opportunity for the development of pharmacological tools and therapeutic agents that target NMDA receptors at a previously unrecognized site and modulate function by a novel mechanism.
Footnotes
This work was supported by National Institutes of Health-National Institute of Neurological Disorders and Stroke (Grants NS036654 and NS065371, to S.F.T.), the Villum Kann Rasmussen Foundation (to K.B.H.), and the Lundbeck Foundation (to K.B.H.). We thank Katie M. Vance and Tim M. Acker for comments on the manuscript. We also thank Cara A. Mosley, Tim M. Acker, and Dennis C. Liotta for synthesizing QNZ46 for these studies.
References
- Balannik V, Menniti FS, Paternain AV, Lerma J, Stern-Bach Y. Molecular mechanism of AMPA receptor noncompetitive antagonism. Neuron. 2005;48:279–288. doi: 10.1016/j.neuron.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Costa BM, Irvine MW, Fang G, Eaves RJ, Mayo-Martin MB, Skifter DA, Jane DE, Monaghan DT. A novel family of negative and positive allosteric modulators of NMDA receptors. J Pharmacol Exp Ther. 2010;335:614–621. doi: 10.1124/jpet.110.174144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- Furukawa H, Singh SK, Mancusso R, Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438:185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- Horak M, Vlcek K, Chodounska H, Vyklicky L., Jr Subtype-dependence of N-methyl-D-aspartate receptor modulation by pregnenolone sulfate. Neuroscience. 2006;137:93–102. doi: 10.1016/j.neuroscience.2005.08.058. [DOI] [PubMed] [Google Scholar]
- Ishii T, Moriyoshi K, Sugihara H, Sakurada K, Kadotani H, Yokoi M, Akazawa C, Shigemoto R, Mizuno N, Masu M. Molecular characterization of the family of the N-methyl-D-aspartate receptor subunits. J Biol Chem. 1993;268:2836–2843. [PubMed] [Google Scholar]
- Jang MK, Mierke DF, Russek SJ, Farb DH. A steroid modulatory domain on NR2B controls N-methyl-D-aspartate receptor proton sensitivity. Proc Natl Acad Sci U S A. 2004;101:8198–8203. doi: 10.1073/pnas.0401838101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, Kalia SK, Salter MW. NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 2008;7:742–755. doi: 10.1016/S1474-4422(08)70165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleckner NW, Dingledine R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science. 1988;241:835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- Kuntal BK, Aparoy P, Reddanna P. EasyModeller: a graphical interface to MODELLER. BMC Res Notes. 2010;3:226. doi: 10.1186/1756-0500-3-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussius CL, Kaur N, Popescu GK. Pregnanolone sulfate promotes desensitization of activated NMDA receptors. J Neurosci. 2009;29:6819–6827. doi: 10.1523/JNEUROSCI.0281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro JT, Paternain AV, Lerma J, Chenard BL, Ewing FE, Huang J, Welch WM, Ganong AH, Menniti FS. Functional characterization of CP-465,022, a selective, noncompetitive AMPA receptor antagonist. Neuropharmacology. 2002;42:143–153. doi: 10.1016/s0028-3908(01)00170-8. [DOI] [PubMed] [Google Scholar]
- Menniti FS, Buchan AM, Chenard BL, Critchett DJ, Ganong AH, Guanowsky V, Seymour PA, Welch WM. CP-465,022, a selective noncompetitive AMPA receptor antagonist, blocks AMPA receptors but is not neuroprotective in vivo. Stroke. 2003;34:171–176. doi: 10.1161/01.str.0000048216.90221.9c. [DOI] [PubMed] [Google Scholar]
- Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mosley CA, Acker TM, Hansen KB, Mullasseril P, Andersen KT, Le P, Vellano KM, Bräuner-Osborne H, Liotta DC, Traynelis SF. Quinazolin-4-one derivatives: a novel class of noncompetitive NR2C/D subunit-selective N-methyl-D-aspartate receptor antagonists. J Med Chem. 2010;53:5476–5490. doi: 10.1021/jm100027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic M, Sedlacek M, Horak M, Chodounska H, Vyklický L., Jr 20-oxo-5beta-pregnan-3alpha-yl sulfate is a use-dependent NMDA receptor inhibitor. J Neurosci. 2005;25:8439–8450. doi: 10.1523/JNEUROSCI.1407-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto ML, Wollmuth LP. Gating modes in AMPA receptors. J Neurosci. 2010;30:4449–4459. doi: 10.1523/JNEUROSCI.5613-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A, Howe JR. How AMPA receptor desensitization depends on receptor occupancy. J Neurosci. 2003;23:847–858. doi: 10.1523/JNEUROSCI.23-03-00847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stern-Bach Y, Stevens CF. The tetrameric structure of a glutamate receptor channel. Science. 1998;280:1596–1599. doi: 10.1126/science.280.5369.1596. [DOI] [PubMed] [Google Scholar]
- Sobolevsky AI, Prodromou ML, Yelshansky MV, Wollmuth LP. Subunit-specific contribution of pore-forming domains to NMDA receptor channel structure and gating. J Gen Physiol. 2007;129:509–525. doi: 10.1085/jgp.200609718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukder I, Borker P, Wollmuth LP. Specific sites within the ligand-binding domain and ion channel linkers modulate NMDA receptor gating. J Neurosci. 2010;30:11792–11804. doi: 10.1523/JNEUROSCI.5382-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL. Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich MH, Isacoff EY. Subunit counting in membrane-bound proteins. Nat Methods. 2007;4:319–321. doi: 10.1038/NMETH1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Inoue Y, Sakimura K, Mishina M. Distinct spatio-temporal distributions of the NMDA receptor channel subunit mRNAs in the brain. Ann N Y Acad Sci. 1993;707:463–466. doi: 10.1111/j.1749-6632.1993.tb38099.x. [DOI] [PubMed] [Google Scholar]
- Welch WM, Ewing FE, Huang J, Menniti FS, Pagnozzi MJ, Kelly K, Seymour PA, Guanowsky V, Guhan S, Guinn MR, Critchett D, Lazzaro J, Ganong AH, DeVries KM, Staigers TL, Chenard BL. Atropisomeric quinazolin-4-one derivatives are potent noncompetitive alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonists. Bioorg Med Chem Lett. 2001;11:177–181. doi: 10.1016/s0960-894x(00)00622-3. [DOI] [PubMed] [Google Scholar]
- Wo ZG, Oswald RE. Unraveling the modular design of glutamate-gated ion channels. Trends Neurosci. 1995;18:161–168. doi: 10.1016/0166-2236(95)93895-5. [DOI] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Vance KM, Ogden KK, Traynelis SF. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J Neurosci. 2009;29:12045–12058. doi: 10.1523/JNEUROSCI.1365-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]