

Abstract
Human genetic studies have demonstrated that polymorphisms in different complement proteins can increase the risk for developing AMD. There are three pathways of complement activation, classical (CP), alternative (AP), and lectin (LP), which all activate a final common pathway. Proteins encoded by the AMD risk genes participate in the AP (CFB), CP/LP (C2), or in the AP and final common pathway (C3). Here we tested which pathway is essential in mouse laser-induced CNV. CNV was analyzed using single complement pathway knockouts (i.e., eliminating one complement pathway at a time), followed by a double knockout in which only the AP is present, and the CP and LP are disabled, using molecular, histological and electrophysiological outcomes. First, single-gene knockouts were analyzed and compared to wild type mice; C1q−/− (no CP), MBL−/− (no LP), and CFB−/− (no AP). Six days after the laser-induced lesion, mice without a functional AP had reduced CNV progression (P<0.001) and preserved ERG amplitudes, whereas those without a functional CP or LP were indistinguishable from the wild type controls (P>0.3). Second, AP-only mice (C1q−/− MBL−/−) were as protected from developing CNV as the CFB−/− mice. The degree of pathology in each strain correlated with protein levels of the angiogenic and anti-angiogenic proteins VEGF and PEDF, respectively, as well as levels of terminal pathway activation product C5a, and C9. The analysis of complement activation pathways in mouse laser-induced CNV allows for the following conclusions. Comparing the single pathway knockouts with those having only a functional AP showed: (1) that AP activation is necessary, but not alone sufficient for injury; and (2) that initial complement activation proceeds via both the LP and CP. Thus, these data indicate an important role for the AP in the generation of complement-dependent injury in the RPE and choroid via amplification of CP- and LP-initiated complement activation. Improving our understanding of the local regulation of this pathway in the eye is essential for developing improved treatment approaches for AMD.
Keywords: complement activation, alternative pathway, age-related macular degeneration, choroidal neovascularization, VEGF, PEDF, mouse model
1. Introduction
Age-related macular degeneration (AMD) is a slowly progressive multifactorial disease involving genetic abnormalities and environmental insults. It is the leading cause of blindness for Americans over the age of sixty (Brown et al., 2005). Dry AMD is characterized by drusen, retinal pigment epithelium (RPE) damage and photoreceptor cell loss (Anderson et al., 2002; Chong et al., 2005; Hageman et al., 2001; Johnson et al., 2001). In some patients, the dry form can transition to wet AMD. Wet AMD is characterized by breakdown of RPE/Bruch’s membrane (Hageman et al., 2001), increased release of the pro-angiogenic factor vascular endothelial growth factor (VEGF, e.g., (van Wijngaarden and Qureshi, 2008)) and development of choroidal neovascularization (CNV, (Hageman et al., 2001)). While mechanistic studies have shown that inflammation (Hageman et al., 2001) and oxidative stress (Snodderly, 1995) are fundamental components of both forms of AMD, genetic studies have demonstrated that polymorphisms in different complement proteins each independently increase the risk for developing AMD (e.g., (Edwards et al., 2008; Hageman et al., 2005; Haines et al., 2005; Klein et al., 2005; Lee et al.; McKay et al., 2009)). There are three pathways of complement activation, classical (CP), alternative (AP), and lectin (LP), which all activate a final common pathway (Sarma and Ward, 2010). The proteins encoded by the AMD risk genes participate in the AP (complement factor B; CFB), CP/LP (complement C2), or in the AP and final common pathway (complement C3). Overall, it has been hypothesized that inadequate control of complement-driven inflammation may be a major factor in disease pathogenesis in AMD. While no effective treatment is available for dry AMD, therapies blocking VEGF ameliorate wet AMD (e.g., (van Wijngaarden and Qureshi, 2008)).
The complement cascade is activated when ligands for one of three pathways engage their respective pattern recognition molecules (Sarma and Ward, 2010). Classical pathway activation is usually antibody-dependent and is initiated when C1q binds to an immune complex, although other C1q ligands have been identified. The lectin pathway is activated when mannose binding protein (MBL) or ficolins bind to conserved carbohydrate structures, and structures on IgM have also been shown to activate the lectin pathway. The alternative pathway is activated by spontaneous hydrolysis of C3 to C3(H2O) that binds factor B (fB), following which cleavage of fB to Bb and Ba by factor D (fD) leads to formation of the alternative pathway C3 convertase [C3(H2O)Bb]. The alternative pathway also provides an amplification loop for the classical and lectin pathways when fB bound to C3b again undergoes cleavage by fD to create the C3bBb amplification C3 convertase which is stabilized by properdin. Thus, all pathways converge at C3 activation with the subsequent cleavage of C5. During this process, the anaphylatoxins C3a and C5a are generated, and C5 cleavage initiates the terminal complement pathway that culminates in the formation of the membrane attack complex (MAC). The MAC can be directly cytolytic and alternatively can stimulate the production of proinflammatory molecules when inserted in cell membranes at sublytic concentrations. Important for our study; since the 3 pathways require unique entry molecules, pathway-specific mutants can be generated (C1q−/−: no CP, MBL−/−: no LP, CFB−/−: no AP and C1q−/− MBL−/−: AP-only mice).
The most studied animal model for wet AMD is the CNV model in rodents. In this model, laser- induced injury is generated by argon laser photocoagulation, rupturing Bruch’s membrane (e.g., (Nozaki et al., 2006)). This injury triggers CNV, which can be visualized by imaging techniques (e.g., (Campa et al., 2008)). Although mouse CNV has been investigated by a number of investigators, it is still not clear how the complement cascade is initiated; and hence which pathways are required for pathology. The strongest argument in support of the involvement of the AP pathway comes from both animal and human data (e.g., (Bora et al., 2006; Rohrer et al., 2009 and Edwards et al., 2005; Gold et al., 2006; Hageman et al., 2005; Haines et al., 2005; Klein et al., 2005)). Conflicting evidence has been published on the involvement of the CP. CP involvement is suggested by the human data, as C2 polymorphisms correlate with AMD (Gold et al., 2006), and autoantibodies are present in patients (Gu et al., 2003; Patel et al., 2005). In the mouse model, blocking C1qα by siRNA does not appear to interfere with CNV development (Bora et al., 2006); however, negative results using in vivo siRNA may be difficult to interpret, and should be repeated using C1qα−/− mice. B- and T-cell infiltrations in mouse CNV appear to be minimal or absent (Tsutsumi-Miyahara et al., 2004), and removing T-cells using antibodies does not interfere with CNV development (Tsutsumi-Miyahara et al., 2004), suggesting that autoantibody formation and cellular immunity do not contribute. No data are available on LP involvement in mouse CNV. Finally, and most importantly, as no information is published on the mechanism underlying activation of the C3 convertase in CNV, it is unclear how the AP is initiated. Here, we use a combination of knockout strategies, to analyze the role of complement activation pathways in CNV development.
2. Material and methods
2.1. Animals
CFB−/− (Matsumoto et al., 1997), MBL-A/C−/− (referred to as MBL−/−) (Takahashi and Ezekowitz, 2005), C1qα−/− (referred to as C1q−/−) (Stuart et al., 2005), and C1qα−/− MBL-A/C−/− (referred to as C1q−/− MBL−/−) mice on a C57BL/6 background were generated from homozygous breeding pairs; C57BL/6 mice also generated from breeding pairs (Harlan Laboratories, Indianapolis, IN) were used as controls. All animals are on a C57BL/6 background and have been backcrossed > 7 generations. Relevant serum complement protein levels are not changed in the different knockout strains used here, eliminating the possibility that the complement deficiency states of these animals might complicate data interpretation (Banda et al., 2007). Mice were housed in the Medical University of South Carolina animal care facility under a 12:12 hour light:dark cycle with access to food and water ad libitum. All experiments were performed in accordance with the Association for Research in Vision and Ophthalmology and were approved by the Institutional Animal Care and Use Committee.
CNV lesions were generated as described previously (Rohrer et al., 2009). In short, 3-month-old mice were anesthetized (xylazine and ketamine, 20 and 80 mg/kg, respectively) and pupils dilated (2.5% phenylephrine HCl and 1% atropine sulfate). Argon laser photocoagulation (532 nm; 100 μm spot size; 0.1 s duration; 100 mW) was used to generate four laser spots in each eye surrounding the optic nerve, while using a handheld coverslip as a contact lens. Bubble formation at the laser spot indicated the rupture of Bruch’s membrane.
2.2. Assessment of CNV lesions
Relative CNV size was determined in flat-mount preparations of RPE-choroid stained with isolectin B (which binds to terminal β-D-galactose residues on endothelial cells and selectively labels the murine vasculature; (Nozaki et al., 2006; Rohrer et al., 2009)). In brief, eyes were collected and immersion-fixed in 4% paraformaldehyde (PFA) for 2 hours at 4°C after which the anterior chamber, lens and retina were removed. The eyecups were incubated in blocking solution (3% bovine serum albumin, 10% normal goat serum, and 0.4% Triton-X in tris-buffered saline) for 1 hour. Isolectin B (1:100 of 1 mg/mL solution; Sigma-Aldrich, St. Louis, MO) was applied to eyecups overnight at 4°C in blocking solution. Following extensive washing, eyecups were flattened using four relaxing cuts, coverslipped using Fluoromount (Southern Biotechnology Associates, Inc., Birmingham, AL), and examined by confocal microscopy (Leica TCS SP2 AOBS, Leica Bannockburn, IL). Fluorescence measurements, taken from 2 μm sections using confocal microscopy (40x oil lens), were used for size determination. A Z-stack of images through the entire depth of the CNV lesion was obtained, using the same laser intensity setting for all experiments. For each slice the overall fluorescence was determined to obtain pixel intensity against depth, from which the area under the curve (indirect volume measurement) was calculated (Rohrer et al., 2009). Data are expressed as mean ±SEM per eye.
2.3. Electroretinography
ERG recordings and data analyses were performed as previously detailed (Gresh et al., 2003; Richards et al., 2006) using the EPIC-2000 system (LKC Technologies, Inc., Gaithersburg, MD). Stimuli to determine overall retinal responsiveness consisted of 10 μsec single-flashes at a fixed intensity (2.48 cd–s/m2) under scotopic conditions. Measurements were performed prior to performing the CNV lesion (baseline ERG) and after the CNV lesion period. Peak a-wave amplitude was measured from baseline to the initial negative-going voltage, whereas peak b-wave amplitude was measured from the trough of the a-wave to the peak of the positive b-wave. ERG responses were found to decline with increasing size of the CNV lesion, affecting a- and b-waves equally (Rohrer et al., 2009).
2.4. Quantitative RT-PCR
RPE-choroid fractions were isolated from control and CNV eyes and stored at -80°C until used. Quantitative RT-PCR analyses were performed as described in detail previously (Lohr et al., 2006). Primers used were: β-actin, forward: 5′-AGCTGAGAGGGAAATCGTGC-3′ and reverse: 5′-ACCAGACAGCACTGTGTTG-3′; C3 forward: 5′-GGAAACGGTGGAGAAAGC-3′ and reverse: 5′-CTCTTGACAGGAATGCCATCGG-3′; CFH forward: 5′-TGGACTTCCTTGTGGACCTC -3′ and reverse: 5′-CCATCAATTCCAAAGCCTGT -3′; CD55 forward: 5′-TAATGCGAGGGGAAAGTGAC -3′ and reverse: 5′-GTGGACTGCTCCATTGTCCT -3′; CD59 forward: 5′-CTGCTTCTGGCTGTGTTCTG -3′ and reverse: 5′-TCCTGGTCAGGAGAGCAAGT -3′; VEGF forward: 5′-CAGGCTGCTGTAACGATGAA-3′ and reverse: 5′-GCATTCACATCTGCTGTGCT-3′; PEDF forward: 5′-CCTCAGCATCCTTCTCCTTG -3′ and reverse: 5′-TGACATCATGGGGACTCTCA -3′. Real-time PCR analyses were performed in triplicate in a GeneAmp® 5700 Sequence Detection System (Applied Biosystems, Foster City, CA) using standard cycling conditions. Quantitative values were obtained by the cycle number.
2.5. ELISA for VEGF and PEDF measurement
To measure production of VEGF and PEDF by the RPE-choroid, eyecups were solubilized in CellLytic MT (mammalian tissue lysis/extraction reagent; Sigma) and centrifuged at 20,000g for 5 min. Microplates were coated with either the anti-mouse VEGF (Antigenix America, Inc.) (Rohrer et al., 2009) or the anti-human PEDF capture antibody (R&D Systems, Minneapolis, MN) (Ma et al., 2009), and 100 μL of the tissue extract was added. The captured proteins were detected with either the same VEGF-specific antibody conjugated to horseradish peroxidase (HRP) or a PEDF-HRP-labeled detection antibody (R&D Systems), followed by development with a chromogenic substrate OPD (Sigma). Product development was assayed by measuring absorbance at 492 nm. Aliquots were assayed in duplicate, and values compared to a VEGF/PEDF dose-response curve. The anti-human PEDF capture antibody was verified to recognize mouse PEDF specifically on a Western blot (data not shown).
2.6. Western blot analysis
RPE-choroid extracts were separated by electrophoresis on a 10% Bis-Tris polyacrylamide gel (Invitrogen), and proteins were transferred to a nitrocellulose membrane. The membrane was probed with polyclonal antibodies to C9 (generously provided by Paul Morgan, University of Cardiff) or C3a and C5a (generously provided by Scott R. Barnum; University of Alabama, Birmingham, AL) and antibody binding was visualized using a chemiluminescence detection kit (Amersham Life Science). The intensity of the bands was quantified using the Alpha Innotech Fluorchem 9900 imaging system running Alpha Ease FC software version 3.3 (Alpha Innotech, San Leandro, CA). As a loading control, blots were stripped and reprobed with an antibody against GAPDH (Stressgen, Ann Arbor, MI).
2.7. Statistics
For data consisting of multiple groups, one-way ANOVA followed by Fisher’s post hoc test (P <0.05) was used; single comparisons were analyzed by t test analysis (P <0.05).
3. Results
3.1. CNV size and photoreceptor cell responses as a function of complement status
Activation of the AP and an associated inflammatory response are involved in the development of CNV in mice and humans. Previously, we have shown that AP activation is required for CNV development (Rohrer et al., 2009). In mice in which the AP is eliminated (CFB−/− mice), CNV was found to be reduced by ~2/3 when compared to control mice. The smaller lesions were associated with better-preserved retinal function. In addition, analysis of VEGF and C3 mRNA levels by QRT-PCR showed that the large increase in message levels produced in the RPE-choroid in control mice was almost entirely eliminated in the CFB−/− mice. Based on these data we formulated a working hypothesis that by eliminating complement, AP activation reduces VEGF required for CNV development. Here, we wished to extend these experiments by asking whether AP activation is alone sufficient for CNV development.
Using a combination of knockout strategies, we analyzed the complement activation pathways involved in CNV development (Fig. 1, and Fig. 2A) and retina function (Fig. 2B), following laser photocoagulation. First, single-knockouts were analyzed; C1q−/− for CP, MBL−/− for LP, and CFB−/− for AP activation (Figs. 1, and 2A). Average CNV sizes in control, MBL−/− and C1q−/− mice did not differ significantly from each other, whereas mice without a functional AP (CFB−/−) were significantly protected (P <0.001). These data reconfirmed the importance of the AP in the pathology of CNV (Bora et al., 2006; Rohrer et al., 2009), and extended the observations using siRNA against C1q (Bora et al., 2006). Furthermore, the data suggest that neither the CP nor the LP alone can activate complement sufficiently to promote CNV development.
Figure 1. CNV development in complement-deficient mice.
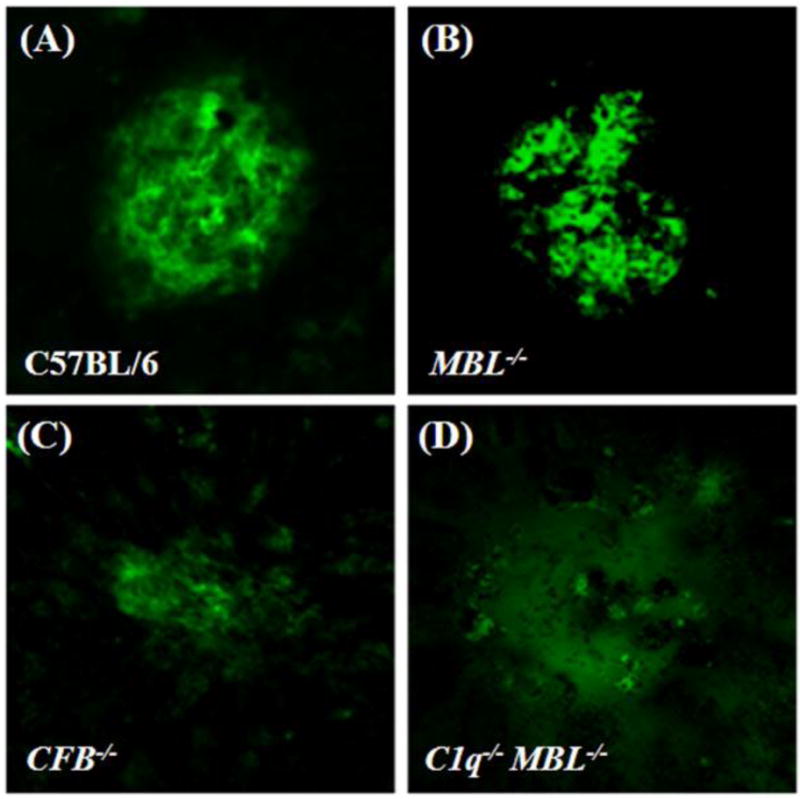
Laser photocoagulation of Bruch’s membrane was used to induce CNV in control (A), MBL−/− (B), CFB−/− (no-AP) (C) and C1q−/− MBL−/− (AP-only) (D) mice. The newly-formed blood vessels that make up the CNV were stained by isolectin–B4 immunohistochemistry and imaged by confocal microscopy, to reveal the size of the CNV lesion 7 days after the induction of CNV. Representative images are shown. Scale bar = 50 μm.
Figure 2. Correlation between CNV size and ERG deterioration.

(A) CNV lesion sizes were determined by capturing Z-stacks of images through the entire depth of individual CNV lesions and integrating the isolectin –B4 signal. Identical laser intensity settings were used for all experiments to allow for relative comparisons. CNV lesions (lesions were normalized to maximum size of C57BL/6 control mice) were indistinguishable between control mice (C57BL/6) and the CP- (C1q−/−) and LP-knockout (MBL−/−) mice; however CFB−/− (no-AP) and C1q−/− MBL−/− (AP-only) mice were equally protected. (B) The effects of the laser lesions on photoreceptor cell function were determined by electroretinography (ERG), measuring a-wave amplitudes and expressing those values as a percentage of baseline amplitudes. Overall, the size of the lesions correlated with the drop in the ERG amplitudes. Unfortunately, ERG analysis was not performed on MBL−/− mice. (n=8-14 animals per group)
We next investigated whether the AP is sufficient for triggering C3 activation and CNV development using double C1q−/− MBL−/− mice (Figs. 1 and 2A). Interestingly, when both the CP and LP were eliminated, CNV development was significantly blunted when compared to control mice (P <0.001); the degree of inhibition was indistinguishable between mice with no AP (CFB−/−) and AP-only mice (C1q−/− MBL−/−). These data together suggest that the AP is required, but not alone sufficient for CNV development.
ERG recordings were assessed as described in CNV by Caicedo and coworkers (Caicedo et al., 2005) and us (Rohrer et al., 2009). The two prominent parameters of the ERG response are the a-wave amplitude, which is a direct reflection of the photoreceptor currents generated by the absorption of light; and the b-wave amplitude, which is a mass potential generated by the bipolar cells that sum photoreceptor output (reviewed by (Pugh et al., 1998)). We have shown previously in C57BL/6 mice, that the reduction in ERG amplitude is correlated with the extent of the lesion when the animals were analyzed at different times after the CNV lesion (Rohrer et al., 2009). Here, we showed that control mice or CP knockout mice exhibited significant rod photoreceptor cell impairment (the data for the LP pathway knockout mice were unfortunately not collected), whereas rod photoreceptor cell function was significantly improved in the no AP (CFB−/−) and the AP-only (C1q−/− MBL −/−) mice (Fig. 2B). Thus, CNV size was found to correlate with photoreceptor cell function in these complement knockout animals (Fig. 2A, B).
3.2. Complement status and CNV-associated changes in complement activation and VEGF activity
We have shown previously that CNV development was associated with C3 activation at the site of the lesion, together with an increase in local expression of VEGF and C3 mRNA within the RPE/choroid (Rohrer et al., 2009). To gain further insight into how complement status may be modulating angiogenesis and complement expression, we examined the local gene expression of C3, C9 and the complement inhibitors CFH, CD55 and CD59 as well as VEGF and the anti-angiogenic protein PEDF (Fig. 3). In addition, protein levels of C3 and C9 together with the split product C5a as well as VEGF and PEDF were examined (Fig. 4, 5). The rationale for examining these angiogenesis and complement components is as follows. C3 is the common C3 convertase target, and its cleavage is required for activation of the common terminal pathway. The terminal pathway culminates with the formation of the cytolytic membrane attack complex (MAC), of which C9 is the terminal component. Since we find C3 deposited at the edge of the CNV lesion, we have proposed that lytic complement activation is involved in increasing the size of the lesion (Rohrer et al., 2009). The complement activation split-products, C3a and C5a, are activators of inflammation and chemotaxis. Eliminating their receptors (C3aR and C5aR) has been found to reduce CNV in mice (Nozaki et al., 2006), although only C5a levels in serum are independently associated with AMD (Reynolds et al., 2009). Protein levels of the membrane-bound complement inhibitors CD55 and CD59 are reduced in a cell-based model of AMD-related RPE damage, as is activity of the soluble inhibitor CFH (Thurman et al., 2009). In addition, recombinant membrane-targeted CD59 can interfere with CNV growth (Bora et al., 2010). Finally, the angiogenic factor, VEGF (e.g., (Hageman et al., 2001; Nozaki et al., 2006)), as well as the anti-angiogenic factor, PEDF (e.g., (Bhutto et al., 2006)), play important roles in the development of CNV in mice and humans, and we have shown that VEGF production and secretion from RPE monolayers can be triggered by sublytic MAC activation (Thurman et al., 2009).
Figure 3. Analysis of mRNA expression for angiogenesis and complement proteins.
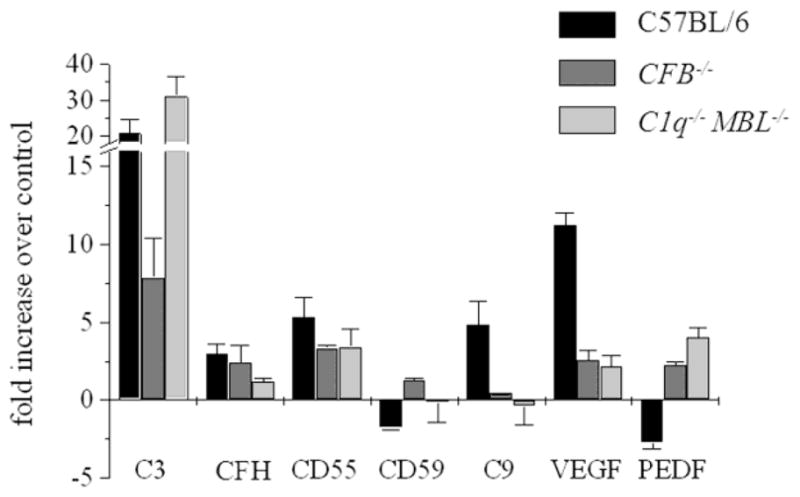
Quantitative RT-PCR was performed on RPE-choroid samples removed 7 days post laser photocoagulation from control (C57BL/6), CFB−/− (no-AP) and C1q−/− MBL−/− (AP-only) mice. Quantitative values were obtained by cycle number (Ct value), determining the difference between the mean experimental (e.g., VEGF) and control (β-actin) ΔCt values. With the exception of C3 mRNA expression, mRNA levels for all the angiogenesis (VEGF and PEDF) and complement markers (C9) were normalized in CFB−/− (no-AP) and C1q−/− MBL−/− (AP-only) mice when compared to the C57BL/6 control. No consistent trends were obtained in the membrane-bound complement inhibitors CD55 and CD659 or the soluble inhibitor CFH. Tissues from 3 animals per group were evaluated.
Figure 4. Analysis of angiogenesis proteins.

Quantitative ELISA assays were performed on RPE-choroid samples removed 7 days post laser photocoagulation from control (C57BL/6), CFB−/− (no-AP) and C1q−/− MBL−/− (AP-only) mice. Quantitative values were obtained by using a standard curve of purified mouse protein (VEGF) or by determining the fold difference between experimental (e.g., CFB−/− and control C57BL/6) values (PEDF). Protein levels for VEGF (A) and PEDF (B) followed the mRNA levels; both were normalized in CFB−/− (no-AP) and C1q−/− MBL−/− (AP-only) mice when compared to the C57BL/6 control. Tissues from 3 animals per group were evaluated.
Figure 5. Analysis of complement proteins expression and split product production.

Western-blot analysis was performed on RPE-choroid samples removed 7 days post laser photocoagulation from control (C57BL/6), CFB−/− (no-AP) and C1q−/− MBL−/− (AP-only) mice. C3 (A), C9 (B), C3a (B) and C5a (D) levels were significantly elevated in CNV of control mice (C57BL/6). Levels of all four complement markers were normalized or dropped to levels below those seen in controls in the RPE-choroid samples derived from CFB−/− (no-AP) or C1q−/− MBL−/− (AP-only) mice. Tissues from 3 animals per group were evaluated, using GAPDH to normalize the values.
Here we found that in wild type control mice, C3 and VEGF mRNA levels were significantly upregulated, confirming our previous data (Fig. 3) (Rohrer et al., 2009). In addition, C9 levels were upregulated ~5-fold. No consistent trend could be established for the mRNA levels of the complement inhibitors. However, mRNA levels for PEDF, the inhibitor of VEGF, were found to be downregulated in wild type mice. In the complement-deficient mice that showed significant reduction of CNV, characteristic differences were observed (Fig. 3). In the no-AP (CFB−/−) mice, C3 and VEGF mRNA levels were significantly reduced as has been reported previously (Rohrer et al., 2009), C9 levels were returned to normal and the drop in PEDF levels was reversed. On the other hand, the AP-only (C1q−/− MBL−/−) mice had elevated levels of C3 mRNA, but almost normal levels of C9, VEGF and PEDF. Again, as in the control mice, no consistent trend could be established for the mRNA levels of the complement inhibitors in the two complement-deficient mice.
We confirmed the above changes at the protein level by ELISA and Western blotting (Figs. 4, 5). VEGF levels were found to be increased ~2.5-fold in the RPE-choroid of wild type animals; this increase was significantly and equally blunted in both no-AP (CFB−/−) and AP-only (C1q−/− MBL−/−) mice (P <0.05) (Fig. 4A). Likewise, while PEDF levels were reduced by ~30% in control animals, both no-AP (CFB−/−) and AP-only (C1q−/− MBL−/−) mice had levels of PEDF comparable to control mice (Fig. 4B). Similar changes were found at the complement level. As with the mRNA levels, C3 protein levels (Fig. 5A) were increased by ~2.5-fold in control mice. This increase was blunted in no-AP (CFB−/−) mice (P <0.05), but remained elevated in AP-only (C1q−/− MBL−/−) mice (P >0.05). C3 protein levels were mirrored by the C3 and C5 split-product levels (Fig. 5C, D). C3a and C5a levels were elevated in control mice by ~40-50% each (P <0.05); however, no-AP (CFB−/−) mice had baseline or below control levels of C3a and C5a, respectively, as did the AP-only (C1q−/− MBL−/−) mice. Finally, mirroring the mRNA levels for C9, protein levels of C9 as assessed by Western blotting (Fig. 5B) revealed that while C9 protein levels were increased by ~2-fold in control mice (P=0.001), both no-AP (CFB−/−) mice and AP-only (C1q−/− MBL−/−) mice had normal levels of C9 protein. This is of particular significance for the AP-only (C1q−/− MBL−/−) mice, since the common C3 convertase was found to be elevated in those mice; a process that, however, did not lead to an increase in C9 protein levels, presumably preventing MAC formation.
4. Discussion
The main findings of this study are: (1) The AP is necessary for the induction of CNV, whereas the CP and the LP, individually, have no effect; (2) AP activation is necessary, but is not alone sufficient for injury; (3) the LP and CP are both activated and contribute to injury since both pathways must be eliminated for protection from CNV and; (4) there is a significant correlation between CNV lesion size and levels of VEGF, PEDF, the terminal pathway activation product C5a, and the terminal pathway component C9.
The involvement of the complement system in mouse CNV has been well documented. The initial experiments focused on the essential contribution of C3 and the anaphlatoxins C3a and C5a (Nozaki et al., 2006) to CNV development, which are common components of all complement activation pathways, but do not distinguish between the initiating events. It was subsequently shown using siRNA against CFB, that complement activation in mouse CNV involves the AP, whereas siRNA against C1qα (the essential component required for CP activation) had no effect (Bora et al., 2006). However, the data using siRNA must be interpreted with caution based on the recent report that siRNA can suppress angiogenesis and CNV via TLR3-activation independent of the target gene (Kleinman et al., 2008). It was subsequently shown that CD59−/− mice (CD59 is an inhibitor of MAC formation) develop CNV significantly faster than wild type mice, and that systemic or intravitreal injections of recombinant soluble CD59a-IgG2a fusion-protein or membrane-targeted CD59 reduced CNV (Bora et al., 2010; Bora et al., 2007). VEGF expression was reduced in CFB siRNA-, rsCD59a-, and C3a or C5a blocking antibody-treated mice, concomitant with a reduction in CNV. In summary, the existing data to date suggest that mouse CNV involves the AP plus the activation of the central C3 convertase, but that the CP may not be essential. Of the downstream components, both the production of anaphlatoxins (Nozaki et al., 2006) and the deposition of MAC (Bora et al., 2006), two important and not mutually exclusive mechanisms, appear to be required for CNV. And finally, an essential downstream molecule required for CNV is VEGF.
The data presented here confirms that the AP is required for CNV development, but is not alone sufficient, and suggests that neither CP nor LP activation alone are sufficient to initiate CNV. As a side note, since MBL mediates coagulation factor-like activities (Takahashi et al., 2010), the negative results of the MBL−/− mice also suggests that this model does not depend on coagulation. Holers and Thurman have recently summarized the importance of the AP pathway in tissue injuries involving immune, ischemic, or infectious insults (Thurman and Holers, 2006). Importantly, the AP is capable of both autoactivation due to a process called "tickover", and of serving as an amplification loop for the other two pathways (reviewed in (Muller-Eberhard, 1988)). Tickover describes a process whereby ~1% of total C3 per hour is spontaneously converted into C3(H2O), that is capable of binding factor B, which triggers the AP pathway. Due to the possibility of autoactivation of the AP pathway, we hypothesized that the AP might be sufficient to create pathology in the mouse CNV model. However, our data revealed that the AP is not sufficient for pathology, indicating that tickover is controlled sufficiently in the eye to prevent complement activation in CNV. Our data differ from those generated by Banda and coworkers in the collagen antibody-induced arthritis (CAIA) model, in which the AP alone is capable of mediating disease (Banda et al.; Banda et al., 2007). In the CAIA model, histopathological scores in CP-only (MBL−/− CFD−/−) and LP-only (C1q−/− CFD−/−) mice were significantly blunted, whereas AP-only mice (C1q−/− MBL−/−) developed disease. Interestingly, while no-AP, CP-only and LP-only mice all exhibited high levels of C3 in the joints after disease induction, C5a was only generated in no-AP mice. It is unclear why the AP alone in the mouse CNV model is not sufficient to deposit large enough amounts of the AP C3 convertase C3bBb on cell surfaces to effectively activate the terminal complement cascade. One possibility is that in CNV no ligands are present that allow for properdin-mediated AP activation. Properdin, which was thought to act only as a positive regulator of AP complement activation, has recently been shown to be able to initiate AP activation (see (Hourcade, 2008)) and has been shown to be involved in AP-mediated autologous tissue injury (Kimura et al., 2010). Several investigators have studied complement pathway requirements in a number of diseases (see (Thurman and Holers, 2006)). While the requirement for AP mediated for pathology has been demonstrated in all diseases investigated, the requirement for LP or CP activity has only been shown in a limited number of disease models, such as intestinal ischemia reperfusion injury (LP; (Hart et al., 2005)), antiphospholipid syndrome (CP; (Girardi et al., 2003)), traumatic brain injury (CP; (Ten et al., 2005)) and cardiac ischemia reperfusion injury (LP; (Busche et al., 2009)) However, since none of the studies that excluded the involvement of the CP or LP pathway used double knockouts (C1q−/− MBL−/−), a redundant activation mechanism as documented herein, in which LP and CP both contribute, may have been overlooked. Thus, the number of diseases in which the AP is involved in amplifying the complement cascade is probably greater than expected. Indeed, Harboe and coworkers have shown recently that the AP contributes up to 80-90% of the activation of the terminal pathway (for CP: (Harboe et al., 2004); for LP: (Harboe et al., 2006)).
Together, our results suggest that both the CP and LP contribute to complement activation in the CNV model, and that pathology is dependent on AP amplification of the cascade. It is not clear what ligands may be responsible for CP or LP activation. In this context, a number of ligands have been identified that can engage both the CP and the LP, such as natural IgM molecules (McMullen et al., 2006) and apoptotic cells (Stuart et al., 2005). Antiretinal IgM autoantibodies have been identified in AMD (Cherepanoff et al., 2006), and both human AMD (Dunaief et al., 2002) as well as CNV in the mouse model (Bora et al., 2010) are associated with cell death in the RPE, choriocapillaris and retina. Interestingly, the majority of the TUNEL-positive cells are located at the edge of the lesions, which is also where we find the majority of the C3d deposition in mouse CNV (Rohrer et al., 2009). Finally, since ficolins can substitute for MBL in activating the LP (Holmskov et al., 2003), it cannot be excluded that an essential role for the LP could be established when both pattern-recognition receptors were eliminated. To test for the additional involvement of the LP pathway, the ficolin A, ficolin B and the MASP-1 and MASP-3 knockout mice should be tested alone and in combination with the MBL−/− mice once they become available.
Finally, a strong correlation was identified between CNV lesion size and terminal pathway activation (C5a), the terminal complement component (C9), VEGF and PEDF. These data further confirm that a tight balance between VEGF as a pro-angiogenic factor (van Wijngaarden and Qureshi, 2008) and PEDF as an anti-angiogenic factor (Holekamp et al., 2002), is required to prevent CNV not only in humans, but also in mice. In addition, the data further suggest that complement activation results in an imbalance of anti- and pro-angiogenic factors in neovascularization. This is in contrast to results in other angiogenesis assays where complement inhibition was found to increase neovascularization in hypoxia-induced retinal neovascularization and in VEGF-triggered Matrigel angiogenesis assays (Langer et al., 2010). The difference between the two findings might be the target cells; in CNV, the main effect of complement is to trigger VEGF-release from the RPE cells, whereas in angiogenesis the inhibitory effect is mediated by macrophages. Finally, however, VEGF is also required as a trophic factor for RPE (Byeon et al., 2010), choriocapillaris (Saint-Geniez et al., 2009) and retina health (Saint-Geniez et al., 2008). Hence, it appears to be essential to identify a strategy for the treatment of wet AMD that only interferes with the pathological increase in VEGF, but does not reduce VEGF levels below normal levels. Here we have shown that eliminating complement activation reduces pathology by interfering with the pathological elevation of VEGF induced by CNV, but does not reduce VEGF below baseline levels. Thus, together, our results suggest that a treatment strategy interfering with AP activation, like the targeted factor-H molecule we have utilized previously (Rohrer et al., 2009), might prove to be a viable strategy for long-term treatment in AMD.
Acknowledgments
This work was supported in part by the National Institutes of Health (B.R. R01EY019320; S.T. HL86576; K.T. UO1 AI074503, R21 AI077081; V.M.H. AR53749), a Veteran’s Affairs grant (B.R. RX000444), Foundation Fighting Blindness (B.R.) and an unrestricted grant to MUSC from Research to Prevent Blindness, Inc. B.R. is a Research to Prevent Blindness Olga Keith Weiss Scholar. Animal studies were conducted in a facility constructed with support from the NIH (C06 RR015455). We thank Luanna Bartholomew, Ph.D. for critical review.
Footnotes
The authors have no financial or proprietary interests in any product(s) mentioned herein.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–31. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- Banda NK, Levitt B, Wood AK, Takahashi K, Stahl GL, Holers VM, Arend WP. Complement activation pathways in murine immune complex-induced arthritis and in C3a and C5a generation in vitro. Clin Exp Immunol. 2010;159:100–8. doi: 10.1111/j.1365-2249.2009.04035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banda NK, Takahashi K, Wood AK, Holers VM, Arend WP. Pathogenic complement activation in collagen antibody-induced arthritis in mice requires amplification by the alternative pathway. J Immunol. 2007;179:4101–9. doi: 10.4049/jimmunol.179.6.4101. [DOI] [PubMed] [Google Scholar]
- Bhutto IA, McLeod DS, Hasegawa T, Kim SY, Merges C, Tong P, Lutty GA. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Exp Eye Res. 2006;82:99–110. doi: 10.1016/j.exer.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bora NS, Jha P, Lyzogubov VV, Kaliappan S, Liu J, Tytarenko RG, Fraser DA, Morgan BP, Bora PS. Recombinant Membrane-targeted Form of CD59 Inhibits the Growth of Choroidal Neovascular Complex in Mice. J Biol Chem. 2010;285:33826–33. doi: 10.1074/jbc.M110.153130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bora NS, Kaliappan S, Jha P, Xu Q, Sivasankar B, Harris CL, Morgan BP, Bora PS. CD59, a complement regulatory protein, controls choroidal neovascularization in a mouse model of wet-type age-related macular degeneration. J Immunol. 2007;178:1783–90. doi: 10.4049/jimmunol.178.3.1783. [DOI] [PubMed] [Google Scholar]
- Bora NS, Kaliappan S, Jha P, Xu Q, Sohn JH, Dhaulakhandi DB, Kaplan HJ, Bora PS. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: role of factor B and factor H. J Immunol. 2006;177:1872–8. doi: 10.4049/jimmunol.177.3.1872. [DOI] [PubMed] [Google Scholar]
- Brown MM, Brown GC, Stein JD, Roth Z, Campanella J, Beauchamp GR. Age-related macular degeneration: economic burden and value-based medicine analysis. Can J Ophthalmol. 2005;40:277–87. doi: 10.1016/S0008-4182(05)80070-5. [DOI] [PubMed] [Google Scholar]
- Busche MN, Pavlov V, Takahashi K, Stahl GL. Myocardial ischemia and reperfusion injury is dependent on both IgM and mannose-binding lectin. Am J Physiol Heart Circ Physiol. 2009;297:H1853–9. doi: 10.1152/ajpheart.00049.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon SH, Lee SC, Choi SH, Lee HK, Lee JH, Chu YK, Kwon OW. Vascular endothelial growth factor as an autocrine survival factor for retinal pigment epithelial cells under oxidative stress via the VEGF-R2/PI3K/Akt. Invest Ophthalmol Vis Sci. 2010;51:1190–7. doi: 10.1167/iovs.09-4144. [DOI] [PubMed] [Google Scholar]
- Caicedo A, Espinosa-Heidmann DG, Hamasaki D, Pina Y, Cousins SW. Photoreceptor synapses degenerate early in experimental choroidal neovascularization. J Comp Neurol. 2005;483:263–77. doi: 10.1002/cne.20413. [DOI] [PubMed] [Google Scholar]
- Campa C, Kasman I, Ye W, Lee WP, Fuh G, Ferrara N. Effects of an anti-VEGF-A monoclonal antibody on laser-induced choroidal neovascularization in mice: optimizing methods to quantify vascular changes. Invest Ophthalmol Vis Sci. 2008;49:1178–83. doi: 10.1167/iovs.07-1194. [DOI] [PubMed] [Google Scholar]
- Cherepanoff S, Mitchell P, Wang JJ, Gillies MC. Retinal autoantibody profile in early age-related macular degeneration: preliminary findings from the Blue Mountains Eye Study. Clin Experiment Ophthalmol. 2006;34:590–5. doi: 10.1111/j.1442-9071.2006.01281.x. [DOI] [PubMed] [Google Scholar]
- Chong NH, Keonin J, Luthert PJ, Frennesson CI, Weingeist DM, Wolf RL, Mullins RF, Hageman GS. Decreased thickness and integrity of the macular elastic layer of Bruch’s membrane correspond to the distribution of lesions associated with age-related macular degeneration. Am J Pathol. 2005;166:241–51. doi: 10.1016/S0002-9440(10)62248-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaief JL, Dentchev T, Ying GS, Milam AH. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–42. doi: 10.1001/archopht.120.11.1435. [DOI] [PubMed] [Google Scholar]
- Edwards AO, Fridley BL, James KM, Sharma AK, Cunningham JM, Tosakulwong N. Evaluation of clustering and genotype distribution for replication in genome wide association studies: the age-related eye disease study. PLoS ONE. 2008;3:e3813. doi: 10.1371/journal.pone.0003813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Suner I, Hernandez EP, Frazier WD, Csaky KG, Cousins SW. Age as an independent risk factor for severity of experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2002;43:1567–73. [PubMed] [Google Scholar]
- Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, Hollmann TJ, Casali P, Caroll MC, Wetsel RA, Lambris JD, Holers VM, Salmon JE. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest. 2003;112:1644–54. doi: 10.1172/JCI18817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, Smith RT, Hageman GS, Dean M, Allikmets R. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–62. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresh J, Goletz PW, Crouch RK, Rohrer B. Structure-function analysis of rods and cones in juvenile, adult, and aged C57bl/6 and Balb/c mice. Vis Neurosci. 2003;20:211–20. doi: 10.1017/s0952523803202108. [DOI] [PubMed] [Google Scholar]
- Gu X, Meer SG, Miyagi M, Rayborn ME, Hollyfield JG, Crabb JW, Salomon RG. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J Biol Chem. 2003;278:42027–35. doi: 10.1074/jbc.M305460200. [DOI] [PubMed] [Google Scholar]
- Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–32. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–21. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- Harboe M, Garred P, Borgen MS, Stahl GL, Roos A, Mollnes TE. Design of a complement mannose-binding lectin pathway-specific activation system applicable at low serum dilutions. Clin Exp Immunol. 2006;144:512–20. doi: 10.1111/j.1365-2249.2006.03072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. 2004;138:439–46. doi: 10.1111/j.1365-2249.2004.02627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart ML, Ceonzo KA, Shaffer LA, Takahashi K, Rother RP, Reenstra WR, Buras JA, Stahl GL. Gastrointestinal ischemia-reperfusion injury is lectin complement pathway dependent without involving C1q. J Immunol. 2005;174:6373–80. doi: 10.4049/jimmunol.174.10.6373. [DOI] [PubMed] [Google Scholar]
- Holekamp NM, Bouck N, Volpert O. Pigment epithelium-derived factor is deficient in the vitreous of patients with choroidal neovascularization due to age-related macular degeneration. Am J Ophthalmol. 2002;134:220–7. doi: 10.1016/s0002-9394(02)01549-0. [DOI] [PubMed] [Google Scholar]
- Holmskov U, Thiel S, Jensenius JC. Collections and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol. 2003;21:547–78. doi: 10.1146/annurev.immunol.21.120601.140954. [DOI] [PubMed] [Google Scholar]
- Hourcade DE. Properdin and complement activation: a fresh perspective. Curr Drug Targets. 2008;9:158–64. doi: 10.2174/138945008783502458. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–96. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Zhou L, Miwa T, Song WC. Genetic and therapeutic targeting of properdin in mice prevents complement-mediated tissue injury. J Clin Invest. 2010;120:3545–54. doi: 10.1172/JCI41782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinman ME, Yamada K, Takeda A, Chandrasekaran V, Nozaki M, Baffi JZ, Albuquerque RJ, Yamasaki S, Itaya M, Pan Y, Appukuttan B, Gibbs D, Yang Z, Kariko K, Ambati BK, Wilgus TA, DiPietro LA, Sakurai E, Zhang K, Smith JR, Taylor EW, Ambati J. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–7. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer HF, Chung KJ, Orlova VV, Choi EY, Kaul S, Kruhlak MJ, Alatsatianos M, Deangelis RA, Roche PA, Magotti P, Li X, Economopoulou M, Rafail S, Lambris JD, Chavakis T. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood. 2010 doi: 10.1182/blood-2010-01-261503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AY, Kulkarni M, Fang AM, Edelstein S, Osborn MP, Brantley MA. The effect of genetic variants in SERPING1 on the risk of neovascular age-related macular degeneration. Br J Ophthalmol. 2010;94:915–7. doi: 10.1136/bjo.2009.172007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr HR, Kuntchithapautham K, Sharma AK, Rohrer B. Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp Eye Res. 2006;83:380–9. doi: 10.1016/j.exer.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Ma W, Zhao L, Fontainhas AM, Fariss RN, Wong WT. Microglia in the mouse retina alter the structure and function of retinal pigmented epithelial cells: a potential cellular interaction relevant to AMD. PLoS ONE. 2009;4:e7945. doi: 10.1371/journal.pone.0007945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Fukuda W, Circolo A, Goellner J, Strauss-Schoenberger J, Wang X, Fujita S, Hidvegi T, Chaplin DD, Colten HR. Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc Natl Acad Sci U S A. 1997;94:8720–5. doi: 10.1073/pnas.94.16.8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay GJ, Silvestri G, Patterson CC, Hogg RE, Chakravarthy U, Hughes AE. Further assessment of the complement component 2 and factor B region associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50:533–9. doi: 10.1167/iovs.08-2275. [DOI] [PubMed] [Google Scholar]
- McMullen ME, Hart ML, Walsh MC, Buras J, Takahashi K, Stahl GL. Mannose-binding lectin binds IgM to activate the lectin complement pathway in vitro and in vivo. Immunobiology. 2006;211:759–66. doi: 10.1016/j.imbio.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Muller-Eberhard HJ. Molecular organization and function of the complement system. Annu Rev Biochem. 1988;57:321–47. doi: 10.1146/annurev.bi.57.070188.001541. [DOI] [PubMed] [Google Scholar]
- Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, Chen Y, Zhang K, Ambati BK, Baffi JZ, Ambati J. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103:2328–33. doi: 10.1073/pnas.0408835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangburn MK. Host recognition and target differentiation by factor H, a regulator of the alternative pathway of complement. Immunopharmacology. 2000;49:149–57. doi: 10.1016/s0162-3109(00)80300-8. [DOI] [PubMed] [Google Scholar]
- Patel N, Ohbayashi M, Nugent AK, Ramchand K, Toda M, Chau KY, Bunce C, Webster A, Bird AC, Ono SJ, Chong V. Circulating anti-retinal antibodies as immune markers in age-related macular degeneration. Immunology. 2005;115:422–30. doi: 10.1111/j.1365-2567.2005.02173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh ENJ, Falsini B, Lyubarsky AL. The origin of the major rod- and cone-driven components of the rodent electroretinogram and the effect of age and light-rearing history on the magnitude of these components. Plenum Press; New York: 1998. [Google Scholar]
- Reynolds R, Hartnett ME, Atkinson JP, Giclas PC, Rosner B, Seddon JM. Plasma complement components and activation fragments: associations with age-related macular degeneration genotypes and phenotypes. Invest Ophthalmol Vis Sci. 2009;50:5818–27. doi: 10.1167/iovs.09-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards A, Emondi AA, Rohrer B. Long-term ERG analysis in the partially light-damaged mouse retina reveals regressive and compensatory changes. Vis Neurosci. 2006;23:91–7. doi: 10.1017/S0952523806231080. [DOI] [PubMed] [Google Scholar]
- Rohrer B, Long Q, Coughlin B, Wilson RB, Huang Y, Qiao F, Tang PH, Kunchithapautham K, Gilkeson GS, Tomlinson S. A Targeted Inhibitor of the Alternative Complement Pathway Reduces Angiogenesis in a Mouse Model of Age-related Macular Degeneration. Invest Ophthalmol Vis Sci. 2009;50:3056–3064. doi: 10.1167/iovs.08-2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado AE, D’Amore PA. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci U S A. 2009;106:18751–6. doi: 10.1073/pnas.0905010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Geniez M, Maharaj AS, Walshe TE, Tucker BA, Sekiyama E, Kurihara T, Darland DC, Young MJ, D’Amore PA. Endogenous VEGF is required for visual function: evidence for a survival role on muller cells and photoreceptors. PLoS ONE. 2008;3:e3554. doi: 10.1371/journal.pone.0003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma JV, Ward PA. The complement system. Cell Tissue Res. 2010 doi: 10.1007/s00441-010-1034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snodderly DM. Evidence for protection against age-related macular degeneration by carotenoids and antioxidant vitamins. Am J Clin Nutr. 1995;62:1448S–1461S. doi: 10.1093/ajcn/62.6.1448S. [DOI] [PubMed] [Google Scholar]
- Stuart LM, Takahashi K, Shi L, Savill J, Ezekowitz RA. Mannose-binding lectin-deficient mice display defective apoptotic cell clearance but no autoimmune phenotype. J Immunol. 2005;174:3220–6. doi: 10.4049/jimmunol.174.6.3220. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Chang WC, Takahashi M, Pavlov V, Ishida Y, La Bonte L, Shi L, Fujita T, Stahl GL, Van Cott EM. Mannose-binding lectin and its associated proteases (MASPs) mediate coagulation and its deficiency is a risk factor in developing complications from infection, including disseminated intravascular coagulation. Immunobiology. 2010 doi: 10.1016/j.imbio.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Ezekowitz RA. The role of the mannose-binding lectin in innate immunity. Clin Infect Dis. 2005;41(Suppl 7):S440–4. doi: 10.1086/431987. [DOI] [PubMed] [Google Scholar]
- Ten VS, Sosunov SA, Mazer SP, Stark RI, Caspersen C, Sughrue ME, Botto M, Connolly ES, Jr, Pinsky DJ. C1q-deficiency is neuroprotective against hypoxic-ischemic brain injury in neonatal mice. Stroke. 2005;36:2244–50. doi: 10.1161/01.STR.0000182237.20807.d0. [DOI] [PubMed] [Google Scholar]
- Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176:1305–10. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- Thurman JM, Renner B, Kunchithapautham K, Ferreira VP, Pangburn MK, Ablonczy Z, Tomlinson S, Holers VM, Rohrer B. Oxidative Stress Renders Retinal Pigment Epithelial Cells Susceptible to Complement-mediated Injury. J Biol Chem. 2009;284:16939–47. doi: 10.1074/jbc.M808166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi-Miyahara C, Sonoda KH, Egashira K, Ishibashi M, Qiao H, Oshima T, Murata T, Miyazaki M, Charo IF, Hamano S, Ishibashi T. The relative contributions of each subset of ocular infiltrated cells in experimental choroidal neovascularisation. Br J Ophthalmol. 2004;88:1217–22. doi: 10.1136/bjo.2003.036392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijngaarden P, Qureshi SH. Inhibitors of vascular endothelial growth factor (VEGF) in the management of neovascular age-related macular degeneration: a review of current practice. Clin Exp Optom. 2008;91:427–37. doi: 10.1111/j.1444-0938.2008.00305.x. [DOI] [PubMed] [Google Scholar]