

Abstract
Recessive mutations in the cartilage-associated protein (CRTAP), leucine proline-enriched proteoglycan 1 (LEPRE1) and peptidyl prolyl cis–trans isomerase B (PPIB) genes result in phenotypes that range from lethal in the perinatal period to severe deforming osteogenesis imperfecta (OI). These genes encode CRTAP (encoded by CRTAP), prolyl 3-hydroxylase 1 (P3H1; encoded by LEPRE1) and cyclophilin B (CYPB; encoded by PPIB), which reside in the rough endoplasmic reticulum (RER) and can form a complex involved in prolyl 3-hydroxylation in type I procollagen. CYPB, a prolyl cis–trans isomerase, has been thought to drive the prolyl-containing peptide bonds to the trans configuration needed for triple helix formation. Here, we describe mutations in PPIB identified in cells from three individuals with OI. Cultured dermal fibroblasts from the most severely affected infant make some overmodified type I procollagen molecules. Proα1(I) chains are slow to assemble into trimers, and abnormal procollagen molecules concentrate in the RER, and bind to protein disulfide isomerase (PDI) and prolyl 4-hydroxylase 1 (P4H1). These findings suggest that although CYPB plays a role in helix formation another effect is on folding of the C-terminal propeptide and trimer formation. The extent of procollagen accumulation and PDI/P4H1 binding differs among cells with mutations in PPIB, CRTAP and LEPRE1 with the greatest amount in PPIB-deficient cells and the least in LEPRE1-deficient cells. These findings suggest that prolyl cis–trans isomerase may be required to effectively fold the proline-rich regions of the C-terminal propeptide to allow proα chain association and suggest an order of action for CRTAP, P3H1 and CYPB in procollagen biosynthesis and pathogenesis of OI.
INTRODUCTION
About 90% of individuals with osteogenesis imperfecta [OI (MIM 166200, 166210, 259420 and 166220)] are heterozygous for mutations in one of the two genes, COL1A1 (MIM120150) and COL1A2 (MIM 120160), that encode the two chains, proα1(I) and proα2(I), respectively, of type I procollagen. In the last few years, it has been recognized that many of the remaining affected individuals are homozygous or compound heterozygous for mutations in genes that encode proteins that provide chaperone functions during molecular assembly or contribute to the complex array of post-translational modifications of type I procollagen. At last count, seven of these genes have been identified in humans with recessively inherited forms of OI [OI with mutations in cartilage-associated protein (CRTAP), MIM 610682; OI with mutations in leucine proline-enriched proteoglycan 1 (LEPRE1), MIM 610915; OI with mutations in peptidyl prolyl cis–trans isomerase B (PPIB), MIM 259440; OI with mutations in serine protease inhibitor 1 (SERPINH1), MIM 600943; OI with mutations in FK506 binding protein 10 (FKBP10), MIM 610968; Bruck syndrome with mutations in procollagen-lysine, 2 oxoglutarate 5 dioxygenase 2 (PLOD2), MIM 609220; and OI with mutations in transcription factor Sp7 (SP7)/osterix (OSX), MIM 606633], and it is likely that there are more in the discovery pipeline. Three of these genes, CRTAP (1,2) (which encodes cartilage-associated protein or CRTAP, MIM 123841), LEPRE1 (3–5) (prolyl 3-hydroxylase 1 or P3H1, MIM 610339) and PPIB (6,7) (cyclophilin B or CYPB, MIM 123841), encode proteins involved in prolyl 3-hydroxylation and prolyl cis–trans isomerization and act in the rough endoplasmic reticulum (RER) during and following the synthesis of the proα chains of type I procollagen. Mutations in these genes, as with those in the dominant genes (COL1A1 and COL1A2) that substitute glycine residues in the triple-helical domain of the chains of type I procollagen, produce in-frame deletions or insertions within the triple-helical domain or result in slow chain association, can produce type I procollagen molecules that undergo excessive post-translational modification, at least in cultured dermal fibroblasts. Three additional genes, FKBP10 (which encodes FK506 binding protein, 65 kDa or FKBP65, MIM 607063) (8), SERPINH1 (heat shock protein 47 HSP47, MIM 600943) (9) and PLOD2 (lysyl hydroxylase 2 LH2, MIM 601856) (10,11), which all encode RER proteins, appear to act later in the pathway so that the procollagens produced by cultured fibroblasts have chains with the normal levels of modification. The recently identified seventh gene, SP7/OSX (12), is a transcription factor involved in the specification of the osteoblast lineage.
The ‘overmodification’ of the triple-helical domain usually represents an increase in the proportion of Y-position (in the canonical Gly-X-Y triplet of the triple-helical domain) lysyl residues that undergo hydroxylation and subsequent glycosylation (13). In the case of mutations that alter sequence within the triple-helical domain, overmodification is asymmetrically distributed N-terminal to the site of the substitution(s) and reflects the C- to N-terminal end winding of the trimer (14,15). Substitutions result in delayed winding and prolonged access of the modifying enzymes to the chains. Mutations that alter residues in the C-terminal propeptide of either proα chain can delay chain association and result in overmodification along the full length of the triple helix.
Most mutations in genes that encode members of the prolyl 3-hydroxylation complex, CRTAP, LEPRE1 and PPIB, result in excess post-translational modification, by fibroblasts in culture, along the length of the triple helix as well as variable hydroxylation of the prolyl residue at position 986 of the triple helix of the proα1(I) chain (1–7). The link between these two processes remains unclear. We have now identified three families in which mutations in PPIB result in increased post-translational modification of the triple-helical domain of the chains of type I procollagen. In cultured dermal fibroblasts from one of these families, we found that overmodification is related to slow incorporation of proα1(I) chains into trimers, which provides a mechanistic explanation for the OI phenotype seen in these families. These findings suggest that another role for the prolyl cis–trans isomerase, CYPB, is to facilitate folding of the proline-rich regions of the C-terminal propeptide to allow proα chain association and is important in procollagen biosynthesis.
RESULTS
Collagens produced by cells with mutations in PPIB are overmodified but have normal thermal stability
Cultured cells from each proband (see Fig. 1 for the pedigrees and radiographs) produced some overmodified type I procollagen, the chains of which had delayed mobility both in the secreted proteins and in the proteins retained in the cells (Fig. 2A), similar to those seen in molecules produced by cells with mutations in CRTAP, LEPRE1 (1–3) and in two (6) but not one other (7) cell strain with mutations in PPIB. The thermal stability of type I collagen molecules made by P2 fibroblasts was similar to the molecules made by control cells (Fig. 2B); this is in contrast to slightly increased thermal stability observed in molecules made by cells from individuals with mutations in LEPRE1 (3) and CRTAP (see Supplementary Material in 7) and decreased stability observed with mutations that alter the triple-helical domains of the proα chains of type I procollagen (16). Mutations that alter sequences in the C-terminal propeptide of proα1(I) chains result in delayed chain assembly, overmodification of the chains but normal thermal stability (17), similar to what we observed with the P2 cells.
Figure 1.

Pedigrees and radiographs for individuals with autosomal recessive OI due to PPIB mutations. (Family 1) II-4 at 3 days: X-rays from the second affected child at 3 days of age show diminished calvarial mineralization; thin, beaded ribs; fractured humerus; short, bowed and undermodeled long bones of the leg. There was no overt platyspondyly. Radiographs from the first affected child (II-3) at 4 months of age demonstrated the lack of calvarial mineralization; undermineralized, broad and beaded ribs; pronounced platyspondyly; short, bowed and undermodeled long bones of the upper and lower extremities. (Family 2) Radiographs of II-2 at 2 weeks of age showed the near absence of calvarial mineralization; very short, beaded ribs; platyspondyly; undermineralized, short and bent femora. (Family 3) At 9 years of age, the femora in II-1 were broad and poorly modeled, and calvarial mineralization was near normal. By 12 years of age, there was thinning of the femoral cortex, which continued to be apparent at 16 years of age. Scoliosis had been present at early ages and was stable by age 16 years.
Figure 2.

Overmodification and normal thermal stability of collagen in the presence of PPIB mutations. (A) SDS–PAGE of medium and cell layer collagens from individuals P1, P2, P3 and a control (C) showed delayed electrophoretic mobility of a population of α1(I) and α2(I) chains, following pepsin treatment of the samples to remove the propeptide extensions. (B) The thermal stability of type I collagen from P2 cells is similar to type I collagen made by controls cells.
Mutations identified in PPIB and consequences for mRNA and protein
We sequenced COL1A1, COL1A2, CRTAP and LEPRE1 from each cell strain and found no mutations. Because CRTAP and P3H1 (encoded by CRTAP and LEPRE1, respectively) can form a complex with CYPB (encoded by PPIB) (1,18) and because type I procollagen produced by cells from these individuals was overmodified, similar to that seen in cells with mutations in CRTAP and LEPRE1, we determined the sequence of the coding regions and flanking intron regions for the PPIB gene in all three individuals. P1 and her sister were homozygous for a 10 bp deletion (c.414_423del, p.Ser139ThrfsX21, Fig. 3P1, A) that resulted in a premature termination codon (PTC) that destabilized the mRNA, leading to significant nonsense-mediated mRNA decay (Fig. 3P1, B). The remaining small amount of mutant mRNA in P1 would result in a shortened protein of 158 amino acids (full length is 216 amino acids) with the last 20 amino acids being different from normal (Fig. 3P1, C, serrated part of the line diagram). No such CYPB species was detected by western blot analysis using a polyclonal antibody (Proteintech) to a full-length recombinant protein (Fig. 3P1, D). Each parent was heterozygous for the mutant allele.
Figure 3.

Molecular basis of autosomal recessive OI due to mutations in PPIB. (P1) (A) Homozygosity for deletion of 10 bp in exon 4 (c.414_423del), shown schematically in the cDNA diagram, led to a reading frame shift and creation of a PTC 61 nt downstream in exon 4 located 41 nt from the final exon–exon boundary in mRNA. The mRNA is predicted to undergo significant NMD. (B) RT–PCR products synthesized with primers in exons 3 and 5 that yield a 343 bp product for the control, after 20, 25 and 30 cycles for a control, P1, and a 1:1 mix of the control and P1 cDNA. After 25 cycles, when the PCR was still in the linear phase, the estimated ratio of control to P1 product was 9:1. (C) The remaining small amount of mutant mRNA in P1 would result in a shortened protein of 158 amino acids (full length is 216 amino acids) with the last 20 amino acids being different from normal (serrated part of the line diagram). (D) Western blot analysis using a polyclonal antibody directed against a full-length recombinant protein of human CYPB did not detect the predicted shortened fragment or a full-length fragment (see Materials and methods). (P2) (A) Compound heterozygosity for c.120delC (maternal allele) and c.313G > A, p.Gly105Arg (paternal allele). The single-nucleotide deletion resulted in a reading frame shift and a PTC 49 nt downstream in exon 2. (B) In genomic DNA (gDNA), P2 is heterozygous A/G at c.313, whereas in cDNA only the A allele is present because mRNA from the G allele that harbors the c.120delC mutation is rapidly degraded. (C) Western blot using a polyclonal antibody directed against a region within residues 150 to the C-terminus of human CYPB showed only a very small amount of CYPB protein derived from the stable mRNA in P2, whereas the amount in the cells from the carrier parents (F2-father, M2-mother) appeared to be close to normal. (P3) (A) Homozygosity for an intron 3 splice donor site mutation (c.343 + 1G > A, IVS3 + 1G > A) yielded two products from each allele. In the first, 27 nucleotides of intron 3 were retained in the mature mRNA due to the use of a strong cryptic splice donor site starting at IVS3 + 28 (gtatgt), which resulted in removal of one glycine residue (p.Gly115) and insertion of 10 new amino acids that are shown in single letter code: DNHRSSGPRR. In the second, exon 3 (94 nt) was skipped, which resulted in a reading frame shift and a PTC in exon 4 that was predicted to lead to NMD (HD, heteroduplex). (B) Western blot analysis using a polyclonal antibody directed against a synthetic peptide corresponding to residues 194–208 of human CYPB did not detect any CYPB protein.
P2 and his sibling were compound heterozygotes for a maternally derived single-nucleotide deletion (c.120delC and p.Val42SerfsX16) that led to a PTC and nonsense-mediated mRNA decay, and for a paternally derived missense mutation (c.313G > A, p.Gly105Arg; Fig. 3P2, A and B). The protein synthesized from the second allele was unstable, shown by western analysis using a polyclonal antibody (Abcam) directed against a region within residues 150 to the C terminus of human CYPB (Fig. 3P2, C), and at least some of the small amount of remaining protein was mislocalized to the Golgi, shown by immunocytochemical analysis with the same antibody (see below).
P3 was apparently homozygous for a splice donor mutation (c.343 + 1G > A, IVS3 + 1G > A, p.Gly115delins10) that led to inclusion of 27 bp of intron 3 in a proportion of the transcripts that produced a stable mRNA. In the remaining transcripts, there was exon 3 skipping (unstable mRNA) (Fig. 3P3, A). The protein produced by the stable mRNA was not detected by western blot using a polyclonal antibody (Thermo Scientific) directed against a synthetic peptide corresponding to residues 194–208 of human CYPB (Fig. 3P3, B). Parental cells were not available to confirm their heterozygosity.
Mutations in PPIB do not destabilize CRTAP or P3H1
Some of the CYPB in the RER is in the form of a stable complex with CRTAP and P3H1 in a 1:1:1 ratio; however, a large portion is not part of this complex when ER-derived proteins are bound to and then eluted from a gelatin-Sepharose column (18) (see also the Supplementary Material in 1). To determine if the same 1:1:1 interaction was present in cellulo, we immunoprecipitated proteins derived from control fibroblasts with an antibody to either P3H1 or CRTAP. Antibodies to P3H1 or CRTAP brought down both proteins, consistent with the recognized interaction (1,18,19), but neither brought down CYPB (Fig. 4A). The discrepancy between our results and those using gelatin-Sepharose could be explained if the interaction of CYPB with CRTAP:P3H1 covers the epitope-binding region of the immunoprecipitation (IP) antibodies or if the interaction is more dynamic within this biological context and is not captured at a level we can visualize by immunostaining of protein complexes. Null mutations in either CRTAP or LEPRE1 led to loss or significant reduction in the interacting protein partner but no reduction in the amount of CYPB in the cell (Fig. 4B) and confirms the observations of others (4,7). Null mutations in PPIB (P1) led to the loss of the PPIB mRNA and protein products, but did not affect the stability of either CRTAP or P3H1 as measured by immunostaining (Fig. 4B); this contrasts with studies in mice which showed a substantial reduction in P3H1 protein but not CRTAP protein in Ppib−/− fibroblasts (20) and in the cell strain reported by Barnes et al. (7) which showed a reduction in both CRTAP and P3H1. In cells from P2, there was little staining of CYPB, consistent with nonsense-mediated mRNA for one allele and protein instability in the other. Most of the small amount of remaining CYPB in those cells was mislocalized to the Golgi where it co-localized with Golgi-specific proteins (Fig. 5B) and would not be available to function as expected in the RER (see below). We could not see the Golgi localization of CYPB in cells from the heterozygous father with the same missense mutation, apparently because of the amount of normal protein from the other allele. Cells from P3 had a very small amount of stable mRNA, but no CYPB protein was detected by western or immunocytochemical analysis (Fig. 5A and B) using a polyclonal antibody directed against a synthetic peptide corresponding to residues 194–208 of human CYPB (the 10 amino acid insertion occurs at glycine 115), which indicated that the inserted peptide probably interfered with stability.
Figure 4.

Mutations in PPIB and loss of CYPB do not destabilize CRTAP or P3H1. (A) Analysis of P3H1, CRTAP and CYPB protein interactions in control cells. The proteins in the entire cell lysate or proteins brought down with antibody to P3H1or CRTAP were separated by SDS–PAGE on 10% polyacrylamide gels and then stained with a pool of antibodies to P3H1 (from Kevin McCarthy), CRTAP (from Roy Morello, see Materials and methods) and CYPB (Abcam). Although CYPB is present in the lysate, it is not brought down at a detectable level by antibodies to the two other proteins. (B) Effects of mutations in PPIB, CRTAP and LEPRE1 on stability of other proteins in the complex. In cells with mutations in PPIB that resulted in loss (P1 and P3) or reduction (P2) in the CYPB protein (antibodies used for CYPB analysis for each proband are indicated in the Results section), the stabilities of CRTAP and P3H1 were unaffected. Parents of P2 (M2 and F2) showed a reduced amount of CYPB protein as expected. In cells with mutations in either CRTAP or LEPRE1, there is a marked reduction or complete loss of the normal protein partner, whereas levels of CYPB remained unaffected. GAPDH was used as a loading control.
Figure 5.

Abnormal CYPB protein in P2 fibroblasts mis-localizes to the Golgi. (A) Immunocytochemistry with antibodies against CYPB and an RER marker showed CYPB protein localized to the RER in control fibroblasts, reduction in CYPB in P2 fibroblasts (antibody used for P2 listed in the Results section) and loss of CYPB in P3 fibroblasts (antibody used for P3 listed in the Results section). (B) Staining with CYPB and a Golgi marker showed the abnormal CYPB protein produced by P2 fibroblasts mis-localized to the Golgi (indicated by a white asterisk). In the P3 fibroblasts, CYPB was not evident.
Effect of PPIB mutation(s) on prolyl 3-hydroxylation at 986 varies
The P3H1:CRTAP:CYPB complex is involved in 3-hydroxylation of the X-position prolyl residue at 986 of the proα1(I) triple-helical domain. Null mutations in either CRTAP or LEPRE1 lead to virtually complete loss of this modification (1–4). We measured Pro986 3-hydroxylation in proα1(I) chains synthesized by fibroblasts from P2 and mesenchymal stem cells (MSCs) from P3 (Supplementary Material, Fig. S1); fibroblasts from P1 were unavailable for this study. In P2, ∼30% of the target residue was hydroxylated in cultured cells; similar levels were reported by van Dijk et al. (6) from cultured fibroblasts from individuals with null mutations in PPIB. In contrast, Ppib−/− mice showed complete absence of Pro986 3-hydroxylation (20). These studies were done with collagen isolated from mouse bone and cartilage samples. In fibroblasts from both parents of P2 (F2 and M2), the extent of 3-hydroxylation of Pro986 was normal (data not shown). The cells from P3 hydroxylated normally, similar to levels reported by Barnes et al. (7).
Mutations in CRTAP, LEPRE1 and PPIB result in differential retention of type I procollagen in the RER bound to protein disulfide isomerase and/or prolyl 4-hydroxylase 1
Some of the type I procollagen molecules produced by cells with mutations in PPIB are retained in the cell layer (Fig. 2A). In P2 cells, the retained procollagen was localized principally in the RER (Fig. 6A), cells from P1 and P3 were not available for this study. Control cells, in contrast, had some staining in the RER but significant accumulation of type I procollagen in the Golgi (Fig. 6B). Cells with null mutations in CRTAP retained less procollagen than the PPIB defective cells and more than those with null mutations in LEPRE1. To identify the manner in which these chains were retained in the RER, we used antibodies to protein disulfide isomerase (PDI) or prolyl 4-hydroxylase 1 (P4H1) to precipitate proteins and used LF9 (21) (directed against an epitope in the N-terminal telopeptide of proα1(I) chains) to measure the proα1(I) chains or type I procollagen molecules bound to PDI and/or P4H1 (Fig. 7A). The proportion of proα1(I) chains brought down by the PDI and P4H1 antibodies differed among cell strains. The amount bound was greatest in the PPIB-deficient cells, less in the CRTAP null cells, even less in the LEPRE1 null cells, and virtually none in control cells (Fig. 7C). These findings suggest that mutations in these genes affect procollagen processing at different stages of assembly and transport from the RER.
Figure 6.

Retention of type I procollagen in the RER differs among cells with mutations in PPIB, CRTAP and LEPRE1 (A and B). Type I procollagen is selectively retained in the RER in cells with mutations in PPIB, whereas cells with mutations in CRTAP or LEPRE1 distribute the molecules between the RER and the Golgi. In contrast, the majority of type I procollagen is located in the Golgi in control cells.
Figure 7.

Type I procollagen binds to PDI and P4H1 in the RER. (A) Proteins in lysates of cells with mutations in PPIB (P2), CRTAP or LEPRE1 were precipitated with antibodies to PDI or P4H1 and then stained with antibodies to proα1(I) (LF9). The greatest proportion of available proα1(I) chains bound by PDI and P4H1 was seen in the PPIB mutant cells with decreasing amounts in CRTAP, LEPRE1 mutant cells and the least bound in control cells, quantitated in (C). (B) PDI precipitated by antibody to P4H1 in cells from individuals with mutations indicated in (A).
Mutation of PPIB results in slow chain association and delayed type I collagen trimer formation and secretion
Steinmann et al. (22) showed that treatment of chick tendon fibroblasts with cyclosporin A (CsA) resulted in the synthesis of type I procollagen molecules that were slightly overmodified and took ∼5 min longer than that in untreated cells to attain a protease-resistant conformation in the triple-helical domain. They suggested that this reflected a delay in the cis–trans isomerization of prolyl-containing peptide bonds in the triple-helical domain. However, a similar outcome would be expected if the nascent proα chains were slow to associate. To determine if the loss of CYPB from the RER of P2 fibroblasts interfered with proα chain association, we pulsed cells with [35S]-cysteine/methionine for 10 min, chased the label for up to 20 min, precipitated type I procollagen and free proα1(I) chains with LF9 (21) and separated the precipitated proteins by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE). We used the antibody to separate the [35S]-cysteine/methionine-labeled procollagen and proα chains from the other labeled proteins. In P2 fibroblasts, the ratio of radiolabel in free proα1(I) chains to that in trimers was higher than in control cells during the first 10 min of chase (Fig. 8A and B). Treatment of control fibroblasts with CsA, an inhibitor of CYPB, delayed chain association and trimer formation in control cells (Fig. 8C and D) using the same experimental protocol. To be sure that antibody precipitation did not skew the molecular population we observed, we repeated the studies using short label with [3H]-proline (see Materials and methods) and examined the ratio of free proα1(I) chains to trimers in control and P2 cells (Fig. 9A and B) and in control cells with and without CsA treatment (Fig. 9C and D). Trimer formation was delayed in P2 cells and in control cells treated with CsA. Cells were not available from P1 and P3 for these studies. Trimer secretion in P2 fibroblasts (which depends on both trimer formation and helix propagation) was delayed in the short chase period (Fig. 8A) and in cells pulsed for 1h and then chased for up to 2 h (Fig. 8E and F). In the latter, only 60% of trimers were secreted into the medium from P2 fibroblasts compared with greater than 90% secreted from control fibroblasts during the study interval. We did not examine the behavior of proα2(I) chains to determine if they followed the same time course of incorporation into trimers.
Figure 8.

Proα1(I) chains in P2 fibroblasts and control cells treated with CsA are slow to assemble and secretion of type I procollagen is delayed. (A and C) Cells were labeled for 10 min with [35S]-methionine and cysteine, immunoprecipitated with the LF9 antibody directed to the N-terminal propeptide of proα1(I) chains, and then analyzed by SDS–PAGE under non-reducing conditions. (B and D) P2 fibroblasts and control fibroblasts treated with 5 µm CsA had more free proα1(I) for up to 10 min compared with controls, and trimers accumulated in the cell over a longer period. (E) Cells were labeled with [3H]-proline for 60 min, chased for up to 2 h with proteins harvested every 20 min. The protein lysates were analyzed by SDS–PAGE under non-reducing conditions. (F) The relative amount of type I procollagen trimers in the medium was far greater in control cells than in the P2 cells. Only the trimers are shown in all four gels.
Figure 9.

Proα1(I) chains in CsA-treated control fibroblasts and P2 fibroblasts are slow to assemble. (A) Control and P2 fibroblasts were labeled for 15 min with [3H]-proline, the cells lysed and the proteins analyzed by SDS–PAGE under non-reducing conditions. Density of the protein bands were quantitated using ImageJ software. (B) P2 fibroblasts had more free proα1(I) at each time point compared with control fibroblasts, and P2 trimers were slow to assemble compared with trimers in control cells. (C) Control fibroblasts were labeled the same as in (A) then treated with 5 µm CsA or EtOH alone. (D) Fibroblasts treated with 5 µm CsA had more free proα1(I) at each time point compared with control fibroblasts with no drug treatment, and trimers were slow to form in the drug-treated cells compared with control cells.
DISCUSSION
These three families bring to six the number of families described with mutations in PPIB, the gene that encodes CYPB. In one of the families described here (P2), the clinical picture is consistent with OI type II; in the second (P1), it is consistent with a very severe OI type III picture; and in the third (P3), the phenotype is similar to a moderately severe deforming OI type III/IV. In the two individuals described by van Dijk et al. (6), the three affected individuals, one from one family and two from the second, all had very severe clinical pictures consistent with the OI type II/III presentations. In the first family (6), the mRNA was unstable in cultured cells, whereas in the second, a small amount of the mRNA was stable but the encoded protein appeared to be unstable. In the family reported by Barnes et al. (7), the clinical picture was moderately severe in one child and more involved in the second. They were both homozygous for a mutation that converted a methionine codon to one for arginine at a position that may be the initiator codon for at least some transcripts (23,24). They reported that although more than half of the mRNA was stable, no protein product could be detected. Thus, in all the reported individuals, the mutations in PPIB lead to a marked reduction in the amount of stable CYPB produced but variation in the amount of 3-hydroxylation in the proα1(I) chains synthesized by skin fibroblasts in vitro. The variation remains unexplained, as do the reported differences in electrophoretic mobilities of the chains of type I procollagen (one possibility is the difference in culture conditions and/or the amount of urea in the electrophoretic gels—the 2 m urea in the system we use enhances the separation). The discrepancy between the human phenotypic spectrum and the consistently milder phenotype in Ppib−/− (20) mice (apparently normal in the perinatal period, reduced body size and weight and development of kyphosis with death at 40–50 weeks of uncertain cause) is also not well understood. Finally, the isolated skin disorder (hereditary equine regional dermal asthenia), which occurs in the absence of a bone phenotype in the American Quarter Horse and apparently results from homozygosity for a missense mutation in equine PPIB, may be explained by the location of the amino substitution near the N-terminal end of the mature protein, but further studies need to be done (25).
The mechanisms by which mutations in PPIB, CRTAP and LEPRE1 result in OI remain poorly understood. Originally, lack of hydroxylation of the single substrate site Pro986 in the triple-helical domain of α1(I) chains was thought to be part of the causative disease mechanism (1,2), but the normal hydroxylation of Pro986 in one of our probands (P3) and in one reported by Barnes et al. (7) with PPIB mutations makes this less likely to be disease causing on its own. Weis and colleagues (26,27) suggest that 3-hydroxyproline plays a role in fibril formation and that additional modified sites in the triple-helical domains of other fibrillar collagens may be important in this function. It is not clear if variation in the extent of prolyl 3-hydroxylation could modify the OI phenotype in individuals with mutations in PPIB as the studies are limited to collagens produced by cultured fibroblasts and do not include bone or other tissue collagens.
Ishikawa et al. (28) found that the three protein complex of CRTAP:P3H1:CYPB prevented aggregation and facilitated refolding of chemically denatured nuclear-encoded mitochondrial proteins, citrate synthase and bovine rhodanese. Those studies provide evidence of a chaperone function that is not altered by the inhibitor of prolyl cis–trans isomerase, CsA. They found, in addition, that the ability of the complex to induce prolyl cis–trans isomerization in a short synthetic peptide is inhibited by the drug. Furthermore, both the complex and the isolated CYPB facilitated refolding of heat-denatured type III collagen, consistent with the concept that prolyl cis–trans isomerization played a role in the increased rate of regain of triple helix. Finally, the three protein complex, but not CYPB alone, prevented fibril formation suggesting that continued presence of the protein complex in the RER could reduce biologically harmful intracellular fibril formation. CYPB is known to be part of other chaperone complexes within the RER including a large multiprotein complex in which CYPB interacts with a number of proteins—that include PDI, binding immunoglobulin protein (BiP) and calreticulin (29). Although this and other chaperone complexes might facilitate folding and protect isolated chains from aggregation, we think that CYPB may play an additional role in early events in proα chain folding and the initial events in trimer formation.
Our data indicate that in the absence of CYPB (but with normal amounts of CRTAP and P3H1) association of proα1(I) chains (we did not study proα2(I) chains separately) into trimers in the very early phases of molecular assembly is slowed. In addition, we found that CsA, an inhibitor of the prolyl cis–trans isomerase function of CYPB, mimicked the effect of the genetic loss of CYPB on incorporation of proα1(I) chains into trimers. These studies suggest that the delay in creating a protease-resistant trimer in the presence of CsA observed by Steinmann et al. (22) is due, at least in part, to delayed chain association. An additional effect on helix propagation cannot be excluded but the similar delay produced by both the drug and the mutations argues for a role for CYPB in facilitating chain association.
Mutations in COL1A1 that alter residues in the C-terminal propeptide region of the proα1(I) chain can result in defective chain association and lead to lethal OI (17,30). These mutations do not involve prolyl residues but induce conformational changes that slow the rate at which the proα chains can be incorporated into trimers. The C-terminal propeptides of the proα chains of type I procollagen have four cysteine residues that form defined pairs of intra-chain disulfide bonds important for the stabilization of the structures involved in chain–chain recognition (31). The folded propeptide, stabilized by disulfide bonds, must form before trimerization can occur. Adjacent to the first and the last of these four cysteines, which form one set of intra-chain disulfide bonds, there are regions of grouped prolyl residues (Fig. 10A; these do not appear adjacent to the other pair of cysteines that stabilize an internal loop). Although the lack of an experimentally determined structure for this region makes structural predictions difficult, the measured delay in chain association that accompanies the loss of CYPB and its prolyl cis–trans isomerase function makes this region a compelling target of interest (see Fig. 10B for proposed model). The longer delay in chain association caused by COL1A1 mutations that alter the C-terminal propeptide is reflected in greater overmodification of type I procollagen but normal thermal stability, like that seen with PPIB mutations. These findings are consistent with the concept that these two classes of mutations act through a common mechanism, i.e. delay in chain association, which may be shorter in the CYPB-deficient cells because of redundancy for cis–trans isomerases and an intrinsic rate of isomerization.
Figure 10.
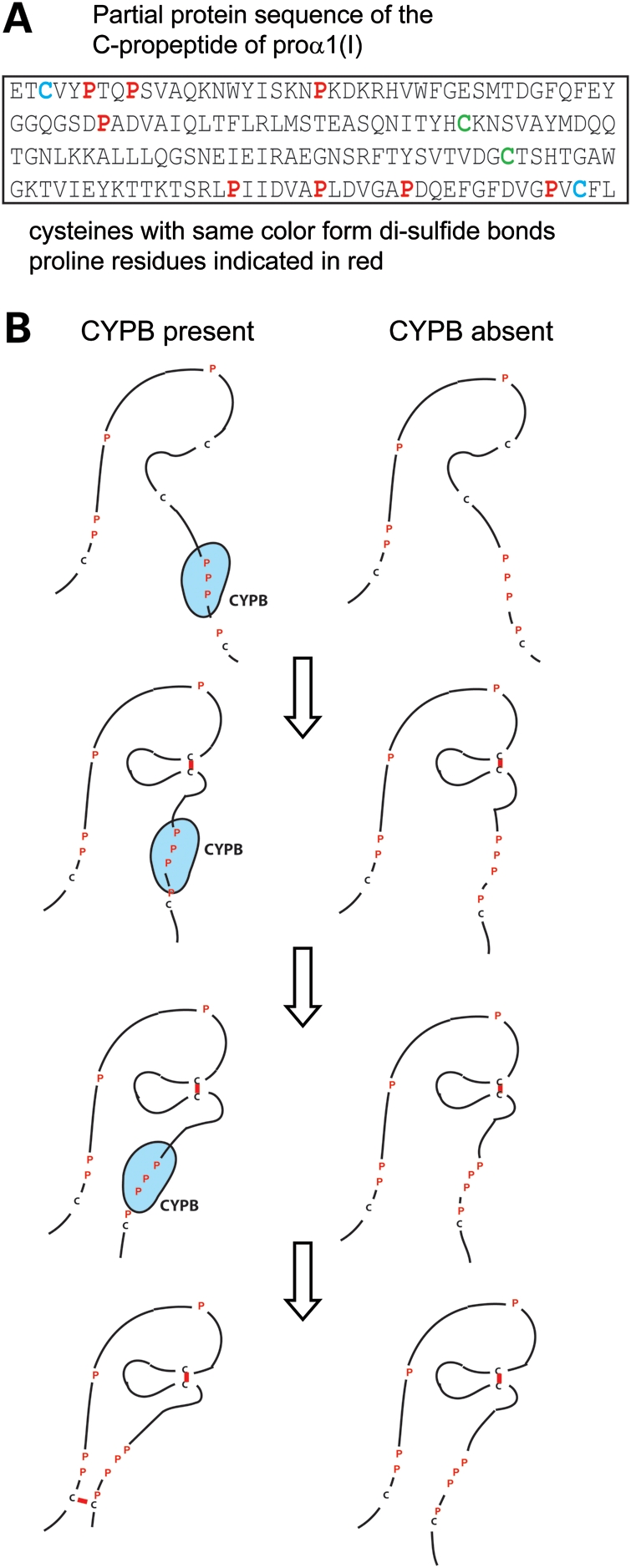
Schematic of proposed model. (A) The sequence of the C-terminal propeptide of proα1(I) chains with interacting cysteine residues and prolyl residues indicated. (B) The rate at which intra-chain disulfide bonds form in the C-terminal propeptide is determined by the folding of the proline-rich region around cysteine residues, a region that is likely a substrate for CYPB.
Neither the genetic loss of CYPB nor its inhibition by CsA abrogate proα chain association but can delay it by several minutes. The cis–trans isomerization of prolyl peptide bonds occurs in the absence of enzymatic assists at a rate estimated to be once per second. The trans configuration is slightly more stable. Both the natural rate of isomerization and the redundancy afforded by the presence of other functional isomerases probably accounts for the observations that overmodification may affect only some type I procollagen molecules in these cells.
This model extends the domains of the chains of type I procollagen in which prolyl cis–trans isomerization may be relevant. Type I procollagen contains a triple-helical region of 1014 amino acids in which glycine is in every third position (Gly-X-Y) and about one-third of the X and Y residues are proline and 4-hydroxyproline, respectively. The ring structure in prolyl residues constrains the adjacent peptide bonds to assume a cis or trans configuration. Following protein synthesis, most prolyl peptide bonds are in the trans conformation, which is also the configuration required in the collagen triple helix (32). Shortly after CYPB was identified, Bächinger et al. (33) showed that the purified enzyme could accelerate refolding of denatured type III collagen peptides. Consistent with this idea, Sarkar et al. (34) found that a small proportion of bonds in type I procollagen was in the cis configuration and so could destabilize individual molecules. Steinmann et al. (22) then showed that inhibition of CYPB with CsA slowed the rate at which protease-resistant triple helix formed in type I procollagen in isolated chick tendon cells and that the stable collagens were overmodified but had normal thermal stability. All groups interpreted these results to indicate that facilitated formation or stabilization of the trans conformation of the prolyl peptide bonds was essential to normal triple helix propagation. Because prolyl-containing peptide bonds in the nascent chains appear to be primarily in the trans configuration, it is not clear that refolding studies are sufficient to determine that cis–trans isomerization alone would be a limiting factor in triple helix formation in cellulo and exclude chain association as a factor.
The key post-translational 4-hydroxylation of the more than 100 Y-position prolyl residues raises the melting temperature of the triple helix from 27°C to 41°C and permits folding in the RER (35). P4H1 appears to have a high preference for substrates in which the target prolyl residues have peptide bonds in the cis conformation (36). In a model proposed by Gorres and Raines (37), the unhydroxylated ring has an endo-pucker that provides the optimal conformation for enzyme activity. Although the traditional view is that CYPB is a cis → trans isomerase, the preferred cis conformation for 4-hydroxylation suggests that in this context CYPB may act as a trans → cis isomerase. Following 4-hydroxylation, the proline ring assumes an exo-pucker that leads to release from the enzyme and, perhaps a non-enzyme dependent, shift to the trans conformation of the peptide bond. The refolding of already hydroxylated chains may benefit from the cis–trans isomerization, whereas the in cellulo function may target trans–cis modification.
In the cells that have mutations in PPIB, not only is proα1(I) chain association delayed, but type I procollagen accumulates in the RER. The retention of type I procollagen is mediated, at least in part, by interaction with PDI and/or its partner, P4H1. PDI catalyzes the formation, reduction and isomerization of disulfide bonds. P4H1 is an enzymatically active moiety responsible for the 4-hydroxylation of prolyl residues in the triple-helical domain of type I procollagen and is stabilized by PDI. Antibodies to both proteins bring down the chains of type I procollagen in cells with mutations in PPIB and CRTAP. Very little is brought down in control cells and the LEPRE1 null cells. Because the amount of type I procollagen brought down by the P4H1 antibody is similar to the amount brought down by PDI in each of the cell types, distinguishing between procollagen molecules that are bound in the triple-helical domain by the P4H1/PDI complex or bound only to PDI at the C-propeptide in molecules that presumably have exposed cysteine residues is difficult and further studies are ongoing. Nonetheless, these results indicate that the processing of proα chains and trimers is affected early in the absence of CYPB and later with mutations in the CRTAP and LEPRE1. Finally, these findings provide a link between disease mechanisms with some mutations in type I procollagen genes and mutations in PPIB, CRTAP and LEPRE1 that may lead to common approaches to therapy.
MATERIALS AND METHODS
Subject selection and clinical features
The three families were identified as part of a systematic search for causative genes for OI. All families met the following criteria: (i) fibroblasts or other mesenchymal cells from affected individuals made overmodified type I collagen, (ii) cells were part of the Repository for Heritable Connective Tissue Disorders at the University of Washington and (iii) a waiver of consent or an appropriate consent for study was on file. In each, no mutations were identified by sequence analysis of both type I collagen genes, CRTAP and LEPRE1. Seven samples were screened for mutations in PPIB, the majority of which was selected because of recurrence in the family or a high likelihood of consanguinity.
Family 1
The parents were from a small town in Mexico but were not aware of being related. During the third pregnancy (Fig. 1, P1), sonographic studies of the fetus were consistent with a severe form of OI or other skeletal dysplasia and cultured fibroblasts taken from the child at birth produced type I collagen molecules with delayed electrophoretic mobility indicating overmodification (Fig. 2A, P1). The infant had a very small chest and died from respiratory failure and pneumonia at 4 months of age (radiographs in Fig. 1, P1). Analysis of type I collagen genes, CRTAP and LEPRE1 identified only normal sequences. A second child (II-4) was similarly affected (radiographs in Fig. 1). Analysis of PPIB identified a causative mutation in both affected siblings. Additional fibroblasts from P1 were not available for further studies.
Family 2
One affected infant (Fig. 1, II-2) born to non-consanguineous parents died shortly after birth. A second pregnancy was identified as affected by analysis of cultured chorionic villi sample cells and electively terminated (P2, II-4). Cultured fetal fibroblasts produced type I collagen molecules with delayed electrophoretic mobility indicating overmodification (Fig. 2A, P2). Analysis of type I collagen genes, CRTAP and LEPRE1 identified only normal sequences. There were three unaffected children born to the parents.
Family 3
The affected child (P3) was adopted from Vietnam, and the initial sample was obtained as part of an analysis of bone marrow-derived MSCs (38). The MSCs produced type I collagen molecules with delayed electrophoretic mobility indicating overmodification (Fig. 2A, P3) but analysis of type I collagen genes, CRTAP and LEPRE1 identified only normal sequences. She had five siblings, two of whom had similar bony alterations but had stayed in Vietnam. Parents were not known to be related and neither parent was affected. Her clinical presentation was consistent with OI type III–IV with broad long bones that were not well modeled and ultimately required intramedullary rodding for stability (radiographs in Fig. 1, P3). She is in her late teens, often uses a wheelchair at school, but is ambulatory at home and is developmentally normal.
Analysis of collagen produced by cultured fibroblasts (P1 and P2) or MSCs (P3)
Cells were cultured and type I collagen was analyzed as previously described by Bonadio et al. (39).
Analysis of candidate genes
Genomic DNA was extracted from cultured dermal fibroblasts of P1 and P2 and from marrow-derived MSCs of P3 using the QIAamp® DNA Mini Kit (Qiagen). The coding sequences and flanking intron regions of COL1A1 and COL1A2 were amplified in 17 and 23 fragments, respectively, and sequenced in multiple reactions (primers and conditions available upon request). The coding sequences of CRTAP and LEPRE1 were amplified and sequenced as described previously (4). The coding sequences and flanking intron regions of PPIB were amplified and sequenced in five fragments. All sequences were analyzed with Mutation Surveyor® software. We used the GenBank reference sequence (NM_000942.4) with the A of the first methionine codon designated +1, which is the same reference sequence used by van Dijk et al. (6) and Tryon et al. (25). Barnes et al. (7) referred to the second methionine codon (c.25_27) as the initiator methionine. Although the sequence around the second methionine codon provides a better Kozak sequence, both may be used as translation start sites (23). Until this point is settled, we have opted to use the GenBank reference nomenclature.
Cell strains with CRTAP and LEPRE1 null mutations
Cultured fibroblasts with homozygous mutations in CRTAP (c.24_31del) and LEPRE1 (c.392C > A, p.Ser131Ter) were used for comparative studies. Both were published in Baldridge et al. (4) (probands 1 and 7).
Western blot analysis of proteins
Cultured fibroblasts or MSCs from affected individuals and controls were plated at confluency (250 K) in 35 mm tissue culture dishes, allowed to attach overnight, incubated for 18 h in the presence of 50 µg/ml ascorbic acid and proteins were harvested from the cell layer by ethanol precipitation. Proteins were resolved by 10 or 15% SDS–PAGE, transferred to nitrocellulose membranes and visualized using the standard western blot techniques with antibodies to CRTAP (1) (polyclonal directed against a full-length recombinant human CRTAP, a gift from Roy Morello), P3H1 (40) (polyclonal directed against a fusion protein representing the carboxyl half of human P3H1, provided by Kevin McCarthy) purified with the Melon™ Gel IgG spin purification kit (Thermo Scientific, 45206), CYPB (polyclonal directed against a region within residues 150 to the C terminus of human CYPB from Abcam, ab16045, polyclonal directed against residues 194–208 of human CYPB from Thermo Scientific, PA1-027A, and a polyclonal directed against a full-length recombinant protein of human CYPB from Proteintech, 11607-1-AP). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Santa Cruz, sc-25778) was used as an internal control.
Immunocytochemical analysis of proteins in cultured fibroblasts
Cultured fibroblasts or MSCs from affected individuals, and controls were plated (75 K) onto sterile coverslips in 12-well tissue culture plates, allowed to attach overnight and then incubated in the presence of 50 µg/ml ascorbic acid for 18 h. Immunofluorescence was performed using a modified protocol from Abcam. Briefly, cells were rinsed with cold 1× phosphate buffered saline (PBS), fixed with 4% paraformaldehyde in 1× Sorenson's for 15 min at room temperature and permeabilized with 1× PBS + 0.25% Triton X-100 at room temperature for 10 min. Following a 1h treatment in block solution at room temperature, primary antibodies to CRTAP (Morello), P3H1 (Abnova H00064175-B01P), CYPB (Abcam ab16045), type I procollagen (LF9) (21), an anti-KDEL RER marker (Abcam ab12223) and Golgi marker (Abcam ab24586, ab6284) were added in sets of two and allowed to hybridize at room temperature. Secondary antibodies conjugated to fluorophores (Invitrogen A11032, A11034 and Jackson Laboratories 112-175-167) were incubated with cells at room temperature; coverslips were mounted in Prolong + 4′,6-diamidino-2-phenylindole, and cells were visualized using a Nikon microphot-SA microscope. Images were captured using a Photometrics sensys monochrome digital camera.
Pulse-chase studies
Pulse-chase studies were performed as described in Christiansen et al. (9).
Thermal stability
Thermal stability of type I collagen molecules was measured as described in Pace et al. (41).
Co-IP of chains of type I procollagen with ER proteins
Fibroblasts from P2, CRTAP null, LEPRE1 null and control cells were plated as described for western blot above. After an 18 h incubation in the presence of 50 µg/ml ascorbic acid, cells were rinsed in cold 1× PBS, lysed in 300 µl of NP-40 lysis buffer, scraped from the dish and placed in a 1.7 ml Eppendorf tube. Cell lysates were gently agitated at 4°C for 30 min, centrifuged for 10 min at 13 4000g and the supernatant moved to a fresh tube. Antibody (anti-PDI, Stressgen SPA-890 or anti-P4H1, Novus Biologicals H00005003-B01) was added to cell lysate at 1:50, and the mixture was gently agitated overnight at 4°C. Fast flow protein A Sepharose beads (70 µl, GE Healthcare) were added, and samples rotated for 4 h at 4°C. Following incubation, samples were spun, washed three times with NP-40 lysis buffer, resuspended in 70 µl of 2× Laemmli sample buffer, heated at 95°C for 5 min and then the protein sample removed from the beads using a 30G needle and placed in a fresh 1.7 ml Eppendorf tube. Disulfide bonds were reduced with dithiothreitol, and the proteins were separated by 5 and 10% SDS–PAGE and transferred to nitrocellulose membranes. For 5% gels, proteins were visualized using a standard western blot detection system. For 10% gels, Clean-Blot™ HRP IP Detection Reagent (Thermo Scientific, 21230) was used at a dilution of 1:2000 in place of the secondary antibody. For the lysate samples, the cells were lysed with 75 µl of lysis buffer, and the supernatant was diluted with 75 µl of 2× sample buffer and 10 µl applied to the gel. The samples for IP were lysed with 300 µl of lysis buffer, and the immunoprecipitates were re-dissolved in 70 µl and 15 µl were applied to the gel. The type I procollagen amounts were quantified using the ImageJ software (http://rsbweb.nih.gov/ij/), and the percentage of type I procollagen bound to PDI was calculated by taking the ratio of bound type I procollagen:total lysate type I procollagen.
Prolyl 3-hydroxylation measurements of type I procollagen
The extent of prolyl 3-hydroxylation in proα1(I) chains was measured as described by Morello et al. (1).
Type I procollagen assembly studies
[35S]-cysteine/methionine labeled
Fibroblasts from P2 and control cells were plated as described above for western blot analysis with one 35 mm plate for each time point (see below). Cells were not available in the appropriate numbers for P1 and P3. After an 18 h incubation in complete medium + 50 µg/ml ascorbic acid, cells were transferred to serum-free modified Eagle's medium without cysteine or methionine (pre-pulse medium) and incubated for 1 h. Cells were then labeled for 10 min with pre-pulse medium + 165 µCi [35S] (Easy tag express protein labeling mix, NEN Life Sciences), rinsed twice with and then chased in pre-pulse medium + 10 mm cysteine + 10 mm methionine for increments of 2.5 min with a final time point at 20 min. Following each chase time, the proteins in the cells were harvested as described in the IP methods above and precipitated using an antibody to the N-terminal propeptide of proα1(I) chain (LF9, Larry Fisher, NIH). Non-reduced proteins were separated by 5% SDS–PAGE, fixed with super-zap (methanol:acetic acid:water), dried at 72°C and visualized by autoradiography. Band density was quantified with the ImageJ software (http://rsbweb.nih.gov/ij/). LF9 brings down trimers, dimers and monomers that contain proα1(I) chains. We measured the density of proα1(I) and trimers, with each measurement adjusted by subtraction of the background intensity for that lane, to assess the proportion of the two and plotted the ratio of free proα1(I):trimers assuming that the free proα1(I) chains had not yet incorporated into higher-order aggregates.
[3H]-proline labeled
Fibroblasts from P2 and control cells were plated as described above for western blot analysis with one 35 mm plate for each time point. Cells were not available in the appropriate numbers for P1 and P3. After an 18 h incubation in complete medium + 50 µg/ml ascorbic acid, cells were transferred to serum-free modified Eagle's medium (pre-pulse medium) and incubated for 1 h. With 15 min remaining during pre-pulse, 5 µm CsA (C1832, Sigma-Aldrich) diluted in ethanol or ethanol alone was added to the appropriate dishes and was maintained throughout the pulse and chase period. Cells were then labeled for 15 min with pre-pulse medium + 250 µCi l-[2,3,4,5-3H] proline (ART0475D, American Radiolabeled Chemicals, Inc.), rinsed twice and then chased with pre-pulse medium + 10 mm proline for increments of 2.5 min with a final time point of 20 min. Following each chase time, the cells were lysed in NP-40 lysis buffer, rotated at 4°C for 30 min and spun at 4°C. The supernatant was transferred to a new tube, 100 µl of 2× sample buffer added and 15 µl of unreduced protein was separated by 5% SDS–PAGE. Gels were processed as described in Christiansen et al. (9). Band density was quantified with the ImageJ software (http://rsbweb.nih.gov/ij/). We measured the density of free proα1(I) chains and trimers, with each measurement adjusted by subtraction of the background intensity for that lane, to assess the proportion of the two and plotted the ratio of free proα1(I):trimers assuming that the free proα1(I) chains had not yet incorporated into higher-order aggregates.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported in part by funds from the National Institutes of Health (AR051582, AR048328), the Osteogenesis Imperfecta Foundation, a student stipend from the HHMI Molecular Medicine Program at the University of Washington and the Freudmann Fund for Translational Research in Ehlers Danlos Syndrome and Related Disorders.
ACKNOWLEDGEMENTS
We thank Roy Morello for providing a CRTAP antibody. We are grateful to the families for their involvement.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Morello R., Bertin T., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F., Vranka J., et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. doi:10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 2.Barnes A., Chang W., Morello R., Cabral W., Weis M., Eyre D., Leikin S., Makareeva E., Kuznetsova N., Uveges T., et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N. Engl. J. Med. 2006;355:2757–2764. doi: 10.1056/NEJMoa063804. doi:10.1056/NEJMoa063804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cabral W., Chang W., Barnes A., Weis M., Scott M., Leikin S., Makareeva E., Kuznetsova N., Rosenbaum K., Tifft C., et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007;39:359–365. doi: 10.1038/ng1968. doi:10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baldridge D., Schwarze U., Morello R., Lennington J., Bertin T., Pace J., Pepin M., Weis M., Eyre D., Walsh J., et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 2008;29:1435–1442. doi: 10.1002/humu.20799. doi:10.1002/humu.20799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willaert A., Malfait F., Symoens S., Gevaert K., Kayserili H., Megarbane A., Mortier G., Leroy J., Coucke P., De Paepe A. Recessive osteogenesis imperfecta caused by LEPRE1 mutations: clinical documentation and identification of the splice form responsible for prolyl 3-hydroxylation. J. Med. Genet. 2009;46:233–241. doi: 10.1136/jmg.2008.062729. doi:10.1136/jmg.2008.062729. [DOI] [PubMed] [Google Scholar]
- 6.van Dijk F., Nesbitt I., Zwikstra E., Nikkels P., Piersma S., Fratantoni S., Jimenez C., Huizer M., Morsman A., Cobben J., et al. PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 2009;85:521–527. doi: 10.1016/j.ajhg.2009.09.001. doi:10.1016/j.ajhg.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnes A., Carter E., Cabral W., Weis M., Chang W., Makareeva E., Leikin S., Rotimi C., Eyre D., Raggio C., et al. Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N. Engl. J. Med. 2010;362:521–528. doi: 10.1056/NEJMoa0907705. doi:10.1056/NEJMoa0907705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alanay Y., Avaygan H., Camacho N., Utine G., Boduroglu K., Aktas D., Alikasifoglu M., Tuncbilek E., Orhan D., Bakar F., et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. doi:10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christiansen H., Schwarze U., Pyott S., AlSwaid A., Al Balwi M., Alrasheed S., Pepin M., Weis M., Eyre D., Byers P. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:389–398. doi: 10.1016/j.ajhg.2010.01.034. doi:10.1016/j.ajhg.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bank R.A., Robins S., Wijmenga C., Breslau-Siderius L.J., Bardoel A.F., van der Sluijs H.A., Pruijs H.E., TeKoppele J.M. Defective collagen crosslinking in bone, but not in ligament or cartilage, in Bruck syndrome: indications for a bone-specific telopeptide lysyl hydroxylase on chromosome 17. Proc. Natl. Acad. Sci. USA. 1999;96:1054–1058. doi: 10.1073/pnas.96.3.1054. doi:10.1073/pnas.96.3.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ha-Vinh R., Alanay Y., Bank R.A., Campos-Xavier A.B., Zankl A., Superti-Furga A., Bonafé L. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am. J. Med. Genet. 2004;131:115–120. doi: 10.1002/ajmg.a.30231. doi:10.1002/ajmg.a.30231. [DOI] [PubMed] [Google Scholar]
- 12.Lapunzina P., Aglan M., Temtamy S., Caparrós-Martín J., Valencia M., Letón R., Martínez-Glez V., Elhossini R., Amr K., Vilaboa N., et al. Identification of a frameshift mutation in osterix in a patient with recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;87:110–114. doi: 10.1016/j.ajhg.2010.05.016. doi:10.1016/j.ajhg.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinmann B., Rao V., Vogel A., Bruckner P., Gitzelmann R., Byers P. Cysteine in the triple-helical domain of one allelic product of the alpha 1(I) gene of type I collagen produces a lethal form of osteogenesis imperfecta. J. Biol. Chem. 1984;259:11129–11138. [PubMed] [Google Scholar]
- 14.Marini J., Forlino A., Cabral W., Barnes A., San Antonio J., Milgrom S., Hyland J., Körkkö J., Prockop D., De Paepe A., et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007;28:209–221. doi: 10.1002/humu.20429. doi:10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bodian D., Chan T., Poon A., Schwarze U., Yang K., Byers P., Kwok P., Klein T. Mutation and polymorphism spectrum in osteogenesis imperfecta type II: implications for genotype-phenotype relationships. Hum. Mol. Genet. 2009;18:463–471. doi: 10.1093/hmg/ddn374. doi:10.1093/hmg/ddn374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bächinger H., Morris N., Davis J. Thermal stability and folding of the collagen triple helix and the effects of mutations in osteogenesis imperfecta on the triple helix of type I collagen. Am. J. Med. Genet. 1993;45:152–162. doi: 10.1002/ajmg.1320450204. doi:10.1002/ajmg.1320450204. [DOI] [PubMed] [Google Scholar]
- 17.Chessler S., Wallis G., Byers P. Mutations in the carboxyl-terminal propeptide of the pro alpha 1(I) chain of type I collagen result in defective chain association and produce lethal osteogenesis imperfecta. J. Biol. Chem. 1993;268:18218–18225. [PubMed] [Google Scholar]
- 18.Vranka J., Sakai L., Bächinger H. Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 2004;279:23615–23621. doi: 10.1074/jbc.M312807200. doi:10.1074/jbc.M312807200. [DOI] [PubMed] [Google Scholar]
- 19.Chang W., Barnes A., Cabral W., Bodurtha J., Marini J. Prolyl 3-hydroxylase 1 and CRTAP are mutually stabilizing in the endoplasmic reticulum collagen prolyl 3-hydroxylation complex. Hum. Mol. Genet. 2010;19:223–234. doi: 10.1093/hmg/ddp481. doi:10.1093/hmg/ddp481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi J., Sutor S., Lindquist L., Evans G., Madden B., Bergen H.R., Hefferan T., Yaszemski M., Bram R. Severe osteogenesis imperfecta in cyclophilin B-deficient mice. PLoS Genet. 2009;5:e1000750. doi: 10.1371/journal.pgen.1000750. doi:10.1371/journal.pgen.1000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher L., Robey P., Tuross N., Otsuka A., Tepen D., Esch F., Shimasaki S., Termine J. The Mr 24,000 phosphoprotein from developing bone is the NH2-terminal propeptide of the alpha 1 chain of type I collagen. J. Biol. Chem. 1987;262:13457–13463. [PubMed] [Google Scholar]
- 22.Steinmann B., Bruckner P., Superti-Furga A. Cyclosporin A slows collagen triple-helix formation in vivo: indirect evidence for a physiologic role of peptidyl-prolyl cis-trans-isomerase. J. Biol. Chem. 1991;266:1299–1303. [PubMed] [Google Scholar]
- 23.Hasel K., Glass J., Godbout M., Sutcliffe J. An endoplasmic reticulum-specific cyclophilin. Mol. Cell. Biol. 1991;11:3484–3491. doi: 10.1128/mcb.11.7.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price E.R., Zydowsky L.D., Jin M.J., Baker C.H., McKeon F.D., Walsh C.T. Human cyclophilin B: a second cyclophilin gene encodes a peptidyl-prolyl isomerase with a signal sequence. Proc. Natl Acad. Sci. USA. 1991;88:1903–1907. doi: 10.1073/pnas.88.5.1903. doi:10.1073/pnas.88.5.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tryon R., White S., Bannasch D. Homozygosity mapping approach identifies a missense mutation in equine cyclophilin B (PPIB) associated with HERDA in the American Quarter Horse. Genomics. 2007;90:93–102. doi: 10.1016/j.ygeno.2007.03.009. doi:10.1016/j.ygeno.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 26.Weis M., Hudson D., Kim L., Scott M., Wu J., Eyre D. Location of 3-hydroxyproline residues in collagen types I, II, III, and V/XI implies a role in fibril supramolecular assembly. J. Biol. Chem. 2010;285:2580–2590. doi: 10.1074/jbc.M109.068726. doi:10.1074/jbc.M109.068726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baldridge D., Lennington J., Weis M., Homan E., Jiang M., Munivez E., Keene D., Hogue W., Pyott S., Byers P., et al. Generalized connective tissue disease in Crtap-/- mouse. PLoS One. 2010;5:e10560. doi: 10.1371/journal.pone.0010560. doi:10.1371/journal.pone.0010560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishikawa Y., Wirz J., Vranka J., Nagata K., Bächinger H. Biochemical characterization of the prolyl 3-hydroxylase 1·cartilage-associated protein·cyclophilin B complex. J. Biol. Chem. 2009;284:17641–17647. doi: 10.1074/jbc.M109.007070. doi:10.1074/jbc.M109.007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kozlov G., Bastos-Aristizabal S., Määttänen P., Rosenauer A., Zheng F., Killikelly A., Trempe J.F., Thomas D.Y., Gehring K. Structural basis of cyclophilin B binding by the calnexin/calreticulin P-domain. J. Biol. Chem. 2010;285:35551–35557. doi: 10.1074/jbc.M110.160101. doi:10.1074/jbc.M110.160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamandé S., Chessler S., Golub S., Byers P., Chan D., Cole W., Sillence D., Bateman J. Endoplasmic reticulum-mediated quality control of type I collagen production by cells from osteogenesis imperfecta patients with mutations in the pro alpha 1 (I) chain carboxyl-terminal propeptide which impair subunit assembly. J. Biol. Chem. 1995;270:8642–8649. doi: 10.1074/jbc.270.15.8642. doi:10.1074/jbc.270.15.8642. [DOI] [PubMed] [Google Scholar]
- 31.Koivu J. Identification of disulfide bonds in carboxy-terminal propeptides of human type I procollagen. FEBS Lett. 1987;212:229–232. doi: 10.1016/0014-5793(87)81350-9. doi:10.1016/0014-5793(87)81350-9. [DOI] [PubMed] [Google Scholar]
- 32.Shoulders M.D., Hodges J.A., Raines R.T. Reciprocity of steric and stereoelectronic effects in the collagen triple helix. J. Am. Chem. Soc. 2006;128:8112–8113. doi: 10.1021/ja061793d. doi:10.1021/ja061793d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bächinger H., Bruckner P., Timpl R., Engel J. The role of cis-trans isomerization of peptide bonds in the coil leads to and comes from triple helix conversion of collagen. Eur. J. Biochem. 1978;90:605–613. doi: 10.1111/j.1432-1033.1978.tb12641.x. doi:10.1111/j.1432-1033.1978.tb12641.x. [DOI] [PubMed] [Google Scholar]
- 34.Sarkar S., Young P., Sullivan C., Torchia D. Detection of cis and trans X-Pro peptide bonds in proteins by 13C NMR: application to collagen. Proc. Natl Acad. Sci. USA. 1984;81:4800–4803. doi: 10.1073/pnas.81.15.4800. doi:10.1073/pnas.81.15.4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berg R., Prockop D. The thermal transition of a non-hydroxylated form of collagen. Evidence for a role for hydroxyproline in stabilizing the triple-helix of collagen. Biochem. Biophys. Res. Commun. 1973;52:115–120. doi: 10.1016/0006-291x(73)90961-3. doi:10.1016/0006-291X(73)90961-3. [DOI] [PubMed] [Google Scholar]
- 36.Gorres K.L., Edupuganti R., Krow G.R., Raines R.T. Conformational preferences of substrates for human prolyl 4-hydroxylase. Biochemistry. 2008;47:9447–9455. doi: 10.1021/bi8009373. doi:10.1021/bi8009373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gorres K., Raines R. Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 2010;45:106–124. doi: 10.3109/10409231003627991. doi:10.3109/10409231003627991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chamberlain J., Schwarze U., Wang P., Hirata R., Hankenson K., Pace J., Underwood R., Song K., Sussman M., Byers P., et al. Gene targeting in stem cells from individuals with osteogenesis imperfecta. Science. 2004;303:1198–1201. doi: 10.1126/science.1088757. doi:10.1126/science.1088757. [DOI] [PubMed] [Google Scholar]
- 39.Bonadio J., Holbrook K., Gelinas R., Jacob J., Byers P. Altered triple helical structure of type I procollagen in lethal perinatal osteogenesis imperfecta. J. Biol. Chem. 1985;260:1734–1742. [PubMed] [Google Scholar]
- 40.Wassenhove-McCarthy D., McCarthy K. Molecular characterization of a novel basement membrane-associated proteoglycan, leprecan. J. Biol. Chem. 1999;274:25004–25017. doi: 10.1074/jbc.274.35.25004. doi:10.1074/jbc.274.35.25004. [DOI] [PubMed] [Google Scholar]
- 41.Pace J., Kuslich C., Willing M., Byers P. Disruption of one intra-chain disulphide bond in the carboxyl-terminal propeptide of the proalpha1(I) chain of type I procollagen permits slow assembly and secretion of overmodified, but stable procollagen trimers and results in mild osteogenesis imperfecta. J. Med. Genet. 2001;38:443–449. doi: 10.1136/jmg.38.7.443. doi:10.1136/jmg.38.7.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.