

Abstract
The metabolic intermediate and endocannabinoid signaling lipid 2-arachidonoylglycerol (2-AG) has not been readily labeled, primarily due to its instability towards rearrangement. We now detail a synthetic method that easily gives tritiated 2-AG from [5,6,8,9,11,12,14,15-3H(N)]arachidonic acid in two steps. We utilized a short chain 1,3-diacylglycerol and proceeded through the “structured lipid” [5″,6″,8″,9″,11″,12″,14″,15″-3H(N)]2-arachidonoyl-1,3-dibutyrylglycerol, a triacylglycerol that was conveniently deprotected in ethanol with acrylic beads containing Candida antarctica lipase B to give [5″,6″,8″,9″,11″,12″,14″,15″-3H(N)]2-arachidonoylglycerol ([3H]2-AG). The flash chromatographic separation necessary to isolate the labeled 2-acylglycerol [3H]2-AG resulted in only 4% of the rearrangement byproducts that have been a particular problem with previous methodologies. This reliable “kit” method to prepare the radiolabeled endocannabinoid as needed gave tritiated 2-arachidonoylglycerol [3H]2-AG with a specific activity of 200 Ci/mmol for enzyme assays, metabolic studies, and tissue imaging. It has been run on unlabeled materials on over 10 mg scales and should be generally applicable to other 2-acylglycerols.
Keywords: 2-arachidonoylglycerol, 2-AG, endocannabinoid, 3H-labeled, triacylglycerol, monoacylglycerol, Candida antarctica lipase B, Novozyme 435, structured lipid
Introduction
2-Acylglycerols are important metabolic intermediates and signaling molecules in plants1,2 and animals.3,4 The endocannabinoid 2-arachidonoylglycerol (1, 2-AG)5,6 has been shown to have important roles in the regulation of a number of mammalian physiological and behavioral effects.7,8 Radiolabeled 2-arachidonoylglycerol (2-AG)9–12 can be a powerful tool with which to probe the endocannabinoid system and arachidonic acid metabolism. However, research relying upon radiolabeled 2-AG has been greatly hindered by 2-AG purification difficulties and the notorious instability of 2-acylglycerols to rearrangement.13 We had already produced analytical quantities of [1″-14C]2-AG enzymatically from 2-[1″-14C]arachidonoyl-1-stearoyl-sn-glycerol14 that was used as the substrate for evaluating human diacylglycerol lipase subtype alpha (hDAGL-α) expression and the inhibitor molecules that we are currently synthesizing. We have since developed a novel protocol that appears to have an even greater potential for application to the preparation of radiolabeled 2-AG due to the relative ease of the two step procedure from arachidonic acid. We now report this simple and convenient methodology for a “kit” to prepare pure radiolabeled 2-AG on demand from a stable precursor without the necessity of reversed-phase high-performance liquid chromatographic (HPLC) purification.
Chemical properties of 2-arachidonoylglycerol (1, 2-AG)
2-Acylglycerols undergo acyl migration,15–17 but have been reported to be stable to rearrangement in hexane18 or toluene/ethanol19 solutions. 2-Arachidonoylglycerol (1, 2-AG, Scheme 1) is stable to rearrangement as a purified frozen liquid below −20 °C, but not in methanol-water, ethanol, or dichloromethane solutions.20 At elevated temperature, in the presence of base, strong mineral acid, or even chloroform solution that can become acidic on standing, rearrangement of 2-AG (1) to a racemic mixture of 1-AG (2) and 3-AG (3) occurs within a day at ambient temperature as an equilibrium of predominantly 1(3)-AG (90%) is reached with 2-AG (10%). Also, the rigorous exclusion of oxygen (argon degassed solvents and atmospheres) is extremely important to prevent the air oxidation of the unsaturated lipid to form oxygenated byproducts21,22 that are observed as lower Rf streaking substances on analytical TLC.
SCHEME 1.

The endocannabinoid 2-AG 1 undergoes rearrangement on standing to 1-AG 2 and 3-AG 3. Treatment with boric acid converts 2-AG 1 to the corresponding six-membered ring borate ester 4 that is chromatographically separable from the five-membered ring borate esters 5 and 6.
The long term storage of radiolabeled 2-AG as a pure substance, even at −30 °C, is likely to result in significant decomposition by autoradiolysis. Storage as a solution in organic solvents would reduce autoradiolysis, but can result in rearrangement to 1(3)-AG within a few hours to a few days, even at low temperature. Thus, our research objective was to develop a stable precursor of radiolabeled 2-AG for immediate point-of-use preparation.
Assessment of the purity of 2-arachidonoylglycerol (1, 2-AG)
The analysis of samples of 2-acylglycerols must not catalyze or perturb the interconversion of 2-acyl and 1(3)-acyl isomers under the analytical conditions. For the analysis of the arachidonate monoacylglycerols, separation of underivatized 2-AG 1 and 1(3)-AG (2 + 3) can be accomplished by reversed-phase high-performance liquid chromatography (HPLC).10,20,23–26 The separation of underivatized 2-AG 1 and 1(3)-AG (2 + 3) by thin layer chromatography (TLC) is not possible on untreated silica gel with any solvent combination. However, underivatized 2-AG 1 and 1(3)-AG (2 + 3) can be administered to boric acid treated silica gel plates and quickly eluted with little opportunity to undergo rearrangements since they are quickly dehydrated to the corresponding six-membered and five-membered boric acid esters 4 and 5 + 6, respectively, that are readily separable. There are reported TLC conditions27,28 utilizing combinations of chloroform/acetone/methanol (sometimes including acetic acid) that we have used for the separation of the less polar 2-acylglycerol borate derivative 4 from the more polar 1(3)-acylglycerol borate derivatives (5 + 6) with no rearrangement interconversion as evidenced by the absence of any off-diagonal spots on two-dimensional TLC plates after visualization with molybdic acid reagent. A possible source of confusion in the previous literature11,29 is the fact that 2-AG 1 and the 2-AG borate ester 4 are not resolvable from each other on either untreated silica gel or boric acid treated silica gel by any commonly used solvent system, nor are 1(3)-AG isomers 2 + 3 and the corresponding borate esters 5 + 6 separable.
Previous syntheses of 2-arachidonoylglycerol (1, 2-AG) and radiolabeled 2-AG
There are several reported preparations of 2-AG 16,12,19,20,23,30–32 and radiolabeled 2-AG.9–12,14 The methodologies that utilize either 1,3-diacylglycerol33–38 or borate ester20,39 synthetic intermediates are superior to the related methods that proceed through the hydrolysis of 1,3-benzylidene40–43 or 1,3-dibenzyloxy12 analogs. Our novel method also required a 1,3-diacylglycerol that can be prepared from the corresponding 2-benzyl compound44,45 or from 1,3-dihydroxyacetone by diacylation followed by ketone reduction with sodium borohydride33,38,46 or diborane.34 1,3-Diacylglycerols have also been prepared directly by enzyme-catalyzed diesterification of glycerol with Rhizomucor miehei lipase,47,48 Mucor mihei lipase,49 Candida antarctica lipase,50–52 and others.53,54
The coupling of arachidonic acid with 1,3-diacylglycerols to give 2-arachidonoyl-1,3- diacylglycerols can utilize carbodiimide23,38,43,48,51,52 or other coupling methods.12,20,34,36 Like the corresponding 2-arachidonoyl-1,3-bistrifluoroacetate analog,31 such triacylglycerols are easily isolated55 by column chromatography or preparative HPLC and represent stable 2-AG precursors.
Our efforts have been directed at the rapid enzymatic hydrolysis of “structured lipids”35,42,56,57 having a 2-arachidonoyl group. Although other enzymes have also been used for the hydrolysis of triacylglycerols to give 2-acylglycerols,6,19,58 enzyme selection has focused on polymer-bound lipases with activities for short chain sn-1- and sn-3-acyl groups that can utilize ethanol for transesterification to release the protecting groups as volatile byproducts. Polymer-bound lipases can have very high activity in spite of the resin volume, are readily removed from reaction mixtures by micropore filtration, and can be recovered for reuse. A number of these immobilized lipases, including immobilized Rhizomucor miehei lipase47,48 and immobilized Candida antarctica lipase,32,59,60 can be used for a final deprotection step that that avoids more extensive workup and purifications, reduces the opportunity for rearrangement byproduct formation, and is both convenient and cost effective.
Preparative chromatographic purification of 2-acylglycerols
A key aspect of any 2-acylglycerol synthesis is preparative isolation without rearrangement. 2-AG 1 is not separable from 1(3)-AG (2 + 3) by column chromatography on untreated silica gel. Large scale chromatographic purification of 2-AG 1 from reaction byproducts will likely lead to a higher percentage of rearrangement 1(3)-AG byproducts (2 + 3). Using boric acid treated silica gel, the column chromatography of 2-acylglycerols still leads to some extent of rearrangement to 1(3)-acylglycerol.32 Reports of 2-AG 1 being purified from boric acid-treated TLC preparative separations11,30 must be due to fortuitous methanolysis or hydrolysis of the corresponding borate ester 4 that forms during chromatography by the washing of the scraped silica gel adsorbant.
Results and Discussion
Preliminary investigations of unlabeled 2-acylglycerol synthesis by this novel method
Preliminary studies were conducted with unlabeled compounds in argon-degassed solvents to develop procedures that could later be utilized in the synthesis of radiolabeled 2-AG. We utilized immobilized Candida antarctica lipase to prepare the required 1,3-diacetylglycerol52,61 and 1,3-dibutyrylglycerol (9)52 analogs (see Scheme 2). For this glycerol diacylation step, the presence of vinyl acetate or vinyl butyrate, respectively, maintained the lipase enzyme in an acylated form in a low-moisture environment, such that the lipase did not have sufficient opportunity to bind and hydrolyze the 1,3-diacylated glycerol product upon formation, which would typically be considered a forward reaction for this enzyme. The lipase function is profoundly altered in organic solvents due to changes in the extent of surface hydration.62,63
SCHEME 2.

Glycerol (7) is converted to 1,3-dibutyrylglycerol (9) that is then coupled to arachidonic acid. The triglyceride BAB 10 is deprotected to give 2-AG 1.
Acylation of the sn-2 position of 1,3-diacylglycerols with arachidonic acid was performed with coupling agent N,N-dimethylaminopropyl-N′-ethylcarbodiimide (EDCI) in the presence of a basic catalyst N,N-dimethylaminopyridine (DMAP). The 2-arachidonoyl-1,3-dibutyrylglycerol (10, BAB) was readily obtained free of the corresponding 1,2(2,3)-dibutyrate isomer. However, the corresponding 2- arachidonoyl-1,3-diacetate was not readily purified free of a small amount of what was presumably the 1,2(2,3)-diacetate isomer that could be seen as small multiplets between δ 4.25–4.27 and as a small triplet at δ 2.34 that was upfield from the α-methylene of the arachidonoyl carbonyl for the 2-arachidonoyl-1,3-diacetylglycerol at δ 2.36.
In the enzymatic ethanolysis step to give the 2-acylglycerol product 2-AG 1, the volatile byproducts were ethyl acetate and ethyl butyrate for the 1,3-diacetate and 1,3-dibutyrate triglycerides, respectively. The enzymatic transesterification of the 2-arachidonoyl-1,3-diacetate was unsatisfactory. The sn-1-acetyl group was readily removed by immobilized Candida antarctica lipase B catalyzed transesterification, but the resulting diglyceride only very slowly underwent further transesterification to give 2-AG 1, thus, the diacetate substrate was not investigated further. However, like the long chain polyunsaturated triglyceride analogs previously studied,32,59,60 2-arachidonoyl-1,3-dibutyrylglycerol (10, BAB) underwent stepwise transesterification of the sn-1 n-butyl ester to rapidly give an intermediate diglyceride-containing mixture that was optically active. When checked at 30 min, the reaction mixture had a highly positive rotation that was not due to the resin-bound lipase itself, as there was no rotation in the control experiment (no triglyceride added). The slower transesterification of the remaining sn-3-butyryl ester of 2-arachidonoyl-3-butyryl-sn-glycerol (11) resulted in the formation of some ethyl arachidonate 12 byproduct from non-specific ethanolysis and the reaction was followed by TLC using 20:80 ethyl acetate/hexane (Rf of ethyl arachidonate 12 = 0.58, Rf of starting triglyceride 10 = 0.42, Rf of diglyceride 11 = 0.10, Rf of 2-AG 1 = 0.01). Ethyl arachidonate 12 could be isolated due to the volatility of the ethyl butyrate byproduct, whereas the separation from ethyl ester byproducts of medium or long chain fatty acids would have been much more difficult. Also, the “structured lipid” BAB 10 from our method allowed for the more efficient use of the costly arachidonic acid than if the 1,2,3-triarachidonoylglycerol32,60 substrate had been used. The immobilized Candida antarctica lipase B was very active and more convenient than the lyophilized powder form of the lipase. The immobilized lipase-catalyzed transesterification of 2-arachidonoyl-1,3-dibutyrylglycerol (10, BAB) was studied in mixtures of ethanol with acetone, diisopropyl ether, methanol, and butanol, but these did not improve the production of 2-arachidonoylglycerol (1, 2-AG) over ester 12 byproduct. Using more of the immobilized lipase (up to four times the mass of triglyceride substrate) or inverse addition (adding triglyceride substrate to immobilized lipase in dry ethanol) only slightly improved the product profile after two hours. Longer reactions, beyond the point where the concentration of intermediate diglyceride 11 has become low, saw the 2-AG 1 undergoing cleavage to ethyl arachidonate (12) and glycerol (7).
Product isolation conditions were developed using our well-established TLC methods to demonstrate the absence or presence of any reaction byproducts including intermediate 2-arachidonoyl-3-butyryl-sn-glycerol (11), ethyl arachidonate (12), or rearrangement byproduct 1(3)-AG (2 + 3). We have carefully investigated column chromatography with boric acid treated silica gel. We first performed flash column chromatography with boric acid treated silica gel prepared according to the reported procedures using aqueous boric acid with heat activation.64,65 We have also utilized methanolic boric acid treatments of silica gel with activation by heating as well as activation by solvent washing according to a reported procedure.66 Proton and carbon NMR analysis of 2-AG 1 chromatographed down any columns containing boric acid by either activation method always detected borate ester 4 impurity, and the more highly activated the adsorbant, the closer to complete conversion to borate ester 4. Since it is possible that borate impurities have biological activities considering the activities of boronic acids,67,68 the use of boric acid in column chromatographic purifications should be avoided. Thus, the use of boric acid treated silica gel is limited to analytical TLC applications of product mixture analysis and not preparative purifications.
Analytical TLC and column purification using untreated silica gel with acetone in either dichloromethane or chloroform did not separate the product 2-AG 1 from a UV-containing higher Rf impurity that comes from solvent washing of the resin. The resin could be washed with ethanol, air-dried overnight, then washed with water, and air-dried overnight to reactivate the resin. This treated resin had considerably less of the UV-containing impurity, although the activity of the enzyme was also reduced. Analytical TLC on boric acid treated silica gel plates eluting with 92.5:25:0.5:2 CHCl3/MeCOMe/HOAc/MeOH also showed the UV-containing impurity eluting (Rf 0.47) just ahead of the 2-AG (1, Rf 0.44) and any 1(3)-AG (2 + 3, Rf 0.33). Also, analytical TLC with 60:40 ethyl acetate/hexane on untreated silica gel separated the UV-containing impurity (Rf 0.48) from the monoacylglycerols (Rf 0.28). Column chromatography with a gradient from 20% to 60% ethyl acetate in hexane eluted the impurity just ahead of the 2-AG 1. Under flash conditions and on scales of sub-milligram to over 10 mg using 0.5 gram silica gel columns, the rearrangement of 2-AG to 1(3)-AG was always well under 5% as determined by analytical TLC using boric acid treated silica gel plates. Spotting and immediately eluting 2-AG 1 with 72.5:25:0.5:2 CHCl3/MeCOMe/HOAc/MeOH is recommended.
Synthesis of radiolabeled endocannabinoid 2-arachidonoylglycerol ([3H]1, [3H]2-AG)
Our efficient methodology to prepare [3H]2-AG [3H]1 first involved preparation of the radiolabeled stable precursor of this radiolabeled endocannabinoid. The yield of [3H]BAB [3H]10 from the reaction of 2 mCi of [5,6,8,9,11,12,14,15-3H(N)]arachidonic acid was significantly lower than cold or tracer experiments that generally went in 70% yield, perhaps due to autoradiolytic degradation of the tritiated arachidonic acid during the initial ethanol evaporation and chases or due to some anhydride formation during the reaction. As a result, the overall radiochemical yield of [3H]2-AG [3H]1 for the two step method was reduced.
Radiolabeled endocannabinoid 2-arachidonoylglycerol ([3H]1, [3H]2-AG) is then made on demand by the easily performed second step of the radiosynthesis (see Scheme 3). The stable precursor BAB [3H]10 was then treated with the immobilized lipase in ethanol, followed by a simple cartridge filtration to give [3H]2-AG [3H]1 that was nearly free of rearrangement 1(3)-AG byproduct(s) (see Figure 1) in about three hours. This method was used to prepare [3H]2-AG [3H]1 with a specific activity of 200 Ci/mmol in two steps from tritiated arachidonic acid in 13% overall radiochemical yield and 96% radiochemical purity (containing 4% of rearrangement byproduct [3H]1(3)-AG).
SCHEME 3.

The radiolabeled “structured lipid” [3H]10 undergoes transesterification in EtOH with Candida antarctica lipase B to first give diglyceride [3H]11 that undergoes further transesterification to give [3H]2-AG [3H]1 as well as some byproduct ethyl ester [3H]12.
FIGURE 1.
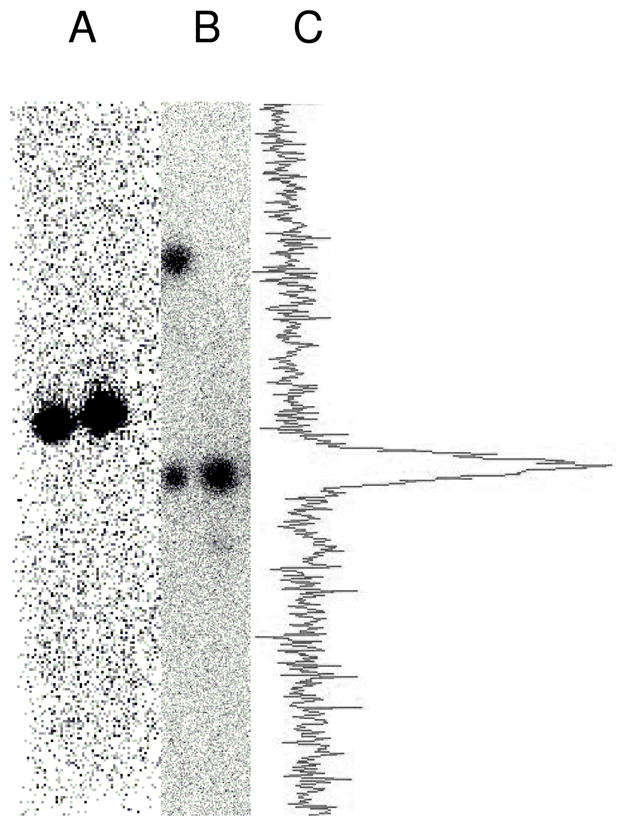
A. [3H]BAB [3H]10 was radiochemically pure by TLC on untreated silica gel (20:80 EtOAc/hex, Rf = 0.51), Left Lane (20K DPM spotted) no carrier, Right Lane (20K DPM spotted) with 0.1 mg unlabeled carrier. The eluted, dried TLC plate was exposed to the PTR screen for 12 h. B. (Left Lane) The lipase-catalyzed deprotection of [3H]10 after 2 h 20 min (30K DPM spotted) showed 50% ester byproduct [3H]12 (Rf = 0.74) and 50% product radiolabeled 2-AG [3H]1 (Rf = 0.41) containing no rearrangement byproduct. (Right Lane) Flash chromatography gave product (27K DPM spotted) that contained 96% radiolabeled 2-AG [3H]1 (Rf = 0.41) and 4% rearrangement byproduct 1(3)-AG ([3H]2 + [3H]3) (Rf = 0.30). TLC was on boric acid treated silica gel (92.5:25:0.5:2 CHCl3/MeCOMe/HOAc/MeOH ) and the eluted, dried TLC plate was exposed to the PTR screen for 3 days. C. Analysis of the contour data from Plate B Right Lane to illustrate the high purity of the product radiolabeled 2-AG [3H]1.
Conclusions
These reactions and purifications were much easier to perform than previously existing methods for the preparation of 2-acylglycerols. Although not likely to be as useful on scales above 100 mg of the precursor 2-arachidonoyl-1,3-dibutyrylglycerol (10, BAB), we have found this method to be very valuable on scales from 10 mg to the sub-microgram scale reactions of radiolabeled analogs. The lipase used did not degrade the tritiated 2-AG [3H]1 product to any greater extent on the sub-microgram scale, even though the conditions represented a much higher ratio of enzyme to radiolabeled substrate than in the preliminary experiments on 10 mg scales of triglyceride substrate. Since the tritiated ethyl arachidonate [3H]13 was chromatographically separable from all other reaction components, it was isolated as a byproduct that will be used for pharmacological studies in the future. We are also currently evaluating the stability of radiolabeled 2-AG [3H]1 in toluene, DMSO, and as a component of hydrated lipid model membranes. The radiolabeled 2-AG is currently being used in radiochemical enzyme assays and for ex-vivo tissue imaging in the extension of our work with [3H]anandamide.69,70 The efficient use of the commercial radiolabeled fatty acid and the relatively few manipulations that are necessary make this an attractive route for also preparing other radiolabeled 2-acylglycerols, including 14C-labeled analogs, essentially free of 1(3)-acylglycerol rearrangement byproducts without utilizing extensive normal-phase chromatography that catalyzes rearrangement or using reversed-phase HPLC that would give aqueous solutions of 2-acylglycerols.
Preparative synthetic approaches to radiolabeled 2-AG 1 must be evaluated on the criteria of regiospecific synthesis of the 2-acylglycerol requiring little purification manipulation. Our novel method represents a “kit” to readily produce the radiolabeled endocannabinoid as required from a stable precursor. We have found this to be the best method for the simple in-house on-demand preparation of pure radiolabeled 2-AG that can then be utilized prior to its subsequent isomerization.
Experimental Section
General Procedures
All solvents were degassed by sparging with argon gas and all reactions were run under argon. Acrylic beads containing active lipase B from Candida antarctica (recombinant, expressed in Aspergillus niger, Sigma L4777, Novozyme® 435, ≥10,000 U/g) were used to prepare 1,3-diacylglycerols and to deprotect 1,3-diacyl-2-arachidonoylglycerols. All solvent ratios are by volume. All column chromatography used silica gel 60 except for the preparation of borate ester 4. The boric acid treated TLC plates were prepared by dipping the silica gel plates (glass-backed silica gel 60, 250 μm, F254) in 9–10% boric acid in methanol. The plates were air dried for an hour and activated at 60 °C for 5 min. After elutions, dried TLC plates were visualized by dipping them in undiluted molybdic acid reagent (Sigma), drying briefly in the air, and then evenly heating using either a heatgun or a hotplate. The radiochemical purity was determined from the TLC plates using Perkin Elmer PTR-type screens with the Perkin Elmer Cyclone phosphoimaging system for quantitative analysis (OptiQuant software version 5.0) after background subtraction.71,72 Using 50,000 DPM for PTR plates required overnight exposure, while using above 300,000 DPM allowed images to be obtained in an hour. Using higher amounts of radioactive material on the TLC plates, and especially having longer times between spotting and elution, gave more origin and streaking artifacts of autoradiolysis for these arachidonic compounds. The 400, 500, or 700 MHz 1H NMR spectra were obtained with internal TMS.
1,3-Dibutyrylglycerol (9)52
To a magnetically stirred solution of glycerol (7) (1.13 g, 12.3 mmol, 100 mol%) in 5 mL anhydrous CH2Cl2 was added vinyl butyrate (8) (3.50 g, 30.7 mmol, 250 mol%) and the mixture was cooled to 0 °C. The immobilized Candida antarctica lipase B (0.463 g) was added and the mixture was stirred for 5 h. The resin was removed from the reaction mixture by filtration through a micropore (Whatman 0.2 μm PVDF 13 mm) filter then the resin and filter cartridge were washed with additional four portions of CH2Cl2. The filtrates were concentrated under vacuum and chromatographed (silica gel, 30:70 EtOAc/hex) to give 1.52 g (6.54 mmol, 53%) of 1,3-dibutyrylglycerol (9) as a clear and colorless liquid that was homogeneous by TLC (30:70 EtOAc/hex, Rf = 0.42): 1H NMR (CDCl3) δ 0.96 (t, J = 7.3 Hz, 6 H, 4′, 4‴), 1.67 (sextuplet, J = 7.4 Hz, 4 H, 3′, 3‴), 2.34 (t, J = 7.3 Hz, 4 H, 2′, 2‴), 4.10 (m,1 H, sn-2), 4.15 (dd, J = 11.7, 5.9 Hz, 2 H) and 4.17 (dd, J = 11.7, 4.4 Hz, 2 H) diastereotopic protons of sn-1 and sn-3; MS (EI) m/z 215.1 ([M − H2O + H]+); HRMS (EI) calcd for C11H19O4 215.12834 ([M − H2O + H]+), found 215.12852.
2-Arachidonoyl-1,3-dibutyrylglycerol (10, BAB)
To a magnetically stirred solution of 1,3-dibutyrylglycerol (9) (340 mg, 1.46 mmol, 100 mol%) in 5 mL of anhydrous CH2Cl2 was added arachidonic acid (489 mg, 1.61 mmol, 110 mol%) and the mixture was cooled to 0 °C. An excess of coupling agent N-dimethylaminopropyl-N′-ethylcarbodiimide (EDCI, 699 mg, 3.65 mmol, 250 mol%) was added followed by the basic catalyst N,N-dimethylaminopyridine (DMAP, 358 mg, 2.93 mmol, 200 mol%) and the reaction mixture was allowed to stir at room temperature for 12 h. The reaction mixture was washed with 0.01 M HCl solution, dried over Na2SO4, concentrated, and chromatographed (silica gel, 20:80 EtOAc/hex) to give 550 mg (1.06 mmol, 72%) of BAB 10 as a clear and colorless liquid that was homogeneous by TLC (20:80 EtOAc/hex, Rf = 0.43): 1H NMR (CDCl3) δ 0.89 (t, J = 6.8 Hz, 3 H, 20″), 0.95 (t, J = 7.3 Hz, 6 H, 4′, 4‴), 1.23 – 1.41 (m, 6 H, 17″, 18″, 19″), 1.53 – 1.79 (m, 6 H, 3′, 3″, 3‴), 2.06 (apparent q, J = 6.8 Hz, 2 H, 16″), 2.12 (apparent q, J = 7.3 Hz, 2 H, 4″), 2.27 – 2.37 (m, 6 H, 2′, 2″, 2‴), 2.75 – 2.90 (m, 6 H, 7″, 10″, 13″), 4.16 (dd, J = 11.7, 5.9 Hz, 2 H) and 4.31 (dd, J = 11.7, 4.4 Hz, 2 H) diastereotopic sn-1and sn-3 protons, 5.27 (quintet, J = 5.0 Hz, 1 H, sn-2), 5.32 – 5.46 (m, 8 H, 5″, 6″, 8″, 9″, 11″, 12″, 14″, 15″); 13C NMR (CDCl3) δ 173.10 (1′, 1‴), 172.65 (1″), 130.51, 128.99, 128.78, 128.62, 128.30, 128.10, 127.84, and 127.54 (olefinics), 69.00 (sn-2), 62.06 (sn-1, sn-3), 35.91 (2′, 2‴), 33.59 (2″), 31.52 (18″), 29.33 (17″), 27.22 (16″), 26.49 (4″), 25.63 (7″,10″,13″), 24.75 (3″), 22.58 (19″), 18.34 (3′, 3‴), 14.07 (20″), 13.62 (4′, 4‴); MS (EI) m/z 518.4 (M+); HRMS (EI) calcd for C31H50O6 518.36074 (M+), found 518.36200. Anal. Calcd for C31H50O6 (518.72): C, 71.78; H, 9.72. Found: C, 71.52; H, 9.48.
Unlabeled 2-Arachidonoylglycerol (1, 2-AG)12,20,23,30,31
To a solution of 2-arachidonoyl-1,3-dibutyrylglycerol (10, BAB) (12.1 mg, 2.33×10−5 mol) in 100 μL of anhydrous EtOH was added 24 mg of immobilized Candida antarctica lipase B. An additional 24 mg of resin was added after 20 min. After an additional 2 h, the resin was removed by filtration through a micropore (Whatman 0.2 μm PVDF 13 mm) filter, then the resin and filter cartridge were washed with an additional four portions of EtOH. The solvent was evaporated in a stream of argon and the residue (9.49 mg) that contained no 1(3)-AG was immediately flash chromatographed. The column consisted of 0.5 g of silica gel in a 3 mL syringe supported on a bed of glass wool and sand having a micropore filter cartridge affixed to the disposable syringe. The column was slurry-packed with 25:75 MeCOMe/CHCl3, washed with 10 mL of 20:80 EtOAc/hex, and then the reaction mixture was loaded. The column was eluted with 7 mL of 20:80 EtOAc/hex to recover the high Rf ethyl arachidonate (12) byproduct as a clear and colorless liquid: 1H NMR (CDCl3) δ 0.90 (t, J = 6.8 Hz, 3 H, 20″), 1.26 (t, J = 7.0 Hz, 3 H, 2), 1.28 – 1.42 (m, 6 H, 17″, 18″, 19″), 1.71 (quintet, J = 7.4 Hz, 2 H, 3″), 2.06 (apparent q, J = 7.2 Hz, 2 H, 16″), 2.12 (apparent q, J = 6.8 Hz, 2 H, 4″), 2.31 (t, J = 7.6 Hz, 2 H, 2″), 2.75 – 2.90 (m, 6 H, 7″, 10″, 13″), 4.13 (q, J = 7.0 Hz, 2 H, 1), 5.26 – 5.51 (m, 8 H, 5″, 6″, 8″, 9″, 11″, 12″, 14″, 15″). Then the product 2-AG 1 was eluted from the column with 1 mL of 40:60 EtOAc/hex followed by 10 mL of 60:40 EtOAc/hex. Fractions after 1.5 mL through 6 mL of elution contained 2-AG 1 that had less than 5% of 1(3)-AG (2 + 3) by analytical TLC on boric acid treated silica gel (92.5:25:0.5:2 CHCl3/MeCOMe/HOAc/MeOH, 2-AG Rf = 0.44, 1(3)-AG Rf = 0.33) and was a clear colorless liquid (4.80 mg, 1.27×10−5 mol, 54%): 1H NMR (CDCl3) δ 0.90 (t, J = 6.8 Hz, 3 H, 20″), 1.21 – 1.45 (m, 6 H, 17″, 18″, 19″), 1.74 (quintet, J = 7.3 Hz, 2 H, 3″), 2.06 (apparent q, J = 7.3 Hz, 2 H, 16″), 2.14 (apparent q, J = 7.1 Hz, 2 H, 4″), 2.40 (t, J = 7.6 Hz, 2 H, 2″), 2.74 – 2.90 (m, 6 H, 7″, 10″, 13″), 3.84 (br d, J = 5.4 Hz, 4 H, sn-1, sn-3), 4.94 (quintet, J = 4.6 Hz, 1 H, sn-2), 5.29 – 5.47 (m, 8 H, 5″, 6″, 8″, 9″, 11″, 12″, 14″, 15″).
2-Hydroxy-1,3,2-dioxaborinan-5-yl (5″Z,8″Z,11″Z,14″Z)-Icosa-5″,8″,11″,14″-tetraenoate (4)
When 2-arachidonoylglycerol (2-AG, 1) is chromatographed under the above conditions except with 10% boric acid treated silica gel prepared by the reported method,64 the corresponding borate ester 4 was eluted as an amorphous white solid: mp 120–130 °C; 1H NMR (CDCl3) δ 0.90 (t, J = 6.8 Hz, 3 H, 20″), 1.20 – 1.44 (m, 6 H, 17″, 18″, 19″), 1.67 – 1.83 (m, 2 H, 3″), 2.06 (apparent q, J = 6.8 Hz, 2 H, 16″), 2.14 (apparent q, J = 7.2 Hz, 2 H, 4″), 2.40 (t, J = 7.6 Hz, 2 H, 2″), 2.69 – 2.92 (m, 6 H, 7″, 10″, 13″), 4.10 (dd, J = 12.0, 1.5 Hz, 2 H) and 4.18 (dd, J = 12.0, 1.5 Hz, 2 H) diastereotopic protons of sn-1 and sn-3, 5.02 – 5.09 (m, 1 H, sn-2), 5.26 – 5.51 (m, 8 H, 5″, 6″, 8″, 9″, 11″, 12″, 14″, 15″); 13C NMR (CDCl3) δ 173.23 (1″), 130.75, 129.29, 128.96, 128.85, 128.51, 128.36, 128.09, and 127.77 (olefinics), 67.53 (sn-2), 65.03 (sn-1, sn-3), 33.86 (2″), 31.75 (18″), 29.55 (17″), 27.45 (16″), 26.70 (4″), 25.87, 25.86, and 25.83 (7″,10″,13″), 24.90 (3″), 22.80 (19″), 14.30 (20″); MS (EI) m/z 402.2 ([M − 2H]+); HRMS (EI) calcd for C23H35O5B 402.25776 ([M − 2H]+), found 402.25929. The borate 4 did not readily release 2-AG 1 in aqueous solution based on competitive binding experiments to hCB2-HEK using radiolabeled CP55940 according to our reported method,73,74 where 2-AG 1 had a Ki of 0.49 micromolar (95% confidence was 300–790) and borate 4 had a Ki that was above 1 micromolar.
[5″,6″,8″,9″,11″,12″,14″,15″-3H(N)]2-Arachidonoyl-1,3-dibutyrylglycerol ([3H]10, [3H]BAB)
The microscale radiosynthesis of triacylglycerol [3H]10 from [5,6,8,9,11,12,14,15-3H(N)]arachidonic acid (2.00 mCi, 200 Ci/mmol, 0.00304 mg, 1.00×10−8 mol, 100 mol%, commercial ethanol solution evaporated with a stream of argon at ambient temperature followed by three chases with dry CH2Cl2) and 1,3-dibutyrylglycerol (1.16 mg, 5.00×10−6 mol, 500 mol%) was performed with a large excess of the coupling agent N-dimethylaminopropyl-N′-ethylcarbodiimide (EDCI, 1.04 mg, 5.42×10−6 mol, 542 mol%) in the presence of N,N-dimethylaminopyridine (DMAP, 0.54 mg, 4.4×10−6 mol, 440 mol%) in 100 μL of argon-degassed anhydrous CH2Cl2. Column chromatography on silica gel eluting with 10:90 EtOAc/hex was used for the purification of the labeled triglyceride [3H]10 to homogeneity (100% radiochemical purity, see Figure 1A, 20:80 EtOAc/hex, Rf = 0.51) in 23% radiochemical yield. This chemically stable triglyceride precursor of radiolabeled 2-AG [3H]1 was stored in the 10:90 EtOAc/hex solution at −78 °C short term over a few months, or can be evaporated under argon and the residue dissolved in toluene and stored at −20 °C pending use.
[5″,6″,8″,9″,11″,12″,14″,15″-3H(N)]2-Arachidonoylglycerol ([3H]1, [3H]2-AG)12
Since [3H]2-AG [3H]1 can isomerize to [3H]1(3)-AG ([3H]2 + [3H]3) in solution, it is necessary to deprotect the stable endocannabinoid precursor [3H]10 immediately before use in biochemical assays and research procedures. The radiolabeled triglyceride [3H]BAB [3H]10 (11 μCi, 200 Ci/mmol) was deprotected enzymatically at neutral pH in argon-degassed absolute ethanol solvent in an argon atmosphere using acrylic beads containing active lipase B from Candida antarctica. The resin-bound lipase (50 mg) was added to a solution of triglyceride in 100 μL of EtOH in a Wheaton vial, the lipase beads were stirred gently with a triangular magnet, and after 2 h were removed by micropore (Whatman 0.2 μm PVDF 13 mm) filtration with the reaction vial, beads, and micropore filter subsequently being washed with four additional 100 μL volumes of EtOH to give a crude product mixture that contained no [3H]1(3)-AG (see Figure 1B). The solvent was removed with a stream of argon and the residue immediately flash chromatographed. The column was prepared and loaded as described for unlabeled 2-AG 1. The column was eluted with 7 mL of 20:80 EtOAc/hex to recover radiolabeled ethyl arachidonate [3H]12. Then the column was eluted with 1 mL of 40:60 EtOAc/hex followed by 10 mL of 60:40 EtOAc/hex. Fractions after 1.5 mL through 6 mL of elution contained [3H]2-AG [3H]1 with very little [3H]1(3)-AG by analytical TLC (see Figure 1B) on boric acid treated silica gel (92.5:25:0.5:2 CHCl3/MeCOMe/HOAc/MeOH, [3H]2-AG Rf = 0.41, [3H]1(3)-AG Rf = 0.30) for a radiochemical purity of 96%. The [3H]2-AG [3H]1 product (6.1 μCi, 55% radiochemical yield) had a specific activity of 200 Ci/mmol.
Supplementary Material
Acknowledgments
This work was supported by the Department of Energy research grant DE-SC0005251 (SJG). We also received support from National Institutes of Health research grants R03-DA-24842 (RID, Jr.), R03-DA-29184 (SKV), K01-DA-21806-01 (STG), and training grant T32-DA-07312 (AM). We are grateful to the office staffs of Northeastern University Environmental Health and Safety, the Department of Pharmaceutical Sciences, and the Center for Drug Discovery, as well as to the CDD analytical biochemistry staff for their support and assistance. We are extremely grateful to Xiaoyu “Ray” Tian, Kyle Whitten, and Roger Kautz for assistance with NMR instrumentation and experimentation.
Footnotes
Supporting Information Available: 1H NMR spectra of 1, 4, 9, 10, and 12. 13C NMR spectra of 4 and 10. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tumaney AW, Shekar S, Rajasekharan R. J Biol Chem. 2001;276:10847–10852. doi: 10.1074/jbc.m100005200. [DOI] [PubMed] [Google Scholar]
- 2.Kim SC, Kang L, Nagaraj S, Blancaflor EB, Mysore KS, Chapman KD. J Biol Chem. 2009;284:34065–34074. doi: 10.1074/jbc.M109.059022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sugiura T, Kishimoto S, Oka S, Gokoh M. Prog Lipid Res. 2006;45:405–446. doi: 10.1016/j.plipres.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Ahn K, McKinney MK, Cravatt BF. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J, Vogel Z. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 6.Sugiura T, Dondo S, Sukagawa A, Nakane S, Shinoda A, Kiyoko I, Yamashita A, Waku K. Biochem Biophys Res Comm. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 7.Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF. Proc Natl Acad Sci USA. 2009;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balsinde J, Diez E, Mollinedo F. J Biol Chem. 1991;266:15638–15643. [PubMed] [Google Scholar]
- 10.Goparaju SK, Ueda H, Yamaguchi H, Yamamoto S. FEBS Lett. 1998;422:69–73. doi: 10.1016/s0014-5793(97)01603-7. [DOI] [PubMed] [Google Scholar]
- 11.Di Marzo V, Bisogno T, Sugiura T, Melck D, De Petrocellis L. Biochem J. 1998;331:15–19. doi: 10.1042/bj3310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cartoni A, Margonelli A, Angelini G, Finazzi-Agro A, Maccarrone M. Tetrahedron Lett. 2004;45:2723–2726. [Google Scholar]
- 13.Mattson FH, Volpenhein RA. J Lipid Res. 1962;3:281–296. [Google Scholar]
- 14.Duclos RI, Jr, Gatley SJ, Bhatt SR, Johnston M. J Label Compd Radiopharm. 2009;52:324–326. doi: 10.1002/jlcr.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boswinkel G, Derksen JTP, van’t Riet K, Cuperus FP. J Am Oil Chem Soc. 1996;73:707–711. [Google Scholar]
- 16.Compton DL, Vermillion KE, Laszlo JA. J Am Oil Chem Soc. 2007;84:343–348. [Google Scholar]
- 17.Laszlo JA, Compton DL, Vermillion KE. J Am Oil Chem Soc. 2008;85:307–312. [Google Scholar]
- 18.Jackson JE, Lundberg WO. J Am Oil Chem Soc. 1963;40:502–504. [Google Scholar]
- 19.Schmid PC, Schwartz KD, Smith CN, Krebsbach RJ, Berdyshev EV, Schmid HHO. Chem Phys Lipids. 2000;104:185–191. doi: 10.1016/s0009-3084(99)00124-3. [DOI] [PubMed] [Google Scholar]
- 20.Seltzman HH, Fleming DN, Hawkins GD, Carroll FI. Tetrahedron Lett. 2000;41:3589–3592. [Google Scholar]
- 21.Porter NA, Lehman LS, Weber BA, Smith KJ. J Am Chem Soc. 1981;103:7447–6455. [Google Scholar]
- 22.Bruna E, Petit W, Beljean-Leymarie M, Huynh S, Nouvelot A. Lipids. 1989;24:970–975. [Google Scholar]
- 23.Han L, Razdan RK. Tetrahedron Lett. 1999;40:1631–1634. [Google Scholar]
- 24.Rouzer CA, Ghebreselasie K, Marnett LJ. Chem Phys Lipids. 2002;119:69–82. doi: 10.1016/s0009-3084(02)00068-3. [DOI] [PubMed] [Google Scholar]
- 25.Saario SM, Savinainen JR, Laitinen JT, Järvinen T, Niemi R. Biochem Pharmacol. 2004;67:1381–1387. doi: 10.1016/j.bcp.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 26.Zhang MY, Gao Y, Btesh J, Kagan N, Kerns E, Samad TA, Chanda PK. J Mass Spectrom. 2010;45:167–177. doi: 10.1002/jms.1701. [DOI] [PubMed] [Google Scholar]
- 27.Thomas AE, III, Scharoun JE, Ralston H. J Am Oil Chem Soc. 1965;42:789–792. [Google Scholar]
- 28.Ueda H, Kobayashi T, Kishimoto M, Tsutsumi T, Okuyama H. J Neurochem. 1993;61:1874–1881. doi: 10.1111/j.1471-4159.1993.tb09829.x. [DOI] [PubMed] [Google Scholar]
- 29.Bisogno T, Sepe N, Melck D, Maurelli S, De Petrocellis L, Di Marzo V. Biochem J. 1997;322:671–677. doi: 10.1042/bj3220671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suhara Y, Takayama H, Nakane S, Miyashita T, Waku K, Sugiura T. Chem Pharm Bull. 2000;48:903–907. doi: 10.1248/cpb.48.903. [DOI] [PubMed] [Google Scholar]
- 31.Stamatov SD, Stawinski J. Tetrahedron Lett. 2002;43:1759–1761. [Google Scholar]
- 32.Pfeffer J, Freund A, Bel-Rhlid R, Hansen CE, Reuss M, Schmid RD, Maurer SC. Lipids. 2007;42:947–953. doi: 10.1007/s11745-007-3084-y. [DOI] [PubMed] [Google Scholar]
- 33.Bentley PH, McCrae W. J Org Chem. 1970;35:2082–2083. [Google Scholar]
- 34.Morrisett JD. Lipids. 1974;9:726–728. doi: 10.1007/BF02532183. [DOI] [PubMed] [Google Scholar]
- 35.Jensen RG, Pitas RE. Adv Lipid Res. 1976;14:213–247. [Google Scholar]
- 36.Paris GY, Cimon DG, Garmaise DL, Swett L, Carter GW, Young PR. Eur J Med Chem. 1982;17:193–195. [Google Scholar]
- 37.Lok CM, Mank APJ, Ward JP. Chem Phys Lipids. 1985;36:329–334. [Google Scholar]
- 38.Lie Ken Jie MSF, Cheung SWH, Ho JCM. Lipids. 2001;36:649–654. [Google Scholar]
- 39.Lie Ken Jie MSF, Lam CC. Chem Phys Lipids. 1995;77:155–171. [Google Scholar]
- 40.Martin JB. J Am Chem Soc. 1953;75:5482–5483. [Google Scholar]
- 41.Paris GY, Garmaise DL, Cimon DG, Swett L, Carter GW, Young P. J Med Chem. 1980;23:9–13. doi: 10.1021/jm00175a003. [DOI] [PubMed] [Google Scholar]
- 42.Paltauf F, Hermetter A. Prog Lipid Res. 1994;33:239–328. doi: 10.1016/0163-7827(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 43.Suhara Y, Takayama H, Nakane S, Miyashita T, Waku K, Sugiura T. Chem Pharm Bull. 2000;48:903–907. doi: 10.1248/cpb.48.903. [DOI] [PubMed] [Google Scholar]
- 44.Porck AJE, Craig BM. Can J Chem. 1955;33:1286–1289. [Google Scholar]
- 45.Breitgoff D, Laumen K, Schneider MP. J Chem Soc, Chem Commun. 1986:1523–1524. [Google Scholar]
- 46.Paris GY, Garmaise DL, Cimon DG, Swett L, Carter GW, Young P. J Med Chem. 1979;22:683–687. doi: 10.1021/jm00192a014. [DOI] [PubMed] [Google Scholar]
- 47.Rosu R, Yasui M, Iwasaki Y, Yamane T. J Am Oil Chem Soc. 1999;76:839–843. [Google Scholar]
- 48.Haraldsson GG, Halldorsson A, Kul E. J Am Oil Chem Soc. 2000;77:1139–1145. [Google Scholar]
- 49.Haftendorn R, Ulbrich-Hofmann R. Tetrahedron. 1995;51:1177–1186. [Google Scholar]
- 50.Robles Medina A, Esteban Cerdán L, Giménez Giménez A, Camacho Páez B, Ibáñez González MJ, Molina Grima E. J Biotech. 1999;70:379–391. [Google Scholar]
- 51.Halldorsson A, Magnusson CD, Haraldsson GG. Tetrahedron. 2003;59:9101–9109. [Google Scholar]
- 52.Magnusson CD, Haraldsson GG. Tetrahedron. 2010;66:2728–2731. [Google Scholar]
- 53.Berger M, Laumen K, Schneider MP. J Am Oil Chem Soc. 1992;69:955–960. [Google Scholar]
- 54.Waldinger C, Schneider M. J Am Oil Chem Soc. 1996;73:1513–1519. [Google Scholar]
- 55.Miura S, Ogawa A, Konishi H. J Am Oil Chem Soc. 1999;76:927–931. [Google Scholar]
- 56.Santaniello E, Ferraboschi P, Grisenti P, Manzocchi A. Chem Rev. 1992;92:1071–1140. [Google Scholar]
- 57.Gunstone FD, Harwood JL, Dijkstra AJ, editors. The Lipid Handbook. 3. CRC Press; Boca Raton: 2007. [Google Scholar]
- 58.Kosugi Y, Oshima A, Koike S, Fukatsu M, Minami K, Miyake Y, Masui K. J Am Oil Chem Soc. 2004;81:235–239. [Google Scholar]
- 59.Irimescu R, Furihata K, Hata K, Iwasaki Y, Yamane T. J Am Oil Chem Soc. 2001;78:285–290. [Google Scholar]
- 60.Irimescu R, Iwasaki Y, Hou CT. J Am Oil Chem Soc. 2002;79:879–883. [Google Scholar]
- 61.Nebel B, Mittelbach M, Uray G. Anal Chem. 2008;80:8712–8716. doi: 10.1021/ac800706s. [DOI] [PubMed] [Google Scholar]
- 62.Trodler P, Pleiss J. BMC Struct Biol. 2008;8:1–10. doi: 10.1186/1472-6807-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li C, Tan T, Zhang H, Feng W. J Biol Chem. 2010;285:28434–28441. doi: 10.1074/jbc.M110.136200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buchnea D. Lipids. 1974;9:55–57. doi: 10.1007/BF02534275. [DOI] [PubMed] [Google Scholar]
- 65.Bergelson LD. Lipid Biochemical Preparations. Elsevier; New York: 1980. [Google Scholar]
- 66.Hirsch J, Ahrens EH., Jr J Biol Chem. 1958;233:311–320. [PubMed] [Google Scholar]
- 67.Yang W, Gao X, Wang B. Med Res Rev. 2003;23:346–368. doi: 10.1002/med.10043. [DOI] [PubMed] [Google Scholar]
- 68.Minkkilä A, Saario SM, Käsnänen H, Leppänen J, Poso A, Nevalainen T. J Med Chem. 2008;51:7057–7060. doi: 10.1021/jm801051t. [DOI] [PubMed] [Google Scholar]
- 69.Glaser ST, Gatley SJ, Gifford AN. J Pharmacol Exp Ther. 2006;316:1088–1097. doi: 10.1124/jpet.105.094748. [DOI] [PubMed] [Google Scholar]
- 70.Glaser ST, Kaczocha M. J Pharmacol Exp Ther. 2010;335:380–388. doi: 10.1124/jpet.110.168831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnston RF, Pickett SC, Barker DL. Electrophoresis. 1990;11:355–360. doi: 10.1002/elps.1150110503. [DOI] [PubMed] [Google Scholar]
- 72.Liberatore GT, Wong JYF, Krenus D, Jeffreys BJ, Porritt MJ, Howells DW. Biotechniques. 1999;26:432–434. doi: 10.2144/99263bm13. [DOI] [PubMed] [Google Scholar]
- 73.Khanolkar AD, Lu D, Ibrahim M, Duclos RI, Jr, Thakur GA, Malan TP, Jr, Porreca F, Veerappan V, Tian X, George C, Parrish DA, Papahatjis DP, Makriyannis A. J Med Chem. 2007;50:6493–6500. doi: 10.1021/jm070441u. [DOI] [PubMed] [Google Scholar]
- 74.Lu D, Guo J, Duclos RI, Jr, Bowman AL, Makriyannis A. J Med Chem. 2008;51:6393–6399. doi: 10.1021/jm8005299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.