

Abstract
A growing body of research supports that members of the vitamin E family are not redundant with respect to their biological function. Palm oil derived from Elaeis guineensis represents the richest source of the lesser characterized vitamin E, α-tocotrienol. One of 8 naturally occurring and chemically distinct vitamin E analogs, α-tocotrienol possesses unique biological activity that is independent of its potent antioxidant capacity. Current developments in α-tocotrienol research demonstrate neuroprotective properties for the lipid-soluble vitamin in brain tissue rich in polyunsaturated fatty acids (PUFAs). Arachidonic acid (AA), one of the most abundant PUFAs of the central nervous system, is highly susceptible to oxidative metabolism under pathologic conditions. Cleaved from the membrane phospholipid bilayer by cytosolic phospholipase A2, AA is metabolized by both enzymatic and nonenzymatic pathways. A number of neurodegenerative conditions in the human brain are associated with disturbed PUFA metabolism of AA, including acute ischemic stroke. Palm oil–derived α-tocotrienol at nanomolar concentrations has been shown to attenuate both enzymatic and nonenzymatic mediators of AA metabolism and neurodegeneration. On a concentration basis, this represents the most potent of all biological functions exhibited by any natural vitamin E molecule. Despite such therapeutic potential, the scientific literature on tocotrienols accounts for roughly 1% of the total literature on vitamin E, thus warranting further investment and investigation.
Keywords: palm oil, vitamin E, tocotrienol, neurodegeneration
Key teaching points.
Palm oil is one of the most abundant natural sources of the lesser characterized vitamin E, α-tocotrienol.
α-Tocotrienol possesses unique neuroprotective properties not shared by other natural vitamin E family members.
Oral supplementation of palm oil–derived α-tocotrienol reaches the brain in sufficient quantity to attenuate stroke-mediated neuropathy.
Antioxidant-independent mechanisms of α-tocotrienol–mediated neuroprotection are discussed in detail.
Introduction
The first evidence of palm oil consumption in human diets dates back as far as 3000 bc, with a long history of use in western Africa [1]. Derived from the fleshy orange-red mesocarp of the fruits of the oil palm tree Elaeis guineensis, oil palm trees were introduced to the West (Brazil, West Indies) in the 15th century by the Portuguese and to the East (Indonesia) by the Dutch in the 19th century [2]. Originally purposed as ornamental landscape, commercial oil palm farming and palm oil production is a relatively recent endeavor of the 20th century, with the first commercial oil palm estate established in Malaysia in 1917 [2]. Of the oil-bearing plants, E. guineensis provides the highest yield of oil, with an average of between 4 and 10 tons of oil per hectare per annum [1,3]. Accounting for ∼30% of the total world production of oils and fats, palm oil has dramatically increased its share of production over the past 10 years and overtaken soybean oil (∼23%) as the most abundant plant-derived oil source worldwide [4]. Throughout the world, 90% of palm oil is purposed for dietary consumption, with the majority of palm oil production and consumption localized in the tropics of South-East Asia [5].
Palm Oil Nutrition and Chemistry
Despite the surge in production and consumption of palm oil over the past decade, incorporation of palm oil into Western diets remains largely unpopular due in part to a higher saturated fatty acid (SFA) content when compared to most other commercially available vegetable oils including olive, canola, and soybean [6]. A positive and significant link between a diet rich in SFA and elevated low-density lipoprotein (LDL) cholesterol and increased risk for cardiovascular disease is widely recognized. However, the scientific evidence for palm oil–rich diets specifically contributing to elevated LDL cholesterol and cardiovascular disease is conflicting [7–10].
Palm oil use in the United States food industry is slowly gaining acceptance as a natural hydrogenated fat substitute [11]. Partial hydrogenation of vegetable oils converts them into semi-solid fats with greater shelf-life, stability during cooking, and palatability [12]. Hydrogenated fats contain high levels of trans-fatty acids, which are thought to have adverse health effects, including increased risk of coronary heart disease [13], increased systemic inflammation [14,15], and impaired endothelial cell function [16]. To promote awareness and discourage their use in food products, the U.S. Food and Drug Administration mandated trans-fatty acid content listing on Nutrition Facts panels in 2006 [17]. With a near 1:1 ratio of saturated to unsaturated fatty acids, palm oil remains in a semi-solid state at room temperature. In many instances, this distinctive property makes it a desirable trans-fatty acid–free alternative to hydrogenated oil for use in food products such as margarine, shortenings, wafers, and candies [11].
Beyond the unique lipid profile of palm oil, there is growing scientific interest in the lipophilic palm oil–associated tocol members of the vitamin E family that possess potent nutritional and therapeutic value. While lipid-soluble vitamin E accounts for less than 1% of total palm oil content, it plays an important role in preserving oil stability and shelf-life while contributing to the health benefit of dietary palm oil consumption. The vitamin E family members are widely recognized and characterized for their antioxidant capacity; a growing body of scientific literature now also recognizes their antioxidant-independent functions, with therapeutic potential in pathologic disorders ranging from cancer to cerebrovascular disease. This review examines the recent body of work on palm oil–derived α-tocotrienol, a lesser characterized vitamin E isomer with therapeutic potential in neuropathology.
The Natural Vitamin E Family
The discovery of vitamin E, as compared to the prior identified vitamins A–D, was uniquely different in that the effects of deficiency in animal models varied from species to species and a specific relevance to human deficiency was not realized for several decades. Herbert M. Evans is credited with the discovery of vitamin E in 1922, when he found that a particular “antisterility factor x” was necessary for reproduction in rats [1]. Specifically, pregnant rodents kept on a rancid lard diet would resorb their embryos, but by enriching their diet with the chlorophyll-rich fraction of oil from lettuce leaves, Evans found that fertility was restored. Knowing that this factor was lipid soluble, he quickly resolved that his supplement was uniquely different from the other 2 known lipid-soluble vitamins of the time: vitamin A and vitamin D. Two years later, a chemistry professor from the University of Arkansas by the name of Barnett Sure named the fertility factor “vitamin E,” suggesting that Evans' antisterility factor was also essential to humans despite no evidence yet to support such a claim. In 1936 Evans published the chemical formula of vitamin E in the Journal of Biological Chemistry [18]. Ascribing the chemical name of the compound was given to a colleague of Evan's at The University of California at Berkeley, professor of Greek, George M. Calhoun. Evan's personal account of the name's origin from Dr. Calhoun gives humorous insight to what would otherwise be considered a prodigious effort:
“Most scientists, medical men especially,” said Calhoun, “have been guilty of coining Greek-Latin terms, bastards, of course, and we might have to do this.” “What does the substance do?” he asked. “It permits an animal to bear off-spring,” I replied. “Well, childbirth in Greek is tocos,” he said, “and if it confers or brings childbirth, we will next employ the Greek verb phero. You have also said that the term must have an ending consonant with its chemical — ‘ol’, it being an alcohol; your substance is ‘tocopherol,’ and the pleasant task assigned me quickly solved and not worth the delightful four-course dinner you have arranged.” [19]
The relevance of “tocopherols” to humans proved to be a difficult problem to solve until the late 1950s with the advent of the free-radical theory of aging and lipid peroxidation pioneered by Denham Harman [20,21]. According to this theory, membrane lipids are susceptible to oxidative damage, and the exact location of damage observed in different animal species depends on specific membrane lipid composition, vitamin E content, and cellular metabolic rate [1]. While it had already been known that vitamin E played an antioxidant role in protecting plant oils from oxidative damage and rancidity, this theory pioneered the concept of oxidative stress and the importance of antioxidants such as vitamin E as related to lipids in mammalian systems. Decades later, the human relevance of vitamin E deficiency became recognized in the medical field in cases of fat malabsorption or the disruption of low-density lipoprotein (LDL) processing, a lipid-rich transporter of vitamin E in the blood [22]. Progressive neuropathy and retinopathy leading to crippling ataxia and impaired vision were prevented by high-dose vitamin E supplementation in patients with abetalipoproteinemia [23,24].
Today, the naturally occurring vitamin E family is known to consist of both tocopherols and tocotrienols (Fig. 1). Tocopherols, as discovered by Evans, are more ubiquitous in plant tissues as compared to tocotrienols and are the principal vitamin E component in the leaves of plants and seeds of most dicots [25]. Tocopherols consist of a chromanol ring and a 15-carbon saturated tail derived from homogentisate (HGA) and phytyl diphosphate, respectively. Tocopherol biosynthesis in plants is catalyzed by homogentisate phytyltransferase which enacts the condensation reaction of HGA and phytyl diphosphate [26]. Unlike tocopherols, the 15-carbon phytyl tail of tocotrienols originates from geranylgeranyl diphosphate and contains 3 trans double bonds at the 3′, 7′, and 11′ positions. Condensation of HGA and geranylgeranyl diphosphate is carried out by homogentisate geranylgeranyl transferase [27]. The position and degree of methylation on the chromanol head defines the α, β, γ, and δ isoform designations within the tocopherol and tocotrienol families.
Fig. 1.
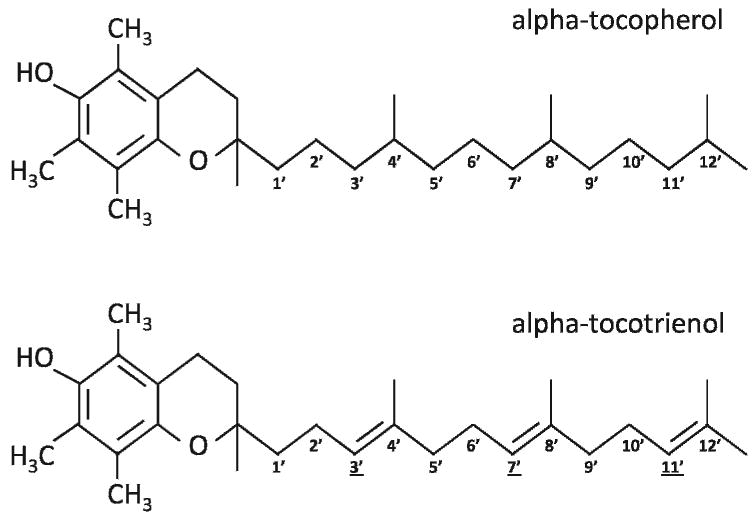
Chemical structure of α-tocopherol and α-tocotrienol. Three unsaturations at carbon positions 3, 7, and 11 in the isoprenoid side chain account for the structural difference in α-tocopherol and α-tocotrienol chemical structure. This seemingly small difference has large implications for bioavailability and the specific biological functions of the vitamin E isomers.
Compared to tocopherols, tocotrienols are considerably less widespread in the plant kingdom [25]. Tocotrienols are the major form of vitamin E in the seeds of most monocots (i.e., oil palm) and a limited number of dicots. A detailed analysis of tocotrienol accumulation revealed that the presence of this natural vitamin E is localized in nonphotosynthetic tissue [25]. Palm oil is one of the most abundant natural sources of tocotrienols, with crude palm oil (also referred to as the “tocotrienol-rich fraction”) containing up to 800 mg/kg weight of α- and γ-tocotrienol isotypes. The distribution of vitamin E in palm oil is 30% tocopherols and 70% tocotrienols. In contrast, other commonly used dietary vegetable oils, including corn, olive, peanut, sesame, soybean, and sunflower, contain tocopherols exclusively [2,28].
Not All Vitamin E Is Created Equal
As the most bioavailable of all natural vitamin E isomers and with a well-characterized and selective transport mechanism in mammals, the vast majority of vitamin E research conducted to date has focused on α-tocopherol (αTOC). It is estimated that only 1% of all vitamin E literature and research during the last 30 years addresses tocotrienols [11]. Consequently, a striking asymmetry exists in characterizing the biological significance of non-αTOC family members to the degree that αTOC and vitamin E are often used synonymously. Recent evidence supports unique biological functions of the lesser-characterized natural vitamin E homologues in mammalian tissue. γ-Tocopherol (γTOC) is the most abundant form of vitamin E in the United States diet [29]. Desmethyl tocopherols, such as γTOC, and specific tocopherol metabolites, such as the carboxyethyl-hyroxychroman products, exhibit functions that are not shared by αTOC. The activities of these tocopherols are not directly related to their chemical antioxidant activity, but rather to anti-inflammatory, antineoplastic, and natriuretic function, possibly mediated through specific binding interactions [30].
Members of the tocotrienol family, known to be enriched in palm oil, also possess biological functions that are not shared by αTOC. Micromolar amounts of tocotrienol suppress the activity of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the hepatic enzyme responsible for cholesterol synthesis [31,32]. HMG-CoA reductase is the same enzyme targeted by the statin class of drugs, one of the most widely prescribed group of drugs in the United States today [33]. Furthermore, tocotrienols, but not tocopherols, have been shown to suppress growth of human breast cancer cells [34]. Finally, dietary supplementation of the α-tocotrienol (αTCT) isoform uniquely protects against acute ischemic stroke injury in vivo [35]. At a nanomolar concentration range achievable by dietary supplementation, αTCT derived from palm oil prevents inducible neurodegeneration by regulating specific mediators of cell death [35–40]. To date, the observed neuroprotective properties of palm oil–derived αTCT are the most potent function of any natural form of vitamin E on a concentration basis.
Bioavailability of Palm Oil–Derived α-Tocotrienol
While mechanisms of αTOC tissue delivery and bioavailability are well characterized, the significance of dietary αTCT bioavailability and tissue distribution has only recently been examined. α-Tocopherol transfer protein (TTP) is a soluble 32-kDa protein expressed in liver that selectively binds αTOC [41]. The TTP transporter is known to bind to αTCT with 8.5-fold lower affinity than that for αTOC [42], and subsequently contributed to the notion that availability of dietary tocotrienol to vital organs is negligible [43]. Recent in vivo evidence, however, suggests otherwise. Despite rodent chow enriched with αTOC, it has been shown that TTP-deficient female mice are infertile due to vitamin E deficiency [44,45]. The placentas of pregnant TTP-deficient females are severely impaired with marked reduction of labyrinthine trophoblasts, and embryos die at midgestation even when fertilized eggs of TTP-sufficient wild-type mice are transferred into TTP-deficient recipients [44]. In 2005 it was reported that oral supplementation of palm oil–derived αTCT in female TTP knockout mice restored fertility, suggesting that αTCT is successfully delivered to relevant tissues in sufficient quantity to support reproductive function under conditions of αTOC deficiency [46]. This observation was consistent with a second line of investigation in rats demonstrating that palm oil–derived αTCT supplementation spared loss of fertility caused by long-term vitamin E deficiency in the diet [46]. Dietary palm oil–derived αTCT supplementation was shown to enhance not only αTCT bioavailability in reproductive tissues, but also in several vital organs, including the brain [46].
Over a decade ago in a study testing the ligand specificity of vitamin E isomers for TTP, it was concluded that the affinity of vitamin E analogues for TTP is one of the critical determinants of their biological activity [42]. This conclusion was based on the assumption that the biological function of natural vitamin E family members is proportionate to their concentration in tissue and that vitamin E isotype function is redundant. Today, however, these assumptions have largely been disproven in light of new knowledge. It is now clear that oral supplementation of palm oil–derived αTCT not only reaches the brain [46,47], but it does so in sufficient quantity to attenuate stroke-mediated neuropathy [35]. Furthermore, αTCT supplementation in humans results in a peak blood plasma level that is over an order of magnitude higher than that required to protect neurons against a range of neurotoxic insults [35,37,46,48].
The Arachidonic Acid Cascade of Oxidative Brain Injury and Neuroprotection by Palm Oil–Derived α-Tocotrienol
Brain tissue is highly susceptible to oxidative stress as it: (1) consumes an inordinate amount of oxygen to meet high metabolic demands, (2) contains high concentrations of polyunsaturated fatty acids (PUFAs) that are vulnerable to lipid peroxidation, and (3) has lower antioxidant capacity as compared to other organ systems. Neurons are particularly vulnerable to oxidative damage in pathologic brain tissue due to lower levels of endogenous antioxidant, glutathione, as compared to resident glial cells [27]. The consequence of increased generation of free radical species in injured brain tissue enriched with PUFAs is an accumulation of lipid peroxidation products that are proven to be culpable neuromodulators of the cell death cascade. Brain tissue is highly enriched with the n-6 PUFA arachidonic acid (AA, 20:4n-6) and the n-3 PUFA docosahexaenoic acid (DHA, 22:6n-3), which are major components of phospholipid membranes. Together, AA and DHA account for ∼20% of all fatty acids in the mammalian brain [49]. Both AA and DHA are nutritionally essential to brain function and structure during early development and influence membrane fluidity, signal transduction, and gene transcription throughout life [50–54]. Neither AA nor DHA is synthesized de novo; rather, they are obtained from the diet and circulated to the brain directly in the plasma [54], or elongated from n-6 and n-3 PUFA linoleic (18:2n-6) and α-linolenic acid (18:3n-3) precursors in the liver [55]. A number of neurodegenerative conditions in the human brain are associated with disturbed PUFA metabolism of AA [56,57]. Recent work supports palm oil–derived αTCT as a potent natural neuroprotective agent in the AA cascade of neurodegeneration with the ability to uniquely target both nonenzymatic and enzymatic mechanisms of brain injury (Fig. 2).
Fig. 2.
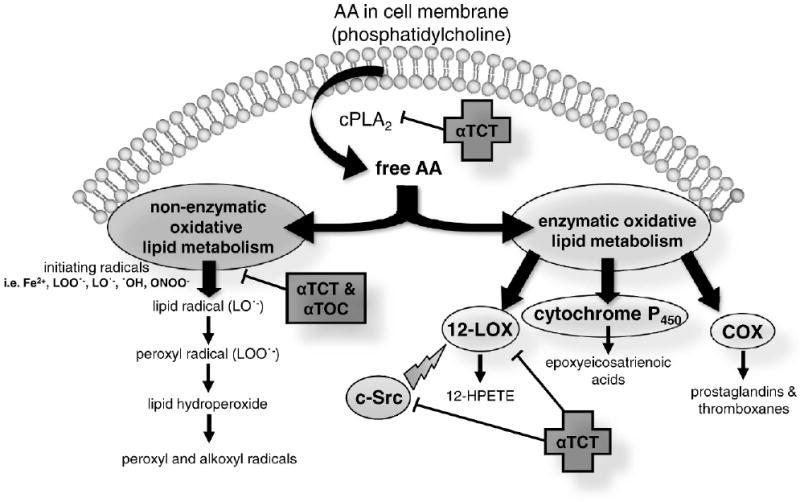
Palm oil–derived natural vitamin E attenuates the arachidonic acid (AA) cascade in brain injury. Following release from the lipid membrane bilayer by phospholipase A2 (PLA2), the polyunsaturated fatty acid AA undergoes oxidative metabolism in nonenzymatic and enzymatic pathways. Well known for their antioxidant function, natural vitamin E isomers α-tocotrienol (αTCT) and α-tocopherol (αTOC) inhibit nonenzymatic oxidative lipid metabolism of AA. Independent of antioxidant function common to all vitamin E isomers, αTCT is a specific and potent inhibitor of cytosolic phospholipase A2 (cPLA2), c-Src kinase (c-Src), and 12-lipoxygenase (12-LOX) at nanomolar concentrations.
Phospholipase A2
Phospholipase A2 (PLA2) isozymes encompass a diverse family of at least 15 different isozyme groups classified into 5 distinct categories: (1) secreted small molecular weight sPLA2, (2) larger cytosolic calcium-dependent cPLA2, (3) calcium-independent iPLA2, (4) platelet-activating factor acetylhydrolases (PAFA), and (5) lysosomal PLA2 isozymes. The entire PLA2 family is characterized by a common function—the enzymatic hydrolysis of the sn-2 ester bond of glycerophospholipids—thereby producing a free fatty acid (i.e., AA) and lysophospholipid (i.e., lysophosphatidylcholine, LPC). Currently, only sPLA2 and cPLA2 have well-defined roles in pathologic AA metabolism [58].
In a central nervous system (CNS) pathologic setting in which reactive oxygen and nitrogen species are abundant, there occurs a rapid accumulation of free fatty acids, due to increases in intracellular Ca2+ and activation of PLA2s [59–62]. The sPLA2s are characterized by the requirement of histidine in their active site, Ca2+ for catalysis, and the presence of 6 conserved disulfide bonds [63]. Under pathologic conditions of ischemic stroke, sPLA2 mRNA and protein expression are significantly upregulated [64,65] and activity is induced by inflammatory cytokine tumor necrosis factor α (TNF-α) [66]. The cPLA2s are the only PLA2 that demonstrate a preference for AA in the sn-2 position of phospholipids [67]. Localized predominantly in grey matter, they lack the disulfide bonding network of sPLA2s and function through the action of a serine/aspartic acid dyad [68]. Under pathologic conditions, cPLA2 subunit mRNA and protein expression are elevated [62]. Intracellular Ca2+ accumulation mediates cPLA2 subunit translocation to the membrane phospholipid bilayer [69] and activity is induced by phosphorylation of serine residue 505 by mitogen-activated protein kinase [70].
Once released, free AA has 3 potential fates: reincorporation into phospholipids, diffusion outside the cell, and metabolism. In a pathologic setting, free AA accumulates and undergoes uncontrolled oxidative metabolism by both nonenzymatic and enzymatic processes. This uncontrolled metabolism, referred to as the “arachidonic acid cascade,” includes the formation of harmful prostaglandins, leukotrienes, thromboxanes, isoprostanes, and nonenzymatic lipid peroxidation products [71]. The AA cascade amplifies the overall production of free radicals, both reactive oxygen and nitrogen species, and subsequently oxidative damage to lipids, proteins, and nucleic acids.
It has recently been shown that palm oil–derived αTCT attenuates cPLA2 activity under conditions of glutamate-mediated toxicity in neural cells [36]. Glutamate is the most abundant neurotransmitter of the CNS. Uncontrolled glutamate release at the synaptic cleft under pathologic conditions induces neurodegeneration in a process known as oxytosis. Glutamate activates cPLA2 in neurons in a calcium-dependent manner, leading to the hydrolysis of AA from phospholipids [36]. Both phosphorylation and translocation of cPLA2 are inhibited with nanomolar concentrations of αTCT, a level previously demonstrated to be readily achievable by dietary supplementation [46,47].
Nonenzymatic AA Oxidative Metabolism
Beyond the initial damage to lipid membranes, reaction of free radical species with double bonds of PUFAs produce alkyl radicals, which in turn react with molecular oxygen to form a peroxyl radical (ROO(x00358)). Peroxyl radical can abstract hydrogen from adjacent PUFAs to produce a lipid hydroperoxide (ROOH) and a second alkyl radical, thereby propagating a chain reaction of lipid oxidation [72]. Lipid peroxides degrade and give rise to α,β-unsaturated aldehydes that include 4-hydroxynonenal, malondialdehyde, and acrolein [73–75]. These aldehydes covalently bind to proteins through reaction with thiol groups and alter their function. They also react with amino groups to form cyclic adducts. Vitamin E is widely known as the major chain-breaking antioxidant and the first line of defense in protecting lipid membranes from peroxidation [76]. The antioxidant properties of vitamin E are thought to be due to the fused heterocyclic ring. In this ring, the p-type lone electron pair of oxygen is kept nearly perpendicular to the aromatic plane. This p-type lone pair orbital overlaps with the semi-occupied molecular orbitals of the lipid radical, stabilizing the radical by conjugative electron delocalization [76].
Compared to tocopherols, the antioxidant capacity of the tocotrienol family is believed to be more potent [77]. Indeed, the tocotrienol-rich fraction of palm oil was found to be significantly more effective than αTOC alone in inhibiting oxidative damage to lipids in isolated mitochondria from rats [78]. Due to the unsaturations found in their phytyl tail, tocotrienols assume a unique conformation in the lipid membrane bilayer with greater flexibility in the side chain that increases curvature stress on phospholipid membranes [43]. It is believed that the unsaturated side chain of tocotrienol also enables more efficient penetration into tissues that have saturated fatty layers, such as brain [79]. These phenomena may contribute to easier access of ascorbate to reduce the α-tocotrienoxyl radical, thereby enhancing antioxidant regeneration of αTCT more effectively than αTOC in brain [80].
Enzymatic AA Oxidative Metabolism
Cyclooxygenase, epoxygenase, and lipoxygenase enzymes are pivotal players in the generation of oxygenated derivatives of AA in pathologic conditions affecting the brain. Cyclooxygenases convert AA to prostaglandins and thromboxanes, epoxygenase activity produces epoxyeicosatrienoic acids, and lipoxygenases catalyze the metabolism of AA into leukotrienes and lipoxins. The functional role of these enzymes and downstream lipid-derived products is largely dependent upon environment and cellular localization. In a pathologic setting of ischemic stroke, the lipoxygenase pathway and derivative products have been identified as key mediators of neurodegeneration and cell death in brain and are the focus of discussion here [35,39].
The first human lipoxygenase (LOX) activity was observed in platelets via the transformation of AA to a prostaglandin-like endoperoxide [81]. Today, the LOX enzyme family includes 4 members that are classified on the basis of the carbon position in which they oxidize AA: 5-, 8-, 12-, and 15-LOX. Despite variances in amino acid sequences and tissue distribution amongst family members, the active site of each isozyme is highly conserved [82]. The structure of LOX at the active site is composed of an N-terminal beta-barrel domain and a C-terminal domain containing a hydrophobic substrate-binding site [83]. A non-heme iron atom is coordinated by 3 histidine residues and the carboxy-terminal isoleucine. The oxidation of ferrous iron (Fe2+) to ferric iron (Fe3+) activates the enzyme, which is then capable of excising a hydrogen atom from a hydrocarbon at 1 of the 4 double bonds of AA. This abstraction generates a radical metabolite of AA that rapidly reorganizes its double bonds to take on a more stable conformation. Next, insertion of molecular oxygen generates a hydroperoxide radical that is reduced to the hydroperoxide anion by the simultaneous oxidation of iron to the ferric state [83]. A proton is then accepted to form a highly reactive fatty acid hydroperoxide; hydroperoxyeicosatetraenoic acid (HPETE). The hydroperoxide derivatives of AA are short-lived and readily metabolized into more stable compounds, including hydroxyeicosatetraenoic acids (HETEs) and leukotrienes.
While short-lived, HPETEs are potent neurotoxins. Highly reactive oxygen radicals are produced during the conversion of HPETEs to HETEs [84], contributing to the overall burden of oxidative stress following stroke. Under conditions of glutathione depletion, as in acute focal stroke, 12-LOX–derived 12-HPETE triggers nitric oxide (NO)–induced neural cell death [85]. Recent evidence supports that the hydroxy-(HETE) derivatives of LOX-mediated metabolism also possess potent biological activity. 12-HETE has been shown to increase mitochondrial NO production, induce cytochrome C release, and subsequently cause mitochondrial dysfunction [86].
The LOX-catalyzed dehydration reaction generates an epoxide intermediate, leukotriene A4 (LTA4) [71]. LTA4 is a highly unstable intermediate that is readily hydrolyzed to LTB4 or conjugated with glutathione by cysteinyl leukotriene C4 synthase to produce leukotriene C4 (LTC4), leukotriene D4 (LTD4), and leukotriene E4 (LTE4) [87]. Together, LTC4, LTD4, and LTE4 are collectively referred to as cysteinyl-leukotrienes (Cys-LTs). Total leukotriene levels accumulate in the mammalian brain and cerebral spinal fluid during and after cerebral ischemia [88,89]. LTB4 is characterized as a powerful chemotactic agent that induces adhesion of pro-inflammatory leukocytes to the endothelium; stimulates phagocytosis, and activates neutrophils and other leukocytes in a paracrine manner [90–92]. The Cys-LTs are potent vasoconstrictors of both venous and arterial smooth muscle [93,94] and are also reported to produce vascular leak and vasogenic edema [95].
While each LOX isoform carries out the same general reaction, namely hydrogen abstraction from AA, each has a unique gene structure, amino acid sequence, and tissue distribution profile. Only 3 forms of LOX are present in brain tissue, 5-, 12-, and 15-LOX [71]. Of these, 12-LOX is the most abundant isoform found in the brain [96], with significant mRNA expression in cortical neurons, astrocytes, and oligodendrocytes [97]. The LOX-mediated metabolites of AA serve as second messengers following stroke by modulating inflammation, apoptosis, and synaptic activity in brain tissue. Using immature cortical neurons and HT cells, it has been shown that a decrease in intracellular glutathione triggers the activation of neuronal 12-LOX, which leads to production of peroxides, influx of Ca2+, and ultimately cell death [98].
Once cleaved from the sn-2 position of glycerophospholipids, AA may be metabolized by 12-LOX to pro-inflammatory HPETEs, HETEs, and leukotrienes. 12-LOX–deficient mice are highly resistant to ischemic stroke injury [35], and palm oil–derived αTCT has been shown to be a potent 12-LOX inhibitor in neural cells subjected to glutamate-mediated neurotoxicity [39]. Furthermore, prophylactic supplementation of palm oil–derived αTCT in stroke-prone spontaneously hypertensive rats significantly attenuated ischemic stroke lesion volume [35]. Studies addressing the effects of αTCT on pure 12-LOX indicate that αTCT directly interacts with the enzyme to suppress AA metabolism. In silico studies examining possible docking sites of αTCT to 12-LOX support the presence of an αTCT-binding solvent cavity close to the active site, potentially blocking AA access [39].
12-LOX catalytic function is believed to be regulated by tyrosine kinases. In response to glutamate-mediated neurodegeneration, 12-LOX is subject to rapid tyrosine phosphorylation in neuronal cells [35]. c-Src kinase is heavily expressed in CNS tissue, with rapid activation demonstrated to play a critical role in executing neurodegeneration [40,99]. Consistently, Src deficiency or blockade of Src activity in mice protects cortical brain tissue from stroke-induced neurodegeneration [100]. Findings from cell biology studies as well as from the study of c-Src kinase and 12-LOX in cell-free systems indicate that in response to glutamate challenge, c-Src is rapidly activated and phosphorylates 12-LOX [35,38]. Importantly, palm oil–derived αTCT prevents glutamate-induced cell death in active c-Src overexpressing cells [40]. Furthermore, glutamate-induced c-Src activity is completely blocked by nanomolar amounts of αTCT. Taken together, the powerful effects of palm oil–derived αTCT on c-Src and 12-LOX suggest that the lesser characterized vitamin E isomer is a potent inhibitor of 12-LOX-mediated AA metabolism and neurodegeneration.
Conclusion
A growing body of literature has only just begun to delineate the unique and potent biological properties of the natural vitamin E αTCT; as evidenced by more than two thirds of the entire PubMed literature on tocotrienols having only been published since 2000 [11]. To date, the neuroprotective qualities of αTCT in neurodegenerative disorders of the CNS are well characterized, with specific molecular targets (cPLA2, 12-LOX, and c-Src) and mechanisms of action identified. Beyond the CNS, αTCT has also demonstrated therapeutic promise in the treatment cancer and hypercholesterolemia. Originating from a dietary source with a long history of safe consumption in humans, the oil palm represents the richest source of αTCT known today. Although tocotrienols are present in edible products such as palm oil, it remains questionable whether a dietary source alone could provide sufficient amounts of αTCT to humans [11]. This is particularly relevant in Western diets that are typically devoid of palm oil and other natural sources of αTCT. Enrichment of αTCT from crude palm oil for dietary supplementation is achievable, and to date represents the most cost effective and readily available source of natural αTCT.
Footnotes
The authors have no conflict of interest to declare.
References
- 1.Kiple KF, Ornelas KC. The Cambridge World History of Food. New York: Cambridge University Press; 2000. [Google Scholar]
- 2.Sundram K, Sambanthamurthi R, Tan YA. Palm fruit chemistry and nutrition. Asia Pac J Clin Nutr. 2003;12:355–362. [PubMed] [Google Scholar]
- 3.Edem DO. Palm oil: biochemical, physiological, nutritional, hematological, and toxicological aspects: a review. Plant Foods Hum Nutr. 2002;57:319–341. doi: 10.1023/a:1021828132707. [DOI] [PubMed] [Google Scholar]
- 4.Sarris A, Food and Agriculture Organization of the United Nations . Medium-Term Prospects for Agricultural Commodities: Projections to the Year 2010. FAO Commodities and Trade Technical Paper, 1. Rome: Food and Agriculture Organization of the United Nations; 2003. [Google Scholar]
- 5.Sundram K, Hayes KC, Siru OH. Dietary palmitic acid results in lower serum cholesterol than does a lauric-myristic acid combination in normolipemic humans. Am J Clin Nutr. 1994;59:841–846. doi: 10.1093/ajcn/59.4.841. [DOI] [PubMed] [Google Scholar]
- 6.Ayorinde FO, Garvin K, Saeed K. Determination of the fatty acid composition of saponified vegetable oils using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2000;14:608–615. doi: 10.1002/(SICI)1097-0231(20000415)14:7<608::AID-RCM918>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 7.Cater NB, Heller HJ, Denke MA. Comparison of the effects of medium-chain triacylglycerols, palm oil, and high oleic acid sunflower oil on plasma triacylglycerol fatty acids and lipid and lipoprotein concentrations in humans. Am J Clin Nutr. 1997;65:41–45. doi: 10.1093/ajcn/65.1.41. [DOI] [PubMed] [Google Scholar]
- 8.Choudhury N, Tan L, Truswell AS. Comparison of palmolein and olive oil: effects on plasma lipids and vitamin E in young adults. Am J Clin Nutr. 1995;61:1043–1051. doi: 10.1093/ajcn/61.4.1043. [DOI] [PubMed] [Google Scholar]
- 9.Katan MB, Zock PL, Mensink RP. Dietary oils, serum lipoproteins, and coronary heart disease. Am J Clin Nutr. 1995;61:1368S–1373S. doi: 10.1093/ajcn/61.6.1368S. [DOI] [PubMed] [Google Scholar]
- 10.Ladeia AM, Costa-Matos E, Barata-Passos R, Costa Guimaraes A. A palm oil–rich diet may reduce serum lipids in healthy young individuals. Nutrition. 2008;24:11–15. doi: 10.1016/j.nut.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 11.Sen CK, Khanna S, Roy S. Tocotrienols in health and disease: the other half of the natural vitamin E family. Mol Aspects Med. 2007;28:692–728. doi: 10.1016/j.mam.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mozaffarian D, Katan MB, Ascherio A, Stampfer MJ, Willett WC. Trans fatty acids and cardiovascular disease. N Engl J Med. 2006;354:1601–1613. doi: 10.1056/NEJMra054035. [DOI] [PubMed] [Google Scholar]
- 13.Ascherio A, Katan MB, Zock PL, Stampfer MJ, Willett WC. Trans fatty acids and coronary heart disease. N Engl J Med. 1999;340:1994–1998. doi: 10.1056/NEJM199906243402511. [DOI] [PubMed] [Google Scholar]
- 14.Mozaffarian D, Pischon T, Hankinson SE, Rifai N, Joshipura K, Willett WC, Rimm EB. Dietary intake of trans fatty acids and systemic inflammation in women. Am J Clin Nutr. 2004;79:606–612. doi: 10.1093/ajcn/79.4.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mozaffarian D, Rimm EB, King IB, Lawler RL, McDonald GB, Levy WC. Trans fatty acids and systemic inflammation in heart failure. Am J Clin Nutr. 2004;80:1521–1525. doi: 10.1093/ajcn/80.6.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Roos NM, Bots ML, Katan MB. Replacement of dietary saturated fatty acids by trans fatty acids lowers serum HDL cholesterol and impairs endothelial function in healthy men and women. Arterioscler Thromb Vasc Biol. 2001;21:1233–1237. doi: 10.1161/hq0701.092161. [DOI] [PubMed] [Google Scholar]
- 17.Remig V, Franklin B, Margolis S, Kostas G, Nece T, Street JC. Trans fats in America: a review of their use, consumption, health implications, and regulation. J Am Diet Assoc. 2010;110:585–592. doi: 10.1016/j.jada.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 18.Evans HM, Emerson OH, Emerson GA. The isolation from wheat germ oil of an alcohol, alpha-tocopherol, having the properties of vitamin E. J Biol Chem. 1936;113:319–332. [Google Scholar]
- 19.Evans HM. Vitamins and Hormones. Vol. 20. Maryland Heights, MO: Elsevier Science and Technology Books; 1963. The pioneer history of vitamin E; pp. 379–387. [Google Scholar]
- 20.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 21.Harman D. Free radical theory of aging: effect of the amount and degree of unsaturation of dietary fat on mortality rate. J Gerontol. 1971;26:451–457. doi: 10.1093/geronj/26.4.451. [DOI] [PubMed] [Google Scholar]
- 22.Kayden HJ, Bjornson L. The dynamics of vitamin E transport in the human erythrocyte. Ann NY Acad Sci. 1972;203:127–140. doi: 10.1111/j.1749-6632.1972.tb27866.x. [DOI] [PubMed] [Google Scholar]
- 23.Azizi E, Zaidman JL, Eshchar J, Szeinberg A. A beta lipoproteinemia treated with parenteral and oral vitamins A and E, and with medium chain triglycerides. Acta Paediatr Scand. 1978;67:796–801. doi: 10.1111/j.1651-2227.1978.tb16264.x. [DOI] [PubMed] [Google Scholar]
- 24.Sokol RJ. Vitamin E deficiency and neurologic disease. Annu Rev Nutr. 1988;8:351–373. doi: 10.1146/annurev.nu.08.070188.002031. [DOI] [PubMed] [Google Scholar]
- 25.Horvath G, Wessjohann L, Bigirimana J, Jansen M, Guisez Y, Caubergs R, Horemans N. Differential distribution of tocopherols and tocotrienols in photosynthetic and non-photosynthetic tissues. Phytochemistry. 2006;67:1185–1195. doi: 10.1016/j.phytochem.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 26.Venkatesh TV, Karunanandaa B, Free DL, Rottnek JM, Baszis SR, Valentin HE. Identification and characterization of an Arabidopsis homogentisate phytyltransferase paralog. Planta. 2006;223:1134–1144. doi: 10.1007/s00425-005-0180-1. [DOI] [PubMed] [Google Scholar]
- 27.Cahoon EB, Hall SE, Ripp KG, Ganzke TS, Hitz WD, Coughlan SJ. Metabolic redesign of vitamin E biosynthesis in plants for tocotrienol production and increased antioxidant content. Nat Biotechnol. 2003;21:1082–1087. doi: 10.1038/nbt853. [DOI] [PubMed] [Google Scholar]
- 28.Heinonen M, Piironen V. The tocopherol, tocotrienol, and vitamin E content of the average Finnish diet. Int J Vitam Nutr Res. 1991;61:27–32. [PubMed] [Google Scholar]
- 29.Jiang Q, Christen S, Shigenaga MK, Ames BN. Gamma-tocopherol, the major form of vitamin E in the US diet, deserves more attention. Am J Clin Nutr. 2001;74:714–722. doi: 10.1093/ajcn/74.6.714. [DOI] [PubMed] [Google Scholar]
- 30.Hensley K, Benaksas EJ, Bolli R, Comp P, Grammas P, Hamdheydari L, Mou S, Pye QN, Stoddard MF, Wallis G, Williamson KS, West M, Wechter WJ, Floyd RA. New perspectives on vitamin E: gamma-tocopherol and carboxyelthyl-hydroxychroman metabolites in biology and medicine. Free Radic Biol Med. 2004;36:1–15. doi: 10.1016/j.freeradbiomed.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Parker RA, Pearce BC, Clark RW, Gordon DA, Wright JJ. Tocotrienols regulate cholesterol production in mammalian cells by post-transcriptional suppression of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 1993;268:11230–11238. [PubMed] [Google Scholar]
- 32.Pearce BC, Parker RA, Deason ME, Qureshi AA, Wright JJ. Hypocholesterolemic activity of synthetic and natural tocotrienols. J Med Chem. 1992;35:3595–3606. doi: 10.1021/jm00098a002. [DOI] [PubMed] [Google Scholar]
- 33.Riley P, Al Bakir M, O'Donohue J, Crook M. Prescribing statins to patients with nonalcoholic fatty liver disease: real cardiovascular benefits outweigh theoretical hepatotoxic risk. Cardiovasc Ther. 2009;27:216–220. doi: 10.1111/j.1755-5922.2009.00088.x. [DOI] [PubMed] [Google Scholar]
- 34.Nesaretnam K, Guthrie N, Chambers AF, Carroll KK. Effect of tocotrienols on the growth of a human breast cancer cell line in culture. Lipids. 1995;30:1139–1143. doi: 10.1007/BF02536615. [DOI] [PubMed] [Google Scholar]
- 35.Khanna S, Roy S, Slivka A, Craft TK, Chaki S, Rink C, Notestine MA, DeVries AC, Parinandi NL, Sen CK. Neuroprotective properties of the natural vitamin E alpha-tocotrienol. Stroke. 2005;36:2258–2264. doi: 10.1161/01.STR.0000181082.70763.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khanna S, Parinandi NL, Kotha SR, Roy S, Rink C, Bibus D, Sen CK. Nanomolar vitamin E alpha-tocotrienol inhibits glutamate-induced activation of phospholipase A2 and causes neuroprotection. J Neurochem. 2010;112:1249–1260. doi: 10.1111/j.1471-4159.2009.06550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khanna S, Roy S, Parinandi NL, Maurer M, Sen CK. Characterization of the potent neuroprotective properties of the natural vitamin E alpha-tocotrienol. J Neurochem. 2006;98:1474–1486. doi: 10.1111/j.1471-4159.2006.04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khanna S, Roy S, Park HA, Sen CK. Regulation of c-Src activity in glutamate-induced neurodegeneration. J Biol Chem. 2007;282:23482–23490. doi: 10.1074/jbc.M611269200. [DOI] [PubMed] [Google Scholar]
- 39.Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, Sen CK. Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J Biol Chem. 2003;278:43508–43515. doi: 10.1074/jbc.M307075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sen CK, Khanna S, Roy S, Packer L. Molecular basis of vitamin E action. Tocotrienol potently inhibits glutamate-induced pp60(c-Src) kinase activation and death of HT4 neuronal cells. J Biol Chem. 2000;275:13049–13055. doi: 10.1074/jbc.275.17.13049. [DOI] [PubMed] [Google Scholar]
- 41.Kaempf-Rotzoll DE, Traber MG, Arai H. Vitamin E and transfer proteins. Curr Opin Lipidol. 2003;14:249–254. doi: 10.1097/00041433-200306000-00004. [DOI] [PubMed] [Google Scholar]
- 42.Hosomi A, Arita M, Sato Y, Kiyose C, Ueda T, Igarashi O, Arai H, Inoue K. Affinity for alpha-tocopherol transfer protein as a determinant of the biological activities of vitamin E analogs. FEBS Lett. 1997;409:105–108. doi: 10.1016/s0014-5793(97)00499-7. [DOI] [PubMed] [Google Scholar]
- 43.Sen CK, Khanna S, Rink C, Roy S. Tocotrienols: the emerging face of natural vitamin E. Vitam Horm. 2007;76:203–261. doi: 10.1016/S0083-6729(07)76008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jishage K, Arita M, Igarashi K, Iwata T, Watanabe M, Ogawa M, Ueda O, Kamada N, Inoue K, Arai H, Suzuki H. Alpha-tocopherol transfer protein is important for the normal development of placental labyrinthine trophoblasts in mice. J Biol Chem. 2001;276:1669–1672. doi: 10.1074/jbc.C000676200. [DOI] [PubMed] [Google Scholar]
- 45.Terasawa Y, Ladha Z, Leonard SW, Morrow JD, Newland D, Sanan D, Packer L, Traber MG, Farese RV., Jr Increased atherosclerosis in hyperlipidemic mice deficient in alpha-tocopherol transfer protein and vitamin E. Proc Natl Acad Sci U S A. 2000;97:13830–13834. doi: 10.1073/pnas.240462697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khanna S, Patel V, Rink C, Roy S, Sen CK. Delivery of orally supplemented alpha-tocotrienol to vital organs of rats and tocopherol-transport protein deficient mice. Free Radic Biol Med. 2005;39:1310–1319. doi: 10.1016/j.freeradbiomed.2005.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patel V, Khanna S, Roy S, Ezziddin O, Sen CK. Natural vitamin E alpha-tocotrienol: retention in vital organs in response to long-term oral supplementation and withdrawal. Free Radic Res. 2006;40:763–771. doi: 10.1080/10715760600672491. [DOI] [PubMed] [Google Scholar]
- 48.Khosla P, Patel V, Whinter JM, Khanna S, Rakhkovskaya M, Roy S, Sen CK. Postprandial levels of the natural vitamin E tocotrienol in human circulation. Antioxid Redox Signal. 2006;8:1059–1068. doi: 10.1089/ars.2006.8.1059. [DOI] [PubMed] [Google Scholar]
- 49.Contreras MA, Greiner RS, Chang MC, Myers CS, Salem N, Jr, Rapoport SI. Nutritional deprivation of alpha-linolenic acid decreases but does not abolish turnover and availability of unacylated docosahexaenoic acid and docosahexaenoyl-CoA in rat brain. J Neurochem. 2000;75:2392–2400. doi: 10.1046/j.1471-4159.2000.0752392.x. [DOI] [PubMed] [Google Scholar]
- 50.Carlson SE, Werkman SH, Peeples JM, Cooke RJ, Tolley EA. Arachidonic acid status correlates with first year growth in preterm infants. Proc Natl Acad Sci U S A. 1993;90:1073–1077. doi: 10.1073/pnas.90.3.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eichberg J. Phospholipids in Nervous Tissues. New York: Wiley; 1985. [Google Scholar]
- 52.Jones CR, Arai T, Rapoport SI. Evidence for the involvement of docosahexaenoic acid in cholinergic stimulated signal transduction at the synapse. Neurochem Res. 1997;22:663–670. doi: 10.1023/a:1027341707837. [DOI] [PubMed] [Google Scholar]
- 53.Neuringer M, Connor WE, Lin DS, Barstad L, Luck S. Biochemical and functional effects of prenatal and postnatal omega 3 fatty acid deficiency on retina and brain in rhesus monkeys. Proc Natl Acad Sci U S A. 1986;83:4021–4025. doi: 10.1073/pnas.83.11.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rapoport SI, Chang MC, Spector AA. Delivery and turnover of plasma-derived essential PUFAs in mammalian brain. J Lipid Res. 2001;42:678–685. [PubMed] [Google Scholar]
- 55.Spector AA. Plasma free fatty acid and lipoproteins as sources of polyunsaturated fatty acid for the brain. J Mol Neurosci. 2001;16:159–165. doi: 10.1385/JMN:16:2-3:159. discussion 215–221. [DOI] [PubMed] [Google Scholar]
- 56.Farooqui AA, Horrocks LA, Farooqui T. Modulation of inflammation in brain: a matter of fat. J Neurochem. 2007;101:577–599. doi: 10.1111/j.1471-4159.2006.04371.x. [DOI] [PubMed] [Google Scholar]
- 57.Rapoport SI. Arachidonic acid and the brain. J Nutr. 2008;138:2515–2520. doi: 10.1093/jn/138.12.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adibhatla RM, Hatcher JF. Phospholipase A(2), reactive oxygen species, and lipid peroxidation in CNS pathologies. BMB Rep. 2008;41:560–567. doi: 10.5483/bmbrep.2008.41.8.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clemens JA, Stephenson DT, Smalstig EB, Roberts EF, Johnstone EM, Sharp JD, Little SP, Kramer RM. Reactive glia express cytosolic phospholipase A2 after transient global forebrain ischemia in the rat. Stroke. 1996;27:527–535. doi: 10.1161/01.str.27.3.527. [DOI] [PubMed] [Google Scholar]
- 60.Lauritzen I, Heurteaux C, Lazdunski M. Expression of group II phospholipase A2 in rat brain after severe forebrain ischemia and in endotoxic shock. Brain Res. 1994;651:353–356. doi: 10.1016/0006-8993(94)90719-6. [DOI] [PubMed] [Google Scholar]
- 61.Saluja I, Song D, O'Regan MH, Phillis JW. Role of phospholipase A2 in the release of free fatty acids during ischemia-reperfusion in the rat cerebral cortex. Neurosci Lett. 1997;233:97–100. doi: 10.1016/s0304-3940(97)00646-0. [DOI] [PubMed] [Google Scholar]
- 62.Stephenson D, Rash K, Smalstig B, Roberts E, Johnstone E, Sharp J, Panetta J, Little S, Kramer R, Clemens J. Cytosolic phospholipase A2 is induced in reactive glia following different forms of neurodegeneration. Glia. 1999;27:110–128. doi: 10.1002/(sici)1098-1136(199908)27:2<110::aid-glia2>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 63.Burke JE, Dennis EA. Phospholipase A2 biochemistry. Cardiovasc Drugs Ther. 2009;23:49–59. doi: 10.1007/s10557-008-6132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adibhatla RM, Hatcher JF, Larsen EC, Chen X, Sun D, Tsao FH. CDP-choline significantly restores phosphatidylcholine levels by differentially affecting phospholipase A2 and CTP: phosphocholine cytidylyltransferase after stroke. J Biol Chem. 2006;281:6718–6725. doi: 10.1074/jbc.M512112200. [DOI] [PubMed] [Google Scholar]
- 65.Lin TN, Wang Q, Simonyi A, Chen JJ, Cheung WM, He YY, Xu J, Sun AY, Hsu CY, Sun GY. Induction of secretory phospholipase A2 in reactive astrocytes in response to transient focal cerebral ischemia in the rat brain. J Neurochem. 2004;90:637–645. doi: 10.1111/j.1471-4159.2004.02540.x. [DOI] [PubMed] [Google Scholar]
- 66.Anthonsen MW, Solhaug A, Johansen B. Functional coupling between secretory and cytosolic phospholipase A2 modulates tumor necrosis factor-alpha- and interleukin-1 beta-induced NF-kappa B activation. J Biol Chem. 2001;276:30527–30536. doi: 10.1074/jbc.M008481200. [DOI] [PubMed] [Google Scholar]
- 67.Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res. 2009;50(Suppl):S237–S242. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stephenson DT, Manetta JV, White DL, Chiou XG, Cox L, Gitter B, May PC, Sharp JD, Kramer RM, Clemens JA. Calcium-sensitive cytosolic phospholipase A2 (cPLA2) is expressed in human brain astrocytes. Brain Res. 1994;637:97–105. doi: 10.1016/0006-8993(94)91221-1. [DOI] [PubMed] [Google Scholar]
- 69.Yoshihara Y, Watanabe Y. Translocation of phospholipase A2 from cytosol to membranes in rat brain induced by calcium ions. Biochem Biophys Res Commun. 1990;170:484–490. doi: 10.1016/0006-291x(90)92117-i. [DOI] [PubMed] [Google Scholar]
- 70.Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 71.Phillis JW, Horrocks LA, Farooqui AA. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: their role and involvement in neurological disorders. Brain Res Rev. 2006;52:201–243. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 72.Beal MF, Howell N, Bodis-Wollner I. Mitochondria and Free Radicals in Neurodegenerative Diseases. New York: Wiley-Liss; 1997. [Google Scholar]
- 73.Adibhatla RM, Hatcher JF, Dempsey RJ. Lipids and lipidomics in brain injury and diseases. AAPS J. 2006;8:E314–E321. doi: 10.1007/BF02854902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 75.Kehrer JP, Biswal SS. The molecular effects of acrolein. Toxicol Sci. 2000;57:6–15. doi: 10.1093/toxsci/57.1.6. [DOI] [PubMed] [Google Scholar]
- 76.Kamal-Eldin A, Appelqvist LA. The chemistry and antioxidant properties of tocopherols and tocotrienols. Lipids. 1996;31:671–701. doi: 10.1007/BF02522884. [DOI] [PubMed] [Google Scholar]
- 77.Serbinova EA, Packer L. Antioxidant properties of alpha-tocopherol and alpha-tocotrienol. Methods Enzymol. 1994;234:354–366. doi: 10.1016/0076-6879(94)34105-2. [DOI] [PubMed] [Google Scholar]
- 78.Kamat JP, Devasagayam TP. Tocotrienols from palm oil as potent inhibitors of lipid peroxidation and protein oxidation in rat brain mitochondria. Neurosci Lett. 1995;195:179–182. doi: 10.1016/0304-3940(95)11812-b. [DOI] [PubMed] [Google Scholar]
- 79.Suzuki YJ, Tsuchiya M, Wassall SR, Choo YM, Govil G, Kagan VE, Packer L. Structural and dynamic membrane properties of alpha-tocopherol and alpha-tocotrienol: implication to the molecular mechanism of their antioxidant potency. Biochemistry. 1993;32:10692–10699. doi: 10.1021/bi00091a020. [DOI] [PubMed] [Google Scholar]
- 80.Atkinson J, Epand RF, Epand RM. Tocopherols and tocotrienols in membranes: a critical review. Free Radic Biol Med. 2008;44:739–764. doi: 10.1016/j.freeradbiomed.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 81.Hamberg M, Samuelsson B. Prostaglandin endoperoxides. Novel transformations of arachidonic acid in human platelets. Proc Natl Acad Sci U S A. 1974;71:3400–3404. doi: 10.1073/pnas.71.9.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aparoy P, Reddy RN, Guruprasad L, Reddy MR, Reddanna P. Homology modeling of 5-lipoxygenase and hints for better inhibitor design. J Comput Aided Mol Des. 2008;22:611–619. doi: 10.1007/s10822-008-9180-0. [DOI] [PubMed] [Google Scholar]
- 83.Glickman MH, Klinman JP. Lipoxygenase reaction mechanism: demonstration that hydrogen abstraction from substrate precedes dioxygen binding during catalytic turnover. Biochemistry. 1996;35:12882–12892. doi: 10.1021/bi960985q. [DOI] [PubMed] [Google Scholar]
- 84.Katsuki H, Okuda S. Arachidonic acid as a neurotoxic and neurotrophic substance. Prog Neurobiol. 1995;46:607–636. doi: 10.1016/0301-0082(95)00016-o. [DOI] [PubMed] [Google Scholar]
- 85.Canals S, Casarejos MJ, de Bernardo S, Rodriguez-Martin E, Mena MA. Nitric oxide triggers the toxicity due to glutathione depletion in midbrain cultures through 12-lipoxygenase. J Biol Chem. 2003;278:21542–21549. doi: 10.1074/jbc.M213174200. [DOI] [PubMed] [Google Scholar]
- 86.Nazarewicz RR, Zenebe WJ, Parihar A, Parihar MS, Vaccaro M, Rink C, Sen CK, Ghafourifar P. 12(S)-hydroperoxyeicosatetraenoic acid (12-HETE) increases mitochondrial nitric oxide by increasing intramitochondrial calcium. Arch Biochem Biophys. 2007;468:114–120. doi: 10.1016/j.abb.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shimizu T, Wolfe LS. Arachidonic acid cascade and signal transduction. J Neurochem. 1990;55:1–15. doi: 10.1111/j.1471-4159.1990.tb08813.x. [DOI] [PubMed] [Google Scholar]
- 88.Rao AM, Hatcher JF, Kindy MS, Dempsey RJ. Arachidonic acid and leukotriene C4: role in transient cerebral ischemia of gerbils. Neurochem Res. 1999;24:1225–1232. doi: 10.1023/a:1020916905312. [DOI] [PubMed] [Google Scholar]
- 89.Unterberg A, Schmidt W, Wahl M, Baethmann A. Role of leukotrienes as mediator compounds in brain edema. Adv Neurol. 1990;52:211–214. [PubMed] [Google Scholar]
- 90.Canetti C, Hu B, Curtis JL, Peters-Golden M. Syk activation is a leukotriene B4–regulated event involved in macrophage phagocytosis of IgG-coated targets but not apoptotic cells. Blood. 2003;102:1877–1883. doi: 10.1182/blood-2003-02-0534. [DOI] [PubMed] [Google Scholar]
- 91.Coffey MJ, Phare SM, Peters-Golden M. Role of leukotrienes in killing of Mycobacterium bovis by neutrophils. Prostaglandins Leukot Essent Fatty Acids. 2004;71:185–190. doi: 10.1016/j.plefa.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 92.Flamand N, Mancuso P, Serezani CH, Brock TG. Leukotrienes: mediators that have been typecast as villains. Cell Mol Life Sci. 2007;64:2657–2670. doi: 10.1007/s00018-007-7228-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Michelassi F, Landa L, Hill RD, Lowenstein E, Watkins WD, Petkau AJ, Zapol WM. Leukotriene D4: a potent coronary artery vasoconstrictor associated with impaired ventricular contraction. Science. 1982;217:841–843. doi: 10.1126/science.6808665. [DOI] [PubMed] [Google Scholar]
- 94.Schellenberg RR, Foster A. Differential activity of leukotrienes upon human pulmonary vein and artery. Prostaglandins. 1984;27:475–482. doi: 10.1016/0090-6980(84)90205-3. [DOI] [PubMed] [Google Scholar]
- 95.Dahlen SE, Bjork J, Hedqvist P, Arfors KE, Hammarstrom S, Lindgren JA, Samuelsson B. Leukotrienes promote plasma leakage and leukocyte adhesion in postcapillary venules: in vivo effects with relevance to the acute inflammatory response. Proc Natl Acad Sci U S A. 1981;78:3887–3891. doi: 10.1073/pnas.78.6.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hambrecht GS, Adesuyi SA, Holt S, Ellis EF. Brain 12-HETE formation in different species, brain regions, and in brain microvessels. Neurochem Res. 1987;12:1029–1033. doi: 10.1007/BF00970932. [DOI] [PubMed] [Google Scholar]
- 97.Bendani MK, Palluy O, Cook-Moreau J, Beneytout JL, Rigaud M, Vallat JM. Localization of 12-lipoxygenase mRNA in cultured oligodendrocytes and astrocytes by in situ reverse transcriptase and polymerase chain reaction. Neurosci Lett. 1995;189:159–162. doi: 10.1016/0304-3940(95)11482-c. [DOI] [PubMed] [Google Scholar]
- 98.Tan S, Schubert D, Maher P. Oxytosis: a novel form of programmed cell death. Curr Top Med Chem. 2001;1:497–506. doi: 10.2174/1568026013394741. [DOI] [PubMed] [Google Scholar]
- 99.Khanna S, Venojarvi M, Roy S, Sen CK. Glutamate-induced c-Src activation in neuronal cells. Methods Enzymol. 2002;352:191–198. doi: 10.1016/s0076-6879(02)52019-x. [DOI] [PubMed] [Google Scholar]
- 100.Paul R, Zhang ZG, Eliceiri BP, Jiang Q, Boccia AD, Zhang RL, Chopp M, Cheresh DA. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat Med. 2001;7:222–227. doi: 10.1038/84675. [DOI] [PubMed] [Google Scholar]