

Abstract
Bacterial histidine kinases (HK) are members of the GHKL superfamily, which share a unique adenosine triphosphate (ATP)-binding Bergerat fold. Our previous studies have shown that Gyrase, Hsp90, MutL (GHL) inhibitors bind to the ATP-binding pocket of HK and may provide lead compounds for the design of novel antibiotics targeting these kinases. In this article, we developed a competition assay using the fluorescent ATP analog, 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate. The method can be used for high-throughput screening of compound libraries targeting HKs or other ATP-binding proteins. We utilized the assay to screen a library of GHL inhibitors targeting the bacterial HK PhoQ, and discuss the applications of the 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate competition assay beyond GHKL inhibitor screening.
Introduction
Bacterial two-component systems (TCS) function as the primary signal transduction system in bacteria. A typical TCS consists of a ligand-responsive histidine kinase (HK) and a response regulator, which acts primarily through transcriptional control to facilitate adaptive responses to numerous environmental stimuli.1–3 Upon activation via extracellular stimuli, HK binds adenosine triphosphate (ATP) and autophosphorylates a conserved histidine residue. The phosphoryl group is then transferred to a conserved aspartic acid on its cognate response regulator. The phosphorylated response regulator can then orchestrate a cellular response, most commonly through binding of downstream DNA or proteins.1–5
A typical bacterial HK consists of a periplasmic sensor domain, flanked by two transmembrane regions, and a catalytic cytoplasmic region. The cytoplasmic region consists of two distinct domains: a four-helical bundle dimerization domain, which houses the conserved His residue, and an ATP-binding catalytic domain.6,7 The ATP-binding motif of bacterial HKs dramatically differs from the typical eukaryotic ATP-binding domains of Ser, Thr, and Tyr kinases. Solution and crystal structures of several catalytic domains, exemplified by EnvZ, CheA, and PhoQ,8–10 reveal a highly conserved domain core that shares a unique Bergerat ATP-binding fold with a diverse set of proteins, which includes DNA gyrase, Hsp90, and MutL, together referred to as the “GHKL superfamily.”11 Despite minimal sequence identity, the structures of the ATP-binding pockets of this superfamily display high topological similarity. The core of the Bergerat fold consists of an α/β sandwich, comprised of a four-stranded antiparallel β-sheet and three α-helices. A highly variable loop, referred to as the “ATP lid,” connects helix α3 and β-strand β3 in HKs, and its conformation and position relative to the bound nucleotide are strikingly different in each member of the GHKL family.8–11
The omnipresent nature of the TCS in bacteria, unconventional phosphorylation substrates, unique Bergerat fold, and notable absence from the animal kingdom make the TCS HK an ideal target for novel antibiotic design.3,12–15 Traditional high-throughput screening (HTS) targeting these kinases has typically utilized random small molecule libraries, screening for differential growth, inhibition of ATPase activity, or decreased TCS-regulated gene expression.12,16 These screens have identified bactericidal compounds; however, their mechanism of inhibition is often TCS independent, and these compounds generally lack potency or display eukaryotic cytotoxicity.12,16 On the other hand, inhibitors targeting the Bergerat fold of GHL family proteins, in particular Hsp90, are extensively developed as anticancer therapeutics.17,18 The Hsp90 inhibitor radicicol, a natural antifungal compound, has been shown to bind to Hsp90's Bergerat fold and inhibit its activity in vitro by directly competing with ATP.17–28 It has also been shown to inhibit the activity of the Saccharomyces cerevisiae Sln1 HK.29 Due to the highly conserved topology of the Bergerat fold, there is potential for the exploitation of such GHL inhibitors as novel bacterial HK inhibitors.30
We have chosen the Salmonella PhoPQ TCS as our model system to explore the possibility of designing inhibitors targeting bacterial HKs. HK PhoQ has been shown to detect extracellular Mg2+, acidic pH, and antimicrobial peptides. In response to these stimuli, the PhoPQ regulon controls ∼3% of the Salmonella genome.33–37 The PhoPQ TCS is critical for Salmonella virulence.33 Salmonella strains with mutations in the phoP or phoQ locus lead to attenuation in virulence, and the median lethal dose of PhoP or PhoQ null mutants in mice are five orders of magnitude higher than that of wild-type Salmonella.38–40 This system is also commonly found in many other gram-negative bacteria, such as Escherichia coli, Shigella, and Yersinia sp., making it an excellent model system to investigate the potential for TCS inhibition in pathogenic species.41,42
Recently, we showed that radicicol binds weakly to the PhoQ ATP-binding pocket, based upon Nuclear Magnetic Resonance (NMR) and crystallographic structure analysis.30 Further, both ATP and radicicol displace a fluorescent ATP analog 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate (TNP-ATP) from the ATP-binding pocket, supporting that radicicol binds in the ATP-binding pocket. These data suggest that GHL inhibitors may indeed be utilized as lead compounds or scaffolds for the development of new antibiotics targeting PhoQ and other bacterial HKs.
Performing HTS using the PhoQ catalytic domain (PhoQcat), which harbors the ATP-binding pocket, with a large number of GHL inhibitors may enable us to identify a much tighter binding inhibitor. Since PhoQcat only binds, but does not hydrolyze ATP,10 we need to develop an assay to identify compounds that inhibit ATP-binding as opposed to hydrolysis. A number of assays have been developed for Ser/Thr or Tyr kinases to identify inhibitors that competitively displace fluorescent-labeled or enzyme-coupled compound bound to the ATP-binding pocket. For example, Vainshtein et al. utilized staurosporine, a potent broad-spectrum kinase inhibitor, conjugated to a small fragment of β-galactosidase.61 Upon displacement from the ATP-binding pocket, conjugated staurosporine is free in solution to form an active enzyme with the complementary fragment of β-galactosidase and produce enzymatic activity. Lebakken et al. uses a fluorophore-labeled staurosporine and time-resolved fluorescence resonance energy transfer to monitor displacement of staurosporine by small molecules from the ATP-binding pocket.63 The assay developed by Kashem et al. takes advantage of a proprietary fluorescent kinase probe and the fluorescence polarization signal reduction upon displacement of this probe.62 Compounds/probes used in these assays typically bind in the ATP-binding pocket of Ser/Thr and Tyr kinases. However, it is unclear whether these compounds bind the ATP-binding pocket of HKs, considering the dramatic differences between the Bergerat ATP-binding fold of HKs and the ATP-binding site of Ser/Thr or Tyr kinases. On the other hand, TNP-ATP, a fluorescent ATP analog, has been shown to bind to the ATP-binding pocket of HKs.8,46,50 In this article, we demonstrate that TNP-ATP binds to PhoQcat and can be competed off by compounds binding to the ATP-binding pocket. We developed an assay based on this for the screening of inhibitors targeting the ATP-binding pocket of PhoQ. Although we have focused upon a bacterial HK as we developed this assay, it holds promise for screening inhibitors targeting other ATP-binding proteins.
Materials and Methods
Purification of PhoQcat
Salmonella typhimurium PhoQ residues 332–487 (PhoQcat) were subcloned into a pGEX-6p1 vector (GE Healthcare) and expressed in E. coli strain XA90 as a glutathione S-transferase (GST) fusion protein. Bacteria were grown at 37°C until OD600 = 0.5–0.6 and were induced with 0.25 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) overnight at room temperature. Bacteria cells were harvested by centrifugation at 5500 rpm for 10 min, resuspended in 50 mM Tris–HCl, pH 7.5, 50 mM NaCl, and lysed via sonication. The fusion protein was first purified using glutathione Sepharose resin (GE Healthcare) and cleaved overnight with PreScission Protease (GE Healthcare) at 4°C to remove glutathione S-transferase (GST). The resultant PhoQcat was further purified using a Superdex-200 gel filtration column (GE Healthcare). Peak fractions were pooled and concentrated using Amicon Ultra centrifugal filter units (Millipore).
Fluorescence Measurements
Fluorescent emission scanning spectra of TNP-ATP (Sigma–Aldrich) and TNP-ATP:PhoQcat complex were obtained using a Fluoromax-3 fluorometer (HORIBA Jobin Yvon) with an excitation wavelength of 403 nm. All other fluorescent measurements were performed using a Biotek Synergy HT plate-reader, equipped with emission and excitation filters of 400/30 and 540/25 nm, respectively (Bio-Tek Instruments). All measurements were obtained in 96-well, black, half-volume, flat-bottom plates (Greiner Bio-One) at 25°C, in 50 μL reaction volumes.
Determination of Dissociation Constants
To examine the interaction between PhoQcat and TNP-ATP, we carried out titration experiments. Increasing concentrations of PhoQcat were added to a constant concentration of TNP-ATP (150 μM) in a buffer containing 50 mM Tris-HCl, pH 7.5, and 50 mM NaCl in 50 μL reaction volumes. Samples were gently shaken for 10 min on an orbital shaker at room temperature before each read.
Relative fluorescent units (RFU) versus PhoQcat concentration was plotted using the program Kaleidagraph (Synergy Software). To obtain the value of the dissociation constant, Kd, we applied the following Langmuir single-site binding equation for a curve fit:
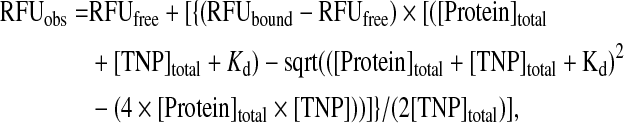 |
where RFU obs, free, and bound are the RFU observed, RFU of free TNP-ATP, and RFU of TNP-ATP when completely bound to protein, respectively, and [Protein] and [TNP] are the concentrations of PhoQcat and TNP-ATP, respectively.
To determine the dissociation constants for competitors (ATP or small molecule compounds), we employed a competitive binding assay. Increasing concentrations of competitor were added to preincubated PhoQcat:TNP-ATP (300 μM:150 μM) complex. Samples were gently shaken at room temperature for 10 min before fluorescence reading. Fraction bound was calculated as
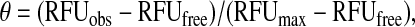 |
where θ is the fraction TNP-ATP-bound protein and RFUmax is the maximal RFU observed at saturation (100% TNP-ATP bound).
Plots of fraction bound versus concentration of inhibitor were analyzed by curve fitting using the following equation as described in Batey and Williamson43:
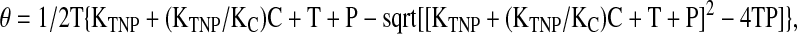 |
where θ is the fraction-bound protein; KTNP and KC are the dissociation constants for TNP-ATP and competitor, respectively; and T, C, and P are the concentrations of TNP-ATP, competitor, and protein, respectively.
Z′-Factor Determination
To determine Z′-factor,44 we measured fluorescence of the TNP-ATP:PhoQcat complex as a negative control. Since there are no known PhoQcat inhibitors, we used high concentration (10 mM) of ATP to mimic the complete ATP-binding inhibition of PhoQcat (positive control) and measured fluorescence of TNP-ATP:PhoQcat + 10 mM ATP. Z′-factor was determined using the following equation:
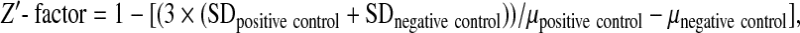 |
where SD and μ are the standard deviations and means, respectively.44
Small Molecule Compound Library Screening
We screened a small molecule library containing 288 Hsp90 inhibitor analogs (geldanamycin-radicicol chimeras and novobiocin analogs) using the TNP-ATP displacement assay (Table 1). Library compounds (100 μM) were incubated with pre-mixed PhoQcat:TNP-ATP complex (100 μM:150 μM) in 96-well, half-volume plates, in a final volume of 50 μL. Positive (10 mM ATP) and negative (dimethyl sulfoxide alone) inhibition control wells were included in every screening plate for quality control purposes. ATP (100 μM) and radicicol (100 μM) were also included so the extent of inhibition of other compounds can be compared with ATP and radicicol. Plates were gently rocked at low speed for 15 min and samples were read using a Biotek Synergy HT as described above. All screens were performed in triplicate. The top hits from the library were then combined with 80 compounds from the National Cancer Institute (NCI) Diversity Set, and screened under the same conditions in triplicate. Protein concentration was tested before incubation with compounds, and again after 15-min incubation and centrifugation, to ensure protein concentration remained constant and observed fluorescence was not affected by protein precipitation.
Table 1.
2′,3′-O-(2,4,6-Trinitrophenyl) Adenosine 5′-Triphosphate Displacement Assay Protocol
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Premix | 100 μM:150 μM | PhoQcat:TNP-ATP |
| 2 | Controls | 0.5 μL | ATP, DMSO, radicicol |
| 3 | Library compounds | 0.5 μL | 10 mM stock |
| 4 | PhoQcat:TNP-ATP | 49.5 μL | Premixed, Step 1 |
| 5 | Incubation Time | 15 min | Ambient temperature, Shaker |
| 6 | Assay readout | 400 nm (Ex)/540 nm (Em) | Fluorescence detection (relative fluorescent units) |
| Step | Notes |
|---|---|
| 1 | Care should be taken to minimize TNP-ATP light exposure. All protein and reagents should be kept on ice before use. Protein storage buffer consisting of 50 mM Tris, pH 7.5, and 50 mM NaCl utilized for dilution of protein. TNP-ATP dissolved in double distilled water (ddH2O), pH adjusted to 7.0. |
| 2 | Columns 1 and 12 reserved for control wells, alternately plated with 100 μM ATP, 100 μM radicicol, and 0.5 μL DMSO. |
| 4 | Multichannel pipette utilized for rapid, uniform dispensing. |
| 5 | Plates covered with adhesive seal. Gently shake (50–100 rpm) on orbital shaker. |
| 6 | Excitation filter of 400/30 and emission filter of 540/25 utilized on a Biotek Synergy HT plate reader, set to fluorescence mode, sensitivity set to 100. |
TNP-ATP, 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate; ATP, adenosine triphosphate; DMSO, dimethyl sulfoxide.
Results and Discussion
Fluorescent ATP Analog TNP-ATP Binds to PhoQcat
The fluorescent ATP analog, TNP-ATP, has been utilized as a valuable probe for examining the nucleotide binding of various eukaryotic and bacterial kinases.45–50 Extensive biochemical and structural characterization of the probe bound to CheA and EnvZ has revealed that TNP-ATP occupies the nucleotide-binding pocket of these bacterial HKs.8,46,50 TNP-ATP displays minimal fluorescence in solution by itself. However, upon binding to protein, a significant fluorescence increase is observed. We found that the relative fluorescence of 200 μM TNP-ATP increases ∼4-fold in the presence of equimolar concentrations of PhoQcat, with a λmax of 538 nm (Fig. 1).
Fig. 1.

Fluorescence emission spectra of TNP-ATP in the absence and presence of PhoQcat. Scanning emission spectra of TNP-ATP alone (gray) and in complex with PhoQcat (black) were collected using an excitation wavelength of 403 nm, and emission wavelength from 500 to 600 nm. TNP-ATP, 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate; PhoQcat, PhoQ catalytic domain.
To determine the binding affinity of the TNP-ATP to PhoQcat, we titrated increasing amounts of PhoQcat into constant concentration of TNP-ATP in a Tris-NaCl buffer. Titrating in the PhoQcat protein while keeping TNP-ATP concentration constant avoided the need for inner filter effect correction due to increasing concentrations of TNP-ATP. Fluorescence intensity versus protein concentration values were plotted and fit into a Langmuir single-site binding equation. We determined the dissociation constant, Kd, for the TNP-ATP:PhoQcat interaction to be 294 ± 33 μM, with a R2 value of 0.99 for curve fit (Fig. 2).
Fig. 2.

Determination of TNP-ATP:PhoQcat dissociation constant. Direct titration of PhoQcat into a TNP-ATP solution leads to the TNP-ATP:PhoQcat complex formation in a concentration-dependent fashion. Curve-fitting of the relative fluorescent units versus PhoQcat concentration to a Langmuir single-site binding equation determined the Kd of the TNP-ATP:PhoQcat interaction to be 294 ± 33 μM. Error bars represent standard deviation of n = 3 values. Kd, dissociation constant.
This dissociation constant between TNP-ATP and PhoQcat is significantly higher than the Kd previously reported for both the TNP-ATP:CheA and TNP-ATP:EnvZ HK interactions (1.9 and 0.5 μM, respectively)8,46,50 Despite the overall structural similarity of the nucleotide-binding pockets of the bacterial HKs, the spacing between conserved motifs, as well as the specific interacting residues within the ATP lid, is unique to each kinase, likely contributing to this differential binding affinity. Indeed, the space-filling models of nucleotides bound to CheA and EnvZ reveal significantly different binding orientations of the adenine ring and triphosphate moieties between the two proteins8,46,50 The distinct nature of nucleotide-binding determinants in the ATP-lid region of GHKL family members may also explain the large affinity differential previously observed between radicicol:Hsp90 and radicicol:PhoQcat binding (19 nm and 715 μM, respectively),17,30 as well as differences in other inhibitor:GHKL family member interactions.17–19,29,30,51,52 As such, although the design of a broad-spectrum antibiotic targeting all HKs may prove challenging, the potential for design of a high-specificity antibiotic targeting specific classes of HKs, but not the eukaryotic GHL family members, is promising. It is worth mentioning that nucleotide specificity may also be influenced by a HK's propensity to dimerize.50 The PhoQcat construct (without the dimerization domain) is monomeric; thus, values obtained herein reflect the binding properties of the isolated catalytic domain, and may not be fully indicative of full-length PhoQ nucleotide binding.
ATP Competes with TNP-ATP for Binding to PhoQcat's Nucleotide-Binding Pocket
ATP has been shown to displace TNP-ATP from the nucleotide-binding pocket in TNP-ATP:protein complexes, resulting in reduction of relative fluorescence.8,46,50 We found that ATP also displaces TNP-ATP from the TNP-ATP:PhoQcat complex, and utilized this displacement assay to determine the Kd of ATP binding to PhoQcat. We titrated ATP into an equilibrated TNP-ATP:PhoQcat complex. Reduction in fluorescence was observed in a concentration-dependent manner, eventually reducing fluorescence to the levels of free TNP-ATP (Fig. 3), indicative of complete displacement of the fluorescent TNP-ATP. Fraction of TNP-ATP bound was plotted against concentration of ATP and fitted to an equation describing the competitive binding (see Materials and Methods for details) to determine the dissociation constant of unmodified ATP for PhoQcat. The Kd was determined to be 412 ± 72 μM, with a R2 value of 0.97 for curve fitting (Fig. 3). This value is significantly higher than that observed for EnvZ (60 μM),50 but is close to that reported for CheA (260 μM).8,46
Fig. 3.

Unmodified ATP competes with TNP-ATP for PhoQcat binding. Titration of unmodified ATP into a TNP-ATP:PhoQcat complex solution displaces TNP-ATP, leading to a decrease in relative fluorescence. Curve-fitting the plot of fraction of TNP-ATP bound versus ATP concentration determined the Kd of the ATP:PhoQcat interaction to be 412 ± 72 μM. Error bars represent standard deviation of n = 3 values. ATP, adenosine triphosphate.
TNP-ATP binds both CheA and EnvZ more tightly than it binds unmodified ATP.8,46,50 We observed a similar trend with the TNP-ATP:PhoQcat interaction. TNP-ATP binds ∼1.4-fold tighter than unmodified ATP. However, the difference in binding between unmodified ATP and TNP-ATP is minimal compared to that of EnvZ and CheA (∼30-fold and 500-fold, respectively),8,46,50 again implicating unique nucleotide-binding determinants within the Bergerat fold.
Although the affinity of ATP to PhoQcat is relatively low (∼412 μM), it does not preclude the development of high affinity inhibitors of the ATP-binding pocket of PhoQcat. Residues in the conserved N, G1, G2, G3 boxes, and the ATP-lid of sensor kinases all participate in ATP binding.11 These conserved boxes and the ATP-lid span over 90 residues on the primary sequence and are located on multiple secondary structures (i.e., α2, β3, α3, and the loops between β3 and α2 as well as between β4 and β5 in PhoQcat). Together they form a large binding pocket for ATP and possibly other compounds. That ATP has a low binding affinity to Salmonella PhoQcat is likely an indication that ATP has not made sufficient interactions with most or all residues in the ATP-binding pocket to reach high affinity. Other compounds may make contacts with different residues in this pocket or make more contacts that enable them to bind PhoQcat with a much higher affinity. For example, ATP binds Hsp90 with a Kd ∼132 μM,64 but radicicol binds to the same ATP-binding pocket with a Kd ∼19 nM.17 ATP binds Gyrase with a Kd of 590 μM.65 However, multiple clorobiocin derivatives bind Gyrase with a Kd around 260 nM.66 There are also many other examples where starting compounds with low affinity (Kd between 500 μM and 1 mM) have been improved to high affinity inhibitors (Kd between 200 nM and 1 μM).67–70 Screening more GHL inhibitors or carrying out an unbiased HTS using a diversity-based compound library and further optimization can potentially identify compounds that make significant interaction with the ATP-binding pocket and has a much tighter binding affinity to PhoQcat.
The GHL Inhibitor Radicicol Competes with TNP-ATP
We recently reported the Kd for the radicicol:PhoQcat interaction to be 715 ± 78 μM, using NMR chemical shift perturbation analysis.30 This provided us with a positive control to validate the fluorescent competition assay. As expected, radicicol displaced TNP-ATP in a concentration-dependent fashion and the dissociation constant determined by the competition assay is 750 ± 111 μM, with a R2 value of 0.91 (Fig. 4), similar to the Kd determined by NMR.
Fig. 4.

Determination of Radicicol:PhoQcat dissociation constant. Titration of radicicol into a TNP-ATP:PhoQcat complex solution displaces TNP-ATP, leading to a decrease in relative fluorescence. Fitting the plot of fraction of TNP-ATP-bound versus Radicicol concentration determined the Kd of the radicicol:PhoQcat interaction to be 750 ± 111 μM. By comparison, the Kd for this interaction was determined to be 715 ± 78 μM via NMR. Error bars represent standard deviation of n = 3 values.
Screening of the Hsp90 Inhibitor Library and the NCI Diversity Set
To evaluate the potential of the TNP-ATP displacement assay as a HTS assay to identify inhibitors that bind to the ATP-binding pocket, we determined the Z′-factor44 of the assay. The complex between TNP-ATP and PhoQcat was utilized as a negative control, and 10 mM ATP was added to mimic total inhibition of TNP-ATP:PhoQcat complex formation (positive control), as indicated by a decrease in fluorescence emission to levels of TNP-ATP alone. Fluorescence from plates of total inhibition and no inhibition controls were measured to calculate the Z′-factor (see Materials and Methods for details), yielding a value of 0.93 (Fig. 5), indicative of an assay feasible for HTS.44
Fig. 5.

Determination of the Z′-factor for the TNP-ATP displacement assay. Negative control samples with no inhibition (TNP-ATP:PhoQcat complex, triangles) and positive control samples with total inhibition (TNP-ATP:PhoQcat + 10 mM ATP, squares) were measured to determine the Z′-factor and the assay's high-throughput screening capability.
We first applied this TNP-ATP displacement assay to a small Hsp90 inhibitor library of 288 compounds (containing geldanamycin/radicicol chimeras and novobiocin analogs). Compounds (100 μM) as well as 100 μM unmodified ATP and radicicol (as controls) were used in the screening. Twelve of the compounds screened were found to decrease fluorescence at levels greater than both radicicol and unmodified ATP (Fig. 6). Compound JH-II-126 was identified as the top hit in the Hsp90 inhibitor library screen. Utilizing our competition assay, the Kd of the compound was determined to be 391 ± 61 μM with an R2 value of 0.97 (Fig. 7). This affinity increases roughly twofold compared to radicicol. Although the affinity of JH-II-126 is still low, this compound may provide a template for a new analog library. There have been examples in which starting compounds with low affinity (Kd between 590 μM and 1 mM) have been improved to high affinity inhibitors (Kds of 200 nM to 10 μM) via structural-guided design.53,54 Therefore, the current affinity of radicicol analog JH-II-126 for PhoQcat suggests that the compound may be a promising candidate for optimization via further screening in correlation with structure-guided approaches.
Fig. 6.

Scatter plot displaying a representative screen of the Hsp90 inhibitor analog library. (A) A compound library consisting of 288 geldanamycin/radicicol chimeras and novobiocin analogs was screened using the TNP-ATP displacement assay. Top hits, displaying greater TNP-ATP displacement than both ATP and Radicicol, are shown in black squares. Other compounds are represented in grey diamonds. Also shown in black are blank (dimethyl sulfoxide control), ATP, and Radicicol. (B) Chemical structures of radicicol (1), novobiocin (2), geldanamycin (3), and JH-II-126 (4).
Fig. 7.

Determination of JH-II-126:PhoQcat dissociation constant. Titration of JH-II-126 into a TNP-ATP:PhoQcat complex solution displaces TNP-ATP, leading to a decrease in relative fluorescence. The Kd of the JH-II-126:PhoQcat interaction determined from this competition assay is 391 ± 61 μM. Error bars represent standard deviation of n = 3 values.
To further validate the ability of the TNP-ATP displacement assay to identify HK inhibitors, we screened 80 randomly selected compounds from the NCI Diversity Set (which contains ∼2000 structurally diverse compounds), combined with the top 12 hits from the radicicol library. None of the small molecules from the NCI library displayed greater TNP-ATP displacement than radicicol, whereas the top hits from the radicicol analog library were clearly identified as top competitors (Fig. 8).
Fig. 8.

Scatter plot displaying a representative screen of the NCI Diversity Library. The top 12 hits from the GHL library screen (black) were combined with randomly selected compounds from the NCI structural diversity set (gray). Also shown in black are ATP, radicicol, and blank (dimethyl sulfoxide control) samples. The top hit from both the GHL and NCI libraries was compound JH-II-126, encircled. NCI, National Cancer Institute.
The Potential of the TNP-ATP Displacement Assay in Drug Development
Resistance to conventional antibiotics has been reported for every major group of bacterial pathogens. Multi-drug and pandrug-resistant strains of Gram-negative bacteria are becoming increasingly prevalent, and there are a rising number of bacteria exhibiting vancomycin resistance, the antibiotic often used as a last resort.55,56 Further, only four new classes of antibiotic have been introduced into clinical practice in the past 40 years.55 Thus, the development of a novel class of antibiotics presents an urgent task for the scientific community.
Bacterial HKs present attractive antibiotic targets due to their absence from the animal kingdom.3 In addition, the bacterial HK ATP-binding fold, the Bergerat fold, and the phosphorylation substrates of the TCS vastly differ from that of conventional eukaryotic Ser, Thr, and Tyr kinases.57 Clinically relevant bacteria, including enterohemorrhagic E. coli 0157, Methicillin-resistant Staphylococcus aureus (MRSA), and vancomycin-resistant MRSA, contain numerous HK-encoding genes,12 and the virulence of several other pathogenic bacteria is also regulated by TCS signaling.41,42 Traditional HTS screens targeting such bacterial HKs have identified bactericidal compounds. However, the bactericidal properties of these compounds are often multi-mechanistic, independent of TCS inhibition, or due to nonspecific protein aggregation.12,15,16,57 One potential approach to more specifically target HKs is to take advantage of the Bergerat fold conservation and combine HTS of known GHL inhibitors with structure-guided design, utilizing GHL inhibitors as scaffolds for optimization. We have recently reported the binding of the GHL inhibitor, radicicol, to the nucleotide-binding pocket of the PhoQcat.30 In this article, we expand upon these findings and explore the potential of using HTS approach to identify a tighter-binding HK inhibitor, using a TNP-ATP displacement based assay.
The TNP-ATP displacement assay we developed can be used to identify compounds capable of inhibiting the nucleotide binding of bacterial HKs using HTS approaches. Although we only screened a library of ∼300 compounds, the screen is scaleable for HTS capabilities. The assay can be used to screen through structurally diverse libraries, as well as structurally focused compound libraries, such as the Hsp90 inhibitor library reported here. Our limited Hsp90 inhibitor library screen has identified compound JH-II-126, which binds with twofold greater affinity than radicicol. There is a wealth of GHL inhibitors, particularly those targeting Hsp90, that have recently been developed for anticancer therapy18–20,23,26,28 and can be screened for their binding to HKs. As such, this compound represents just one of many potential lead compounds based upon known GHL inhibitor scaffolds. Structural analysis will likely provide a critical complementary approach to HTS of GHL inhibitors, especially in cases where initial binding affinities are weak. A detailed analysis of the JH-II-126:PhoQcat structure, for example, may help to identify key binding moieties and aid in the design of compounds with enhanced affinities for the nucleotide-binding pocket of PhoQcat.
The current TNP-ATP displacement assay has some limitations. Due to the relatively weak binding affinity between TNP-ATP and PhoQcat (Kd ∼294 μM), we need to use large quantity of protein (100 μM) and TNP-ATP (150 μM) to obtain sufficient fluorescent signal. The concentration of the screening compounds therefore has to be rather high (we have used 100 μM in our pilot screens) to demonstrate significant inhibition. A higher affinity fluorescent ATP analog would likely circumvent this problem, allowing for HTS at lower compound concentrations, in addition to requiring less protein and ATP analogs. There are currently a number of fluorescent ATP analogs available other than trinitrophenol analogs.71 For example, MANT-ATP, a methyl anthraniloyl-substituted nucleotide, has been shown to bind HK PrrB from Mycobacterium tuberculosis with an affinity of 0.17 μM.72 It will be interesting to test in the future whether these other fluorescent ATP analogs can bind PhoQcat and whether the affinity is higher than that of TNP-ATP bound to PhoQcat.
Fluorescent TNP-ATP displacement presents a useful screening method for identifying inhibitors of proteins or protein domains that bind ATP, but do not hydrolyze ATP. For example, the C-terminal domain of Hsp90 houses an alternative, non-Bergerat ATP-binding site, which becomes accessible upon N-terminal Bergerat pocket occupation.19,58 Protein domains such as the isolated H4-H5 loop of the Na + /K + ATPase, recently identified as another novel target for anti-cancer therapeutics, also present ideal targets for TNP-ATP displacement screening.59 In addition, many proteins capable of hydrolyzing unmodified ATP are incapable of hydrolyzing TNP-ATP. For example, full-length HK CheA does not hydrolyze TNP-ATP, despite binding the probe significantly tighter than unmodified ATP, which it readily hydrolyzes.8,46 A similar occurrence is observed for ATP-dependent DNA helicases.48,60 TNP-ATP displacement has also been reported for unconventional nucleotide-interacting proteins such as ATP-sensitive K+ channels.48,49 In cases where proteins are capable of hydrolyzing ATP, but incapable of hydrolyzing TNP-ATP, TNP-ATP displacement can also be utilized as a screening tool.
Abbreviations
- ATP
adenosine triphosphate
- ddH2O
double distilled water
- GHL
Gyrase, Hsp90, MutL
- GST
glutathione S-transferase
- HK
histidine kinase
- HTS
high-throughput screening
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- Kd
dissociation constant
- MRSA
Methicillin-resistant Staphylococcus aureus
- NCI
National Cancer Institute
- NMR
Nuclear Magnetic Resonance
- PhoQcat
PhoQ catalytic domain
- TCS
two-component system
- TNP-ATP
2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate
- RFU
relative fluorescent units
Acknowledgments
We thank Melanie Blevins for help with PhoQcat purification and radicicol library screening; Changwei Liu, Brook Hirsch, and the Biophysics core at the University of Colorado Denver for help with fluorescence experiments; and Drs. Jeff Kieft, David Bain, and Robert Batey for helpful discussions. This work was supported by grants from the University of Colorado Cancer Center, Cancer League of Colorado, and in part by an American Cancer Society research scholar grant (RSG-06-165-01-GMC) and an NIH grant (5R01GM080334) to R.Z.
Disclosure Statement
No competing financial interests exist.
References
- 1.Hoch JA. Two-Component Signal Transduction. American Society for Microbiology; Washington, DC: 1995. [Google Scholar]
- 2.Hoch JA. Two-component and phosphorelay signal transduction. Curr Opin Microbiol. 2000;3:165–170. doi: 10.1016/s1369-5274(00)00070-9. [DOI] [PubMed] [Google Scholar]
- 3.Stock AM. Robinson VL. Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 4.Stock JB. Stock AM. Mottonen JM. Signal transduction in bacteria. Nature. 1990;344:395–400. doi: 10.1038/344395a0. [DOI] [PubMed] [Google Scholar]
- 5.Eguchi Y. Utsumi R. Introduction to bacterial signal transduction networks. Adv Exp Med Biol. 2008;631:1–6. doi: 10.1007/978-0-387-78885-2_1. [DOI] [PubMed] [Google Scholar]
- 6.Tomomori C, et al. Solution structure of the homodimeric core domain of Escherichia coli histidine kinase EnvZ. Nat Struct Biol. 1999;6:729–734. doi: 10.1038/11495. [DOI] [PubMed] [Google Scholar]
- 7.Yamada S. Shiro Y. Structural basis of the signal transduction in the two-component system. Adv Exp Med Biol. 2008;631:22–39. doi: 10.1007/978-0-387-78885-2_3. [DOI] [PubMed] [Google Scholar]
- 8.Bilwes AM. Quezada CM. Croal LR. Crane BR. Simon MI. Nucleotide binding by the histidine kinase CheA. Nat Struct Biol. 2001;8:353–360. doi: 10.1038/86243. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka T, et al. NMR structure of the histidine kinase domain of the E. coli osmosensor EnvZ. Nature. 1998;396:88–92. doi: 10.1038/23968. [DOI] [PubMed] [Google Scholar]
- 10.Marina A. Mott C. Auyzenberg A. Hendrickson WA. Waldburger CD. Structural and mutational analysis of the PhoQ histidine kinase catalytic domain. Insight into the reaction mechanism. J Biol Chem. 2001;276:41182–41190. doi: 10.1074/jbc.M106080200. [DOI] [PubMed] [Google Scholar]
- 11.Dutta R. Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci. 2000;25:24–28. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- 12.Matsushita M. Janda KD. Histidine kinases as targets for new antimicrobial agents. Bioorg Med Chem. 2002;10:855–867. doi: 10.1016/s0968-0896(01)00355-8. [DOI] [PubMed] [Google Scholar]
- 13.Barrett JF. Hoch JA. Two-component signal transduction as a target for microbial anti-infective therapy. Antimicrob Agents Chemother. 1998;42:1529–1536. doi: 10.1128/aac.42.7.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barrett JF, et al. Antibacterial agents that inhibit two-component signal transduction systems. Proc Natl Acad Sci USA. 1998;95:5317–5322. doi: 10.1073/pnas.95.9.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stephenson K. Hoch JA. Developing inhibitors to selectively target two-component and phosphorelay signal transduction systems of pathogenic microorganisms. Curr Med Chem. 2004;11:765–773. doi: 10.2174/0929867043455765. [DOI] [PubMed] [Google Scholar]
- 16.Rowland SL. King GF. Histidine kinases as antimicrobial targets: prospects and pitfalls. Mini Rev Med Chem. 2007;7:1144–1154. doi: 10.2174/138955707782331759. [DOI] [PubMed] [Google Scholar]
- 17.Roe SM, et al. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 18.Hadden MK. Lubbers DJ. Blagg BS. Geldanamycin, radicicol, and chimeric inhibitors of the Hsp90 N-terminal ATP binding site. Curr Top Med Chem. 2006;6:1173–1182. doi: 10.2174/156802606777812031. [DOI] [PubMed] [Google Scholar]
- 19.Marcu MG. Chadli A. Bouhouche I. Catelli M. Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 20.Chiosis G, et al. Development of purine-scaffold small molecule inhibitors of Hsp90. Curr Cancer Drug Targets. 2003;3:371–376. doi: 10.2174/1568009033481778. [DOI] [PubMed] [Google Scholar]
- 21.Soga S. Shiotsu Y. Akinaga S. Sharma SV. Development of radicicol analogues. Curr Cancer Drug Targets. 2003;3:359–369. doi: 10.2174/1568009033481859. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto K, et al. Total synthesis as a resource in the discovery of potentially valuable antitumor agents: cycloproparadicicol. Angew Chem Int Ed Engl. 2003;42:1280–1284. doi: 10.1002/anie.200390329. [DOI] [PubMed] [Google Scholar]
- 23.Yang ZQ, et al. New efficient synthesis of resorcinylic macrolides via ynolides: establishment of cycloproparadicicol as synthetically feasible preclinical anticancer agent based on Hsp90 as the target. J Am Chem Soc. 2004;126:7881–7889. doi: 10.1021/ja0484348. [DOI] [PubMed] [Google Scholar]
- 24.Janin YL. Heat shock protein 90 inhibitors. A text book example of medicinal chemistry? J Med Chem. 2005;48:7503–7512. doi: 10.1021/jm050759r. [DOI] [PubMed] [Google Scholar]
- 25.Moulin E. Zoete V. Barluenga S. Karplus M. Winssinger N. Design, synthesis, and biological evaluation of HSP90 inhibitors based on conformational analysis of radicicol and its analogues. J Am Chem Soc. 2005;127:6999–7004. doi: 10.1021/ja043101w. [DOI] [PubMed] [Google Scholar]
- 26.McDonald E. Jones K. Brough PA. Drysdale MJ. Workman P. Discovery and development of pyrazole-scaffold Hsp90 inhibitors. Curr Top Med Chem. 2006;6:1193–1203. doi: 10.2174/156802606777812086. [DOI] [PubMed] [Google Scholar]
- 27.Proisy N, et al. Inhibition of Hsp90 with synthetic macrolactones: synthesis and structural and biological evaluation of ring and conformational analogs of radicicol. Chem Biol. 2006;13:1203–1215. doi: 10.1016/j.chembiol.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 28.Workman P. Burrows F. Neckers L. Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann NY Acad Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- 29.Besant PG. Lasker MV. Bui CD. Turck CW. Inhibition of branched-chain alpha-keto acid dehydrogenase kinase and Sln1 yeast histidine kinase by the antifungal antibiotic radicicol. Mol Pharmacol. 2002;62:289–296. doi: 10.1124/mol.62.2.289. [DOI] [PubMed] [Google Scholar]
- 30.Guarnieri MT. Zhang L. Shen J. Zhao R. The Hsp90 inhibitor radicicol interacts with the ATP-binding pocket of bacterial sensor kinase PhoQ. J Mol Biol. 2008;379:82–93. doi: 10.1016/j.jmb.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 31.Stephenson K. Hoch JA. Virulence- and antibiotic resistance-associated two-component signal transduction systems of Gram-positive pathogenic bacteria as targets for antimicrobial therapy. Pharmacol Ther. 2002;93:293–305. doi: 10.1016/s0163-7258(02)00198-5. [DOI] [PubMed] [Google Scholar]
- 32.Stephenson K. Hoch JA. Two-component and phosphorelay signal-transduction systems as therapeutic targets. Curr Opin Pharmacol. 2002;2:507–512. doi: 10.1016/s1471-4892(02)00194-7. [DOI] [PubMed] [Google Scholar]
- 33.Groisman EA. The pleiotropic two-component regulatory system PhoP-PhoQ. J Bacteriol. 2001;183:1835–1842. doi: 10.1128/JB.183.6.1835-1842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prost LR. Miller SI. The Salmonellae PhoQ sensor: mechanisms of detection of phagosome signals. Cell Microbiol. 2008;10:576–582. doi: 10.1111/j.1462-5822.2007.01111.x. [DOI] [PubMed] [Google Scholar]
- 35.Prost LR. Sanowar S. Miller SI. Salmonella sensing of anti-microbial mechanisms to promote survival within macrophages. Immunol Rev. 2007;219:55–65. doi: 10.1111/j.1600-065X.2007.00557.x. [DOI] [PubMed] [Google Scholar]
- 36.Soncini FC. Garcia Vescovi E. Solomon F. Groisman EA. Molecular basis of the magnesium deprivation response in Salmonella typhimurium: identification of PhoP-regulated genes. J Bacteriol. 1996;178:5092–5099. doi: 10.1128/jb.178.17.5092-5099.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vescovi EG. Ayala YM. Di Cera E. Groisman EA. Characterization of the bacterial sensor protein PhoQ. Evidence for distinct binding sites for Mg2+ and Ca2+ J Biol Chem. 1997;272:1440–1443. doi: 10.1074/jbc.272.3.1440. [DOI] [PubMed] [Google Scholar]
- 38.Galan JE. Curtiss R., 3rd Virulence and vaccine potential of phoP mutants of Salmonella typhimurium. Microb Pathog. 1989;6:433–443. doi: 10.1016/0882-4010(89)90085-5. [DOI] [PubMed] [Google Scholar]
- 39.Fields PI. Groisman EA. Heffron F. A Salmonella locus that controls resistance to microbicidal proteins from phagocytic cells. Science. 1989;243:1059–1062. doi: 10.1126/science.2646710. [DOI] [PubMed] [Google Scholar]
- 40.Miller SI. Kukral AM. Mekalanos JJ. A two-component regulatory system (phoP phoQ) controls Salmonella typhimurium virulence. Proc Natl Acad Sci USA. 1989;86:5054–5058. doi: 10.1073/pnas.86.13.5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oyston PC, et al. The response regulator PhoP is important for survival under conditions of macrophage-induced stress and virulence in Yersinia pestis. Infect Immun. 2000;68:3419–3425. doi: 10.1128/iai.68.6.3419-3425.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moss JE. Fisher PE. Vick B. Groisman EA. Zychlinsky A. The regulatory protein PhoP controls susceptibility to the host inflammatory response in Shigella flexneri. Cell Microbiol. 2000;2:443–452. doi: 10.1046/j.1462-5822.2000.00065.x. [DOI] [PubMed] [Google Scholar]
- 43.Batey RT. Williamson JR. Interaction of the Bacillus stearothermophilus ribosomal protein S15 with 16 S rRNA: I. Defining the minimal RNA site. J Mol Biol. 1996;261:536–549. doi: 10.1006/jmbi.1996.0481. [DOI] [PubMed] [Google Scholar]
- 44.Zhang JH. Chung TD. Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 45.Vas M. Merli A. Rossi GL. Antagonistic binding of substrates to 3-phosphoglycerate kinase monitored by the fluorescent analogue 2'(3')-O-(2,4,6-trinitrophenyl)adenosine 5'-triphosphate. Biochem J. 1994;301(Pt 3):885–891. doi: 10.1042/bj3010885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stewart RC. VanBruggen R. Ellefson DD. Wolfe AJ. TNP-ATP and TNP-ADP as probes of the nucleotide binding site of CheA, the histidine protein kinase in the chemotaxis signal transduction pathway of Escherichia coli. Biochemistry. 1998;37:12269–12279. doi: 10.1021/bi980970n. [DOI] [PubMed] [Google Scholar]
- 47.Soti C. Vermes A. Haystead TA. Csermely P. Comparative analysis of the ATP-binding sites of Hsp90 by nucleotide affinity cleavage: a distinct nucleotide specificity of the C-terminal ATP-binding site. Eur J Biochem. 2003;270:2421–2428. doi: 10.1046/j.1432-1033.2003.03610.x. [DOI] [PubMed] [Google Scholar]
- 48.Hiratsuka T. Fluorescent and colored trinitrophenylated analogs of ATP and GTP. Eur J Biochem. 2003;270:3479–3485. doi: 10.1046/j.1432-1033.2003.03748.x. [DOI] [PubMed] [Google Scholar]
- 49.Vanoye CG, et al. The carboxyl termini of K(ATP) channels bind nucleotides. J Biol Chem. 2002;277:23260–23270. doi: 10.1074/jbc.M112004200. [DOI] [PubMed] [Google Scholar]
- 50.Plesniak L, et al. Probing the nucleotide binding domain of the osmoregulator EnvZ using fluorescent nucleotide derivatives. Biochemistry. 2002;41:13876–13882. doi: 10.1021/bi020331j. [DOI] [PubMed] [Google Scholar]
- 51.Kim OK. Ohemeng K. Barrett JF. Advances in DNA gyrase inhibitors. Expert Opin Investig Drugs. 2001;10:199–212. doi: 10.1517/13543784.10.2.199. [DOI] [PubMed] [Google Scholar]
- 52.Lamour V. Hoermann L. Jeltsch JM. Oudet P. Moras D. Crystallization of the 43 kDa ATPase domain of Thermus thermophilus gyrase B in complex with novobiocin. Acta Crystallogr D Biol Crystallogr. 2002;58:1376–1378. doi: 10.1107/s0907444902010429. [DOI] [PubMed] [Google Scholar]
- 53.Carr R. Jhoti H. Structure-based screening of low-affinity compounds. Drug Discov Today. 2002;7:522–527. doi: 10.1016/s1359-6446(02)02245-6. [DOI] [PubMed] [Google Scholar]
- 54.Rees DC. Congreve M. Murray CW. Carr R. Fragment-based lead discovery. Nat Rev Drug Discov. 2004;3:660–672. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]
- 55.Fischbach MA. Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Y, et al. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob Agents Chemother. 2007;51:1398–1406. doi: 10.1128/AAC.00925-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stephenson K. Yamaguchi Y. Hoch JA. The mechanism of action of inhibitors of bacterial two-component signal transduction systems. J Biol Chem. 2000;275:38900–38904. doi: 10.1074/jbc.M006633200. [DOI] [PubMed] [Google Scholar]
- 58.Soti C. Racz A. Csermely P. A nucleotide-dependent molecular switch controls ATP binding at the C-terminal domain of Hsp90. N-terminal nucleotide binding unmasks a C-terminal binding pocket. J Biol Chem. 2002;277:7066–7075. doi: 10.1074/jbc.M105568200. [DOI] [PubMed] [Google Scholar]
- 59.Yang P, et al. Oleandrin-mediated inhibition of human tumor cell proliferation: importance of Na,K-ATPase alpha subunits as drug targets. Mol Cancer Ther. 2009;8:2319–2328. doi: 10.1158/1535-7163.MCT-08-1085. [DOI] [PubMed] [Google Scholar]
- 60.Huang SG. Weisshart K. Fanning E. Characterization of the nucleotide binding properties of SV40 T antigen using fluorescent 3'(2')-O-(2,4,6-trinitrophenyl)adenine nucleotide analogues. Biochemistry. 1998;37:15336–15344. doi: 10.1021/bi981094g. [DOI] [PubMed] [Google Scholar]
- 61.Inna Vainshtein I. Scott Silveria S. Kaul P. Rouhani R. Eglen R. Wang J. A high-throughput, nonisotopic, competitive binding assay for kinases using nonselective inhibitor probes (ED-NSIPTM) J Biomol Screen. 2002;7:507–514. doi: 10.1177/1087057102238624. [DOI] [PubMed] [Google Scholar]
- 62.Kashem MA. Nelson RM. Yingling JD. Pullen SS. Prokopowicz AS., III Jones JW. Wolak JP. Rogers GR. Morelock MM. Snow RJ. Homon CA. Jakes S. Three mechanistically distinct kinase assays compared: measurement of intrinsic ATPase activity identified the most comprehensive set of ITK inhibitors. J Biomol Screen. 2007;12:70–83. doi: 10.1177/1087057106296047. [DOI] [PubMed] [Google Scholar]
- 63.Lebakken CS. Riddle SM. Singh U. Frazee WJ. Eliason HC. Gao Y. Reichling LJ. Marks BD. Vogel KW. Development and applications of a broad-coverage, TR-FRET-based kinase binding assay platform. J Biomol Screen. 2009;14:924–935. doi: 10.1177/1087057109339207. [DOI] [PubMed] [Google Scholar]
- 64.Prodromou C. Roe SM. O'Brien R. Ladbury J. Piper PW. Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 65.Kampranis S. Maxwell A. The DNA gyrase-quinolone complex. J Biol Chem. 1998;273:2615–22626. doi: 10.1074/jbc.273.35.22615. [DOI] [PubMed] [Google Scholar]
- 66.Galm U. Heller S. Shapiro S. Page M. Li SM. Heide L. Antimicrobial and DNA gyrase-inhibitory activities of novel chlorobiocin derivatives produced by mutasynthesis. Antimicrob Agents Chemother. 2004;48:1307–1312. doi: 10.1128/AAC.48.4.1307-1312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Erlanson DA. Braisted AC. Raphael DR. Randal M. Stroud RM. Gordon EM. Wells JA. Site-directed ligand discovery. Proc Natl Acad Sci USA. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fejzo J. Lepre CA. Peng JW. Bemis GW. Ajay Murcko MA. Moore JM. The SHAPES strategy: an NMR-based approach for lead generation in drug discovery. Chem Biol. 1999;6:755–769. doi: 10.1016/s1074-5521(00)80022-8. [DOI] [PubMed] [Google Scholar]
- 69.van Dongen MJ. Uppenberg J. Svensson S. Lundbäck T. Akerud T. Wikström M. Schultz J. Structure-based screening as applied to human FABP4: a highly efficient alternative to HTS for hit generation. J Am Chem Soc. 2002;124:11874–11880. doi: 10.1021/ja017830c. [DOI] [PubMed] [Google Scholar]
- 70.Hajduk PJ. Dinges J. Schkeryantz JM. Janowick D. Kaminski M. Tufano M. Augeri DJ. Petros A. Nienaber V. Zhong P. Hammond R. Coen M. Beutel B. Katz L. Fesik SW. Novel inhibitors of Erm methyltransferases from NMR and parallel synthesis. J Med Chem. 1999;42:3852–3859. doi: 10.1021/jm990293a. [DOI] [PubMed] [Google Scholar]
- 71.Jameson DM. Eccleston JF. Fluorescent nucleotide analogs: Synthesis and applications. Methods Enzymol. 1997;278:363–390. doi: 10.1016/s0076-6879(97)78020-0. [DOI] [PubMed] [Google Scholar]
- 72.Nowak E. Panjikar S. Morth JP. Jodanova R. Svergun DI. Tucker PA. Structural and functional aspects of the sensor histidine kinase PrrB from Mycobacterium tuberculosis. Structure. 2006;14:275–285. doi: 10.1016/j.str.2005.10.006. [DOI] [PubMed] [Google Scholar]