

Abstract
Neurofibromatosis type 1 (NF1) is a common genetic disorder characterized by multiple neurofibromas, peripheral nerve tumors containing mainly Schwann cells and fibroblasts. The NF1 gene encodes neurofibromin, a tumor suppressor postulated to function in part as a Ras GTPase-activating protein. The roles of different cell types and of elevated Ras-GTP in neurofibroma formation are unclear. To determine which neurofibroma cell type has altered Ras-GTP regulation, we developed an immunocytochemical assay for active, GTP-bound Ras. In NIH 3T3 cells, the assay detected overexpressed, constitutively activated K-, N-, and Ha-Ras and insulin-induced endogenous Ras-GTP. In dissociated neurofibroma cells from NF1 patients, Ras-GTP was elevated in Schwann cells but not fibroblasts. Twelve to 62% of tumor Schwann cells showed elevated Ras-GTP, unexpectedly revealing neurofibroma Schwann cell heterogeneity. Increased basal Ras-GTP did not correlate with increased cell proliferation. Normal human Schwann cells, however, did not demonstrate elevated basal Ras activity. Furthermore, compared with cells from wild type littermates, Ras-GTP was elevated in all mouse Nf1−/− Schwann cells but never in Nf1−/− mouse fibroblasts. Our results indicate that Ras activity is detectably increased in only some neurofibroma Schwann cells and suggest that neurofibromin is not an essential regulator of Ras activity in fibroblasts.
Type 1 neurofibromatosis (NF1)1 is a common, autosomal dominant disease affecting 1 in 3500 individuals worldwide (1). NF1 patients develop multiple, frequently disfiguring peripheral nerve tumors, called neurofibromas. These benign tumors contain mainly Schwann cells and fibroblasts, with smaller numbers of axons, mast cells, and sometimes glandular elements (2). Other diagnostic features of NF1 include café-au-lait macules, iris Lisch nodules, and benign optic nerve gliomas. Loss of heterozygosity at the NF1 locus in humans has been demonstrated in malignant peripheral nerve sheath tumors (3), in myeloid disease (4), and in neurofibromas (5, 6), indicating that NF1 functions as a tumor suppressor gene. Furthermore, chimeric mice bearing Nf1−/− cells also develop neurofibromas, consistent with the idea that loss of the wild type Nf1 allele is critical for tumor formation (7).
The NF1 gene encodes neurofibromin, a large protein with a central Ras GTPase-activating protein (Ras-GAP)-related domain (8). Neurofibromin can function as a Ras-GAP, reducing the amount of active, GTP-bound Ras (9–11). Loss of neurofibromin is correlated with increased levels of Ras-GTP in some cell types in vitro (12–16). Neurofibromin may also have functions that are not related to Ras regulation. The Drosophila homologue of neurofibromin, for example, appears to regulate a cyclic AMP-dependent protein kinase A pathway in a Ras-Raf-independent manner (17, 18). The functional consequences of Nf1 mutations in neurofibroma cell types could therefore occur through Ras-dependent and/or Ras-independent mechanisms.
Loss of neurofibromin correlates with increases in Ras-GTP in lysates from NF1 patient neurofibromas (19). Due to the multiple cell types comprising neurofibromas, however, it is not known whether elevated Ras-GTP in neurofibroma lysates can be ascribed to Schwann cells, fibroblasts, and/or other cells. Furthermore, dissociated neurofibroma cultures yield only small numbers of viable Schwann cells, and even Schwann cell-enriched cultures typically contain some fibroblasts (20, 21). Standard assays of Ras-GTP cannot, therefore, reveal the origins of elevated Ras activity in these tumors.
Both neurofibroma Schwann cells and fibroblasts have abnormal phenotypes in vitro. Schwann cells isolated from neurofibromas stimulate angiogenic responses in the chicken chorioallantoic membrane model, and are invasive, while normal human Schwann cells are not (21). Fibroblasts from neurofibromas secrete excess collagens as compared with normal skin fibroblasts (22) and sometimes demonstrate abnormal growth in vitro (reviewed in Ref. 2; see Ref. 23). The extent to which these phenotypes are due to aberrant Ras activation has not been determined.
Unlike Nf1−/− chimeras, mice with heterozygous targeted mutations in the Nf1 gene do not spontaneously develop neurofibromas (24, 25) but are at increased risk to develop fibrosarcomas, pheochromocytomas, and myeloid leukemias that show loss of both Nf1 alleles (15, 25, 26). Nf1 null embryos die in utero between embryonic days 11 and 14 (24, 25), so adult null cells are unavailable for analysis. However, it is possible to isolate both Schwann cells and fibroblasts from mutant embryos prior to embryo death and to analyze the purified cell populations. Based on levels of [32P]orthophosphate incorporation into GTP bound to Ras, embryonic Nf1−/− Schwann cells have 2-fold elevated levels of Ras-GTP compared with wild type Schwann cells in vitro (14). Furthermore, these neurofibromin-deficient cells are growth-inhibited, angiogenic, and invasive (27). Some of these phenotypes are mimicked when normal Schwann cells express a constitutively activated Ras allele (14, 28), and some phenotypes of Nf1−/− Schwann cells are inhibited by a farnesyl transferase inhibitor (27) that blocks Ras signaling (29). Collectively, these data suggest that neurofibromin is a key Ras-GAP in mouse Schwann cells.
Fibroblasts from Nf1−/− mouse embryos also show abnormalities in vitro, including increased proliferation, increased collagen deposition, and an inability to form normal perineurium in Schwann cell-neuron co-cultures (30, 31). Ras-GTP levels in Nf1−/− fibroblasts from embryonic day 12.5 embryos have not previously been investigated. Van der Geer et al. (32) demonstrated a variable increase in basal Ras-GTP and mitogen-activated protein kinase activation in cells from whole embryonic day 9.5 Nf1−/− embryos, but the mitogen-activated protein kinase activation was lost after cells were passaged. The cultures from these early Nf1−/− mouse embryos may have contained cell types in addition to fibroblasts that could account for the elevated Ras-GTP. Johnson et al. (33) showed no change in Ras-GTP in mouse NIH 3T3 fibroblasts after introduction of full-length NF1, suggesting that neurofibromin may not regulate Ras-GTP in fibroblasts.
Here, we have determined which cells in neurofibromas have elevated Ras activity using a new in situ assay for Ras-GTP. Active GTP-bound Ras associates with the Raf1 serine/threonine kinase, a key effector of Ras signaling (34). The Ras-binding domain (RBD) of Raf1 kinase binds active, GTP-bound Ras with an affinity that is 3 orders of magnitude higher than for inactive, GDP-bound Ras (35). Recently, it was demonstrated that Ras activity could be measured by incubating cell lysates with a Raf1-RBD-GST fusion protein immobilized on glutathione-agarose and then detecting the bound Ras-GTP by Western blotting with a Ras antibody (36, 37). We have utilized Raf1-RBD-GST in an immunocytochemical assay to demonstrate that aberrant Ras activity is a characteristic of only a unique subpopulation of neurofibroma Schwann cells but not of fibroblasts.
EXPERIMENTAL PROCEDURES
DNA Constructs
Ha(61L)-, K(12V)-, and N(12D)-ras cDNAs were cloned into pCGN-hyg as in frame BamHI fragments and are described elsewhere (38). pGEX 2T-RBD, encoding the Raf1-RBD-GST (amino acids 51–131 of Raf1) was constructed as described previously (36). The PZIPneoSV(X)1 construct encoding constitutively active Rap1a (39) was generously provided by Geoff Clark (National Institutes of Health).
Cell Culture and Transient Transfections
Parental NIH 3T3 cells were grown in DMEM plus 10% fetal bovine serum. Before transfections, cells were switched to DMEM alone and transfected with 5 μg of plasmid DNA in DOTAP (Roche Molecular Biochemicals) according to the manufacturer’s instructions. After 4 h, cells were switched back to serum-containing medium and grown for 48 h. Cells were then switched again to serum-free medium for 48 h, fixed, and assayed for hemagglutinin (HA) immunofluorescence and Ras-GTP as described below.
NIH 3T3 cells containing dexamethasone-inducible activated Ha-Ras (NIH-pJ5W-Ha-Ras(61L); previously described (40)) were grown in DMEM plus 10% calf serum. To induce Ha-Ras expression, cells were treated with 5 or 10 μM dexamethasone for 24 h. Ras activity was determined using a nonradioactive Ras-GTP assay (Upstate Biotech-nology, Inc., Lake Placid, NY) according to the manufacturer’s instructions (36). Following immobilization on glutathione-agarose beads, Raf1-RBD-GST-Ras-GTP complexes were analyzed by Western blotting with a Ras-specific antibody as described previously (36) and quantified using a Molecular Dynamics Storm 860 PhosphorImager with Image-Quant software.
Primary cultures of mouse Schwann cells were grown from the dorsal root ganglia of embryonic day 12.5 embryos as described previously (14). The genotypes of embryos were determined by polymerase chain reaction as described previously (24). Primary human neurofibromas were dissociated using enzymes as described previously (41). Fibroblasts from these tumors were grown in DMEM plus 10% fetal bovine serum on 100-mm plastic tissue culture dishes. Schwann cell enriched cultures were grown in serum-free N2 medium supplemented with 5 ng/ml recombinant human glial growth factor 2 (Cambridge Neuroscience) and 2 μM forskolin (Calbiochem) on poly-L-lysine-coated 100-mm tissue culture dishes as described previously (41). Once cultures had reached near confluency, 20,000 cells were replated in the same medium into each well of eight-well chamber slides (LabTek) coated with poly-L-lysine. Cells were grown for 24 h and then switched to N2 (Schwann cells) or DMEM (fibroblasts) only for an additional 48 h and examined as described below.
Immunocytochemistry for Influenza HA and S100
Cells were fixed in an ice-cold mixture of ethanol, glacial acetic acid, and water (90:5:5) for 15 min and then washed three times with phosphate-buffered saline. Fixed cells were blocked in 3% normal goat serum in phosphate-buffered saline and then incubated for 1 h with a 1:500 dilution of rabbit-anti-HA (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), washed, then incubated with fluorescein isothiocyanate-conjugated goat anti-rabbit IgG (Jackson Immunoresearch Laboratories, Inc.) for 30 min. Cells were then washed three times with blocking buffer and either processed further for Ras-GTP or mounted and examined by fluorescence microscopy. For S100 immunocytochemistry, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and then processed as described previously (14).
5-Bromo-2′-deoxyuridine (BrdUrd) Incorporation Assay
Neurofibroma-derived Schwann cells were seeded onto poly-L-lysine and laminin-coated LabTek slides (20,000 cells/well) and were allowed to attach to culture surfaces in rh-GGF2/forskolin-supplemented N2 medium (as above) overnight before medium was changed to N2 alone for 2 days. Cultures were incubated with 10 mM BrdUrd for 18 h and then fixed with 4% paraformaldehyde for 15 min. Immunocytochemical detection of BrdUrd incorporation was performed as described previously (42). Five hundred Schwann cells were counted in duplicate experiments, and the percentage of BrdUrd-positive Schwann cells was determined.
Purification of Raf1-RBD-GST
The Raf1-RBD-GST protein was synthesized and purified by glutathione-agarose chromatography as described previously (36), except that after thrombin cleavage, the protein was subjected to two rounds of purification over SP-Sepharose fast flow (Amersham Pharmacia Biotech). Protein was concentrated to 5 mg/ml and kept frozen at −80 °C in the elution buffer as described (36). We found that under these conditions, the Raf1-RBD-GST is stable in the Ras-GTP assay for up to 2 weeks (43).
In Situ Ras-GTP Assay
Cells were fixed as described above for HA staining and then blocked in ice-cold phosphate-buffered saline with 5% normal goat serum and 0.5% bovine serum albumin for 1 h at 4 °C. Cells were then incubated with 10 μg/ml of Raf1-RBD-GST for 3 h at 4 °C, washed three times in blocking buffer without albumin, and then incubated for 1 h with a 1:250 dilution of a mouse anti-GST monoclonal antibody (that only recognizes bacterial GST; Santa Cruz Biotechnology) at room temperature. The procedures for optimizing this protocol are described elsewhere (43). Signal was then detected using a tyramide signal amplification kit (NEN Life Science Products) as follows. Cells were washed three times in blocking buffer and then incubated in the blocking buffer for an additional 30 min. The blocking buffer was drained and replaced with wash buffer (0.1 M Tris-HCl, pH 7.5, 0.15 M NaCl, 0.05% Tween 20), and then cells were incubated for 1 h with a 1:1000 dilution of goat anti-mouse-horseradish peroxidase secondary antibody (Bio-Rad). Cells were then washed three times for 5 min each in wash buffer and processed for tyramide fluorophore immunofluorescence following the kit manufacturer’s instructions. Labeled cells were mounted and examined by fluorescence microscopy, as above.
RESULTS
Raf1-RBD-GST Detects Increased Ras-GTP in Situ
To evaluate Ras-GTP levels in individual Schwann cells and/or fibroblasts from NF1 patient neurofibromas, we established an assay whereby a Raf1-RBD-GST fusion protein is added to fixed, permeabilized cells and binds Ras-GTP in situ (43). Raf1-RBD-GST-Ras-GTP complexes are then visualized using fluorescence immunocytochemistry to detect GST.
To test both the specificity and sensitivity of this assay, we utilized NIH-pJ5W-Ha-Ras(61L) cells that can be induced to express activated Ha-Ras by treatment with dexamethasone. Using RBD-GST bound to glutathione-agarose beads to precipitate Ras-GTP (36, 37), these cells expressed a low level of Ras-GTP when grown in 10% calf serum alone (Fig. 1A). After 24 h in the presence of 5 μM dexamethasone, we observed a weak (0.8–1.2-fold as determined by PhosphorImager analysis) increase in Ras-GTP, while in the presence of 10 μM dexamethasone, we saw a 3.2–3.5-fold increase in Ras-GTP (ranges from three separate experiments; see Fig. 1A). Using our immunocytochemical assay, we found that we could easily detect Ras-GTP in cells treated with 10 μM dexamethasone but not in cells treated with lower concentrations of the drug or untreated controls (Fig. 1, B–G). The specificity of this signal was verified by incubating cells treated with 10 μM dexamethasone with GST in place of Raf1-RBD-GST. In this case, only background signal could be detected (Fig. 1, H and I). These data indicate that this immunocytochemical assay can detect between approximately 1- and 3-fold increases of Ras-GTP in situ.
Fig. 1. The Ras binding domain of Raf1 can be used as an immunocytochemical probe for Ras-GTP in situ.

NIH-pJ5W-Ha-Ras(61L) cells were grown in 0, 5, or 10 μM dexamethasone for 24 h and then assayed for levels of Ras-GTP using either a biochemical (A) or immunocytochemical (B–I) assay. A, cells treated with 5 μM dexamethasone demonstrated a 0.8–1.2-fold increase in Ras-GTP (range over three experiments; 0.8-fold shown here), while cells treated with 10 μM dexamethasone had a 3.2–3.5-fold increase (3.2-fold shown here). B and C, cells grown in the absence of dexamethasone; D and E, cells grown in 5 μM dexamethasone; F and G, cells grown in 10 μM dexamethasone; H and I, cells grown in 10 μM dexamethasone but probed with GST in place of Raf1-RBD-GST.
Raf1-RBD-GST Recognizes Activated Ha-, K-, and N-Ras
Raf1-RBD binds three major Ras proteins (Ha-, K-, and N-Ras) (36), one or more of which could be activated in cells lacking neurofibromin. We tested whether RBD-GST can detect activated forms of these Ras proteins using immunocytochemistry by transiently transfecting NIH 3T3 cells with HA-tagged constitutively active Ha(61L)-, K(12V)- or N(12D)-ras. Twenty-four hours after transfection, cells were switched to serum-free medium for 48 h and then assayed for HA expression and Ras-GTP. We found that Raf1-RBD-GST detected elevated Ras-GTP in cells expressing each of the three ras constructs but not in cells expressing HA alone (Fig. 2, A–H). Furthermore, our assay failed to detect increased Rap1a activity in cells transfected with Rap1a(63E) (not shown) that is constitutively bound to GTP and binds Raf1-RBD with low affinity (44). These data are consistent with previous findings indicating that Raf1-RBD-GST could not precipitate GTP-bound Rap1a (36). Thus, Raf1-RBD-GST detects constitutively active, GTP-bound, forms of Ha-, K-, and N-Ras in fixed cells.
Fig. 2. Raf1-RBD-GST recognizes GTP-bound Ha-, K-, and N-Ras.
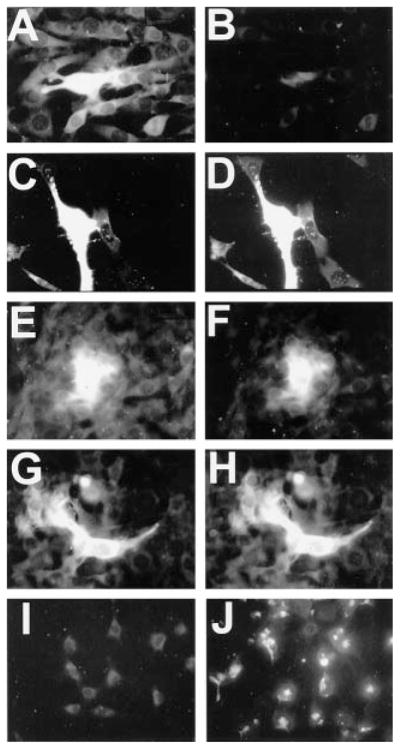
NIH 3T3 cells were transfected with pCGN-hyg constructs encoding HA-tagged Ha-Ras (C and D), K-Ras (E and F), or N-Ras (G and H) or the HA tag alone (A and B) and then grown for 24 h. Cells were then switched to serum-free medium for 48 h, processed for HA immunocytochemistry (A, C, E, and G) and Ras-GTP analysis (B, D, F, and H), and then examined by fluorescence microscopy. Note that cells transfected with all three HA-tagged Ras species show high levels of Raf1-RBD-GST immunofluorescence, compared with cells transfected with HA tag alone. Untransfected NIH 3T3 cells grown in the absence of serum for 48 h demonstrated only background staining when examined by Raf1-RBD-GST immunocytochemistry (I). However, after treatment for 2 min with 20 ng/ml insulin (J), these cells demonstrated increased, punctate immunofluorescence that was localized in patches, apparently at the cell membrane (see “Results”). These data indicate that Raf1-RBD-GST can detect elevated Ras-GTP in situ.
Raf1-RBD-GST Recognizes Endogenous Ras-GTP
To test if Raf1-RBD-GST detects alterations in levels of endogenous Ras-GTP in this immunocytochemical assay and to verify that the signal is not due to overexpression of active Ras proteins, NIH 3T3 cells were serum-starved for 48 h and then treated for 2 min with 20 ng/ml insulin, a potent stimulus of Ras-GTP (45). Insulin treatment resulted in a dramatic increase in Raf1-RBD-GST binding compared with untreated cells (Fig. 2, I and J). Interestingly, this signal was compartmentalized within a distinct region of the cell (Fig. 2J). This compartment did not co-localize with markers for the nucleus (Hoechst 33342) or the Golgi apparatus (C5-ceramide) but rather appeared to be restricted to regions of the plasma membrane as determined by scanning laser confocal microscopy (not shown). The active Ras signal became weaker over time in cultures treated with insulin for 30 min and was not detectable after 2 h. (not shown).
Raf1-RBD-GST Detects Elevated Ras Activity in Nf1−/− Schwann Cells
Having demonstrated that Raf1-RBD-GST is an effective immunocytochemical probe for endogenous Ras-GTP in NIH 3T3 cells, we tested whether this technique could detect Ras-GTP in cultured Schwann cells from Nf1−/− mouse embryos. We cultured Schwann cells from Nf1−/− mice and their wild type littermates in serum-free medium in the absence of added growth factors for 48 h and then analyzed their basal Ras-GTP levels using the Raf1-RBD-GST probe. Compared with wild type Schwann cells, neurofibromin-deficient Schwann cells demonstrated constitutively elevated Ras-GTP (Fig. 3, A and B). This result was confirmed in Schwann cell cultures isolated from five separate Nf1−/− embryos. We previously found that Ras-GTP in Nf1+/− Schwann cells is variably elevated by as much as 1.5-fold over wild type controls (14). However, we were unable to detect increased basal Ras-GTP in Nf1+/− Schwann cells from three separate embryos using the Raf1-RBD-GST probe (not shown). The Ras-GTP signal in Nf1−/− Schwann cells was dramatically reduced when cells were treated for 48 h. with 1 μM L-744,832 (46), an inhibitor of farnesyl transferase (Fig. 3C). The effectiveness of this low dose of drug may reflect predominant usage of Ha-Ras in Schwann cells; a similar farnesyl protein transferase inhibitor with some specificity toward Ha-Ras was previously shown to inhibit Ras processing and to correct some abnormal phenotypes of Nf1−/− Schwann cells (27). The observed elevated Ras-GTP levels in these cells cannot simply be a reflection of increased proliferation rates, since neither wild type nor Nf1−/− Schwann cells proliferate in serum-free, mitogen-free N2 medium. In a typical experiment, 0.16% of wild type and 0.38% of Nf1−/− cells incorporated [3H]thymidine as determined by autoradiography following an 18-h pulse, as described previously (14). These data confirm that Ras-GTP is elevated in Nf1−/− Schwann cells and demonstrate that our assay can be used to probe for endogenous Ras-GTP in cells lacking neurofibromin. These data also indicate that Raf1-RBD-GST can be used to track the effectiveness of drugs that influence Ras-GTP levels.
Fig. 3. Schwann cells from Nf1 null mice have constitutively elevated Ras-GTP.

Schwann cells from wild type (A) and Nf1−/− mouse embryos (B and C) were grown in serum-free N2 medium supplemented with rhGG2 and forskolin as described previously and then switched to N2 alone for 48 h and processed for Ras-GTP immunocytochemistry. Compared with wild type Schwann cells, Nf1−/− Schwann cells show significantly increased basal levels of Ras-GTP. This basal Ras activity dropped to levels comparable with those of wild type cells in cultures grown in the presence of 1 μM L-744,832 for 48 h. (c).
Nf1−/− Fibroblasts Demonstrate Normal Basal Levels of Ras-GTP
To determine if Nf1−/− fibroblasts have elevated Ras-GTP, fibroblast cultures from four individual Nf1−/− and three individual wild type embryos were studied following 48 h in the absence of serum. Both wild type and Nf1−/− fibroblasts demonstrated equally low basal Ras-GTP signals (Fig. 4, A and D). Furthermore, Ras-GTP in wild type and Nf1−/− fibroblasts increased and decreased with similar kinetics following treatment with 20 ng/ml insulin (Fig. 4, A–F). These data indicate that Nf1−/− fibroblasts either do not have elevated Ras-GTP or only have small increases in Ras-GTP that cannot be detected using this assay. Neurofibromin, therefore, may not function as an essential Ras-GAP in mouse fibroblasts.
Fig. 4. Ras-GTP is unaltered in fibroblasts from Nf1 null mice.
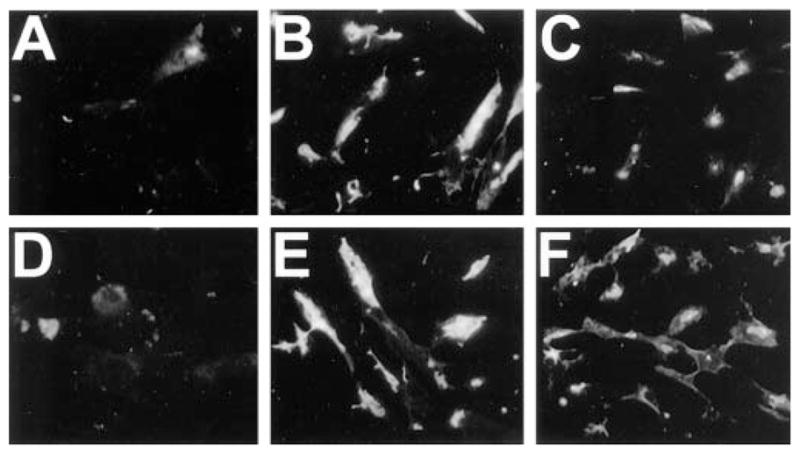
Fibroblasts from wild type (A–C) or Nf1−/− mice (D and E) were grown in DMEM in the absence of serum for 24 h and then treated with 20 ng/ml insulin for 30 min (B and E) or 1 h (C and F) and examined by Ras-GTP immunocytochemistry. Fibroblasts from these same Nf1−/− embryos demonstrated increased proliferation and collagen deposition (not shown), characteristic features of mouse fibroblasts lacking neurofibromin. Note that Ras-GTP levels were similar in Nf1−/− and wild type fibroblasts and that Ras-GTP levels decreased over a similar time course in both cell populations.
A Subpopulation of NF1 Patient Neurofibroma Schwann Cells, but Not Fibroblasts, Have Elevated Ras-GTP
Since Nf1−/− mouse Schwann cells but not fibroblasts showed elevated Ras activity, we next aimed to determine if Schwann cells or fibroblasts from NF1 patient neurofibromas have elevated Ras-GTP. We dissociated neurofibromas from five NF1 patients and established short term primary cultures enriched for fibroblasts or Schwann cells from each patient. As expected, cultures initially contained various proportions of Schwann cells and fibroblasts. We found no increased basal Ras-GTP in any of the neurofibroma fibroblast cultures (Table I; Fig. 5, G–J). However, increased Ras-GTP was evident in a subpopulation of Schwann cells from each tumor (Table I; Fig. 5, C–F). Immunostaining with an anti-S100 antibody, a marker for Schwann cells, confirmed that most of the cells were Schwann cells (see Table I). The proportion of cells with increased Ras-GTP ranged from 12 to 62% in individual tumor cultures. Normal human Schwann cells grown under identical culture conditions never demonstrated increased Ras-GTP levels (Fig. 5, A and B).
Table I. Percentage of cells with high Ras-GTP in dissociated neurofibroma Schwann cell and fibroblast cultures.
High Ras-GTP was defined as signal over the level observed when neurofibroma Schwann cells were stained with GST alone or when normal human Schwann cells were stained with RBD-GST (not shown). All patients whose tumors were studied had been diagnosed with NF1 based on family history, café-au-lait spots, neurofibromas and/or other criteria described by Gutmann et al. (57). Percentages are shown of positive cells out of 200 counted. S100-Ras-GTP double labeling was not possible due to the distinct fixation requirements of the Ras-GTP assay.
| Patient age, sex | Tumor type | Schwann cell cultures |
Fibroblast cultures |
||
|---|---|---|---|---|---|
| Ras-GTP+ cells | S100+ cells | Ras-GTP+ cells | S100+ cells | ||
| % | % | % | % | ||
| 10, female | Plexiform | 26 | 87 | 0 | 0 |
| 46, female | Neurofibroma | 15 | 92 | 0 | 0 |
| 40, male | Neurofibroma | 62 | 85 | 0 | 0 |
| 55, female | Neurofibroma | 12 | 91 | 0 | 0 |
| 5, male | Neurofibroma | 18 | 80 | 0 | 0 |
Fig. 5. A subpopulation of Schwann cells, but not fibroblasts, from NF1 patient neurofibromas has constitutively elevated Ras-GTP.

A and B, phase-contrast and Ras-GTP staining in normal human Schwann cells. Note that none of the cells from normal nerve express detectable levels of Ras-GTP. Schwann cells (C–F) and fibroblasts (G–J) from a neurofibroma of a 40-year-old male NF1 patient (C, D, G, and H) and a plexiform neurofibroma of a 10-year-old female NF1 patient (E, F, I, and J) were analyzed for Ras-GTP in first passage cultures. C, E, G, and I, phase-contrast images of cells; D, F, H, and J, Ras-GTP immunofluorescence. Tumors were dissociated and split into two cultures: one grown on a poly-L-lysine-coated dish in N2 plus rhGG2 and forskolin (conditions favoring Schwann cell growth and survival) and one grown on an uncoated dish in DMEM plus 10% fetal bovine serum (conditions favoring fibroblast growth and survival). After 1 week, cells were passaged by trypsinization, grown for an additional 2 days, and then processed for S100 protein and Ras-GTP immunocyto-chemistry. Note that Ras-GTP is elevated in Schwann cell cultures but not in fibroblasts. The arrowheads show cells with elevated Ras-GTP, while arrows indicate cells with basal Ras-GTP.
We previously found that normal human Schwann cells do not show significant rates of proliferation (labeling index range from 1 to 3%; n = 3) when grown in N2 medium (43). To determine whether the relatively high percentage of neurofibroma Schwann cells with elevated Ras activity reflected a proliferating subpopulation, we assayed neurofibroma Schwann cells for BrdUrd incorporation. Consistent with the data shown above for mouse Schwann cells, in cultures derived from three additional NF1 patients neurofibroma Schwann cells pulsed with BrdUrd for 18 h in N2 medium incorporated 0, 1, and 7% BrdUrd, respectively. At least 98% of the cells in each of these cultures were stained positive for S100 protein. This range of BrdUrd incorporation is far below the range of cells demonstrating elevated Ras-GTP, excluding the hypothesis that aberrant Schwann cell proliferation correlates with the observed increases in Ras-GTP signal. Collectively, these data indicate that Ras-GTP is constitutively elevated in only a sub-population of neurofibroma Schwann cells and not in neurofibroma fibroblasts.
DISCUSSION
Both clinical and experimental data have implicated elevated Ras activity in the development of neurofibromas in NF1 patients. A key question has been whether all cells comprising neurofibromas have elevated Ras-GTP. Using a novel in situ assay for Ras-GTP, we have directly shown for the first time that only a subpopulation of Schwann cells from NF1 patient neurofibromas has constitutively elevated Ras-GTP. In contrast, we never observed elevated Ras activity in neurofibroma fibroblasts, consistent with the idea that high Ras activity contributes to abnormal Schwann cell but not fibroblast phenotypes in these tumors.
Consistent with the findings of de Rooij and Bos (36), we found that Raf1-RBD-GST recognizes GTP-bound forms of Ha-, K-, and N-Ras but not Rap1a, demonstrating the specificity of our assay. Interestingly, we observed endogenous Ras-GTP-Raf1-GST complexes in punctate regions within NIH 3T3 cells following stimulation with insulin. We are presently attempting to identify the precise cellular compartments where active Ras proteins are localized. One possibility is that these compartments represent caveolae. Previous studies indicated that Ras localizes to caveolae and that this localization is critical for Ras function (47, 48). This idea is especially tantalizing in light of a recent report suggesting that caveolae may also be critical for signaling in Schwann cells (49).
Using a standard biochemical assay, we previously found that > 99% pure Schwann cell cultures from Nf1−/− mouse embryos have a 2-fold increase in Ras-GTP compared with wild type Schwann cells (14). Some of the aberrant phenotypes of these Nf1−/− Schwann cells were inhibited by a farnesyl transferase inhibitor and could be mimicked by overexpression of a constitutively active Ras allele (27, 28). Our novel assay confirms that Schwann cells from these mutant embryos have elevated Ras-GTP and that Ras-GTP levels are dramatically reduced by treatment with a farnesyl transferase inhibitor (27), which blocks Ras activation by preventing Ras association with the plasma membrane (reviewed in Ref. 50). This result also indirectly confirms previous data regarding Ras processing in Nf1−/− Schwann cells (27) and demonstrates the remarkable sensitivity of mouse Schwann cell Ras proteins to this class of drug.
We showed that lack of neurofibromin does not result in detectably elevated Ras-GTP in Nf1−/− mouse fibroblasts or neurofibroma fibroblasts that are probably heterozygous at NF1 (51). This is consistent with data suggesting that Schwann cells, but not fibroblasts, from at least some neurofibromas have sustained somatic Nf1 mutations (51) and the finding that human neurofibroma fibroblasts progress from G0 to S phase normally in response to serum (52). Mouse Nf1+/− and Nf1−/− fibroblasts and human neurofibroma fibroblasts, nonetheless, have abnormal phenotypes (22, 23, 30, 31). Thus, although lack of neurofibromin can influence fibroblast phenotypes, these phenotypes may be mediated by signaling cascades other than the Ras-mitogen-activated protein kinase pathway. Based on experimental evidence, cyclic AMP-dependent signaling or other related pathways are likely candidates (17, 18). It should be noted, however, that we cannot exclude the possibility that there are small changes (e.g. less than 2-fold over basal levels) in Ras-GTP in Nf1−/− fibroblasts that could also contribute to fibroblast abnormalities.
Ras activity may not be influenced by loss of neurofibromin in numerous other cells types. For example, melanoma and neuroblastoma cell lines lacking neurofibromin also fail to show significantly increased Ras-GTP, although neurofibromin modulates growth in those cells (53). Melanocytes cultured from NF1 patient café-au-lait patches are abnormal but have normal Ras-GTP levels (54). Our findings are consistent with previous data suggesting that neurofibromin has non-Ras-related functions in some mammalian cell types (33, 53) and in Drosophila, where neurofibromin regulates a protein kinase A pathway that is independent of the Ras-Raf1-mitogen-activated protein kinase pathway (17, 18).
Neurofibromas contain cells with mutations in both NF1 alleles (5–7), and recent data suggest that while additional cytogenetic changes occur in cells comprising other NF1-related lesions, including plexiform neurofibromas (55), loss of heterozygosity at NF1 is a limiting step in neurofibroma formation. We have shown that only subpopulations of Schwann cells in neurofibromas from NF1 patients have high Ras-GTP. It is likely that the cells with high Ras-GTP are those with two mutant NF1 alleles. However, we cannot exclude the possibility that changes in Ras-GTP levels reflect other alterations these cells encountered within the tumors. It is interesting to speculate that the neurofibroma Schwann cells with lower Ras-GTP might be recruited into the tumor by the high Ras-GTP cells. Ras activation results in increased synthesis of various growth factors that could contribute to this process.
This model has important consequences for our understanding of NF1 pathogenesis and for the development of therapeutic strategies. Anti-Ras therapies including use of farnesyl transferase inhibitors are in phase I clinical trials that include NF1 patients. These drugs reverse growth of malignant peripheral nerve sheath tumor cells (56) that have increased Ras-GTP (12, 13). Based on our results, drug treatments that target Ras in neurofibromas are predicted to affect those cells with elevated Ras-GTP and cells whose presence in the tumor depends on the high Ras-GTP population.
Acknowledgments
We thank G. Bollag, G. Martin, and F. McCormick (Onyx Pharmaceuticals, Richmond, CA) for the Raf1-RBD-GST construct; M. Marchionni (Cambridge Neuroscience, Cambridge, MA) for rh-GGF2; J. Gibbs and N. Kohl (Merck Sharpe and Dohme Research Laboratories) for L-744,832; M. Kobayashi, J. Sinsky, A. Crawford, S. Schroeder, and their patients for neurofibroma tissue; and the University of Miami organ procurement team (Les Olson, Director) for human nerves.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS28840 (to N. R.).
The abbreviations used are: NF1, neurofibromatosis type 1; RBD, Raf1 Ras binding domain; GST, glutathione S-transferase; GAP, GT-Pase-activating protein; DMEM, Dulbecco’s modified Eagle’s medium; HA, hemagglutinin; BrdUrd, 5-bromo-2′-deoxyuridine.
References
- 1.Riccardi VM. Neurofibromatosis: Phenotype, Natural History and Pathogenesis. The Johns Hopkins Press; Baltimore: 1992. [Google Scholar]
- 2.Rosenbaum T, Patrie K, Ratner N. Neuroscientist. 1997;3:412–420. [Google Scholar]
- 3.Legius E, Marchuk DA, Collins FS, Glover TW. Nat Genet. 1993;3:122–126. doi: 10.1038/ng0293-122. [DOI] [PubMed] [Google Scholar]
- 4.Side L, Taylor B, Cayouette M, Conner E, Thompson P, Luce M, Shannon K. N Engl J Med. 1997;336:1713–1720. doi: 10.1056/NEJM199706123362404. [DOI] [PubMed] [Google Scholar]
- 5.Sawada S, Florell S, Purandare SM, Ota M, Stephens K, Viskochil D. Nat Genet. 1996;14:110–112. doi: 10.1038/ng0996-110. [DOI] [PubMed] [Google Scholar]
- 6.Serra E, Puig S, Otero D, Gaona A, Kruyer H, Ars E, Estivill X, Lazaro C. Am J Hum Genet. 1997;61:512–519. doi: 10.1086/515504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cichowski K, Shih T, Schmitt E, Santiago S, Reilly K, McLaughlin M, Bronson R, Jacks T. Science. 1999;286:2172–2176. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 8.Kim MR, Tamanoi F. In: Neurofibromatosis Type 1: from Genotype to Phenotype. Upadhyaya M, Cooper DN, editors. BIOS Scientific Publishers Ltd; Oxford: 1998. pp. 89–112. [Google Scholar]
- 9.Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, Collins F. Cell. 1990;63:851–859. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- 10.Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, Conroy L, Clark R, O’Connell P, Cawthon RM, Innis MA, McCormick F. Cell. 1990;63:843–849. doi: 10.1016/0092-8674(90)90150-d. [DOI] [PubMed] [Google Scholar]
- 11.Xu GF, Lin B, Tanaka K, Dunn D, Wood D, Gesteland R, White R, Weiss R, Tamanoi F. Cell. 1990;63:835–841. doi: 10.1016/0092-8674(90)90149-9. [DOI] [PubMed] [Google Scholar]
- 12.DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, Lowy DR. Cell. 1992;69:265–273. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- 13.Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Nature. 1992;356:713–715. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 14.Kim HA, Rosenbaum T, Marchionni MA, Ratner N, DeClue JE. Oncogene. 1995;11:325–335. [PubMed] [Google Scholar]
- 15.Largaespada DA, Brannan CI, Jenkins NA, Copeland NG. Nat Genet. 1996;12:137–143. doi: 10.1038/ng0296-137. [DOI] [PubMed] [Google Scholar]
- 16.Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, Lange BJ, Freedman MH, McCormick F, Jacks T, Shannon K. Nat Genet. 1996;12:144–148. doi: 10.1038/ng0296-144. [DOI] [PubMed] [Google Scholar]
- 17.The I, Hannigan GE, Cowley GS, Reginald S, Zhong Y, Gusella JF, Hariharan IK, Bernards A. Science. 1997;276:791–794. doi: 10.1126/science.276.5313.791. [DOI] [PubMed] [Google Scholar]
- 18.Guo HF, The I, Hannan F, Bernards A, Zhong Y. Science. 1997;276:795–798. doi: 10.1126/science.276.5313.795. [DOI] [PubMed] [Google Scholar]
- 19.Guha A, Lau N, Huvar I, Gutmann D, Provias J, Pawson T, Boss G. Oncogene. 1996;12:507–513. [PubMed] [Google Scholar]
- 20.Pleasure D, Kreider B, Sobue G, Ross AH, Koprowski H, Sonnenfeld KH, Rubenstein AE. Ann N Y Acad Sci. 1986;486:227–240. doi: 10.1111/j.1749-6632.1986.tb48076.x. [DOI] [PubMed] [Google Scholar]
- 21.Sheela S, Riccardi VM, Ratner N. J Cell Biol. 1990;111:645–653. doi: 10.1083/jcb.111.2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peltonen J, Nanto-Salonen K, Aho HJ, Kouri T, Virtanen I, Penttinen R. Acta Neuropathol. 1984;63:269–275. doi: 10.1007/BF00687332. [DOI] [PubMed] [Google Scholar]
- 23.Zelkowitz M, Stambouly J. Pediatr Res. 1981;15:290–293. doi: 10.1203/00006450-198103000-00018. [DOI] [PubMed] [Google Scholar]
- 24.Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, Buchberg AM, Jenkins NA, Parada LF, Copeland NG. Genes Dev. 1994;8:1019–1029. doi: 10.1101/gad.8.9.1019. [DOI] [PubMed] [Google Scholar]
- 25.Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Nat Genet. 1994;7:353–361. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- 26.Shannon KM, O’Connell P, Martin GA, Paderanga D, Olson K, Dinndorf P, McCormick F. N Engl J Med. 1994;330:597–601. doi: 10.1056/NEJM199403033300903. [DOI] [PubMed] [Google Scholar]
- 27.Kim HA, Ling B, Ratner N. Mol Cell Biol. 1997;17:862–872. doi: 10.1128/mcb.17.2.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ridley AJ, Paterson HF, Noble M, Land H. EMBO J. 1988;7:1635–1645. doi: 10.1002/j.1460-2075.1988.tb02990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohl NE, Mosser SD, deSolms SJ, Giuliani EA, Pompliano DL, Graham SL, Smith RL, Scolnick EM, Oliff A, Gibbs JB. Science. 1993;260:1934–1937. doi: 10.1126/science.8316833. [DOI] [PubMed] [Google Scholar]
- 30.Rosenbaum T, Boissy YL, Kombrinck K, Brannan CI, Jenkins NA, Copeland NG, Ratner N. Development. 1995;121:3583–3592. doi: 10.1242/dev.121.11.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atit RP, Crowe M, Greenhalgh D, Wenstrup R, Ratner N. J Invest Dermatol. 1999;112:835–842. doi: 10.1046/j.1523-1747.1999.00609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Geer P, Henkemeyer M, Jacks T, Pawson T. Mol Cell Biol. 1997;17:1840–1847. doi: 10.1128/mcb.17.4.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson MR, DeClue JE, Felzmann S, Vass WC, Xu G, White R, Lowy DR. Mol Cell Biol. 1994;14:641–645. doi: 10.1128/mcb.14.1.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vojtek AB, Der CJ. J Biol Chem. 1998;273:19925–19928. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- 35.Herrmann C, Martin G, Wittinghofer A. J Biol Chem. 1995;270:2901–2905. doi: 10.1074/jbc.270.7.2901. [DOI] [PubMed] [Google Scholar]
- 36.de Rooij J, Bos JL. Oncogene. 1997;14:623–625. doi: 10.1038/sj.onc.1201005. [DOI] [PubMed] [Google Scholar]
- 37.Taylor SJ, Shalloway D. Curr Biol. 1996;6:1621–1627. doi: 10.1016/s0960-9822(02)70785-9. [DOI] [PubMed] [Google Scholar]
- 38.Fiordalisi JJ, Johnson RJ, Ulku AS, Cox AD. Methods Enzymol. 2000;332 doi: 10.1016/s0076-6879(01)32189-4. in press. [DOI] [PubMed] [Google Scholar]
- 39.Clark GJ, Quilliam LA, Hisaka MM, Der CJ. Proc Natl Acad Sci U S A. 1993;90:4887–4891. doi: 10.1073/pnas.90.11.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cox AD, Solski PA, Jordan DJ, Der CJ. Methods Enzymol. 1995;255:195–220. doi: 10.1016/s0076-6879(95)55023-2. [DOI] [PubMed] [Google Scholar]
- 41.Pelton PD, Sherman LS, Rizvi TA, Marchionni MA, Wood P, Friedman RA, Ratner N. Oncogene. 1998;17:2195–2209. doi: 10.1038/sj.onc.1202141. [DOI] [PubMed] [Google Scholar]
- 42.Bosse F, Zoidl G, Wilms S, Gillen CP, Kuhn HG, Muller HW. J Neurosci Res. 1994;37:529–537. doi: 10.1002/jnr.490370412. [DOI] [PubMed] [Google Scholar]
- 43.Sherman LS, Ratner N. Methods Enzymol. 2000;332 doi: 10.1016/s0076-6879(01)33069-0. in press. [DOI] [PubMed] [Google Scholar]
- 44.Nassar N, Horn G, Herrmann C, Scherer A, McCormick F, Wittinghofer A. Nature. 1995;375:554–560. doi: 10.1038/375554a0. [DOI] [PubMed] [Google Scholar]
- 45.Burgering BM, Medema RH, Maassen JA, van de Wetering ML, van der Eb AJ, McCormick F, Bos JL. EMBO J. 1991;10:1103–1109. doi: 10.1002/j.1460-2075.1991.tb08050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kohl NE, Omer CA, Conner MW, Anthony NJ, Davide JP, deSolms SJ, Giuliani EA, Gomez RP, Graham SL, Hamilton K, Handt LK, Hartman GD, Koblan KS, Kral AM, Miller PJ, Mosser SD, Oneill TJ, Rands E, Schaber MD, Gibbs JB, Oliff A. Nat Med. 1995;1:792–797. doi: 10.1038/nm0895-792. [DOI] [PubMed] [Google Scholar]
- 47.Song KS, Li S, Okamoto T, Quilliam L, Sargiacomo M, Lisanti M. J Biol Chem. 1996;271:9690–9697. doi: 10.1074/jbc.271.16.9690. [DOI] [PubMed] [Google Scholar]
- 48.Roy S, Luetterforst R, Harding A, Apolloni A, Etheridge A, Stang E, Rolls B, Hancock J, Parton R. Nat Cell Biol. 1999;1:98–105. doi: 10.1038/10067. [DOI] [PubMed] [Google Scholar]
- 49.Mikol D, Hong H, Cheng H, Feldman E. Glia. 1999;27:39–52. [PubMed] [Google Scholar]
- 50.Prendergast GC. Curr Opin Cell Biol. 2000;12:166–173. doi: 10.1016/s0955-0674(99)00072-1. [DOI] [PubMed] [Google Scholar]
- 51.Kluwe L, Friedrich R, Mautner VF. Genes Chromosomes Cancer. 1999;24:283–285. doi: 10.1002/(sici)1098-2264(199903)24:3<283::aid-gcc15>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 52.Kitano Y, Saito K, Okamoto E, Hashizume K. J Dermatol Sci. 1994;7:96–99. doi: 10.1016/0923-1811(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 53.Johnson MR, Look AT, DeClue JE, Valentine MB, Lowy DR. Proc Natl Acad Sci U S A. 1993;90:5539–5543. doi: 10.1073/pnas.90.12.5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Griesser J, Kaufmann D, Eisenbarth I, Bauerle C, Krone W. Biol Chem Hoppe Seyler. 1995;376:91–101. doi: 10.1515/bchm3.1995.376.2.91. [DOI] [PubMed] [Google Scholar]
- 55.Wallace MR, Rasmussen SA, Lim IT, Gray BA, Zori RT, Muir D. Genes Chromosomes Cancer. 2000;27:117–23. [PubMed] [Google Scholar]
- 56.Yan N, Ricca C, Fletcher J, Glover T, Seizinger BR, Manne V. Cancer Res. 1995;55:3569–3575. [PubMed] [Google Scholar]
- 57.Gutmann DH, Aylsworth H, Carey JC, Korf B, Marks J, Pyeritz RE, Rubenstein A, Viskochil D. J Am Med Assoc. 1997;278:51–57. [PubMed] [Google Scholar]