

Abstract
A microscopy-based endospore viability assay (micro-EVA) capable of enumerating germinable Clostridium endospores (GCEs) in less than 30 min has been validated and employed to determine GCE concentrations in Greenland ices and Atacama Desert soils. Inoculation onto agarose doped with Tb3+ and d-alanine triggers Clostridium spore germination and the concomitant release of ∼108 molecules of dipicolinic acid (DPA) per endospore, which, under pulsed UV excitation, enables enumeration of resultant green Tb3+-DPA luminescent spots as GCEs with time-gated luminescence microscopy. The intensity time courses of the luminescent spots were characteristic of stage I Clostridium spore germination dynamics. Micro-EVA was validated against traditional CFU cultivation from 0 to 1,000 total endospores/ml (i.e., phase-bright bodies/ml), yielding 56.4% ± 1.5% GCEs and 43.0% ± 1.0% CFU. We also show that d-alanine serves as a Clostridium-specific germinant (three species tested) that inhibits Bacillus germination of spores (five species tested) in that endospore concentration regime. Finally, GCE concentrations in Greenland ice cores and Atacama Desert soils were determined with micro-EVA, yielding 1 to 2 GCEs/ml of Greenland ice (versus <1 CFU/ml after 6 months of incubation) and 66 to 157 GCEs/g of Atacama Desert soil (versus 40 CFU/g soil).
Bacterial endospores are dormant microbial structures that are highly resistant to chemical, physical, and radiation sterilization processes (20, 31). They represent one of the most successful survival strategies of microorganisms and are formed when members of spore-forming genera (e.g., Bacillus and Clostridium) face unfavorable conditions, such as environmental extremes or starvation (19, 34). Once they are formed, endospores can stay dormant for extended periods of time, from thousands (8, 13, 20, 25, 33) to millions (3, 35) of years, although for the more extreme claims of longevity it is difficult to rule out modern contamination (37).
Anaerobic spore-forming clostridia include numerous pathogenic species that are dangerous contaminants. For example, Clostridium botulinum and C. perfringens are common food-poisoning agents that produce toxins which cause diseases such as botulism and human necrotic enteritis (9, 17). C. perfringens, C. difficile, and C. tetani are causative agents of gas gangrene, pseudomembranous colitis, and tetanus (9, 30). Some psychrotrophic clostridia are also responsible for the spoilage of chilled vacuum-packed meat (9). In addition, C. perfringens has been used as an indicator of fecal contamination, because it is present in large numbers in human and animal wastes (4, 6). Due to their resistance to various extreme conditions, Clostridium endospores are also employed as biological indicators to monitor the effectiveness of various sterilization processes (11, 12).
Currently, the standard method for quantifying viable Clostridium endospores is measuring CFU after heat shock killing of vegetative cells. This method requires several days of incubation and a tedious anaerobic culturing technique, and it is amenable for culturing only fewer than 1% of environmental species (24). Other molecular endospore viability assays include ATP assay (26) and quantitative PCR (qPCR) coupled with propidium monoazide (PMA) (28), which, unlike microscopy-based endospore viability assay (micro-EVA), require extensive sample preparation and are labor-intensive.
Previously, we described a spectroscopy-based endospore viability assay (spectro-EVA) to quantify dipicolinic acid (DPA) released from germinating Clostridium spores in liquid suspension (39). Germination was triggered by various germinants, such as l-alanine/NaHCO3, l-lactate, or d-alanine, which cause the release of approximately 108 molecules of a unique biomarker, DPA, from the spore core. Spectro-EVA is based on the detection of DPA in bulk solution via Tb3+-DPA luminescence spectroscopy, with a limit of detection (LOD) of 1,000 spores/ml. Unfortunately, spectro-EVA has even lower detection limits when environmental extracts are analyzed due to sensitivity to interference from contaminants. Previously, this limitation was overcome for the case of Bacillus endospores by employing a microscopy-based EVA (41), where individual spores are enumerated as they germinate in a microscope field of view.
Here we report details of a rapid microscopy-based endospore viability assay (micro-EVA) that enables enumeration of single germinating Clostridium endospores on Tb3+- and d-alanine-doped agarose. d-Alanine was used as a germinant, which serves to trigger Clostridium spore germination while inhibiting Bacillus spore germination (2, 14, 38). Germination releases DPA from endospores, and subsequent Tb3+-DPA binding results in green luminescent spots under pulsed UV excitation in a field of view of a time-gated microscope. These were enumerated as germinable Clostridium endospores (GCEs) using time-gated Tb3+-DPA luminescence microscopy (i.e., micro-EVA). A parallel comparison of micro-EVA data with culturing data validated this method. Finally, we compared micro-EVA to culturing methods to quantify GCEs from two Mars analog environments, Greenland ice core and Atacama Desert.
MATERIALS AND METHODS
Materials.
Terbium(III) chloride hexahydrate (99.999%), dipicolinic acid (2,6-pyridinedicarboxylic acid) (DPA) (99%), l-alanine, and d-alanine were purchased from Sigma-Aldrich (Milwaukee, WI) and used without further purification. Sodium pyrophosphate was purchased from Mallinckrodt (Paris, KY). Two types of Clostridium growth media, reinforced clostridial medium (RCM) and ATCC 2135 broth (GS-2CB medium), were purchased from the American Type Culture Collection (ATCC) (Manassas, VA). Agarose for culturing experiments was obtained from MacConnell Research (San Diego, CA). Agarose for micro-EVA experiments was obtained from Invitrogen (Carlsbad, CA). Clostridium sporogenes (ATCC 7955) and Clostridium hungatei (ATCC 700212), from ATCC, were obtained as freeze-dried pellets. Bacillus atrophaeus (ATCC 9372) and Bacillus cereus endospores were purchased from Raven Biological Laboratories.
Endospore production and purification.
C. sporogenes and C. hungatei were revived from frozen, dry pellets in a small volume (5 to 6 ml) of growth medium at the optimal growth temperatures of 37°C and 30°C, respectively. The growth media for C. sporogenes and C. hungatei were RCM and GS-2CB medium, respectively. Incubation was commenced under strict anaerobic conditions with a 100% N2 headspace. The culture was transferred to fresh medium after development of visible turbidity. The production and purification of various Clostridium spores were performed using a protocol developed specifically for Clostridium species (40). The spore suspension contained less than 1% vegetative cell material as determined by phase-contrast microscopy.
Bacillus spores were obtained using the following protocol. Bacillus vegetative cells were grown on tryptic soy agar (TSA) and inoculated onto a sporulation medium after reaching exponential growth phase. The sporulation medium contained 1.6% nutrient broth, 1.6% agar, 0.2% KCl, 0.05% MgSO4, 1 mM Ca(NO3)2, 100 μM MnCl24H2O, 1 μM FeSO4, and 0.1% glucose (pH 7.0). After incubation at 37°C for 1 week, cells were suspended into sterile deionized water. With phase-contrast microscopy, 95% of the cells formed endospores free of sporangia. Endospores were harvested and separated from vegetative cells and debris by centrifugation at 6,300 × g, washing 10 times with sterile deionized water, and sonication (25 kHz) for 5 min. The endospore suspension was incubated in lysozyme (0.2 mg/ml) and trypsin (0.1 mg/ml) at 30°C with constant stirring overnight to lyse and degrade any remaining vegetative cells. Endospores were purified by eight cycles of centrifugation (6,300 × g) and washed with sterile deionized water until >99.9% of the cells were fully refractile with no noticeable cellular debris.
Both Clostridium and Bacillus spore suspensions were stored at 4°C in the dark before use. Total spore concentrations were determined using a Petroff-Hausser hemocytometer (model 3900; Hausser Scientific, Horsham, PA), and CFU concentrations were determined using spread plating measurement in triplicate.
Determination of total endospore concentrations using phase-contrast microscopy.
Aliquots (5 μl) of spore suspension were placed in a Petroff-Hausser counting chamber, and spores were observed at a magnification of ×400 using a phase-contrast microscope (Nikon Eclipse 80i; AG Heinze Co, Lake Forest, CA) mounted with a digital camera (Nikon Digital Sight DS-5 M). The smallest squares in the counting chamber are 0.05 mm by 0.05 mm, of which 80 were analyzed to obtain the average spore count per square. To obtain statistically significant counts, the spore concentration was held above 107 spores/ml, which resulted in at least 10 spores per 16 squares.
Spore culturability.
To determine the endospore culturability, heat shock treatment at 80°C for 15 min was applied to a purified spore suspension with a known concentration determined by microscopy as described above. A series of dilutions was made to reach an expected concentration range of 0 to 1,000 spores/ml. For concentrations of 1,000 to 333 spores/ml, 100-μl aliquots from each dilution were plated in triplicate on solidified RCM (15 g agar per liter). For concentrations of 100 spores/ml or below, 1-ml aliquots from each dilution were plated in triplicate on the same solidified medium. Plates were incubated in a vinyl anaerobic chamber containing 97.5% N2 and 2.5% H2 (type C; Coy Laboratory Products Inc., Grass Lake, MI) at room temperature for 10 days. Colonies were counted, and the average number was designated the CFU per volume of original sample. The culturability was determined as the percentage of spores capable of forming colonies.
Spectroscopy.
Tb3+-DPA luminescence excitation spectra (λex = 250 to 360 nm; λem = 544 nm) and emission spectra (λex = 278 nm; λem = 450 to 650 nm) were recorded with a Fluorolog-3 model FL3-22 spectrofluorometer (Horiba Jobin-Yvon, Edison, NJ) consisting of a 450-W xenon short-arc lamp for excitation, two Czerny-Turner double-grating monochromators (with all-reflective optics and 0.5-nm accuracy), a temperature-controlled sample chamber, and a R928P photomultiplier tube (PMT) as a detector (Products for Research Inc., Danvers, MA). The PMT is thermoelectrically cooled with a Peltier cooling unit, and the reference detector is a photodiode. A 350-nm-cutoff filter (Melles Griot, Covina, CA) was placed at the entrance of the emission monochromator to prevent second-order diffraction. Endospores immobilized on agarose were measured in front-face configuration.
Sample preparation for micro-EVA experiments.
An endospore suspension of known concentration was filtered onto a 1.5-mm2 spot on a 0.2-μm polycarbonate membrane filter (Whatman, Florham Park, NJ) using a 96-well microsample filtration manifold (Schleicher & Schuell, Keene, NH). To ensure that the endospore surface density was optimal for a given initial endospore concentration, the concentration was adjusted so that each microscopic field of view contains fewer than 300 endospores. Endospores concentrated on the filter were transferred to an ∼0.5-mm-thick, 9-mm-diameter slab of 1.5% agarose substrate containing 5 mM TbCl3 and 100 mM l-alanine (or d-alanine) mounted in a silicone isolator (Molecular Probes, Eugene, OR) on a quartz microscope slide. After endospore transfer, the agarose surface was covered with a piece of 0.2-mm-thick polydimethylsiloxane (PDMS). The PDMS was prepared by mixing the polymer base and curing agent (Sylgard; Dow Corning) at a 10-to-1 ratio. After degassing, the mixture was cast over a 0.2-mm-thick stainless steel mold and cured in an oven for 2 h at 65°C. The agarose, silicone isolator, and PDMS were autoclaved at 121°C for 15 min before use. A piece of PDMS was peeled off and attached on top of an endospore-laden agarose surface for sealing.
The micro-EVA instrument.
The micro-EVA instrument consists of a time-gated camera (Photonics Research Systems, Salford, United Kingdom) mounted on a Nikon SMZ800 stereoscopic microscope (large working distance for xenon lamp), a xenon flash lamp (Perkin-Elmer, Waltham, MA) mounted at 45 degrees with respect to the sample, and a temperature-controlled microscope slide holder (Thermal) (Fig. 1) (41). The slide holder enables endospores to germinate at 37°C. The charge-coupled-device (CCD) camera has a resolution of 752 by 582 pixels at 14 bits with a chip size of 2/3 in. The camera has 50% sensitivity at 430 to 730 nm, with peak sensitivity at 550 nm, and was Peltier cooled to 40°C below ambient temperature and synchronized to the xenon lamp via TTL pulses (300 Hz with tail time up to 50 μs). A high-pass filter (03FCG067; Melles Griot) centered at 500 nm was placed along the light path on the emission side before reaching the microscope objective. We collected time stacks of time-gated images by real-time streaming with a delay of 100 microseconds and an exposure time of 5 s in each frame.
FIG. 1.

(a) Configuration of the micro-EVA instrument used in this investigation, consisting of a stereomicroscope mounted with a time-gated camera and a xenon flash lamp for UV excitation. (b) Sample well on a quartz microscope slide containing Tb3+- and d-alanine-doped agarose. (c) Schematic representation of the sample slide, consisting of a quartz slide on which Tb3+- and d-alanine-doped agarose is confined by a red rubber gasket well. Endospores (brown circles) are inoculated onto an agarose substrate and subsequently covered with a thin layer of PDMS. (d) Inoculated endospores germinate due to d-alanine, causing the release of ∼108 molecules of DPA and subsequent formation of highly luminescent Tb3+-DPA complexes that appear as discrete bright spots in the microscope field of view. (e) Absorption-energy transfer-emission photophysics of the Tb3+-DPA luminescence assay. DPA acts as a light harvester that transfers excitation energy to luminescent terbium ion. (f) Energy (Jablonski) diagram of the Tb3+-DPA photophysics.
Assignment of germinable endospores using micro-EVA.
Endospore germination on the agarose surface followed the reported microgermination dynamics. DPA released from single endospores manifested as individual bright spots in 30 min under time-gated microscopy due to local formation of a Tb3+-DPA complex. Assignment of germinable endospores was made based on intensity and size. Adaptive thresholding was applied to segment pixels that were 20% brighter than the background with a characteristic rising intensity. Each bright spot must exhibit a continuous rising intensity over the course of germination in order to register an endospore count. This criterion eliminated false positives by not counting sporadic bright spots and long-lived luminescent interference. The 8-connected adjacent pixels were analyzed to screen for endospore clumps. The number of endospores present was calculated by dividing the squared sum of neighboring pixel brightness by the mean brightness of an individual endospore determined empirically. This was done in a recursive way until all of the pixels were counted and marked.
GISP2 ice core handling and analysis.
The ice cores were donated from the Greenland Ice Sheet Project (GISP2). Initially stored at −80°C, the GISP2 ice cores (MCA.02 no. 158 [depth, 157.45 to 157.70 m; age, 600 years], MCA.02 no. 480 [depth, 480.15 to 480.40 m; age, 2,000 years], and MCA.02 no. 835 [depth, 834.10 to 834.35 m; age, 4,000 years]) were transitioned to −25°C for 8 h and then to 0°C prior to decontamination. The melted volume of each ice core was ∼500 ml. Temperature equilibration of the ice core was necessary to prevent the decontamination solutions from freezing onto the outside of the ice core.
Decontamination was performed according to the modified decontamination protocol of Rogers et al. (28a). Briefly, under aseptic conditions within a biological hood (Sterilgard III Advance; The Baker Company, Sanford, ME), the ice core(s) was soaked for 10 s in a solution of 1,600 ml cold (4°C) 6.25% sodium hypochlorite (NaOCl) and then rinsed three times with 1,600 ml of cold (4°C) sterile water (18.2 MΩ, <1 ppb total organic carbon [TOC], DNase free and RNase free). Each rinse lasted for 10 s. The rinsed ice core(s) was then melted within a sterile beaker at room temperature. The decontamination procedure was validated using synthetic ice core sections (a cylinder 5 cm in diameter and 15 cm in length) consisting of frozen suspensions of 104 B. atrophaeus endospores in the interior and 105 B. megaterium endospores smeared on the exterior surfaces.
The melted ice core(s) was vacuum filtered through a 45-mm, 0.1-μm membrane filter (Nuclepore Track-Etch membrane; Sterlitech Corporation, Kent, WA) backed with a 0.45-μm backing filter. The filter(s) was resuspended in 4.4 ml of cold (4°C) sterile water. The suspension and filter(s) were vortexed for 5 min, with a 1-min chilling increment in between to prevent cells from overheating. The concentrated suspension(s) was used for subsequent endospore viability assays. A 100-μl aliquot of the concentrated sample was filtered onto a 1.5-mm2 spot on a 0.2-μm polycarbonate membrane filter (Whatman, Florham Park, NJ) using a 96-well microsample filtration manifold (Schleicher & Schuell, Keene, NH). The sample was transferred aseptically to 1.5% agarose doped with 5 mM TbCl3 and 100 mM d-alanine, which was contained in a 9-mm silicone well (Molecular Probes, Eugene, OR) on a quartz slide. The sample was transferred by streaking the membrane filter across the agarose surface. The d-alanine within the agarose triggered Clostridium spores present in the sample to germinate and release DPA. A piece of 4.5-μm-thick polydimethylsiloxane (PDMS) film was placed on top of the agarose to prevent aerial contamination and to minimize water evaporation and DPA diffusion across the agarose substrate. Images were taken with a time-gated microscope (described above). Over a germination course of 120 min, formation of luminescent spots could be observed under time-gated microscopy, which was indicative of the localized formation of the Tb3+-DPA complex due to DPA release during endospore germination. Background fluorescence and autofluorescence were minimized because of time gating, rendering a low-intensity background. The characteristic germination time course allows unambiguous assignment of germinating endospores.
Atacama soil handling and analysis.
An expedition of our research group to the Atacama Desert in northern Chile brought back soil samples from three different locations (7, 13a): the most arid zone, the Yungay area (site A), where another research group has performed experiments (18); site E, 5 km northeast of site A; and a depth transect in a soil pit 80 to 220 cm underneath the surface soil. We selected three samples for this study: a subsurface soil sample from site E and the bottom and top samples from the soil pit. A field-compatible Pawkit water activity meter (Decagon, Pullman, WA) was used to measure water activities (aw) of soil samples in the field. The meter was calibrated using standards of 6.0 M NaCl (aw = 0.76) and 13.41 M LiCl (aw = 0.25). Following calibration, a thin layer of homogenized soil was placed along the bottom of a disposable sample cup and placed in the instrument for measurement. Soil hydrogen ion activity (pH) was recorded in the field on a 1:5 solids-deionized water suspension. An IQ150 hand-held pH/mV/temperature meter (I.Q. Scientific Instruments) was used to measure pH and redox potential.
Cells and spores were extracted following a previously developed protocol. A 30-ml aliquot of autoclaved phosphate-buffered saline (PBS) (EMD Chemicals Inc., Gibbstown, NJ) with 10 mM sodium pyrophosphate and 0.1% Tween 80 (Sigma-Aldrich, St. Louis, MO) was added to 15 g of soil in a 50-ml centrifuge tube. The tube was vortexed at maximum speed for 5 min, with intervening 1-min cooling in ice. The tube contents were then allowed to sediment for 20 to 40 min until a clear layer of supernatant was visible (7). The supernatant was aliquoted for micro-EVA measurement. Five hundred microliters of soil extract was filtered onto 13-mm2 spots using a 96-well microsample filtration manifold (Schleicher & Schuell, Keene, NH). All of these steps were carried out inside a biohazard cabinet hood (Sterilgard III Advance; The Baker Company, Sanford, ME). Micro-EVA measurements to quantify viable Clostridium spores were conducted following the procedures described in the previous section.
RESULTS AND DISCUSSION
We have developed a microscopy-based endospore viability assay (micro-EVA) to rapidly measure single GCEs extracted from extreme environments. We first validated the micro-EVA with respect to traditional culturing methods over a concentration range of 0 to 1,000 spores/ml. Second, we demonstrated that d-alanine is a Clostridium-specific germinant that inhibits Bacillus germination. Third, we successfully applied micro-EVA to quantify GCEs from two Mars analog environments, Greenland ices and Atacama Desert soils.
Observations of single Clostridium spore germination.
As a spore germinates, ∼108 DPA molecules are released into the immediate volume surrounding the spore. DPA then combines with Tb3+ in the agarose matrix to form the Tb3+-DPA complex, which can be visualized as a bright luminescence halo under UV excitation. The germinating endospores manifest as bright spots that are enumerated in a microscope field of view. The germination time course showed an increase in spot intensity as DPA molecules were released, reaching a plateau after 15 min that indicates the completion of germination. Germination of C. sporogenes spores on Tb3+- and germinant (l-alanine or d-alanine)-doped agarose was confirmed by the detection of DPA release using a Fluorolog-3 model FL3-22 spectrofluorometer in a front-face configuration (see Fig. S1 in the supplemental material). Figure 2 shows the micro-EVA time-lapse images of a germinating C. sporogenes spore. Figure S2 in the supplemental material shows single germinated spores under micro-EVA in one field of view. Negative-control data showed that Tb3+ does not luminescence in the absence of either spores or germinant (data not shown). In combination, these data establish the assignment of micro-EVA luminescent spots as single germinating endospores.
FIG. 2.

Germination time courses of single C. sporogenes spores at 37°C, monitored by Tb3+-DPA luminescence using micro-EVA.
Validation of micro-EVA against culturing.
To validate micro-EVA, we performed parallel germination and culturing experiments with from 0 to 1,000 spores/ml. Figure 3 shows the GCE concentrations measured with micro-EVA and the culturable endospore concentrations measured with plate counting plotted against total endospore concentrations as determined with phase-contrast microscopy. Sterile samples did not yield counts (i.e., there were no false positives), which enabled us to achieve the ultimate sensitivity of one germinable endospore per micro-EVA field of view. Of the total endospore population (i.e., phase-bright endospore bodies), micro-EVA revealed that 56.4% ± 1.5% of the population were germinable within 60 min, while 43.0% ± 1.0% were culturable within 10 days of incubation. The germinable/culturable ratio was 1.31, which is consistent with the fact that a subset of the total endospore population is germinable but not culturable (24). Specifically, micro-EVA measurements probe DPA release during stage 1 germination, while CFU measurements probe the cell division, which is the last stage of the germination process and requires proper growth conditions (e.g., enough nutrient and an anaerobic environment for Clostridium spores). It is expected, then, that the additional requirements for achieving the observable colonies will inherently yield lower culturable versus germinable populations in a given endospore sample. Our results indicate that micro-EVA was a faster method to assess spore viability, as germinability has been proposed as the indicator of spore viability.
FIG. 3.

Comparison of germinable endospore concentrations (closed circles, left y axis) determined by micro-EVA and culturable endospore concentrations (open circles, right y axis) versus total endospore concentration as determined by phase-contrast microscopy.
Specificity for Clostridium spores.
d-Alanine has been reported as a germination inhibitor for various Bacillus spores (2, 14, 21, 38); conversely, for a number of Clostridium species, d-alanine has shown no inhibitory effect on spore germination (23, 27, 36). In our investigation, d-alanine was used as the Clostridium-specific germinant to distinguish Clostridium spores from Bacillus spores. Previously, we had tested the effect of d-alanine on germination with various Clostridium and Bacillus spores using spectro-EVA and found that in liquid suspension, d-alanine germinated all Clostridium spores tested, while it inhibited the germination of all Bacillus spores. This showed that d-alanine may be used to selectively germinate Clostridium in the presence of Bacillus endospores.
We conducted micro-EVA experiments with d-alanine on five different Bacillus spores and three Clostridium spores. The Bacillus spores used in this study were B. subtilis isolated from the Atacama Desert, B. simplex isolated from Kilimanjaro, B. atrophaeus and B. cereus from ATCC, and B. longisporus isolated from Jet Propulsion Laboratory (JPL) soil samples. The Clostridium spores used in this study were C. sporogenes and C. hungatei from ATCC and Clostridium G5A-1 isolated from a Greenland ice core. None of the Bacillus spores tested germinated with d-alanine as the sole germinant on agarose, including the Kilimanjaro strain, which germinated with d-alanine in liquid suspension. All three Clostridium spores germinated with d-alanine, as indicated by bright spots under UV light on agarose after 1 hour of germination at 37°C. Figure S3 in the supplemental material shows the time-gated images of four selected strains.
Quantification of germinable Clostridium endospores from Greenland ice cores.
Greenland ice cores have been considered unique repositories of microbes frozen at various points in the geologic past. Previously, several efforts to culture sporeformers from cold environments have been reported. For example, spore-forming bacteria have been isolated from 750,000-year-old Guliya ice (5) and from a Malan Glacier ice core (0 to 102 m) (42, 43). Miteva and colleagues found sporeformers among the isolates acquired from GISP2 at 3,043 m, a visibly silty layer near the bottom, which had a total microbial concentration of 1 to 9 × 107 cells/ml (15, 16, 32). The ice core samples used in this study were donated from GISP2 and ranged from 157 m to 834 m in depth. They are from shallower locations than the GISP2 3,043-m sample, and no silty layer was observed in any of our samples. The micro-EVA experiments reported here quantify germinable Clostridium spores from Greenland ice cores, which show about 1 to 2 GCEs/ml ice meltwater, while culturing on R2A medium over 9 months yielded no growth.
Figure S4a in the supplemental material shows representative micro-EVA images of GCEs from a Greenland ice core sample, and Fig. 4 a shows the corresponding germination time course plots for two Clostridium spores. DPA release during germination resulted in bright luminescent spots due to Tb3+-DPA complex formation. While individual endospores, with reported sizes of between 0.8 and 4 μm, could not be spatially resolved with our time-gated microscope, the intense Tb3+-DPA luminescence emanating from the location of germinating endospores enabled rapid enumeration. From micro-EVA experiments, we determined that the numbers of GCEs from the three ice core depths (arranged in ascending depth order) are 2.0 ± 0.3 GCEs/ml, 1.2 ± 0.4 GCEs/ml, and 2.0 ± 0.8 GCEs/ml (Table 1). Due to the low spore concentrations in ice cores and the limited sample volume, the entire ice core samples from the three depths reported here were used to conduct micro-EVA experiments. However, cultivation experiments recovered fewer than 1 CFU/ml from other ice core samples at similar depths (data not shown). From the germination time course (Fig. 4a), we observed that the spore from a depth of 834 m (4,000 years old) started to germinate within the first 5 min, and the luminescence intensity of the spot reached a plateau in 50 min. However, another spore from 158 m (600 years old) showed a different germination profile. It started with a long lag phase (∼20 min), which was the time period when the luminescence intensity stayed unchanged, and then the intensity started to increase gradually. The rapid release of DPA started after 40 min, and the intensity reached a plateau after 100 min. Distinctive germination time courses between species may enable species-specific analysis.
FIG. 4.
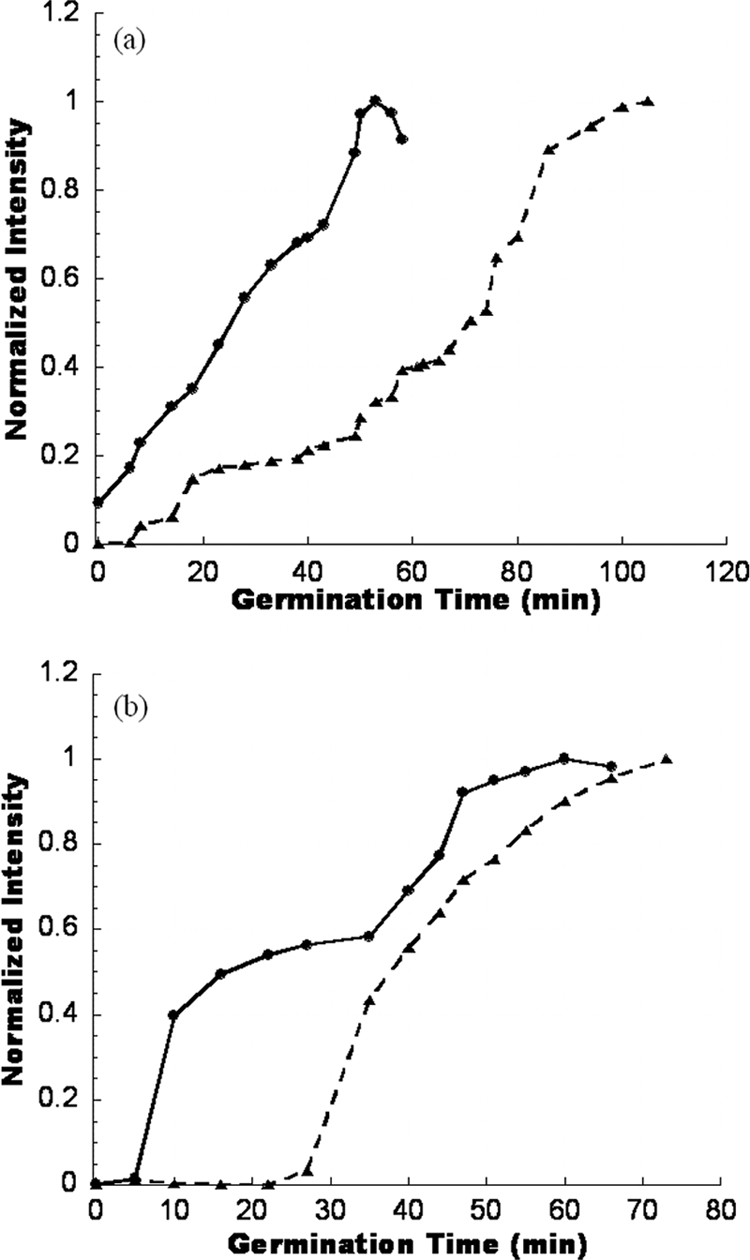
(a) Germination time course plots for two germinating spores from ice core samples, aged 600 years and 4,000 years. (b) Germination time course plots for two germinating spores from Atacama soil samples.
TABLE 1.
Summary of micro-EVA results for Greenland ice core samples
| Depth (m) | Age (yr) | Time to filter (min)a | pH | No. of GCEs/ ml ice core water (mean ± SD) |
|---|---|---|---|---|
| 158 | 600 | 14 | 5.0 | 2.0 ± 0.3 |
| 480 | 2,000 | 23 | 5.0 | 1.2 ± 0.4 |
| 834 | 40,00 | 35 | 5.0 | 2.0 ± 0.8 |
The amount of time it takes to finish filtering 450 ml ice core meltwater. This parameter indicates the turbidity of ice core samples.
Quantification of germinable Clostridium endospores from Atacama Desert soils.
We also applied micro-EVA to investigate endospore viability in one of the driest and oldest deserts on Earth, the Atacama Desert in Chile. This desert serves as a proving ground for NASA's future life detection instrumentation. Endospore-forming bacteria are extremely common in soils due to the high resistance of bacterial spores to both dehydration and high temperatures. Past efforts have shown a near-sterile region (site A in the Yungay region) with no recoverable DNA and extremely low culturable cell counts (7, 13a, 18). However, micro-EVA has been used to measure both aerobic and anaerobic sporeformers from multiple sites in the Atacama Desert. Our data suggest that life's most resilient representative can survive in much harsher conditions than previously thought, which has implications for the probability of growth in special regions (i.e., where the water activity is above 0.5 and the temperature is above −20°C) on another planet, such as the subsurface with less UV radiation and higher water activity.
Figure S4b in the supplemental material shows a representative micro-EVA image of GCEs in one soil sample, and Fig. 4b shows the corresponding germination time course plots for two different Clostridium spores. From micro-EVA experiments, we determined that the number of GCEs from all the sites tested ranged from 66 to 157 GCEs/g soil (Table 2), while the CFU cultivation recovered only 40 spores/g soil from the subsurface of site E. Our data suggested a correlation of higher GCE counts and increased water activity. From the germination time course (Fig. 4b), we observed that, as in the case of ice core samples, there is a large variance in germination time courses. In one case, germination started after 5-min lag time, and the luminescence intensity of the spot reached a plateau in 60 min in a biphasic manner. In another example, the lag phase was ∼30 min, after which the intensity increased continuously and reached a plateau at around 70 min.
TABLE 2.
Summary of micro-EVA results for Atacama Desert soil samples
| Sample description | pHa | awa | No. of GCEs/ g soil (mean ± SD) |
|---|---|---|---|
| Site E subsurface | 6.73 | 0.07 | 66 ± 23 |
| Depth profile 6, top | 8.67 | 0.25 | 98 ± 19 |
| Depth profile 1, bottom | 8.14 | 0.37 | 157 ± 18 |
pH and water activity (aw) were measured in the field when samples were collected.
Comparison of micro-EVA and spectro-EVA.
In a previous study, we reported a related method where germinating spores were enumerated in bulk suspension by luminescence spectroscopy (i.e., spectro-EVA), where Tb3+-DPA luminescence intensities were tabulated against a C. sporogenes spore calibration curve. The micro-EVA approach is superior to spectro-EVA, because micro-EVA is capable of enumerating single spores, while the limit of detection of spectro-EVA is 1,000 spores/ml. This advantage is gained because in micro-EVA experiments the millimolar DPA halos surrounding single germinated endospores are readily imaged with high contrast, whereas germination of single endospores in bulk suspension (∼1 ml) gives rise to mere femtomolar DPA concentrations, which are far below the LOD for spectro-EVA. The lower detection limit of micro-EVA enables the quantification of viable spores from diluted samples, such as Greenland ice core (fewer than 10 GCEs/ml). With micro-EVA, we take advantage of the long luminescence lifetime (τ = 0.5 to ∼2 ms) of Tb3+-DPA, enabling the use of time gating to effectively remove background fluorescence (i.e., interfering fluorophores with nanosecond lifetimes). Time gating eliminates potential features causing false-positive results and renders the image background dark. Elimination of this background enables a striking increase in image contrast and detection sensitivity even for the most challenging environmental extracts, such as Atacama Desert soil samples.
Summary.
Endospores are the most resilient form of microbial life. We obtained samples from two extreme environments: Greenland, which is considered an analog to icy worlds (e.g., Europa and Enceladus) (1, 10), and the Atacama Desert, Chile, which is considered a Mars analog site (18, 29). The validated micro-EVA approach was used successfully to measure germinable Clostridium endospores from samples of these extreme environments, while culturing yielded mostly negative growth results. This underscores the utility of the micro-EVA approach and instrumentation, as it enables viability assessment of endospores in environments that approximate those of other solar system bodies.
Supplementary Material
Acknowledgments
We thank Morgan L. Cable for editing and the NSF National Ice Core Laboratory for GISP2 samples.
We are grateful to the DHS Science and Technology Directorate, NASA Astrobiology Institute, and Planetary Protection for funding.
The research described in this paper was carried out at the Jet Propulsion Laboratory, California Institute of Technology, under a contract with the National Aeronautics and Space Administration.
Footnotes
Published ahead of print on 4 February 2011.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Abyzov, S., et al. 1999. Antarctic ice sheet as an object for solving some methodological problems of exobiology. Adv. Space Res. 23:371-376. [Google Scholar]
- 2.Barlass, P. J., C. W. Houston, M. O. Clements, and A. Moir. 2002. Germination of Bacillus cereus spores in response to l-alanine and to inosine: the roles of gerL and gerQ operons. Microbiology 148:2089-2095. [DOI] [PubMed] [Google Scholar]
- 3.Cano, R. J., and M. K. Borucki. 1995. Revival and identification of bacterial spores in 25-million-year-old to 40-million-year-old Dominican amber. Science 268:1060-1064. [DOI] [PubMed] [Google Scholar]
- 4.Chauret, C. P., C. Z. Radziminski, M. Lepuil, R. Creason, and R. C. Andrews. 2001. Chlorine dioxide inactivation of Cryptosporidium parvum oocysts and bacterial spore indicators. Appl. Environ. Microbiol. 67:2993-3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christner, B. C., E. Mosley-Thompson, L. G. Thompson, and J. N. Reeve. 2003. Bacterial recovery from ancient glacial ice. Environ. Microbiol. 5:433-436. [DOI] [PubMed] [Google Scholar]
- 6.Committee on Indicators for Waterborne Pathogens, National Research Council. 2004. Indicators for water borne pathogens. National Academy Press, Washington, DC.
- 7.Connon, S. A., E. D. Lester, H. S. Shafaat, D. C. Obenhuber, and A. Ponce. 2007. Bacterial diversity in hyperarid Atacama Desert soils. J. Geophys. Res. 112:G04S17. [Google Scholar]
- 8.Gest, H., and J. Mandelstam. 1987. Longevity of microorganisms in natural environments. Microbiol. Sci. 4:69-71. [PubMed] [Google Scholar]
- 9.Hatheway, C. L. 1990. Toxigenic clostridia. Clin. Microbiol. Rev. 3:66-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Homeck, G. 2000. The microbial world and the case for Mars. Planet. Space Sci. 48:1053-1063. [Google Scholar]
- 11.Johnston, M. D., S. Lawson, and J. A. Otter. 2005. Evaluation of hydrogen peroxide vapour as a method for the decontamination of surface contaminated with Clostridium botulinum spores. J. Microbiol. Methods 60:403-411. [DOI] [PubMed] [Google Scholar]
- 12.Kanemitsu, K., et al. 2005. A comparative study of ethylene oxide gas, hydrogen peroxide gas plasma, and low-temperature steam formaldehyde sterilization. Infect. Control Hosp. Epidemiol. 26:486-489. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy, M. J., S. L. Reader, and L. M. Swierczynski. 1994. Preservation records of microorganisms—evidence of the tenacity of life. Microbiology 140:2513-2529. [DOI] [PubMed] [Google Scholar]
- 13a.Lester, E. D., M. Satomi, and A. Ponce. 2007. Microflora of extreme arid Atacama Desert soils. Soil Biol. Biochem. 39:704-708. [Google Scholar]
- 14.McKevitt, M. T., et al. 2007. Effect of endogenous d-alanine synthesis and autoinhibition of Bacillus anthracis germination on in vitro and in vivo infections. Infect. Immun. 75:5726-5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miteva, V. I., and J. E. Brenchley. 2005. Detection and isolation of ultrasmall microorganisms from a 120,000-year-old Greenland glacier ice core. Appl. Environ. Microbiol. 71:7806-7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miteva, V. I., P. P. Sheridan, and J. E. Brenchley. 2004. Phylogenetic and physiological diversity of microorganisms isolated from a deep Greenland glacier ice core. Appl. Environ. Microbiol. 70:202-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murray, P., E. Baron, M. Pfaller, J. Jorgensen, and R. Yolken (ed.). 2003. Manual of clinical microbiology, 8th ed., vol. 1. ASM Press, Washington, DC.
- 18.Navarro-González, R., et al. 2003. Mars-like soils in the Atacama Desert, Chile, and the dry limit of microbial life. Science 302:1018-1021. [DOI] [PubMed] [Google Scholar]
- 19.Nicholson, W. L., and P. Setlow. 1990. Sporulation, germination and outgrowth, p. 391-450. In C. R. Harwood and S. M. Cutting (ed.), Molecular biology methods for Bacillus. John Wiley and Sons, Sussex, England.
- 20.Nicholson, W. L., N. Munakata, G. Horneck, H. J. Melosh, and P. Setlow. 2000. Resistance of Bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiol. Mol. Biol. Rev. 64:548-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paidhungat, M., and P. Setlow. 1999. Isolation and characterization of mutations in Bacillus subtilis that allow spore germination in the novel germinant d-alanine. J. Bacteriol. 181:3341-3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reference deleted.
- 23.Plowman, J., and M. W. Peck. 2002. Use of a novel method to characterize the response of spores of non-proteolytic Clostridium botulinum types B, E and F to a wide range of germinants and conditions. J. Appl. Microbiol. 92:681-694.11966909 [Google Scholar]
- 24.Ponce, A., S. A. Connon, and P. T. Yung. 2008. Detection and viability assessment of endospore-forming pathogens, p. 481-523. In M. Zourob, S. Elwary, and A. Turner (ed.), Principles of bacterial detection: biosensors, recognition receptors and microsystems. Springer, New York, NY.
- 25.Potts, M. 1994. Desiccation tolerance of prokaryotes. Microbiol. Rev. 58:755-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rakotonirainy, M. S., C. Héraud, and B. Lavédrine. 2003. Detection of viable fungal spores contaminant on documents and rapid control of the effectiveness of an ethylene oxide disinfection using ATP assay. Luminescence 18:113-121. [DOI] [PubMed] [Google Scholar]
- 27.Ramirez, N., and E. Abel-Santos. 2010. Requirements for germination of Clostridium sordellii spores in vitro. J. Bacteriol. 192:418-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rawsthorne, H., C. N. Dock, and L. A. Jaykus. 2009. PCR-based method using propidium monoazide to distinguish viable from nonviable Bacillus subtilis spores. Appl. Environ. Microbiol. 75:2936-2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28a.Rogers, S. O., et al. 2004. Comparisons of protocols for decontamination of environmental ice samples for biological and molecular examinations. Appl. Environ. Microbiol. 70:2540-2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rothschild, L. J., and R. L. Mancinelli. 2001. Life in extreme environments. Nature 409:1092-1101. [DOI] [PubMed] [Google Scholar]
- 30.Russell, A. D. 1990. Bacterial spores and chemical sporicidal agents. Clin. Microbiol. Rev. 3:99-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Setlow, P. 2000. Resistence of bacterial spores, p. 217-230. In G. Storz and R. Hengge-Aronis (ed.), Bacterial stress responses. ASM Press, Washington, DC.
- 32.Sheridan, P. P., V. I. Miteva, and J. E. Brenchley. 2003. Phylogenetic analysis of anaerobic psychrophilic enrichment cultures obtained from a Greenland glacier ice core. Appl. Environ. Microbiol. 69:2153-2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sneath, P. H. 1962. Longevity of micro-organisms. Nature 195:643-646. [DOI] [PubMed] [Google Scholar]
- 34.Sonenshein, A. L. 2000. Endospore-forming bacteria: an overview, p. 133-150. In Y. V. Brun and L. J. Shimkets (ed.), Prokaryotic development. American Society for Microbiology, Washington, DC.
- 35.Vreeland, R. H., W. D. Rosenzweig, and D. W. Powers. 2000. Isolation of a 250 million-year-old halotolerant bacterium from a primary salt crystal. Nature 407:897-900. [DOI] [PubMed] [Google Scholar]
- 36.Waites, W. M., and L. R. Wyatt. 1971. Germination of spores of Clostridium bifermentans by certain amino acids, lactate and pyruvate in the presence of sodium or potassium ions. J. Gen. Microbiol. 67:215-222. [DOI] [PubMed] [Google Scholar]
- 37.Willerslev, E., A. J. Hansen, and H. N. Poinar. 2004. Isolation of nucleic acids and cultures from fossil ice and permafrost. Trends Ecol. Evol. 19:141-147. [DOI] [PubMed] [Google Scholar]
- 38.Wolf, J., and S. A. Z. Mahmoud. 1957. The effect of l- and d-alanine on the germination of some Bacillus spores. J. Appl. Bacteriol. 20:373-383. [Google Scholar]
- 39.Yang, W.-W., and A. Ponce. 2009. Rapid endospore viability assay of Clostridium sporogenes spores Int. J. Food Microbiol. 133:213-216. [DOI] [PubMed] [Google Scholar]
- 40.Yang, W.-W., E. N. Crow-Willard, and A. Ponce. 2009. Production and characterization of pure Clostridium spore suspensions. J. Appl. Microbiol. 106:27-33. [DOI] [PubMed] [Google Scholar]
- 41.Yung, P. T., and A. Ponce. 2008. Fast sterility assessment by germinable-endospore biodosimetry. Appl. Environ. Microbiol. 74:7669-7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang, X. J., T. D. Yao, X. J. Ma, and N. L. Wang. 2002. Microorganisms in a high altitude glacier ice in Tibet. Folia Microbiol. 47:241-245. [DOI] [PubMed] [Google Scholar]
- 43.Zhang, X. J., T. D. Yao, X. J. Ma, and N. L. Wang. 2001. Analysis of the characteristics of microorganisms packed in the ice core of Malan Glacier, Tibet, China. Sci. China Ser. D Earth Sci. 44:369-374. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.