

Abstract
Porphyromonas gingivalis is an etiological agent that is strongly associated with periodontal disease, and it correlates with numerous inflammatory disorders, such as cardiovascular disease. Circulating bacteria may contribute to atherogenesis by promoting CD11b/CD18-mediated interactions between neutrophils and platelets, causing reactive oxygen species (ROS) production and aggregation. Lipoxin A4 (LXA4) is an endogenous anti-inflammatory and proresolving mediator that is protective of inflammatory disorders. The aim of this study was to investigate the effect of LXA4 on the P. gingivalis-induced activation of neutrophils and platelets and the possible involvement of Rho GTPases and CD11b/CD18 integrins. Platelet/leukocyte aggregation and ROS production was examined by lumiaggregometry and fluorescence microscopy. Integrin activity was studied by flow cytometry, detecting the surface expression of CD11b/CD18 as well as the exposure of the high-affinity integrin epitope, whereas the activation of Rac2/Cdc42 was examined using a glutathione S-transferase pulldown assay. The study shows that P. gingivalis activates Rac2 and Cdc42 and upregulates CD11b/CD18 and its high-affinity epitope on neutrophils, and that these effects are diminished by LXA4. Furthermore, we found that LXA4 significantly inhibits P. gingivalis-induced aggregation and ROS generation in whole blood. However, in platelet-depleted blood and in isolated neutrophils and platelets, LXA4 was unable to inhibit either aggregation or ROS production, respectively. In conclusion, this study suggests that LXA4 antagonizes P. gingivalis-induced cell activation in a manner that is dependent on leukocyte-platelet interaction, likely via the inhibition of Rho GTPase signaling and the downregulation of CD11b/CD18. These findings may contribute to new strategies in the prevention and treatment of periodontitis-induced inflammatory disorders, such as atherosclerosis.
Periodontitis is one of the most prevalent inflammatory diseases in humans, the key etiologic agent being the Gram-negative anaerobic rod Porphyromonas gingivalis (54). This bacterium not only is involved in tooth loss but also may cause recurrent bacteremias and contribute to systemic disorders, such as cardiovascular disease (10, 22, 23, 39, 46, 65). P. gingivalis expresses a broad range of virulence factors, such as cysteine proteinases (gingipains), fimbriae, lipopolysaccharide (LPS), and capsular polysaccharide. Infection with the bacterium may lead to chronic inflammation in which hyperresponsive neutrophils contribute to host-mediated tissue destruction. P. gingivalis has been found in human atherosclerotic plaques (15, 27) and has been shown to promote the phenotypic switch of murine monocytes into foam cells, e.g., by inducing reactive oxygen species (ROS) generation and the oxidation of low-density lipoprotein (LDL) (31, 38, 57).
We have recently reported that the exposure of human blood to P. gingivalis causes the formation of atherogenic LDL through a gingipain-mediated cleavage of apoB-100 (5). Furthermore, P. gingivalis, unlike other periodontopathic bacteria, has been shown to trigger platelet aggregation in vitro (55, 66), mainly through the interaction between bacterial gingipains and protease-activating receptors (PARs) on the platelets (49). Since platelet aggregation precedes thromboembolic events, this is an important pathogenic feature of the bacterium (1, 32).
CD11b/CD18 (complement receptor 3 or Mac-1), the main β2 integrin expressed on leukocytes, plays an important role in inflammation by promoting leukocyte adhesion and transmigration to sites of infection and by stimulating iC3b-mediated phagocytosis and cytokine production (21). In neutrophils, CD11b/CD18 binds to the platelet GPIIb/IIIa receptor via fibrinogen, thereby mediating neutrophil-platelet interaction and ROS production (11). In accordance with this, we have shown previously that platelet-leukocyte aggregation and ROS production in whole blood are mediated through selectin- and integrin-dependent interactions involving P-selectin and CD11b/CD18 (4).
CD11b/CD18 requires inside-out signaling to expose and activate its high-affinity epitope and to enable ligand binding (6). P. gingivalis has been shown to induce inside-out activation of CD11b/CD18 in monocytes/macrophages (25, 29) and to upregulate the CD11b/CD18 receptors on human neutrophils via LPS (68). The P. gingivalis-induced activation of CD11b/CD18 has been most extensively studied in monocytes/macrophages, where two main signaling pathways have been implicated. First, the CD14-mediated binding of fimbriae and LPS to toll-like receptor 2 (TLR-2) stimulates CD11b/CD18 activation through a Rac1- and phosphatidylinositol 3-kinase (PI3K)-mediated pathway (26, 28, 29). Second, the bacteria can bind and activate PAR2 via gingipains, which induces CD11b/CD18 activation (34), possibly via a Rho-dependent pathway (69). Interestingly, these two pathways have been suggested to work synergistically (67). P. gingivalis also mediates platelet and neutrophil activation by acting on platelet TLR-2 and the P13K/Akt pathway (7, 35). Harokopakis and Hajishengallis (29) have shown previously that fimbriae of P. gingivalis induce CD11b/CD18 activation in human neutrophils; however, the mechanisms by which the whole bacterium interacts with CD11b/CD18 and the associated intracellular signaling in neutrophils need to be clarified.
In neutrophils, Rac2 accounts for >96% of the Rac protein expressed (33, 58) and is involved in oxidase activity (13). Upon the binding of GTP, Rac and the closely related Rho GTPase Cdc42 interact with the downstream effector p21-activated kinase (PAK) (43). In human neutrophils, CD11b/CD18-mediated adhesion and phagocytosis activates Rac2 as well as Cdc42, which correlates with ROS production (9). The involvement of Rac2 in ROS generation has been demonstrated repeatedly (13), whereas Cdc42 is suggested to have an antagonistic role in oxidative activation (14).
Lipoxins (LXs) are endogenously produced eicosanoids with potent anti-inflammatory and proresolving effects (41, 63). Merched et al. (44) proposed that a failure in the endogenous synthesis of LXA4 underlies the unremitting inflammation that fuels atherosclerosis. LXA4 functions mainly through the G protein-coupled receptor ALXR (18) and has been shown repeatedly to be protective in periodontal disease (36, 37, 61). Mouse models demonstrate that the administration of stable LXA4 analogues significantly inhibits P. gingivalis-induced neutrophil influx, cyclooxygenase-2 expression, and prostaglandin E2 secretion, which is done without promoting any further spreading of the infection (56). Moreover, 15-lipoxygenase transgenic rabbits, overexpressing LXA4, show significantly diminished bone loss upon infection with P. gingivalis compared to the bone loss of control animals (64). Lipoxins also have been shown to downregulate CD11b/CD18 expression on whole-blood leukocytes (17). We have shown previously that LXA4 modulates the inside-out activation of CD11b/CD18 in neutrophils (51), and Godson and coworkers showed that LXA4 may influence the activation of integrins in monocytes/macrophages by modulating Rho GTPases (40, 59).
In this study, we demonstrate that LXA4 inhibits P. gingivalis-induced platelet/leukocyte aggregation and ROS production in whole blood. Furthermore, LXA4 blocks the bacterium-induced expression and function of CD11b/CD18 on neutrophils, possibly by inhibiting Rac2 and Cdc42 signaling pathways. Interestingly, the effects of LXA4 in P. gingivalis-induced cellular responses appear to be dependent on platelet-neutrophil interactions.
MATERIALS AND METHODS
Materials and chemicals.
The materials and their sources were (S),6(R),15-trihydroxyeicosa-7E,9E,11Z,13E-tetraenoic acid (LXA4) (BioMol Research Lab Inc., Plymouth Meeting, PA); 5-amino-2,3-dihydro-1,4-phthalazinedione (Luminol) and horseradish peroxidase (HRP); fMet-Leu-Phe (fMLF); EDTA, Tween 20, and fluorescein isothiocyanate (FITC) (Sigma Chemical Co., St. Louis, MO); rhodamine phalloidin (Molecular Probes, Eugene, OR); phenylmethanesulfonyl fluoride (PMSF), leupeptin, aprotinin, guanosine 5′-0-(thio)triphosphate (GTPγS), dithiothreitol (DTT), Nonidet P-40 (NP-40), and Pefablock SC [4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF)] (Roche Diagnostics Corporation, Indianapolis, IN); Ficoll-Paque, glutathione-Sepharose beads, and enhanced chemiluminescence (ECL) (GE-Healthcare GmbH, Uppsala, Sweden); and Polymorphprep (Axis-Shield PoC AS, Oslo, Norway).
Buffers.
Phosphate-buffered saline (PBS) consisted of 137 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, and 6.7 mM Na2HPO4 × 2H2O, pH 7.3; Krebs-Ringer glucose buffer (KRG) consisted of 120 mM NaCl, 4.9 mM KCl, 1.2 mM MgSO4, 1.7 mM KH2PO4, 8.3 mM Na2HPO4, and 10 mM glucose, pH 7.3; acid citrate dextrose (ACD) contained 85 mM trisodiumcitrate × 2H2O, 71 mM citric acid, and 111 mM glucose; Tris-buffer saline (TBS) consisted of 25 mM Tris base, 0.15 M NaCl, pH 7.4; TEDG buffer contained 50 mM Tris, pH 7.4, 1.5 mM EDTA, 10% glycerol, 0.4% NaCl, 0.2 M DTT, 1 mM MgCl2, 0.5% PMSF, 1 mM benzamidine, and 1 μg/ml aprotinin; and lysis buffer for pulldown consisted of 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 200 mM NaCl, 2% NP-40, 10% glycerol, 1 mM DTT, 2 mM PMSF, 20 μg/ml leupeptin, 20 μg/ml aprotinin, 2.8 μg/ml pepstatin, and 2 mM Na3VO4.
Porphyromonas gingivalis.
ATCC 33277 (American Type Culture Collection, Manassas, VA) was grown under anaerobic conditions (80% N2, 10% CO2, and 10% H2) at 37°C in an anaerobic chamber (Concept 400 Anaerobic Workstation; Ruskinn Technology Ltd., Leeds, United Kingdom). The bacteria were cultured for 3 to 4 days in fastidious anaerobe broth (29.7 g/liter, pH 7.2) before being washed and resuspended in KRG supplemented with 1.1 mM CaCl2. To estimate the bacterial concentration, the optical density (OD) was measured at 600 nm in a Hitachi U2000 spectrophotometer (KEBO Lab AB, Stockholm, Sweden) and set to an OD of approximately 2. Through viable count, where the bacteria were grown on fastidious anaerobe agar (46.0 g/liter supplemented with l-tryptophan 0.1 g/liter, pH 7.2; Lab M, Lancashire, United Kingdom) for 5 to 7 days, an OD of 2 was determined to correlate with approximately 109 CFU/ml.
Labeling of P. gingivalis with FITC.
Bacteria, suspended in KRG supplemented with 1.1 mM CaCl2, were incubated with 10 μg/ml FITC for 1 h at room temperature (RT) during gentle agitation. The bacteria were washed in PBS-Tween (1%, vol/vol), resuspended in KRG supplemented with 1.1 mM CaCl2, and stored at −70°C. The bacteria were sonicated before further use.
Whole and platelet-depleted blood.
Peripheral venous blood was drawn from healthy donors and collected in heparin-containing (20 U/ml) Vacutainer tubes. The donors had no allergies and were not undergoing any medical treatment. To obtain platelet-depleted blood, whole blood was centrifuged at 220 × g for 20 min, after which platelet-rich plasma (PRP) was removed and replaced with the same volume of 0.9% NaCl.
Heparin, which was used as an anticoagulant, has been reported to inhibit complement activation. This effect of heparin may have a role in our study, since different factors of the complement system stimulate ROS production in neutrophils, e.g., through CD11b/CD18. However, work by Bexborn et al. (6a) shows that although heparin activates the complement system, the activation is relatively mild.
Preparation of neutrophils.
Polymorphonuclear cells (PMN) were isolated from whole blood by density centrifugation as previously described (24). Briefly, whole blood was layered upon Polymorphprep, and PMN were isolated through density centrifugation (480 × g, 40 min, RT). Contaminating erythrocytes were lysed using hypotonic water, after which the osmotic pressure was restored. The cells were washed (400 × g, 5 min, 4°C), counted, and resuspended in KRG supplemented with 1.1 mmol/liter CaCl2 and kept on ice until further use.
Preparation of platelets.
Platelets were isolated from heparinized whole blood as previously described (25). Briefly, blood was mixed with RT ACD at a ratio of 5:1. Following centrifugation (220 × g, 20 min, 22°C), PRP was obtained and carefully collected. A final centrifugation (400 × g, 20 min, 22°C) resulted in a platelet-rich pellet, which was carefully washed before being resuspended in KRG. The cell concentration was calculated by using a Bürker chamber, and the suspension was kept in plastic tubes at RT until use. Immediately before an experiment, the extracellular calcium concentration was set to 1 mM. Since no platelet inhibitors were used during the preparation, extra care was taken when handling the cells to avoid any preactivation. Previous evaluation has shown that platelets isolated in this manner are not activated, and that the contamination of other blood cells is negligible.
Whole-blood lumiaggregometry.
Aggregation and ROS production was measured in heparinized whole or platelet-depleted blood using a lumiaggregometer model 560 (Chrono-Log Corp., Havertown, PA). Cell aggregation was measured as increased impedance (Ω) between two platinum electrodes, and ROS production was determined simultaneously through luminol-amplified chemiluminescence, as previously described (5). Briefly, heparinized whole or platelet-depleted blood, drawn no more than 20 min before the onset of an experiment, was diluted in a ratio of 1:1 in physiological sodium chloride (0.9% NaCl) containing 100 μM luminol. The samples were preincubated for 15 min at 37°C, either in the presence or absence of 500 nM LXA4, in plastic cuvettes with siliconized stirring bars rotating at 800 rpm. P. gingivalis (1 × 107 CFU/ml blood) subsequently was added and incubated for 25 min, initiating cellular aggregation and ROS production. The instrument was calibrated before each experiment so that a 5-Ω change in impedance determined 7.5 mm deflection.
Neutrophil ROS production.
ROS production in isolated neutrophils was analyzed using a six-channel Biolumat LB 9505 (Berthold Co., Wildbaden, Germany). Briefly, PMN (1 × 106 cells/ml) were suspended in KRG containing 50 μM luminol and 4 U/ml HRP, and extracellular calcium was set to 1 mM. The cells were incubated for 15 min at 37°C, either in the presence or absence of LXA4 (1 or 100 nM, respectively) before being stimulated with P. gingivalis (1 × 107 CFU/ml) for 1 h, during which time chemiluminescence was registered. The mean integral value of the chemiluminescence curve was taken as a measurement of ROS production and calculated as fold induction compared to levels for the control. Care was taken to change the position of the samples between different experiments so that the channels were used randomly.
Platelet aggregation.
The aggregation of isolated platelets was analyzed under stirring conditions using a calibrated two-sample lumiaggregometer model 560 (ChronoLog Corp., Havertown, PA). Briefly, platelets (2 × 108 cells/ml) were suspended in KRG with extracellular calcium set to 1 mM, incubated for 15 min at 37°C in either the presence or absence of LXA4 (1 or 100 nM), and then stimulated with P. gingivalis (1 × 107 CFU/ml). Aggregation was measured as the change in light transmission, where the unstimulated platelet suspension was set to 0% and the buffer (KRG) to 100%.
Fluorescence microscopy.
Whole blood was preincubated for 15 min in the presence or absence of 500 nM LXA4 prior to incubation with FITC-labeled P. gingivalis (1 × 107 CFU/ml blood) for 25 min and fixation overnight in 4% paraformaldehyde (PFA) at 4°C. To visualize F-actin in leukocytes and platelets, the cells were washed and incubated in a mixture of 600 μg/ml rhodamine phalloidin and 100 μg/ml lysophosphatidylcholine in darkness for 30 min and, after further washes, were mounted on a coverslip. The samples were analyzed by inverted fluorescence microscopy (Axiovert 200; Carl Zeiss, Germany), where the number and the size of aggregates on a set area were counted (aggregates were included if located on a cross line drawn in the center of the coverslip) using Scion Image software.
Flow cytometry.
Immunolabeling was performed as described previously (51, 60). In short, whole blood was incubated at 37°C in the presence or absence of LXA4 (1, 100, 250, or 500 nM) for 15 min and subsequently stimulated with P. gingivalis (1 × 107 CFU/ml blood) for 10 min. To detect the total CD11b expression, an R-phycoerythrin (RPE)-conjugated anti-human CD11b antibody (mouse monoclonal antibody; clone 2LPM19c; Dako, Glostrup, Denmark) was added 5 min after the onset of bacterial stimulation. To detect the high-affinity epitope of CD11b, an FITC-conjugated anti-human CD11b antibody (mouse monoclonal antibody; clone CBRM1/5 [12]) diluted 1:13 was added 1 min prior to the addition of the bacteria. The antibodies were added to separate samples to avoid steric hindrance. Stimulation was stopped by incubation for 30 min on ice, after which erythrocytes were lysed for 5 min at 15°C using lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 100 μM EDTA), and the remaining cells were kept in 0.1% PFA until flow cytometry analysis. Granulocytes (predominantly neutrophils), monocytes, and lymphocytes were identified and gated by plotting forward scatter (FSC) versus side scatter (SSC), excluding cell debris. Unspecific binding was determined through the use of isotypic antibodies. For each sample, the mean fluorescence intensity (MFI) values of 15,000 events were determined, representing roughly 7,000 granulocytes, 4,000 lymphocytes, and 700 monocytes.
Pulldown and Western blotting.
To assess the activation of Rac2 and Cdc42, a pulldown assay was used, employing a fusion protein of glutathione-S-transferase (GST) and the p21-binding domain (PBD) of PAK, as described previously (3, 19, 42). Briefly, the cDNA encoding residues 67 to 150 of PAK1 was cloned into the expression vector pGEX-4T3, kindly provided by the late Gary M. Bokoch (Scripps Research Institute, La Jolla, CA) and expressed in Escherichia coli. Neutrophils (5 × 106/sample) were preincubated in the presence or absence of 5 nM LXA4 for 5 min at 37°C and stimulated with P. gingivalis (6 × 105 CFU/ml) for 10 min. The cells subsequently were lysed in ice-cold lysis buffer for 15 min at 4°C during gentle agitation. Cellular debris was cleared by centrifugation (10,000 × g, 10 min, 4°C), after which the supernatants were incubated for 45 min at 4°C with glutathione-Sepharose beads, which had been precoupled with GST-PBD. The beads were washed three times in lysis buffer and boiled in Laemmli sample buffer (98°C for 5 min) to extract the proteins. The proteins were separated using standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred to a polyvinylidene difluoride membrane.
Unspecific binding was blocked by incubating the membranes in 5% milk-TBS (for 1 h at RT) prior to incubation with the mouse monoclonal anti-Cdc42 antibody (1:250; clone 44; BD Transduction Laboratories, San Diego, CA) or a rabbit polyclonal anti-Rac2 antibody (1:200; clone C-11; Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at RT. The membranes were thereafter incubated with an HRP-conjugated goat anti-mouse antibody (Santa Cruz Biotechnology) or an HRP-conjugated donkey anti-rabbit antibody (GE-Healthcare GmbH), respectively. To ascertain the specificity of the assay, 100 μM GTPγS (a nonhydrolyzable GTP analogue that arrests Cdc42 and Rac2 in their active state) and 1 mM EDTA were added to the cell lysate of fMLF (1 μM)-stimulated cells (15 min, 30°C), thereby serving as a positive control. Unspecific binding was excluded by the use of uncoupled GST beads (beads without the PBD domain of PAK1).
Statistical analysis.
Data were statistically analyzed by paired Student t tests or by one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc test. Data were expressed as means ± standard errors of the means (SEM), and P < 0.05 was considered statistically significant.
RESULTS
Effect of LXA4 on P. gingivalis-induced aggregation and ROS production in whole blood.
Using lumiaggregometry, P. gingivalis-induced aggregation and ROS production were examined in heparinized whole blood. No spontaneous aggregation or ROS production was seen when whole blood was incubated for 15 min at 37°C, and incubation with LXA4 did not per se induce any detectable responses (Fig. 1 A). The addition of P. gingivalis potently triggered both the aggregation and ROS production of whole-blood cells, as measured by changes in impedance and luminol-amplified chemiluminescence (Fig. 1). The bacterial response was initiated after 5 to 6 min and reached an irreversible maximum after approximately 15 min. LXA4 inhibited the aggregation and ROS generation in a concentration-dependent manner (Fig. 1B). Preincubation with 500 nM LXA4 significantly decreased both the P. gingivalis-induced aggregation (63%; P < 0.01) and ROS production (31%; P < 0.01) (Fig. 1B). At a cellular level, LXA4 usually is potent at concentrations ranging from 1 to 10 nM. In our experimental setup, the use of whole blood required a higher concentration of LXA4 in concordance with previous findings (16), most likely due to the interaction of LXA4 with blood components, e.g., albumin. The effect of LXA4 is not due to the metabolic inactivation of the bacteria, since we found that LXA4 did not per se affect P. gingivalis viability in buffer (data not shown).
FIG. 1.

LXA4 inhibits P. gingivalis-induced aggregation and ROS production in whole blood. Whole blood was incubated for 15 min in the absence or presence of lipoxin A4 (LX; 1 to 500 nM) prior to stimulation with P. gingivalis (P.g; 1 × 107 CFU/ml blood) for 25 min. Aggregation was measured as changes in impedance, and ROS production was detected by luminol-amplified chemiluminescence. (A) Representative experiment (500 nM lipoxin A4). (B) P. gingivalis-induced aggregation and ROS production, presented as percentage of P. gingivalis-stimulated control. Data are presented as means ± SEM of n = 7. *, P < 0.05; **, P < 0.01.
Effect of LXA4 on P. gingivalis-induced formation of cellular and bacterial aggregates.
The effect of LXA4 on P. gingivalis-induced aggregate formation in whole blood was visualized with fluorescence microscopy by using FITC-labeled P. gingivalis and the rhodamine phalloidin staining of F-actin in leukocytes and platelets. The stimulation of whole blood with P. gingivalis induced the formation of large mixed aggregates of platelets, leukocytes, and bacteria (Fig. 2). The majority of the leukocytes had internalized a high number of bacteria (Fig. 2C and E). The preincubation of whole blood with 500 nM LXA4 decreased the size of aggregates by 67% (P = 0.018) after 20 min of stimulation with the bacteria (Fig. 2D, F, and G). There was no spontaneous formation of aggregates in whole blood when incubated at 37°C and after incubation with LXA4 per se (Fig. 2A and B). Accumulations of rhodamine-stained neutrophils and platelets, together with FITC-labeled bacteria, appear yellow when the colors are merged.
FIG. 2.

LXA4 inhibits P. gingivalis-induced formation of cellular aggregates. Whole blood was incubated in the absence or presence of 500 nM LXA4 for 15 min before being stimulated with P. gingivalis (1 × 107 CFU/ml blood) for 25 min. An aliquot of each sample was fixed in 4% PFA overnight and analyzed by fluorescence microscopy. The figure shows samples in the absence (A and B) or presence (C to F) of P. gingivalis and preincubated with (B, D, and F) or without (A, C, and E) LXA4 as indicated. Rhodamine phalloidin-labeled leukocytes and platelets appear red, and FITC-labeled bacteria appear green. (A) Unstimulated whole blood. (B) Whole blood in the presence of LXA4. Magnifications are ×63 (A to D) and ×10 (E and F). To analyze the microscopy data, the size of 30 aggregates over a set area was assessed using Scion Image software (G). A paired Student t test was used for statistical analysis, and data are presented as representative images (A to F) or means ± SEM of n = 6. *, P < 0.05.
Effect of LXA4 on P. gingivalis-induced ROS production in isolated neutrophils.
P. gingivalis-induced ROS production in isolated neutrophils was determined through luminol-amplified chemiluminescence during the course of 1 h. P. gingivalis induced a marked increase in ROS production. However, preincubation with LXA4, at either 1 or 100 nM concentration, did not alter the P. gingivalis-induced ROS production (Fig. 3). Unstimulated neutrophils did not produce any significant levels of ROS, and preincubation with LXA4 did not per se induce any detectable ROS (data now shown).
FIG. 3.
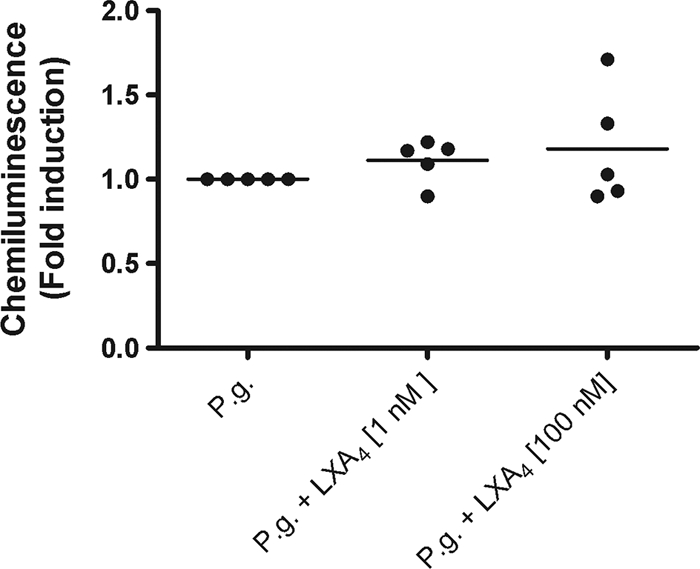
LXA4 does not affect P. gingivalis-induced ROS production in isolated neutrophils. Isolated polymorphonuclear cells (PMN), consisting predominantly of neutrophils, were preincubated for 15 min at 37°C in the presence or absence of LXA4 (1 or 100 nM). Following subsequent stimulation with P. gingivalis (1 × 107 CFU/ml), luminol-amplified chemiluminescence was measured during the course of 1 h. PMN stimulated with vehicle did not produce any detectable ROS, and LXA4 did not induce a chemiluminescence response. ANOVA was used for statistical analysis, and data are presented as fold induction (means ± SEM of n = 5).
Effect of LXA4 on P. gingivalis-induced aggregation of isolated platelets.
Platelet aggregation was measured to clarify the effects of P. gingivalis on the activation of isolated platelets. We found that P. gingivalis caused an extensive platelet aggregation, and that this response was not inhibited by LXA4 (1 or 100 nM) (Fig. 4). Unstimulated platelets did not spontaneously aggregate, and LXA4 did not induce an aggregatory response by itself (data not shown).
FIG. 4.

LXA4 does not affect P. gingivalis-induced aggregation of isolated platelets. Isolated platelets were preincubated for 15 min at 37°C in the presence or absence of LXA4 (1 or 100 nM) and then monitored for aggregation upon stimulation with P. gingivalis (P.g; 1 × 107 CFU/ml). (A) Representative aggregation traces of P. gingivalis with or without LXA4 (1 or 100 nM). (B) P. gingivalis-induced aggregation presented as fold induction. The vehicle did not cause platelet aggregation, and LXA4 did not induce an aggregatory response. Data are presented as percentage of P. gingivalis-stimulated control (means ± SEM of n = 3).
Effect of LXA4 on P. gingivalis-induced aggregation and ROS production in platelet-depleted whole blood.
P. gingivalis-induced aggregation and ROS production was studied in platelet-depleted whole blood using lumiaggregometry. The addition of P. gingivalis triggered a considerable production of ROS but just a marginal aggregatory response. Neither the P. gingivalis-induced ROS production nor the minor P. gingivalis-induced aggregation was affected by preincubation with LXA4 (Fig. 5).
FIG. 5.

Effect of LXA4 on P. gingivalis-induced aggregation and ROS production in platelet-depleted whole blood. Platelet-depleted whole blood was incubated in the absence or presence of LXA4 (500 nM). Aggregation was measured as changes in impedance and ROS production as luminol-amplified chemiluminescence during 25 min of incubation with P. gingivalis (1.5 × 107 CFU/ml blood). (A) Representative experiment. (B) P. gingivalis-induced ROS production presented as percentage of P. gingivalis-stimulated control. Paired student t test was used for statistical analysis. Data are presented as means ± SEM of n = 3. AU, arbitrary units.
Effect of LXA4 on P. gingivalis-induced upregulation of CD11b and exposure of the high-affinity integrin epitope on leukocytes.
The P. gingivalis-induced upregulation of CD11b and exposure of the high-affinity epitope on neutrophils were analyzed by flow cytometry. The expression of CD11b on the cell surface of neutrophils was increased by 25% upon stimulation with P. gingivalis and was significantly decreased to basal levels when the cells were preincubated with 500 nM LXA4 (P < 0.05) (Fig. 6 A and C). Similarly, the surface expression of the CD11b high-affinity epitope increased by 59% upon stimulation with P. gingivalis and was significantly reduced by 28% by preincubation with 500 nM LXA4 (P < 0.01) (Fig. 6B and D). A small and possibly dose-dependent reduction in CD11b expression and high-affinity epitope exposure was seen when cells were incubated with only LXA4. However, this reduction was not statistically significant and therefore was disregarded. It is noteworthy that a small subset of the PMN population appeared to express reduced CD11b when stimulated with P. gingivalis (Fig. 6C). A possible explanation for this is that the RPE-conjugated antibody was added after bacterial stimulation, unlike the FITC-conjugated antibody, and that a portion of the CD11b receptor had already been internalized and therefore was inaccessible to the antibody. P. gingivalis also induced the surface expression of CD11b/CD18 in the monocytic population, which furthermore was inhibited by LXA4 (see Fig. S1 in the supplemental material). Lymphocytes expressed very low levels of CD11b/CD18, and neither P. gingivalis nor PMA (which was used as a positive control) affected the expression in our protocol setup (data not shown).
FIG. 6.

LXA4 inhibits P. gingivalis-induced upregulation of CD11b/CD18 and exposure of the CD11b/CD18 high-affinity epitope on neutrophils. Whole blood was incubated in the presence or absence of LXA4 (100 or 500 nM) for 15 min and subsequently stimulated with P. gingivalis (P.g; 1 × 107 CFU/ml blood) for 10 min. Following the lysis of erythrocytes, the samples were fixed in 0.1% PFA and analyzed by flow cytometry. The granulocyte population (consisting predominantly of neutrophils) was gated by plotting forward scatter (FSC) versus side scatter (SSC), and CD11b/CD18 expression was determined by mean fluorescence intensity (MFI). Total CD11b surface expression was detected by use of RPE-conjugated mouse anti-human CD11b clone 2LPM19c (A and C), and the high-affinity domain was recognized through the use of an FITC-conjugated mouse anti-human CD11b antibody, clone CBRM1/5 (B and D). ANOVA was used for statistical analysis, and data are presented as means ± SEM of n = 5. *, P < 0.05; **, P < 0.01.
Effect of LXA4 on P. gingivalis-induced activation of Rac2 and Cdc42.
The P. gingivalis activation of Rac2 and Cdc42 in neutrophils was analyzed by GST pulldown and immunoblotting. GTP-bound forms of Rac2 and Cdc42 were precipitated from the 10,000 × g fraction of lysed cells using a fusion protein consisting of the PBD of PAK1 and GST (GST-PBD). The activation of both Rac2 and Cdc42 from isolated neutrophils was detected as early as 0.5 min after the addition of P. gingivalis and persisted up to 10 min (Fig. 7 A). Preincubation with 5 nM LXA4 for 5 min markedly inhibited the P. gingivalis-induced activation of both Rac2 and Cdc42 (Fig. 7A). No unspecific binding using uncoupled GST beads was detected (Fig. 7B).
FIG. 7.

LXA4 inhibits P. gingivalis-induced activation of Rac2 and Cdc42 in neutrophils. Neutrophils (5 × 106/sample) were preincubated in the absence or presence of 5 nM LXA4 for 5 min at 37°C and stimulated with P. gingivalis (6 × 105 CFU/ml) for various times as indicated in the figure. The stimulation was stopped, and activated forms of Rac2 and Cdc42 were precipitated from the lysates using a GST pulldown assay. (A) Neutrophils were stimulated by P. gingivalis for 0.5, 3, and 10 min with and without preincubation with LXA4, a GST pulldown assay was performed with beads lacking the GST-PBD (Neg; negative control), or neutrophils were incubated at 0 or 37°C (basal level). (B) Neutrophils were stimulated with fMLF plus GTPγS (Pos; positive control) or whole-cell lysate (Wc; positive control), or a GST-pulldown assay was performed with beads lacking the GST-PBD (Neg; negative control). An equivalent volume was loaded in each lane, and the electrophoretically separated proteins were detected with Western blotting using a polyclonal rabbit anti-human Rac2 antibody (1:200) and a mouse monoclonal anti-human Cdc42 antibody (1:250), clone 44.
DISCUSSION
In the present study, the anti-inflammatory and proresolving eicosanoid LXA4 is shown to effectively inhibit P. gingivalis-induced aggregation and ROS production in whole blood. Our data suggest that this effect is mediated by an LXA4-induced impediment of P. gingivalis-triggered activation of the small GTPases Rac2 and Cdc42 in neutrophils and the upregulation of CD11b/CD18 and its high-affinity epitope. Interestingly, the effect of LXA4 on P. gingivalis-induced responses is dependent on platelet-neutrophil interaction.
To investigate the effect of P. gingivalis on cell activation, we examined the expression of CD11b/CD18 integrins on the cell surface of neutrophils. Interestingly, we found that P. gingivalis not only increased the surface expression of CD11b/CD18 but also enhanced the expression of integrins in the high-affinity state. Furthermore, both of these effects were significantly suppressed by LXA4. The P. gingivalis-induced surface expression of CD11b/CD18 in monocytes also was inhibited by LXA4, although the high-affinity epitope remained unaffected. This is noteworthy, since P. gingivalis-induced CD11b/CD18 upregulation on monocytes has been shown to have a crucial role in stimulating monocyte adhesion to endothelial cells and transendothelial migration (28). LXA4 thereby could play an important role in inhibiting integrin activation on monocytes and limiting P. gingivalis infection. Lymphocytes, on the other hand, expressed very low levels of this integrin, and consequently we were not able, in our system, to observe any effects of P. gingivalis on lymphocyte CD11b/CD18 expression. CD11b/CD18 requires inside-out signaling to expose and activate its high-affinity epitope and enable ligand binding (7). The activation of CD11b/CD18 by P. gingivalis has been shown in monocytes and macrophages, through a Rac1 and PI3K-mediated (28, 30, 31), and possibly a Rho-dependent, pathway (64), and we have shown previously that the ligation of CD11b/CD18 activates Rac2 and Cdc42 (21). Lipoxins have been shown to modulate the inside-out activation of CD11b/CD18 (48) and to downregulate CD11b/CD18 expression in neutrophils (18). In monocytes/macrophages, LXA4 also has been shown to influence integrins by modulating the functions of Rho GTPases (40, 54). Our study demonstrates that LXA4 modifies P. gingivalis-induced Rac2 and/or Cdc42 activation, which could be the mechanism by which it inhibits CD11b/CD18 upregulation and activation. Another possible mechanism of how LXA4 modulates CD11b/CD18 integrins is through impeding Akt activation, since CD11b upregulation in PMN is Akt dependent (47) and LXA4 has been shown to modulate PI3K/Akt activation (45).
Resolvins and protectins are mediators endogenously produced from ω-3 polyunsaturated fatty acids that share many of the proresolving and anti-inflammatory properties of lipoxins (8, 62). The topical application of resolvins protects against inflammation-induced tissue and bone loss associated with periodontitis in vivo (30). Thus, to include these compounds in future experiments and investigate their effects on the P. gingivalis-induced modulation of CD11b integrins would be of great interest.
Activated CD11b/CD18 has a crucial role in platelet-leukocyte aggregate formation and the associated ROS production by the binding of the integrin to the platelet GPIIb/IIIa receptor via fibrinogen (5, 62). Our data clearly demonstrate that neutrophil-platelet interaction is essential for LXA4 to inhibit P. gingivalis-induced effects. We found that P. gingivalis-induced ROS production in platelet-depleted blood is unaffected by LXA4. The crucial role of interplay between platelets and neutrophils is further strengthened by our finding that neither P. gingivalis-induced ROS production in isolated neutrophils nor P. gingivalis-induced aggregation in isolated platelets was affected by LXA4. It therefore is possible that LXA4 inhibits P. gingivalis-induced ROS via intercepting neutrophil-platelet interaction through the downregulation of CD11b/CD18.
The microscopic evaluation of the aggregates in whole blood formed by P. gingivalis infection revealed that they contained a mixture of platelets, leukocytes, and bacteria, and that many leukocytes had internalized a high number of P. gingivalis. Hajishengallis et al. (26) have shown that fimbriae of P. gingivalis induce CD11b/CD18 binding and the internalization of the bacteria within macrophages through CD14/TLR2 signaling. Internalized P. gingivalis replicates within the host cell by activating cellular autophagy while suppressing apoptosis (2), which is a mechanism of bacterial survival and the spreading of the infection. Thus, our finding that LXA4 inhibits CD11b/CD18 expression as well as the formation of aggregates in whole blood suggests a role of LXA4 in inhibiting the propagation of the infection induced by the bacteria.
Patients with stable and unstable angina pectoris show an increased number of circulating neutrophil-platelet aggregates, which are suggested as an early and sensitive marker of myocardial infarction and an important component of systemic inflammation (20, 48, 50, 52, 53). Periodontitis may contribute to cardiovascular disease by recurrent and transient bacteremias, where periodontal pathogens, including P. gingivalis, interact with blood cells, form neutrophil-platelets conjugates, and trigger inflammatory processes in the vessels. In this study, we show that LXA4 effectively inhibits neutrophil-platelet interaction and the formation of aggregates, thus supporting a role of lipoxin in the prevention and treatment of cardiovascular disease.
In summary, we have demonstrated that P. gingivalis-induced ROS generation and aggregation in whole blood is inhibited by LXA4, possibly by mechanisms dependent on leukocyte-platelet interaction and involving the downregulation of the P. gingivalis-induced Rac2 and Cdc42 activation and the inside-out signaling of CD11b/CD18. Consequently, we suggest that a supplement of exogenous LXA4 facilitates anti-inflammatory actions and reduces the progression of chronic inflammatory disorders, such as periodontitis and atherosclerosis.
Supplementary Material
Acknowledgments
This work was supported by grants from the Swedish Research Council, the Swedish Heart-Lung Foundation, the Swedish Fund for Research without Animal Experiments, the Swedish Heart and Lung Association, the Foundation of Olle Engkvist, the Emil & Maria Palm Foundation, and the Filip Lundberg Foundation.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 24 January 2011.
The authors have paid a fee to allow immediate free access to this article.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Amar, S., et al. 2003. Periodontal disease is associated with brachial artery endothelial dysfunction and systemic inflammation. Arterioscler. Thromb. Vasc. Biol. 23:1245-1249. [DOI] [PubMed] [Google Scholar]
- 2.Bélanger, M., P. H. Rodrigues, W. A. Dunn, Jr., and A. Progulske-Fox. 2006. Autophagy: a highway for Porphyromonas gingivalis in endothelial cells. Autophagy 2:165-170. [DOI] [PubMed] [Google Scholar]
- 3.Benard, V., B. P. Bohl, and G. M. Bokoch. 1999. Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J. Biol. Chem. 274:13198-13204. [DOI] [PubMed] [Google Scholar]
- 4.Bengtsson, T., and M. Grenegard. 2002. Leucocyte activation by collagen-stimulated platelets in whole blood. Scand. J. Clin. Lab. Investig. 62:451-461. [DOI] [PubMed] [Google Scholar]
- 5.Bengtsson, T., et al. 2008. The periodontal pathogen Porphyromonas gingivalis cleaves apoB-100 and increases the expression of apoM in LDL in whole blood leading to cell proliferation. J. Intern. Med. 263:558-571. [DOI] [PubMed] [Google Scholar]
- 6.Berton, G., and C. A. Lowell. 1999. Integrin signalling in neutrophils and macrophages. Cell. Signal. 11:621-635. [DOI] [PubMed] [Google Scholar]
- 6a.Bexborn, F., et al. 2009. Hirudin versus heparin for use in whole blood in vitro compatibility models. J. Biomed. Mater. Res. A 89:951-959. [DOI] [PubMed] [Google Scholar]
- 7.Blair, P., et al. 2009. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ. Res. 104:346-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Börgeson, E., and C. Godson. 2010. Molecular circuits of resolution in renal disease. ScientificWorldJournal 10:1370-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang, L. C., et al. 2009. Inhibition of superoxide anion generation by CHS-111 via blockade of the p21-activated kinase, protein kinase B/Akt and protein kinase C signaling pathways in rat neutrophils. Eur. J. Pharmacol. 615:207-217. [DOI] [PubMed] [Google Scholar]
- 10.Chun, Y. H., K. R. Chun, D. Olguin, and H. L. Wang. 2005. Biological foundation for periodontitis as a potential risk factor for atherosclerosis. J. Periodont. Res. 40:87-95. [DOI] [PubMed] [Google Scholar]
- 11.de Gaetano, G., C. Cerletti, and V. Evangelista. 1999. Recent advances in platelet-polymorphonuclear leukocyte interaction. Haemostasis 29:41-49. [DOI] [PubMed] [Google Scholar]
- 12.Diamond, M. S., and T. A. Springer. 1993. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J. Cell Biol. 120:545-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diebold, B. A., and G. M. Bokoch. 2005. Rho GTPases and the control of the oxidative burst in polymorphonuclear leukocytes. Curr. Topics Microbiol. Immunol. 291:91-111. [DOI] [PubMed] [Google Scholar]
- 14.Diebold, B. A., B. Fowler, J. Lu, M. C. Dinauer, and G. M. Bokoch. 2004. Antagonistic cross-talk between Rac and Cdc42 GTPases regulates generation of reactive oxygen species. J. Biol. Chem. 279:28136-28142. [DOI] [PubMed] [Google Scholar]
- 15.Dorn, B. R., W. A. Dunn, Jr., and A. Progulske-Fox. 1999. Invasion of human coronary artery cells by periodontal pathogens. Infect. Immun. 67:5792-5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Filep, J. G., T. Khreiss, and L. Jozsef. 2005. Lipoxins and aspirin-triggered lipoxins in neutrophil adhesion and signal transduction. Prostaglandins Leukot. Essent. Fatty Acids 73:257-262. [DOI] [PubMed] [Google Scholar]
- 17.Filep, J. G., C. Zouki, N. A. Petasis, M. Hachicha, and C. N. Serhan. 1999. Anti-inflammatory actions of lipoxin A(4) stable analogs are demonstrable in human whole blood: modulation of leukocyte adhesion molecules and inhibition of neutrophil-endothelial interactions. Blood 94:4132-4142. [PubMed] [Google Scholar]
- 18.Fiore, S., J. F. Maddox, H. D. Perez, and C. N. Serhan. 1994. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J. Exp. Med. 180:253-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forsberg, M., P. Druid, L. Zheng, O. Stendahl, and E. Sarndahl. 2003. Activation of Rac2 and Cdc42 on Fc and complement receptor ligation in human neutrophils. J. Leukoc. Biol. 74:611-619. [DOI] [PubMed] [Google Scholar]
- 20.Furman, M. I., et al. 1998. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J. Am. College Cardiol. 31:352-358. [DOI] [PubMed] [Google Scholar]
- 21.Gahmberg, C. G. 1997. Leukocyte adhesion: CD11/CD18 integrins and intercellular adhesion molecules. Curr. Opin. Cell Biol. 9:643-650. [DOI] [PubMed] [Google Scholar]
- 22.Gibson, F. C., III, et al. 2004. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation 109:2801-2806. [DOI] [PubMed] [Google Scholar]
- 23.Gibson, F. C., III, H. Yumoto, Y. Takahashi, H. H. Chou, and C. A. Genco. 2006. Innate immune signaling and Porphyromonas gingivalis-accelerated atherosclerosis. J. Dental Res. 85:106-121. [DOI] [PubMed] [Google Scholar]
- 24.Gunnarsson, P., L. Fornander, P. Pahlsson, and M. Grenegard. Sialic acid residues play a pivotal role in alpha(1)-acid glycoprotein (AGP)-induced generation of reactive oxygen species in chemotactic peptide pre-activated neutrophil granulocytes. Inflamm. Res. 59:89-95. [DOI] [PubMed]
- 25.Hajishengallis, G., and E. Harokopakis. 2007. Porphyromonas gingivalis interactions with complement receptor 3 (CR3): innate immunity or immune evasion? Front. Biosci. 12:4547-4557. [DOI] [PubMed] [Google Scholar]
- 26.Hajishengallis, G., M. Wang, E. Harokopakis, M. Triantafilou, and K. Triantafilou. 2006. Porphyromonas gingivalis fimbriae proactively modulate beta2 integrin adhesive activity and promote binding to and internalization by macrophages. Infect. Immun. 74:5658-5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haraszthy, V. I., J. J. Zambon, M. Trevisan, M. Zeid, and R. J. Genco. 2000. Identification of periodontal pathogens in atheromatous plaques. J. Periodontol. 71:1554-1560. [DOI] [PubMed] [Google Scholar]
- 28.Harokopakis, E., M. H. Albzreh, M. H. Martin, and G. Hajishengallis. 2006. TLR2 transmodulates monocyte adhesion and transmigration via Rac1- and PI3K-mediated inside-out signaling in response to Porphyromonas gingivalis fimbriae. J. Immunol. 176:7645-7656. [DOI] [PubMed] [Google Scholar]
- 29.Harokopakis, E., and G. Hajishengallis. 2005. Integrin activation by bacterial fimbriae through a pathway involving CD14, Toll-like receptor 2, and phosphatidylinositol-3-kinase. Eur. J. Immunol. 35:1201-1210. [DOI] [PubMed] [Google Scholar]
- 30.Hasturk, H., et al. 2006. RvE1 protects from local inflammation and osteoclast-mediated bone destruction in periodontitis. FASEB J. 20:401-403. [DOI] [PubMed] [Google Scholar]
- 31.He, J., et al. 2006. Role of Porphyromonas gingivalis FeoB2 in metal uptake and oxidative stress protection. Infect. Immun. 74:4214-4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herzberg, M. C., and M. W. Weyer. 1998. Dental plaque, platelets, and cardiovascular diseases. Ann. Periodontol. 3:151-160. [DOI] [PubMed] [Google Scholar]
- 33.Heyworth, P. G., B. P. Bohl, G. M. Bokoch, and J. T. Curnutte. 1994. Rac translocates independently of the neutrophil NADPH oxidase components p47phox and p67phox. Evidence for its interaction with flavocytochrome b558. J. Biol. Chem. 269:30749-30752. [PubMed] [Google Scholar]
- 34.Howells, G. L., et al. 1997. Proteinase-activated receptor-2: expression by human neutrophils. J. Cell Sci. 110(Pt 7):881-887. [DOI] [PubMed] [Google Scholar]
- 35.Kalvegren, H., C. Skoglund, C. Helldahl, M. Lerm, M. Grenegard, and T. Bengtsson. Toll-like receptor 2 stimulation of platelets is mediated by purinergic P2X1-dependent Ca2+ mobilisation, cyclooxygenase and purinergic P2Y1 and P2Y12 receptor activation. Thrombosis Haemostasis 103:398-407. [DOI] [PubMed]
- 36.Kantarci, A., H. Hasturk, and T. E. Van Dyke. 2006. Host-mediated resolution of inflammation in periodontal diseases. Periodontol. 2000 40:144-163. [DOI] [PubMed] [Google Scholar]
- 37.Kantarci, A., and T. E. Van Dyke. 2005. Lipoxin signaling in neutrophils and their role in periodontal disease. Prostaglandins Leukot. Essent. Fatty Acids 73:289-299. [DOI] [PubMed] [Google Scholar]
- 38.Kuramitsu, H. K., I. C. Kang, and M. Qi. 2003. Interactions of Porphyromonas gingivalis with host cells: implications for cardiovascular diseases. J. Periodontol. 74:85-89. [DOI] [PubMed] [Google Scholar]
- 39.Li, L., E. Messas, E. L. Batista, Jr., R. A. Levine, and S. Amar. 2002. Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model. Circulation 105:861-867. [DOI] [PubMed] [Google Scholar]
- 40.Maderna, P., et al. 2002. Lipoxins induce actin reorganization in monocytes and macrophages but not in neutrophils: differential involvement of rho GTPases. Am. J. Pathol. 160:2275-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maderna, P., and C. Godson. 2009. Lipoxins: resolutionary road. Br. J. Pharmacol. 158:947-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madesh, M., O. Benard, and K. A. Balasubramanian. 1999. Apoptotic process in the monkey small intestinal epithelium. 2. Possible role of oxidative stress. Free Radic. Biol. Med. 26:431-438. [DOI] [PubMed] [Google Scholar]
- 43.Manser, E., T. Leung, H. Salihuddin, Z. S. Zhao, and L. Lim. 1994. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 367:40-46. [DOI] [PubMed] [Google Scholar]
- 44.Merched, A. J., K. Ko, K. H. Gotlinger, C. N. Serhan, and L. Chan. 2008. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 22:3595-3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitchell, D., et al. 2004. Lipoxins inhibit Akt/PKB activation and cell cycle progression in human mesangial cells. Am. J. Pathol. 164:937-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyakawa, H., K. Honma, M. Qi, and H. K. Kuramitsu. 2004. Interaction of Porphyromonas gingivalis with low-density lipoproteins: implications for a role for periodontitis in atherosclerosis. J. Periodont. Res. 39:1-9. [DOI] [PubMed] [Google Scholar]
- 47.Montecucco, F., et al. 2008. Tumor necrosis factor-alpha (TNF-alpha) induces integrin CD11b/CD18 (Mac-1) up-regulation and migration to the CC chemokine CCL3 (MIP-1alpha) on human neutrophils through defined signalling pathways. Cell. Signal. 20:557-568. [DOI] [PubMed] [Google Scholar]
- 48.Nijm, J., A. Wikby, A. Tompa, A. G. Olsson, and L. Jonasson. 2005. Circulating levels of proinflammatory cytokines and neutrophil-platelet aggregates in patients with coronary artery disease. Am. J. Cardiol. 95:452-456. [DOI] [PubMed] [Google Scholar]
- 49.Nylander, M., T. L. Lindahl, T. Bengtsson, and M. Grenegard. 2008. The periodontal pathogen Porphyromonas gingivalis sensitises human blood platelets to epinephrine. Platelets 19:352-358. [DOI] [PubMed] [Google Scholar]
- 50.Ott, I., F. J. Neumann, M. Gawaz, M. Schmitt, and A. Schomig. 1996. Increased neutrophil-platelet adhesion in patients with unstable angina. Circulation 94:1239-1246. [DOI] [PubMed] [Google Scholar]
- 51.Patcha, V., et al. 2004. Differential inside-out activation of beta2-integrins by leukotriene B4 and fMLP in human neutrophils. Exp. Cell. Res. 300:308-319. [DOI] [PubMed] [Google Scholar]
- 52.Patel, P. B., et al. 2004. Comparison of coronary artery specific leukocyte-platelet conjugate formation in unstable versus stable angina pectoris. Am. J. Cardiol. 93:410-413. [DOI] [PubMed] [Google Scholar]
- 53.Patel, Y., et al. 1998. Functional characterization of PM6/13, a beta3-specific (GPIIIa/CD61) monoclonal antibody that shows preferential inhibition of fibrinogen binding over fibronectin binding to activated human platelets. Thrombosis Haemostasis 79:177-185. [PubMed] [Google Scholar]
- 54.Petersen, P. E., and H. Ogawa. 2005. Strengthening the prevention of periodontal disease: the WHO approach. J. Periodontol. 76:2187-2193. [DOI] [PubMed] [Google Scholar]
- 55.Pham, K., D. Feik, B. F. Hammond, T. E. Rams, and E. J. Whitaker. 2002. Aggregation of human platelets by gingipain-R from Porphyromonas gingivalis cells and membrane vesicles. Platelets 13:21-30. [DOI] [PubMed] [Google Scholar]
- 56.Pouliot, M., C. B. Clish, N. A. Petasis, T. E. Van Dyke, and C. N. Serhan. 2000. Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: a role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry 39:4761-4768. [DOI] [PubMed] [Google Scholar]
- 57.Qi, M., H. Miyakawa, and H. K. Kuramitsu. 2003. Porphyromonas gingivalis induces murine macrophage foam cell formation. Microb. Pathog. 35:259-267. [DOI] [PubMed] [Google Scholar]
- 58.Quinn, M. T., T. Evans, L. R. Loetterle, A. J. Jesaitis, and G. M. Bokoch. 1993. Translocation of Rac correlates with NADPH oxidase activation. Evidence for equimolar translocation of oxidase components. J. Biol. Chem. 268:20983-20987. [PubMed] [Google Scholar]
- 59.Reville, K., J. K. Crean, S. Vivers, I. Dransfield, and C. Godson. 2006. Lipoxin A4 redistributes myosin IIA and Cdc42 in macrophages: implications for phagocytosis of apoptotic leukocytes. J. Immunol. 176:1878-1888. [DOI] [PubMed] [Google Scholar]
- 60.Sarndahl, E., et al. 2007. Neutrophil activation status in stable coronary artery disease. PLoS One 2:e1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Serhan, C. N. 2004. Clues for new therapeutics in osteoporosis and periodontal disease: new roles for lipoxygenases? Expert Opin. Ther. Targets 8:643-652. [DOI] [PubMed] [Google Scholar]
- 62.Serhan, C. N., N. Chiang, and T. E. Van Dyke. 2008. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. 8:349-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Serhan, C. N., M. Hamberg, and B. Samuelsson. 1984. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. U. S. A. 81:5335-5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Serhan, C. N., et al. 2003. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J. Immunol. 171:6856-6865. [DOI] [PubMed] [Google Scholar]
- 65.Seymour, G. J., P. J. Ford, M. P. Cullinan, S. Leishman, and K. Yamazaki. 2007. Relationship between periodontal infections and systemic disease. Clin. Microbiol. Infect. 13(Suppl. 4):3-10. [DOI] [PubMed] [Google Scholar]
- 66.Sharma, A., et al. 2000. Porphyromonas gingivalis platelet aggregation activity: outer membrane vesicles are potent activators of murine platelets. Oral Microbiol. Immunol. 15:393-396. [DOI] [PubMed] [Google Scholar]
- 67.Uehara, A., T. Imamura, J. Potempa, J. Travis, and H. Takada. 2008. Gingipains from Porphyromonas gingivalis synergistically induce the production of proinflammatory cytokines through protease-activated receptors with Toll-like receptor and NOD1/2 ligands in human monocytic cells. Cell. Microbiol. 10:1181-1189. [DOI] [PubMed] [Google Scholar]
- 68.Wilton, J. M., T. J. Hurst, R. J. Carman, and M. G. Macey. 1990. Effects of Porphyromonas gingivalis culture products on human polymorphonuclear leukocyte function. FEMS Microbiol. Immunol. 2:285-293. [DOI] [PubMed] [Google Scholar]
- 69.Yagi, Y., et al. 2006. Involvement of Rho signaling in PAR2-mediated regulation of neutrophil adhesion to lung epithelial cells. Eur. J. Pharmacol. 536:19-27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.