

Abstract
The Sindbis virus RNA-dependent RNA polymerase nsP4 possesses an amino-terminal region that is unique to alphaviruses and is predicted to be disordered. To determine the importance of this region during alphavirus replication, 29 mutations were introduced, and resultant viruses were assessed for growth defects. Three small plaque mutants, D41A, G83L, and the triple mutant GPG(8-10)VAV, had defects in subgenome synthesis, minus-strand synthesis, and overall levels of viral RNA synthesis, respectively. Large plaque viruses were selected following passage in BHK-21 cells, and the genomes of these were sequenced. Suppressor mutations in nsP1, nsP2, and nsP3 that restored viral RNA synthesis were identified. An nsP2 change from M282 to L and an nsP3 change from H99 to N corrected the D41A-induced defect in subgenomic RNA synthesis. Three changes in nsP1, I351 to V, I388 to V, or the previously identified change, N374 to H (C. L. Fata, S. G. Sawicki, and D. L. Sawicki, J. Virol. 76:8641-8649, 2002), suppressed the minus-strand synthetic defect. A direct reversion back to G at position 8 reduced the RNA synthesis defect of the GPG(8-10)VAV virus. These results imply that nsP4's amino-terminal domain participates in distinct interactions with other nsPs in the context of differentially functioning RNA synthetic complexes, and flexibility in this domain is important for viral RNA synthesis. Additionally, the inability of the mutant viruses to efficiently inhibit host protein synthesis suggests a role for nsP4 in the regulation of host cell gene expression.
The Alphavirus genus of the Togaviridae contains almost 30 species of viruses. These viruses are globally distributed and cause a variety of disease, ranging from mild febrile illness to encephalitis and death. Alphaviruses are arthropod borne and enzootically maintained in a cycle between mosquitoes and birds or small mammals. In vertebrate hosts, infection is acute, producing high viral yields prior to clearance or death. In cultured vertebrate, cells with large amounts of viral RNA are rapidly generated prior to apoptotic cell death. Factors likely contributing to the efficiency of RNA synthesis include the sequestration of machinery in cytoplasmic membrane-associated centers (8, 12, 13, 19) and specific regulation of the viral RNA-dependent RNA polymerase (RdRp) activity. The morphogenesis of viral RNA synthetic complexes and regulation of RNA synthetic activity are integrally linked to programmed proteolytic processing of the viral components of the complex over the course of virus infection.
Sindbis virus (SIN) is the type species of the Alphavirus genus and serves as a model for the study of viral RNA synthesis. The SIN genome is 11.7 kb of message-sense RNA that is capped and polyadenylated (49, 51), allowing for efficient translation. Following viral entry and release of the genome to the cytoplasm, the large 5′ open reading frame of the genomic RNA is translated into the nonstructural polyproteins P123 or P1234, required for viral RNA synthesis. P1234 results from a readthrough event at the 3′ end of the nsP3-coding region (22, 31, 50). Approximately 10% of translating ribosomes read through the stop codon to give rise to P1234. Cleavage of the nonstructural polyproteins is catalyzed by the nsP2-associated proteinase activity (3, 16), ultimately resulting in four mature proteins, nsP1, nsP2, nsP3, and nsP4. Cleavage of the polyproteins is sequential and temporally regulated, creating viral RNA synthetic complexes with distinct functions over the course of the single-cell replication cycle (21, 23, 25, 46). Initial cleavage of P1234 releases mature nsP4 and occurs rapidly (2, 45). nsP4 contains the RdRp catalytic core but requires the other nsP proteins for activity. P123 and nsP4 form the early RNA synthetic complex that recognizes the promoter at the 3′ end of the genomic RNA and synthesizes the complementary minus-strand RNA. Further cleavage of P123 releases nsP1, yielding an intermediate complex of nsP1, P23, and nsP4. This complex is active for synthesis of minus-strand as well as the two plus-strand RNAs, the genome and a subgenomic mRNA synthesized from an internal promoter on the minus-strand template. Late in infection, fully processed nsP1, nsP2, nsP3, and nsP4 form a complex active for plus-strand synthesis, while minus-strand synthesis is essentially shutoff (21, 23, 25, 46). Additionally, mature nsP2 rapidly cleaves the nsp1-nsp2 and nsp2-nsp3 junctions but not nsp3-nsp4, resulting in an accumulation of P34, which lacks polymerase activity, later in infection (2, 24).
The characterization of the cleavage cascade of the nonstructural polyprotein and the corresponding changes in the activity of the RNA synthetic complex have made it apparent that the changing nature of the nsPs throughout infection alter the promoter usage of the RNA synthetic complex. Notably, the RdRp nsP4 is stoichiometrically scarce and fully processed throughout due to its production by translational readthrough and rapid cleavage of the 3-4 junction. The questions therefore arise as to how this relatively low-abundance essential catalytic subunit of the viral RNA synthetic complex maintains the appropriate contact with the other components of the complex as they undergo structural modifications and how these interactions determine cis element recognition.
It is known that recognition of cis-acting elements by the RNA synthetic complex is determined by the cleavage state of the nsPs. Prior work by Frolov et al. (7) demonstrated a requirement for the 5′ end of the genome during minus-strand synthesis (14, 15). Work produced by our lab has shown that nsP4 isolated away from P123 is capable of copying a genomic RNA lacking the authentic 5′ end in a cell-free system, suggesting that template specificity during minus-strand synthesis is, at least in part, bestowed by P123 (53). Additional in vitro analyses by Li and Stollar have shown that while nsP4 binds directly to the promoter for subgenomic RNA synthesis on minus-strand RNA, it requires nsP1, nsP2, and nsP3 to do so (27). It is therefore apparent that nsP4 must maintain the appropriate molecular contacts with the other nonstructural proteins in order to recognize viral promoters and maintain template specificity; however, it is not apparent how this is accomplished. As the other viral components of the RNA synthetic complex are changing as a consequence of proteolytic processing, we assume that contacts with nsP4 are also changing.
Mutational analyses of nsP4 have resulted in evidence for specific interactions within the RNA synthetic complexes. Replacing the strictly conserved tyrosine at the amino terminus of nsP4 results in defects in RNA synthesis (47). Second-site suppressor mutations arose in nsP1 and the 5′ end of the genomic RNA that are suggestive of interactions with nsP4's amino terminus (43, 44). The nsP1 suppressor is adjacent to a residue known to affect minus-strand synthesis (41, 55), implying that this interaction is important for minus-strand synthesis (44). Add to these findings the fact that purified, amino-terminally truncated nsP4 demonstrates terminal transferase activity but lacks copying activity and the importance of the amino-terminal domain is apparent (39, 54).
The amino-terminal domain of nsP4 is predicted by modeling software to be disordered and has less similarity to RdRps of viruses outside the Alphavirus genus. The multiple interactions suggested by the mutation and suppressor data could then be explained by the amino-terminal domain assuming multiple conformations for distinct interactions, as has been seen for naturally disordered domains in other systems (4). We hypothesize a role for this region as an adaptor domain, mediating interactions with other factors within the RNA synthetic complexes. Disrupting such interactions may be a viable approach for the development of antiviral interventions.
This study tests the hypothesis that the inherent flexibility of the amino-terminal domain allows multiple interactions to be maintained between the polymerase and the other components of the RNA synthetic complex as the complex composition changes throughout virus infection. Using charged-to-alanine mutagenesis or mutations predicted to reduce the flexibility of this novel domain, we have identified residues in this region of nsP4 necessary for efficient viral RNA synthesis and shown that different mutations in this region affect different viral RNA synthetic processes. Second-site mutations in each of the other three nsPs that led to the direct correction of the original RNA synthetic defect were identified. This is additional genetic evidence for interactions between the amino-terminal domain of nsP4 and the other nsPs and supports the hypothesis that this domain functions as an adaptor domain. Additionally, we present evidence that this domain may play a role in the inhibition of host cell gene expression following virus infection.
MATERIALS AND METHODS
Cultured cells and media.
BHK-21 cells (American Type Culture Collection, Rockville, MD) were cultured in alpha minimal essential medium (αMEM; Invitrogen) supplemented with 5% fetal bovine serum (FBS) and penicillin-streptomycin. BSC-40 cells (ATCC) were used for vaccinia virus amplification and were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% FBS (Premium; Atlanta Biologicals), nonessential amino acids, vitamins, and penicillin-streptomycin.
DNA constructs.
SINs were derived from the full-length cDNA clone pToto1101 (36), and bacterial expression of nsP4 was derived from pSUMOnsP4 (39). Point mutations were introduced via PCR mutagenesis (QuikChange; Stratagene). Whenever possible, multiple bases per codon were substituted to reduce the likelihood of direct reversion. Mutagenic oligonucleotide primers used for construction of the more extensively studied mutants are as follows, and primers for the other mutants are available upon request (lowercase letters indicate non-wt [wild-type] bases): GPG(8-10)VAV (GGC CCT GGG to GtC gCa GtG), 5′-CGACGGACACAGtCgCaGtGCACTTGCAAAAGAAGTCCG-3′, and the complement; D41A (GAC to Gca), 5′-CATGCCCCGGTGCTCGcaACGTCGAAAGAGGAAC-3′, and the complement; D41K (GAC to aAg), 5′-CATGCCCCGGTGCTCaAgACGTCGAAAGAGGAA-3′, and the complement; and G83L (GGA to ctA), 5′-ATAACCACTGAGCGACTACTGTCActACTACGACTGTATAAC-3′, and the complement.
The firefly luciferase (FFLuc) gene was cloned into pUC19 following a T7 RNA polymerase promoter and short nonviral 5′ untranslated region (5′UTR) sequence. The FFLuc open reading frame was followed by a short nonviral 3′UTR and poly(A) sequence and an XhoI site for linearization. Details of the construct may be obtained upon request.
In vitro RNA transcriptions and transfections.
SIN- or luciferase-carrying constructs were purified with commercial spin columns (Zymo) and transcribed as described previously (36), with modification. Templates were linearized with XhoI (New England Biolabs [NEB]) and transcribed in the presence of the RNA cap analog mGpppG (NEB) with Sp6 or T7 RNA polymerase (NEB) for SIN or luciferase, respectively. Transcription reactions were used directly for transfection into BHK-21 cells in serum-free DMEM using Lipofectamine 2000 (Invitrogen). For virus stock generation, medium was harvested after the cytopathic effect (CPE) was observed, at between 24 and 48 h. The titers of the recovered virus stocks were determined by plaque assay using BHK-21 cells.
Viral RNA synthesis in cultured cells.
BHK-21 cells in 6-well dishes were infected at a multiplicity of infection (MOI) of 10. Thirty minutes before being labeled, the medium was replaced with complete medium containing actinomycin D (ActD) at 10 μg/ml. At the indicated times, medium was replaced with 1 ml medium plus 50 μCi/ml [3H]uridine (Moravek Biochemicals) and ActD. After 2 h of labeling, RNAs were harvested by TRIzol (Invitrogen) extraction. RNAs were denatured at 55°C with glyoxal in dimethyl sulfoxide (DMSO) and separated by 1% agarose gel electrophoresis in sodium phosphate buffer. Gels were dehydrated in methanol and impregnated with 2,5-diphenyloxazole (PPO). Dried gels were exposed to film at −80°C, and developed films were scanned and quantitated using ImageQuant software.
Minus-strand synthesis in infected cells.
Cells were infected as described above for RNA synthesis. RNAs were harvested by TRIzol extraction. cDNAs were synthesized from 100 ng of total RNA using primers specific to the SIN minus strand and the host 18S rRNA with ImProm-II reverse transcriptase (Promega). cDNAs were quantitated by real-time PCR analysis in a Stratagene MX3000P thermocycler using SYBR green (Stratagene) chemistry. Mutant minus-strand values were analyzed by the cycle threshold (ΔΔCT) method (30). This method calculates the relative changes in RNA levels based on changes in the number of PCR cycles needed to reach the threshold of detection, relative to the number of cycles necessary to detect an internal control RNA of constant abundance (18S rRNA).
Viral protein synthesis.
Cells were infected as described above for RNA synthesis. Thirty minutes before being labeled, cells were rinsed with phosphate-buffered saline, and the medium was replaced with Cys- and Met-free DMEM (Gibco). At the indicated times, the medium was replaced with 1 ml Cys/Met-free DMEM plus 30 μCi/ml [35S]Cys/Met mix (Perkin-Elmer). After 1 h of labeling, cells were washed with 0°C phosphate-buffered saline and lysed in a mild detergent buffer (10 mM Tris-HCl at pH 7.4, 140 mM NaCl, 2 mM EDTA, 0.5% NP-40, 0.4% sodium deoxycholate). Nuclei were removed by centrifugation at 13,000 × g for 30 s, and SDS was added to the postnuclear extracts to a concentration of 0.1%. Proteins were separated by denaturing SDS-PAGE, and the gels were dried and visualized by phosphorimaging.
Western blotting.
Cells were infected, and cytoplasmic lysates were generated as described above for the protein synthesis assays. Proteins were separated by 8% SDS-PAGE and transferred to nitrocellulose (Whatman). Blots were blocked and probed in Tris-buffered saline (TBS) with 5% nonfat dry milk and 0.1% Tween 20. Blots were probed with anti-nsP1 rabbit polyclonal antiserum, anti-nsP2 rabbit polyclonal antiserum, or anti-actin polyclonal IgG fraction (Sigma) followed by an anti-rabbit goat antiserum conjugated to horseradish peroxidase (HRP). Bands were visualized by chemiluminescence (SuperSignal West Pico; Pierce).
Isolation of recombinant nsP4.
Wild-type and mutant nsP4s were isolated from Escherichia coli as described previously (39), with modification. Expression vectors were transformed into Rosetta 2(DE3) (Novagen) chemically competent cells. Cells were grown in terrific broth supplemented with kanamycin (50 μg/ml) and chloramphenicol (37 μg/ml) to an optical density at 600 nm of 0.4 and then induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 18°C for 18 h. Cells were then harvested and lysed by lysozyme (Sigma) treatment and sonication in buffer A (50 mM Tris at pH 8, 200 mM Li2SO4, 50 mM arginine, 50 mM glutamate, 5% glycerol, 0.2% CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate}, 1 mM TCEP [Tris(2-carboxyethyl)phosphine]) with 1 mM MgCl2, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor cocktail (Sigma). Clarified sonicate was applied to nickel (GE Healthcare) or cobalt (Clontech) resin and eluted in buffer A plus a stepped gradient of imidazole. Fractions containing nsP4 were pooled, and the buffer was exchanged with storage buffer (20 mM Tris-HCl at pH 8, 200 mM NaCl, 50 mM arginine, 50 mM glutamate, 20% glycerol, 1 mM TCEP) using 10-kDa molecular mass cutoff centrifuge concentrators (Millipore). The 6His-SUMO tag was cleaved from nsP4 with SUMO protease (courtesy of Thomas Bernhardt, Harvard Medical School, Boston, MA) for 12 h at 4°C. The released nsP4 was again passed over cobalt resin to remove the cleaved histidine tag, uncleaved protein, and protease that also possesses a 6His tag. The concentrations of the isolated nsP4s were determined by Bradford protein assay (Bio-Rad), and isolation and enrichment were confirmed by SDS-PAGE.
Cell-free minus-strand synthesis.
RNA template for in vitro assays was transcribed from the pMini1(+) plasmid (15) linearized with BsgI (NEB). This RNA template is analogous to the SIN genome, with the majority of the coding regions removed. Membranous cell fractions (P15s) were generated as described previously (21, 39). Briefly, BHK-21 cells were infected with vaccinia virus expressing the T7 polymerase and SIN polyprotein P123 with the nsP2 C481S mutation, which abolishes nsP2's proteolytic activity (48). Membranes from the postnuclear cytoplasmic fraction were pelleted at 15,000 × g and resuspended in storage buffer.
Minus-strand synthesis assays were performed as described previously (39) with the isolated mutant polymerases. Briefly, reaction mixtures containing purified nsP4, P123 containing P15s, and RNA template were labeled with [α-32P]CTP. RNA products were denatured at 55°C with glyoxal in DMSO and separated by 1.5% agarose gel electrophoresis in sodium phosphate buffer. Gels were dried and visualized by phosphorimaging.
In vivo luciferase assay.
BHK-21 cells in 24-well dishes were infected at an MOI of 10. At 5 h postinfection (hpi), cells were rinsed and transfected with in vitro-transcribed luciferase carrying mRNA at 0.2 μg per well. Cells were returned to 37°C and remained in the transfection mix until harvesting. At 8 h postinfection, cells were rinsed and lysed in cell culture lysis buffer (Promega). Luciferase levels in each sample were assayed in triplicate in half-area opaque 96-well plates (Corning). Luciferase assay reagent (Promega) was added just before reading in a BioTek Synergy 2 plate reader. Relative light units were normalized by setting the mock transfection value to 0 and the mock-infected value to 100 in order to compare at least 3 biological replicates.
Plaque purification and RT-PCR sequencing.
Small plaque mutant viruses were passaged in BHK-21 cells. Cells were infected at an MOI of 0.1, and released virus particles were collected after the onset of the CPE, at around 16 h. Plaque size revertants were observed after three passages. Large plaquing revertants were isolated from single plaques and amplified in BHK-21 cells. RNA was harvested from cells infected by plaque-purified revertant stocks at an MOI of 10 using TRIzol reagent. cDNAs complementary to polyadenylated RNAs were synthesized from oligo(dT) primers using ImProm-II reverse transcriptase. Overlapping double-stranded DNA (dsDNA) fragments were amplified by PCR using SIN-specific primers. Fragments were sequenced on an Applied Biosystems 3730 automated capillary sequencer.
RESULTS
Mutagenesis of the nsP4 amino-terminal domain.
When the primary sequence is aligned to other known RdRps, SIN nsP4's approximately 460-residue carboxy-terminal region shows appreciable similarity to those of the other polymerases, while the 150-residue amino-terminal region fails to demonstrate similarity to those of known RdRps (Fig. 1 A) (33, 54). The amino-terminal region also contains a span of more than 60 residues that are predicted by modeling programs to be disordered (residues 23 to 82 predicted by Predictor of Naturally Disordered Regions [PONDR; Molecular Kinetics] VL-XT [WSU Research Foundation] [29, 37, 38]; residues 24 to 81 predicted by VL3H VL3 [35]) (Fig. 1B). To examine the role of the nsP4 amino-terminal domain in viral replication, we generated a suite of mutants within the coding region for this domain in the infectious SIN clone Toto1101 (36). Twenty-nine mutants were constructed at 24 sites in the amino-terminal region. Multiple approaches to mutant design were taken; charged residues were changed to alanine or an oppositely charged residue, and glycines and prolines were changed to larger or less flexible residues. The residues targeted for mutagenesis are shown in Fig. 1C.
FIG. 1.

Mutations introduced into the amino-terminal domain of nsP4. (A) Schematic representation of the primary sequence of nsP4. Shading indicates the region without similarity to nonalphavirus RNA-dependent RNA polymerases (RdRps). The GDD active-site motif and amino-terminal truncation site, residue 97 (54), are indicated. (B) Predictor of Naturally Disordered Regions (PONDR) output (VL-XT) for nsP4's primary sequence. The thick black bar indicates a significant region of disorder according to the algorithm. (C) Primary sequence of the amino-terminal domain, with residues targeted for mutagenesis highlighted in gray. Sites at which two or three residues were simultaneously targeted are underlined.
Transfection of in vitro-transcribed genomic RNAs into BHK-21 cells yielded viable, plaque-forming virus for each of the 29 mutants. Titers and plaque morphology were determined for the recovered virus stocks (Table 1). Mutants displayed a range of plaque sizes and titers, with more severe effects observed mainly in viruses in which glycines and prolines were changed.
TABLE 1.
Titers and plaque morphology of nsP4 amino-terminal mutants
| Virus | Plaque size (mm) | Titer (PFU/ml) |
|---|---|---|
| wt | 4 | 1 × 109 |
| D6A | 3.5 | 8 × 108 |
| D6K | 3.5 | 1 × 108 |
| P9A | 4 | 1 × 109 |
| GPG(8-10)VAVa | 0.5 | 3 × 107 |
| H11A | 3.5 | 1 × 109 |
| H11E | 3 | 5 × 108 |
| K15A | 4 | 1 × 109 |
| QKK(13-15)AAA | 4 | 5 × 108 |
| E24R | 4 | 1 × 109 |
| E33A | 4 | 1 × 109 |
| E33R | 4 | 1 × 109 |
| H36A | 4 | 1 × 109 |
| APV(37-39)ERE | 2 | 5 × 108 |
| D41Aa | 1 | 2 × 108 |
| D41Ka | 4 | 1 × 109 |
| K44A | 4 | 1 × 109 |
| KEE(44-46)AAA | 4 | 4 × 108 |
| R51A | 4 | 1 × 109 |
| MMP(54-56)KKN | 2 | 5 × 108 |
| R63A | 4 | 1 × 109 |
| KSR(61-63)AAA | 4 | 1 × 109 |
| RK(67-68)AA | 3.5 | 1 × 109 |
| K73A | 3.5 | 8 × 108 |
| K73D | 3 | 4 × 108 |
| R79A | 4 | 1 × 109 |
| G83La | 2.5 | 5 × 108 |
| Y87A | 3 | 8 × 108 |
| P94V | 4 | 1 × 109 |
| Y97A | 3 | 5 × 108 |
Mutants used in subsequent studies.
Growth defects of select mutants.
Preliminary assays testing the growth kinetics, RNA synthesis, and protein synthesis of the 29 mutants were performed (data not shown), and on the basis of displaying different phenotypes in these preliminary assays, the following three mutants were chosen for further analyses: GPG(8-10)VAV, D41A, and G83L. D41K was also examined for comparison with D41A. The presence of the original mutation in virus stocks was confirmed by sequencing of reverse transcription-PCR (RT-PCR) products (data not shown). The plaque sizes of mutant virus stocks were confirmed, in triplicate, on BHK-21 cells. Plaque size reduction from these mutations varied from moderate for G83L to severe for GPG(8-10)VAV and D41A, with D41K showing wt-sized plaques (Fig. 2 A).
FIG. 2.

nsP4 amino-terminal domain mutations affect viral growth. (A) Plaque morphology of select mutants. BHK-21 cells were infected with virus stocks rescued from transfections and stained with crystal violet at 40 hpi. (B) Growth kinetics in vertebrate cells. BHK-21 cells were infected at an MOI of 10 PFU/cell, media were removed and replaced at 2-hour intervals, and titers were determined by plaque assay.
The growth kinetics of these viruses corresponded with the plaque phenotypes (Fig. 2B). GPG(8-10)VAV and D41A show at least a 10-fold reduction in released infectious units relative to that of the wild type at each time point. G83L also shows reduced growth at early time points but by 10 h postinfection (hpi) had achieved wild-type levels of virus production. Thus, we have three mutations showing various degrees of growth inhibition.
Viral RNA synthesis in cells.
If the amino-terminal domain of nsP4 does mediate multiple interactions during RNA synthetic complex maturation, we hypothesized that mutations should affect one or more viral RNA synthetic processes.
To test the mutants for RNA synthetic defects, viral RNA synthesis was monitored in infected BHK-21 cells. RNAs were labeled by addition of [3H]uridine in the presence of actinomycin D. Cells were labeled for 2-h periods starting at 4 and 8 hpi, and RNAs were visualized by fluorography (Fig. 3 A and B). Genome length and subgenomic bands were quantitated by image densitometry for total signal as well as the ratio of subgenome to full-length RNA. The three small plaque mutants each showed a decrease in total RNA synthesis relative to that of the wild type and each displayed a distinct RNA synthetic defect.
FIG. 3.
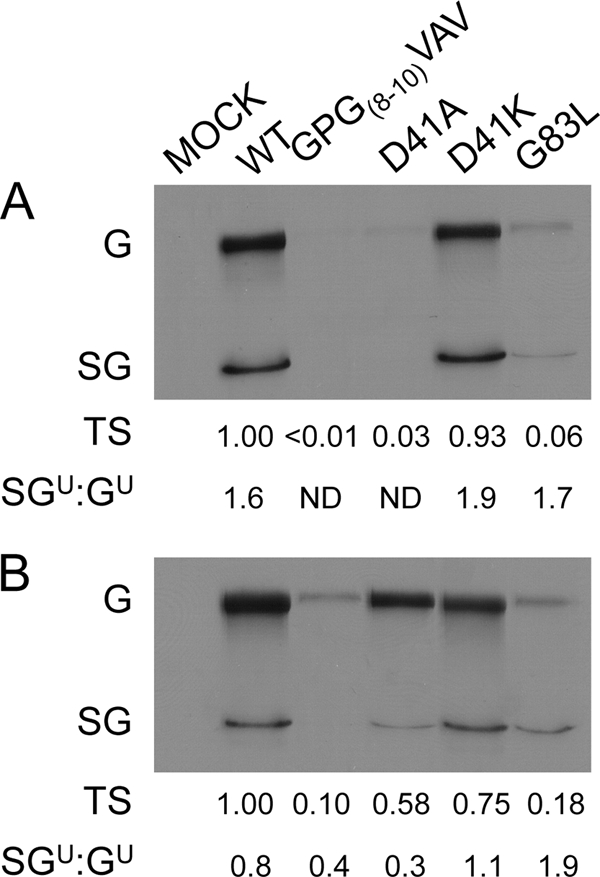
Select amino-terminal mutations each affect the synthesis of different viral RNAs. BHK-21 cells were infected at an MOI of 10 PFU/cell, treated with ActD, and labeled with [3H]uridine from 4 to 6 hpi (A) or 8 to 10 hpi (B), after which RNA was harvested. RNAs were separated by denaturing agarose gel electrophoresis and visualized by fluorography. The signal was quantified by image densitometry. Values for total signal (TS) relative to that of the wild type and the molar ratios of subgenome (SG) to genome (G) RNA were determined. Ratios were corrected for the following numbers of uridine (U) moieties per molecule: genome, 2,438; subgenome, 856. ND, not determined.
As expected, the virus that made the smallest plaques, GPG(8-10)VAV, synthesized the least genomic and subgenomic RNA at both the early and later time points. Also of note, this mutant synthesized reduced levels of subgenomic RNA, resulting in a subgenome-to-genome (SG/G) ratio of 0.4. D41A also synthesized a very low level of total RNA early in infection, but by 8 h, was synthesizing greater than 50% of that seen for the wt. It was also apparent that the ability of D41A to synthesize subgenomic RNA was compromised. The D41K virus displayed approximately wild-type levels of RNA synthesis, confirming the importance of a residue of either charge at this position. G83L virus showed an intermediate level of RNA synthesis, corresponding with its plaque phenotype. Interestingly, G83L seemed to show a greater defect in the synthesis of genome-length RNAs than that of subgenomic RNAs, resulting in an increased SG/G ratio of 1.9 at 8 hpi, more than double that of the wild type. An alternate explanation may be that the reduced growth of this virus leads to an SG/G ratio late in infection that is similar to that of the wt early in infection. Thus, we see greater effects at distinct stages of RNA synthesis from three mutations in nsP4's amino-terminal domain. These results add weight to the idea that this region of nsP4 is involved in regulating the specific RNA synthetic functions of the enzyme.
In vitro minus-strand RNA synthesis.
Since genomic and subgenomic RNA synthesis is dependent on minus-strand synthesis, we performed assays to test the mutant polymerases' minus-strand synthetic abilities in isolation. Mutant nsP4s were expressed in bacteria and isolated as described previously, with minor modification (39) (Fig. 4 A). These enzymes were tested for minus-strand synthetic activity in the cell-free assay, which includes P123 and host cell membranes isolated from BHK-21 cells. P123 contains the nsP2 C481S mutation (48), preventing polyprotein processing and keeping the RNA synthetic complexes in the minus-strand synthetic state. The minus-strand synthetic ability of each polymerase was assayed as described in Materials and Methods and is shown in Fig. 4B. NsP4 with the GPG(8-10)VAV change (Fig. 4B) shows significantly reduced RNA synthetic ability. In contrast to the results of the in vivo RNA synthesis assay examining plus-strand synthesis, D41A synthesizes minus-strand RNA at approximately wt levels, as does D41K. G83L nsP4 (Fig. 4B) is more significantly compromised, synthesizing minus-strand RNA less efficiently than GPG(8-10)VAV. As this assay eliminates differences in RNA and protein amounts and polyprotein cleavage, we can infer that the effects are due to defects within the minus-strand synthetic complex. Also, as the relative magnitudes of the defects for the four mutants are different than that of the respective defects seen when examining plus-strand RNA synthesis in cells, this is further evidence that the mutations in the amino-terminal domain of nsP4 are each affecting specific RNA synthetic complexes.
FIG. 4.

Minus-strand RNA synthetic activity of purified nsP4s. Bacterially expressed nsP4 was purified by immobilized metal affinity chromatography. (A) Purified nsP4s were separated by SDS-polyacrylamide gel electrophoresis and stained with Coomassie brilliant blue. (B) In vitro RNA synthesis assays were performed using the purified enzymes in the presence of [α-32P]CTP, with polyprotein P123, host membranes, and a synthetic SIN RNA template supplied in trans. RNA products were separated by denaturing agarose gel electrophoresis and visualized by a phosphorimager. Three independent experiments were quantified by image densitometry, and the average signal relative to that of the wild type with standard deviation is shown.
Protein synthesis phenotypes.
SIN infection inhibits the transcription and translation of vertebrate host cells, with at least nsP2 playing a direct role (9, 10, 34). To assess any effects of the amino-terminal mutants on viral and host translation, we labeled proteins synthesized in infected cells. Infected BHK-21 cells were labeled with [35S]Cys and Met, and cytoplasmic fractions were analyzed by SDS-PAGE (Fig. 5 A and B). Each mutant virus showed delayed onset of host protein synthesis shutoff. In addition, GPG(8-10)VAV and D41A showed altered translation of viral proteins. In wild-type-infected cells by 7 hpi, host protein synthesis, most clearly visualized by the actin band, was no longer detectable. Likewise, viral genomic translation, visible as nsP2 and polyprotein bands, had also nearly ceased, and subgenomic translation had become maximal. GPG(8-10)VAV and D41A showed a delay in host cell shutoff, with incomplete shutoff later in infection. GPG(8-10)VAV and D41A also showed sustained genome translation late in infection (as indicated by nsP2 levels), and production of structural proteins failed to reach wild-type levels, presumably as a consequence of decreased subgenomic RNA synthesis. Increased translation of viral genomic RNA was seen to result in a marked increase in the accumulation of nsP1 and nsP2, particularly for D41A compared to the wild type, based on Western blotting (Fig. 5C).
FIG. 5.

Viral and host protein syntheses are affected by the amino-terminal mutations. BHK-21 cells infected at an MOI of 10 PFU/cell were labeled with [35S]cysteine and methionine at 3, 7, and 11 h (A) or 7, 17, and 23 h (B). After 1 h of labeling, cells were harvested and postnuclear lysates were separated by SDS-polyacrylamide gel electrophoresis and visualized by a phosphorimager. (C) Viral protein accumulation. Unlabeled postnuclear lysates were harvested at 8 hpi. Separated proteins were transferred to nitrocellulose and probed with the indicated antisera. Bands were visualized by chemiluminescence. α, anti.
G83L virus-infected cells showed a pattern of incomplete host shutoff similar to those of cells infected with GPG(8-10)VAV and D41A; however, viral translation more closely resembled that of the wild type. Structural protein synthesis levels are only slightly lower than those of the wild type, and genome translation shutoff occurs normally. However, host protein synthesis continues at low levels to 23 hpi. The minus-strand synthesis defect of G83L does not readily explain this lack of complete shutoff, leaving a possibility that nsP4 has an as-yet-undefined function related to the regulation of host cell gene expression.
Suppressor mutations and virus growth.
The three small plaque mutants, GPG(8-10)VAV, D41A, and G83L, were passaged in BHK-21 cells, and large plaque revertants were selected. Revertants were isolated from single plaques and amplified in BHK-21 cells. RNA was isolated from these plaque-purified viruses, amplified by RT-PCR, and sequenced. Eight genomes were analyzed, yielding several silent changes and 6 independent missense changes, with 2 of which found twice (see below). The missense mutations were reintroduced into the full-length SIN clone (Toto1101) with and without their respective parental mutations (Fig. 6 A). Large plaque virus originating from the parental GPG(8-10)VAV mutant virus had a change of the V residue at position 8 back to a G residue (this change was found in two independently isolated viruses). This is a partial direct reversion reintroducing a flexible residue at this position and indicates the importance of this region for nsP4 function. Mutations identified in large plaque viruses originating from D41A were found in both nsP2 and nsP3 at residues M282 (found in two independently isolated viruses) and H99, respectively. Mutations were identified in nsP1, at I351, N374, and I388, in large plaque viruses originating from G83L.
FIG. 6.

Second-site mutations in nsP1, -2, and -3 suppress the growth defects of nsP4 amino-terminal domain mutants. (A) Schematic representation of the suppressor mutations identified. The amino-terminal mutant viruses were successively passaged, and large plaque descendants were plaque purified. These genomes were sequenced, and identified changes were reintroduced into the cDNA SIN constructs. (B to D) Growth kinetics of the recombinant and parental viruses were determined as described in the legend to Fig. 2, with an additional 12-hour interval, and are shown, with the wt curve included in each panel.
Growth kinetics of viruses with these changes combined with parental mutations were determined in BHK-21 cells (Fig. 6B to D). Mutations identified in revertant viruses resulted in clear restoration of growth to wild-type rates for both GPG(8-10)VAV and D41A. The original G83L mutation had a more subtle effect on virus growth, causing decreased virus production early in infection but catching up to wt levels later. The second-site mutations coupled with G83L conferred plaque morphology closer to that of the wild type (data not shown); however, the second-site mutations corrected the mild G83L growth defect to various degrees, with only the N374I change appearing to completely restore virus growth. These data provide genetic evidence for interactions between the amino-terminal domain of nsP4 and those of each of the other nsPs.
Effects of suppressor mutations on plus-strand RNA synthesis.
Viruses with the suppressor mutations were analyzed for their ability to synthesize viral RNA in infected cells (Fig. 7 A). Cells were infected and labeled as shown in Fig. 3 from 8 to 10 hpi. For the GPG(8-10)VAV revertant, with a single change from V to G at position 8, viral RNA synthesis was still significantly below wild-type levels despite growth being near that of the wild type. However, the level of subgenomic RNA was increased in the revertant compared to that in the parental mutant.
FIG. 7.

Suppressor mutations restore the synthesis of specific viral RNAs. (A) Cells were infected and labeled with [3H]uridine, as described in the legend to Fig. 3, from 8 to 10 hpi. The image is representative of four independent experiments. (B) Quantitative PCR analysis of minus-strand synthesis. Duplicate sets of equal numbers of BHK-21 cells were infected at an MOI of 10 PFU/cell, and RNAs were extracted at 4 and 9 hpi. SIN minus-strand and host 18S rRNAs were reverse transcribed using gene-specific primers. cDNAs were quantitated by real-time PCR using SYBR green chemistry. At least 3 experiments per time point with standard deviations are shown.
Interestingly, for the D41A second-site changes, the amount of viral RNA synthesized by viruses with either the nsP2 M282L or nsP3 H99N changes alone was greater than that by the wild-type virus. When combined with the D41A change, each of the second-site changes resulted in RNA synthesis, similar to that of the wild type. Significantly, the impairment of subgenomic RNA synthesis caused by the parental D41A mutation was corrected by the second-site changes.
Second-site changes found for the G83L virus were all located in nsP1. In combination with wild-type nsP4, the I351V virus synthesized RNA at levels equivalent to or better than the wt, whereas both the N374I and I388V viruses synthesized RNA less efficiently than the wt virus. None of these changes in combination with the G83L nsP4 mutation had an obvious restorative effect on genomic or subgenomic RNA synthesis compared to the parental G83L virus. As these second-site changes were seen to suppress a defect in viral growth (Fig. 6), this suggests that they suppress a defect in minus-strand synthesis or potentially a separate function of nsP4.
Effects of suppressor mutations on minus-strand RNA synthesis.
In order to determine the effects of the suppressor mutations on viral minus-strand synthesis, we performed quantitative RT-PCR on RNA extracted from infected cells using primers specific for SIN minus-strand RNA. The trend for minus-strand synthesis by the three parental mutants was similar to that observed using the cell-free minus-strand synthesis assay; that is, D41A was least affected, and GPG(8-10)VAV and G83L were more compromised in their ability to synthesize minus-strand RNA (Fig. 7B). It should be noted that the overall level of minus-strand synthesis by the D41A virus was approximately 20% of that of the wt in this assay compared to approximately 80% in the cell-free assay. We believe the differences in the supply of template for minus-strand synthesis between the two assays may account for this. In the cell-free assay, the plus-strand RNA template is supplied in a virus-independent manner in the form of a set amount of in vitro-transcribed RNA, whereas the plus-strand template supplied in vivo is virus derived and is amplified over the course of infection. Differences in the ability of the viruses to make genomic RNA in vivo (at 4 hpi) (Fig. 3A) may result in a decrease in minus-strand synthesis that is not observed in the cell-free system.
The suppressor mutations partially or completely suppressed the minus-strand synthesis defects of the parental mutations in all cases. The viruses with D41A and its suppressors showed wild-type levels of minus-strand synthesis at 9 hpi, but as discussed above, we suspect that this is due to the increase in template for minus-strand synthesis, i.e., genome. The G83L suppressor mutations were observed to suppress the minus-strand RNA synthetic defect. The greatest fold recovery of minus-strand synthesis was observed in G83L/N374I, which did not show altered genome and subgenome synthesis.
Overall, these data from both plus- and minus-strand analyses indicate that the amino-terminal domain of nsP4 mediates interactions, either directly or indirectly, with each of the other three nsPs and that these interactions are critical for viral RNA synthesis.
Effects of suppressor mutations on virus and host protein synthesis.
The viral and host protein syntheses in cells infected with recombinant revertant viruses were assayed as described in the legend to Fig. 5 at 7 hpi (Fig. 8 A) and more quantitatively by measuring luciferase activity derived from a nonviral mRNA-carrying FFLuc transfected into virus-infected cells (Fig. 8B). The partial direct reversion of GPG(8-10)VAV to GPG(8-10)GAV displayed viral and host translation levels much closer to those of the wild type than the original mutation when levels of labeled cellular actin and luciferase activity were examined. The lower levels of viral RNA seen in this mutant do not keep it from expressing viral proteins or from shutting off host gene expression.
FIG. 8.

Second-site changes restore viral protein expression and host translation shutoff. (A) BHK-21 cells infected at an MOI of 10 PFU/cell were labeled with [35S]cysteine and methionine at 7 hpi. After 1 h of labeling, cells were harvested and postnuclear lysates were separated by SDS-polyacrylamide gel electrophoresis and visualized by a phosphorimager. (B) Quantification of luciferase activity produced by a reporter mRNA. BHK-21 cells were infected at an MOI of 10 PFU/cell and transfected with a luciferase mRNA at 5 hpi (see Materials and Methods for details). At 8 hpi, cells were lysed, and luciferase activity was measured in microtiter plates. The results and standard deviations obtained from three separate experiments are shown.
For D41A, both M282L (nsP2) and H99N (nsP3) significantly suppress the D41A translation phenotype. Most clearly, host cell translation is nearly shutoff, and structural protein synthesis is near wild-type levels (Fig. 8A). Luciferase activity from mRNA transfected into infected cells was reduced to levels seen in wt-infected cells for both the D41A/M282L and D41A/H99N viruses, indicating that the second-site changes restore shutoff host cell translation (Fig. 8B).
I351V, N374I, and I388V each suppress G83L's slower growth and small plaque phenotypes; however, the second-site changes had no observable effect on the RNA synthetic phenotype of the G83L virus. When protein synthesis was examined, it was found that all 3 second-site changes suppressed the G83L translation phenotype (characterized by a lack of host translational shutoff) to different degrees. With the original G83L change, I351V partially restores shutoff host cell translation. Actin is still labeled at 7 hpi, and luciferase activity is lower than that in G83L virus-infected cells but higher than that in wt-infected cells. The G83L/N374I virus shuts off host cell translation in a manner indistinguishable from that of the wt virus (Fig. 8A and B). The I388V change is least effective in restoring host cell translational shutoff. Actin labeling is slightly reduced in the G83L/I388V virus-infected cells but still readily observable, and luciferase activity is slightly lower than that in G83L virus-infected cells.
Together, these data imply a role for nsP4, either directly or as a consequence of one of its activities, in the shutoff host cell translation.
DISCUSSION
SIN RNA synthesis is intricately regulated over the course of infection. Minus-strand synthesis peaks early and is subsequently shutoff, while genome and subgenome syntheses are highest late in infection (40). The RNA synthetic machinery undergoes structural and conformational changes during infection that correlate with these shifts in RNA synthetic activity. Previous work utilizing vector-expressed nsPs with cleavage-deficient protease or cleavage-resistant junctions and SIN variants with cleavage-resistant junctions has shown that nonstructural polyprotein cleavage confers functional changes to the RNA synthetic complexes (23-25, 46). Other work has found that host factors, including RNase L, play a role in the regulation of viral RNA synthesis (11, 42).
In this work, we targeted the catalytic subunit nsP4 of each of the RNA synthetic complexes and found three changes in its putative flexible amino-terminal domain that affect different aspects of RNA synthetic function. Interestingly, when phenotypic revertant viruses were selected, each of the three original mutant viruses had employed a different strategy of reversion. The GPG(8-10)VAV virus reverted through a direct change back to a G residue at position 8 that partially restored minus-strand synthesis and subgenomic RNA synthesis. Second-site changes in nsP2 and nsP3 were found to restore subgenomic RNA synthesis of the D41A virus. Finally, second-site changes in nsP1 restored the plaque and growth phenotype of G83L, but this suppression was not at the level of plus-strand RNA synthesis; rather, it was through the restoration of minus-strand RNA synthesis and/or host cell translational shutoff.
One hypothesis with which we had begun this study was that the amino-terminal domain of nsP4 functioned as a flexible adaptor domain capable of maintaining contact(s) within a changing complex over the course of a single-cell infectious cycle. In order to examine whether the predicted flexibility of this domain was important for nsP4 function and virus replication, we targeted amino acids or groups of amino acids predicted to inhibit the formation of α-helices and beta-sheets and/or introduce rotational freedom in the polypeptide. One such group of amino acids was GPG at positions 8 to 10. These residues were mutated to VAV, and the resulting virus was severely attenuated in growth, had defects in minus-strand RNA synthesis and subgenomic RNA synthesis, and failed to inhibit host cell protein production efficiently. Reversion at position 8 back to G partially restored minus-strand, subgenomic RNA synthesis and host cell shutoff. Correction of RNA synthetic defects as a consequence of the V-to-G change suggests that flexibility in this domain is important for the function of nsP4. The differential effects of the original mutation on RNA synthesis (genomic RNA synthesis being less affected than minus-strand or subgenomic RNA synthesis) imply that this mutation disrupts the interaction of nsP4 with some forms of the viral RNA synthetic complex but not others.
Further evidence that this domain within nsP4 may play a significant role in mediating interactions within the changing viral RNA synthetic complex was found through the D41A mutant. The growth of D41A was severely impaired; however, the effect on minus-strand RNA synthesis was relatively minor (Fig. 4). Genomic RNA synthesis was delayed but caught up with that of the wt virus by 8 hpi, while subgenomic RNA synthesis was significantly compromised. Since the basic polymerase function of the D41A nsP4 does not appear to be compromised, as it functions well in the minus-strand synthesis assay, the data indicate that this mutation disrupted the appropriate formation of the subgenomic RNA synthetic complex and/or recognition of the subgenomic RNA promoter.
Second-site suppressors of D41A arose in nsP2 and nsP3, implying a functional interaction between the amino-terminal domain of nsP4 and those of nsP2 and nsP3 for the restored functions, primarily subgenome synthesis. The D41A suppressor in nsP2, M282L, is within motif 3 of the helicase domain. Previous work has implicated the nsP2 C-terminal protease domain in subgenomic synthesis (52), and processing itself is important for subgenomic synthesis (17). This suppressor found in the helicase domain implies that there is an additional functional partnership between nsP4 and nsP2 in subgenomic RNA synthesis. The M282L change alone increases RNA synthesis above wild-type levels but decreases host translational shutoff. Combined with D41A, both of these functions more closely resemble those of the wild type, suggesting that RNA levels are at optimal levels in wild-type infections and that higher levels of viral RNA synthesis are not necessarily beneficial, in agreement with previous suggestions (6).
The D41A suppressor in nsP3, H99N, also displayed greater-than-wild-type levels of RNA synthesis. This change is within the broadly conserved macrodomain that is involved in RNA synthesis but for which a specific function has not been identified (1, 20). Mutations in this region of nsP3 have been found to affect minus-strand synthesis (1), and the macrodomain has been found to bind ADP-ribose and have phosphatase activity (32). The His-99 residue is not conserved across alphaviruses, and indeed, nsP3 residue 99 is asparagine in Venezuelan equine encephalitis virus (18). That this substitution is made in a closely related virus could indicate that the functional adjustment conferred by the change is advantageous under certain conditions. H99 is not part of the RNA, poly-ADP-ribose (PAR), and ADP-ribose binding site of the domain (32) and could feasibly be a feature of a binding surface for interaction with nsP4 or the host factor. If there were direct interaction with the nsP4 amino-terminal domain based on these data, we might expect a charge-charge interaction with D41 of nsP4. However, the reversed-charge mutation D41K, which would interfere with such an interaction, was near levels of the wild type in all of our assays, suggesting that this residue is solvent exposed and may play a role in the folding of the amino-terminal domain induced by interactions with other proteins.
The data obtained from the D41A mutation further supported the idea that the amino-terminal domain functions to make multiple contacts with other components of the RNA synthetic complex during infection. These observations are also in line with previously published data showing the requirement for nsP1, nsP2, and nsP3, in addition to nsP4, for appropriate subgenomic promoter recognition and efficient subgenomic RNA synthesis (26-28).
The G83L nsP4 was compromised in its ability to synthesize minus-strand RNA (Fig. 4 and 7), and suppressors of the G83L mutation were found in nsP1 as follows: I351V, N374I, and I388V. Each of these mutations restored minus-strand synthesis that was severely compromised by the parental mutation. This is in line with previously published studies that identified the amino-terminal Y and R183 in nsP4 as critical residues for minus-strand synthetic activity of the virus and showed that the effect of these mutations was suppressed by changes in nsP1 (5, 47). In fact second-site changes were identified in nsP1 at N374 (6) (Fig. 6) and T349 (44), demonstrating the importance of the 349-to-388 region of nsP1 in minus-strand RNA synthesis. Interestingly, these second-site changes that we identified (I351V, N374I, and I388V) failed to restore plus-strand RNA synthesis levels (Fig. 7). Both genomic and subgenomic RNA levels in the revertant viruses remained low. This implies that G83 is essential for the formation or maintenance of plus-strand RNA synthetic complexes or possibly involved in promoter recognition. Regardless of the function of this residue in viral RNA synthesis, it is apparent that the restoration of virus growth in the revertant viruses was not a consequence of an increase in plus-sense RNA synthesis.
The G83L mutation in the amino-terminal domain of nsP4 demonstrated an interesting phenotype that implies a function for nsP4 in shutoff host cell gene expression. When viral and host cell translation was examined, it was apparent that the G83L virus was unable to efficiently shut off host cell translation. This was also true of the GPG(8-10)VAV and D41A viruses, but these viruses also showed distinct differences in translation of viral proteins compared to that in the wt (Fig. 5). For GPG(8-10)VAV and D41A, translation of the genomic RNA continued late in infection, as indicated by nonstructural protein production, whereas in a wt infection, translation of genomic RNA is shutoff, and subgenomic RNA predominates late in infection. The fact that the mutant viruses produce more nsP2, a known inhibitor of host cell transcription (9), and yet fail to turn off host cell protein production efficiently suggests that it is translational shutoff that is affected by these changes. We believe this phenotype for GPG(8-10)VAV and D41A is a consequence of low levels of subgenomic RNA, resulting from defects in RNA synthesis for these mutant viruses. One hypothesis is that the subgenomic RNA is capable of outcompeting both cellular and virus genomic RNA for translational machinery, and if levels of this RNA are diminished, host cell translation is not appropriately inhibited (A. Burnham and R. W. Hardy, unpublished data). In contrast, the G83L virus, while delayed in protein expression, showed a pattern of viral gene expression similar to that of the wt virus. However, host cell protein synthesis was not shut off efficiently, and the deficit in host cell shutoff persisted throughout infection, with actin still being labeled at up to 23 hpi.
NsP2 is known to inhibit host cell transcription and has been implicated in host cell translational shutoff (34). Our data suggest that nsP4 is involved in the inhibition of host cell gene expression. Preliminary examination indicates that host cell transcription is efficiently inhibited during an infection with the G83L virus (data not shown), further supporting the idea that a complex containing nsP4 and nsP1 plays a role in the inhibition of host cell translation.
Overall, our data show that the amino-terminal domain of nsP4 is important for viral RNA synthesis, that it plays a role in multiple viral RNA synthetic events, that flexibility within this region is important for function, and that this domain mediates multiple functional interactions with other viral nonstructural proteins. These findings also raise the possibility that this domain expressed in trans may be inhibitory to the formation of active RNA synthetic complexes. While this domain is not highly conserved at the level of sequence, its presence and apparent flexibility are conserved across the Alphavirus genus; thus, provision of inhibitory peptides or peptide mimics of this domain may represent a broadly applicable means of antialphavirus therapy.
Acknowledgments
We thank Suchetana Mukhopadhyay for critical comments about the manuscript. Access to PONDR was provided by Molecular Kinetics, Indianapolis, IN.
This work was supported by grant MCB0749482 from the National Science Foundation to R.W.H. J.C.R was supported by the Genetics, Cellular and Molecular Sciences Training Grant (T32GM007757) funded by the National Institutes of Health.
Footnotes
Published ahead of print on 19 January 2011.
REFERENCES
- 1.Dé, I., C. Fata-Hartley, S. G. Sawicki, and D. L. Sawicki. 2003. Functional analysis of nsP3 phosphoprotein mutants of Sindbis virus. J. Virol. 77:13106-13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Groot, R. J., W. R. Hardy, Y. Shirako, and J. H. Strauss. 1990. Cleavage-site preferences of Sindbis virus polyproteins containing the non-structural proteinase. Evidence for temporal regulation of polyprotein processing in vivo. EMBO J. 9:2631-2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ding, M. X., and M. J. Schlesinger. 1989. Evidence that Sindbis virus NSP2 is an autoprotease which processes the virus nonstructural polyprotein. Virology 171:280-284. [DOI] [PubMed] [Google Scholar]
- 4.Dunker, A. K., M. S. Cortese, P. Romero, L. M. Iakoucheva, and V. N. Uversky. 2005. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 272:5129-5148. [DOI] [PubMed] [Google Scholar]
- 5.Fata, C. L., S. G. Sawicki, and D. L. Sawicki. 2002. Alphavirus minus-strand RNA synthesis: identification of a role for Arg183 of the nsP4 polymerase. J. Virol. 76:8632-8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fata, C. L., S. G. Sawicki, and D. L. Sawicki. 2002. Modification of Asn374 of nsP1 suppresses a Sindbis virus nsP4 minus-strand polymerase mutant. J. Virol. 76:8641-8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frolov, I., R. Hardy, and C. M. Rice. 2001. Cis-acting RNA elements at the 5′ end of Sindbis virus genome RNA regulate minus- and plus-strand RNA synthesis. RNA 7:1638-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frolova, E. I., R. Gorchakov, L. Pereboeva, S. Atasheva, and I. Frolov. 2010. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J. Virol. 84:11679-11695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garmashova, N., R. Gorchakov, E. Frolova, and I. Frolov. 2006. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J. Virol. 80:5686-5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gorchakov, R., E. Frolova, and I. Frolov. 2005. Inhibition of transcription and translation in Sindbis virus-infected cells. J. Virol. 79:9397-9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorchakov, R., et al. 2008. A new role for ns polyprotein cleavage in Sindbis virus replication. J. Virol. 82:6218-6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorchakov, R., N. Garmashova, E. Frolova, and I. Frolov. 2008. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J. Virol. 82:10088-10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grimley, P. M., I. K. Berezesky, and R. M. Friedman. 1968. Cytoplasmic structures associated with an arbovirus infection: loci of viral ribonucleic acid synthesis. J. Virol. 2:1326-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardy, R. W. 2006. The role of the 3′ terminus of the Sindbis virus genome in minus-strand initiation site selection. Virology 345:520-531. [DOI] [PubMed] [Google Scholar]
- 15.Hardy, R. W., and C. M. Rice. 2005. Requirements at the 3′ end of the Sindbis virus genome for efficient synthesis of minus-strand RNA. J. Virol. 79:4630-4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hardy, W. R., and J. H. Strauss. 1989. Processing the nonstructural polyproteins of Sindbis virus: nonstructural proteinase is in the C-terminal half of nsP2 and functions both in cis and in trans. J. Virol. 63:4653-4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heise, M. T., et al. 2003. An attenuating mutation in nsP1 of the Sindbis-group virus S.A.AR86 accelerates nonstructural protein processing and up-regulates viral 26S RNA synthesis. J. Virol. 77:1149-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinney, R. M., K. R. Tsuchiya, J. M. Sneider, and D. W. Trent. 1992. Genetic evidence that epizootic Venezuelan equine encephalitis (VEE) viruses may have evolved from enzootic VEE subtype I-D virus. Virology 191:569-580. [DOI] [PubMed] [Google Scholar]
- 19.Kujala, P., et al. 2001. Biogenesis of the Semliki Forest virus RNA replication complex. J. Virol. 75:3873-3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LaStarza, M. W., J. A. Lemm, and C. M. Rice. 1994. Genetic analysis of the nsP3 region of Sindbis virus: evidence for roles in minus-strand and subgenomic RNA synthesis. J. Virol. 68:5781-5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemm, J. A., A. Bergqvist, C. M. Read, and C. M. Rice. 1998. Template-dependent initiation of Sindbis virus RNA replication in vitro. J. Virol. 72:6546-6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lemm, J. A., R. K. Durbin, V. Stollar, and C. M. Rice. 1990. Mutations which alter the level or structure of nsP4 can affect the efficiency of Sindbis virus replication in a host-dependent manner. J. Virol. 64:3001-3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemm, J. A., and C. M. Rice. 1993. Assembly of functional Sindbis virus RNA replication complexes: requirement for coexpression of P123 and P34. J. Virol. 67:1905-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemm, J. A., and C. M. Rice. 1993. Roles of nonstructural polyproteins and cleavage products in regulating Sindbis virus RNA replication and transcription. J. Virol. 67:1916-1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lemm, J. A., T. Rumenapf, E. G. Strauss, J. H. Strauss, and C. M. Rice. 1994. Polypeptide requirements for assembly of functional Sindbis virus replication complexes: a model for the temporal regulation of minus- and plus-strand RNA synthesis. EMBO J. 13:2925-2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li, M. L., and V. Stollar. 2007. Distinct sites on the Sindbis virus RNA-dependent RNA polymerase for binding to the promoters for the synthesis of genomic and subgenomic RNA. J. Virol. 81:4371-4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li, M. L., and V. Stollar. 2004. Identification of the amino acid sequence in Sindbis virus nsP4 that binds to the promoter for the synthesis of the subgenomic RNA. Proc. Natl. Acad. Sci. U. S. A. 101:9429-9434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, M. L., H. Wang, and V. Stollar. 2010. In vitro synthesis of Sindbis virus genomic and subgenomic RNAs: influence of nsP4 mutations and nucleoside triphosphate concentrations. J. Virol. 84:2732-2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li, X., P. Romero, M. Rani, A. K. Dunker, and Z. Obradovic. 1999. Predicting protein disorder for N-, C-, and internal regions. Genome Inform. Ser. Workshop Genome Inform. 10:30-40. [PubMed] [Google Scholar]
- 30.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 31.Lopez, S., J. R. Bell, E. G. Strauss, and J. H. Strauss. 1985. The nonstructural proteins of Sindbis virus as studied with an antibody specific for the C terminus of the nonstructural readthrough polyprotein. Virology 141:235-247. [DOI] [PubMed] [Google Scholar]
- 32.Malet, H., et al. 2009. The crystal structures of Chikungunya and Venezuelan equine encephalitis virus nsP3 macro domains define a conserved adenosine binding pocket. J. Virol. 83:6534-6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marchler-Bauer, A., et al. 2009. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 37:D205-D210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayuri, T. W. Geders, J. L. Smith, and R. J. Kuhn. 2008. Role for conserved residues of sindbis virus nonstructural protein 2 methyltransferase-like domain in regulation of minus-strand synthesis and development of cytopathic infection. J. Virol. 82:7284-7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Obradovic, Z., et al. 2003. Predicting intrinsic disorder from amino acid sequence. Proteins 53(Suppl. 6):566-572. [DOI] [PubMed] [Google Scholar]
- 36.Rice, C. M., R. Levis, J. H. Strauss, and H. V. Huang. 1987. Production of infectious RNA transcripts from Sindbis virus cDNA clones: mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J. Virol. 61:3809-3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Romero, Obradovic, and K. Dunker. 1997. Sequence data analysis for long disordered regions prediction in the calcineurin family. Genome Inform. Ser. Workshop Genome Inform. 8:110-124. [PubMed] [Google Scholar]
- 38.Romero, P., et al. 2001. Sequence complexity of disordered protein. Proteins Struct. Funct. Gen. 42:38-48. [DOI] [PubMed] [Google Scholar]
- 39.Rubach, J. K., et al. 2009. Characterization of purified Sindbis virus nsP4 RNA-dependent RNA polymerase activity in vitro. Virology 384:201-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sawicki, D. L., and S. G. Sawicki. 1994. Alphavirus positive and negative strand RNA synthesis and the role of polyproteins in formation of viral replication complexes. Arch. Virol. Suppl. 9:393-405. [DOI] [PubMed] [Google Scholar]
- 41.Sawicki, D. L., S. G. Sawicki, S. Keränen, and L. Kääriäinen. 1981. Specific Sindbis virus-coded function for minus-strand RNA synthesis. J. Virol. 39:348-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawicki, D. L., R. H. Silverman, B. R. Williams, and S. G. Sawicki. 2003. Alphavirus minus-strand synthesis and persistence in mouse embryo fibroblasts derived from mice lacking RNase L and protein kinase R. J. Virol. 77:1801-1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shirako, Y., E. G. Strauss, and J. H. Strauss. 2003. Modification of the 5′ terminus of Sindbis virus genomic RNA allows nsP4 RNA polymerases with nonaromatic amino acids at the N terminus to function in RNA replication. J. Virol. 77:2301-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shirako, Y., E. G. Strauss, and J. H. Strauss. 2000. Suppressor mutations that allow sindbis virus RNA polymerase to function with nonaromatic amino acids at the N-terminus: evidence for interaction between nsP1 and nsP4 in minus-strand RNA synthesis. Virology 276:148-160. [DOI] [PubMed] [Google Scholar]
- 45.Shirako, Y., and J. H. Strauss. 1990. Cleavage between nsP1 and nsP2 initiates the processing pathway of Sindbis virus nonstructural polyprotein P123. Virology 177:54-64. [DOI] [PubMed] [Google Scholar]
- 46.Shirako, Y., and J. H. Strauss. 1994. Regulation of Sindbis virus RNA replication: uncleaved P123 and nsP4 function in minus-strand RNA synthesis, whereas cleaved products from P123 are required for efficient plus-strand RNA synthesis. J. Virol. 68:1874-1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shirako, Y., and J. H. Strauss. 1998. Requirement for an aromatic amino acid or histidine at the N terminus of Sindbis virus RNA polymerase. J. Virol. 72:2310-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strauss, E. G., R. J. de Groot, R. Levinson, and J. H. Strauss. 1992. Identification of the active site residues in the nsP2 proteinase of Sindbis virus. Virology 191:932-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strauss, E. G., C. M. Rice, and J. H. Strauss. 1984. Complete nucleotide sequence of the genomic RNA of Sindbis virus. Virology 133:92-110. [DOI] [PubMed] [Google Scholar]
- 50.Strauss, E. G., C. M. Rice, and J. H. Strauss. 1983. Sequence coding for the alphavirus nonstructural proteins is interrupted by an opal termination codon. Proc. Natl. Acad. Sci. U. S. A. 80:5271-5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strauss, J. H., and E. G. Strauss. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suopanki, J., D. L. Sawicki, S. G. Sawicki, and L. Kääriäinen. 1998. Regulation of alphavirus 26S mRNA transcription by replicase component nsP2. J. Gen. Virol. 79(Pt. 2):309-319. [DOI] [PubMed] [Google Scholar]
- 53.Thal, M. A., B. R. Wasik, J. Posto, and R. W. Hardy. 2007. Template requirements for recognition and copying by Sindbis virus RNA-dependent RNA polymerase. Virology 358:221-232. [DOI] [PubMed] [Google Scholar]
- 54.Tomar, S., R. W. Hardy, J. L. Smith, and R. J. Kuhn. 2006. Catalytic core of alphavirus nonstructural protein nsP4 possesses terminal adenylyltransferase activity. J. Virol. 80:9962-9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang, Y. F., S. G. Sawicki, and D. L. Sawicki. 1991. Sindbis virus nsP1 functions in negative-strand RNA synthesis. J. Virol. 65:985-988. [DOI] [PMC free article] [PubMed] [Google Scholar]