

Abstract
Cells that are enzymatically detached from a solid substrate rapidly round up as the tensile prestress in the cytoskeleton is suddenly unopposed by cell–ECM adhesions. We recently showed that this retraction follows sigmoidal kinetics with time constants that correlate closely with cortical stiffness values. This raises the promising prospect that these de-adhesion measurements may be used for high-throughput screening of cell mechanical properties; however, an important limitation to doing so is the possibility that the retraction kinetics may also be influenced and potentially rate-limited by the time needed to sever matrix adhesions. In this study, we address this open question by separating contributions of contractility and adhesion to cellular de-adhesion and retraction kinetics. We first develop serum-free conditions under which U373 MG glioma cells can be cultured on substrates of fixed fibronectin density without direct matrix contributions from the medium. We show that while spreading area increases with ECM protein density, cortical stiffness and the time constants of retraction do not. Conversely, addition of lysophosphatidic acid (LPA) to stimulate cell contractility strongly speeds retraction, independent of the initial matrix protein density and LPA’s contributions to spreading area. All of these trends hold in serum-rich medium commonly used in tissue culture, with the time constants of retraction much more closely tracking cortical stiffness than adhesive ligand density or cell spreading. These results support the use of cellular de-adhesion measurements to track cellular mechanical properties.
Keywords: Adhesion, Contractility, Extracellular matrix (ECM), Ligand density, Serum, Myosin, Cell mechanics, Atomic force microscopy, Cell stiffness
Introduction
Cells adherent to planar extracellular matrix (ECM) substrates exist in a force balance in which tensile forces in the cytoskeleton are opposed by the mechanical rigidity of the ECM. This “tensional homeostasis” plays key regulatory roles in a wide variety of cellular phenomena, including shape determination, migration, tissue assembly, and fate choice.3,6,27 Moreover, there is growing evidence that changes in cellular mechanical properties relevant to this force balance may serve as biomarkers for disease, including various types of cancer.17,24 Many methods have been introduced to measure single-cell mechanical properties, including atomic force microscopy,4,5 micropipette aspiration,10,15 traction force microscopy,22,28 optical tweezers,14,32,33 and intracellular particle tracking.8,31 While all of these methods have proven extremely powerful in producing high-resolution measurements of cellular viscoelastic properties, efforts to incorporate these methods into high-throughput technologies that might be used to diagnose disease or screen drug libraries based on changes in cell mechanics have been limited in part by these methods’ inherently low throughput and low proclivity toward automation. This in turn has spawned a new generation of high-throughput cell mechanics technologies including optical stretchers 7,12 and controlled detachment assays.30
We recently introduced an assay in which cellular tensional homeostasis could be gauged by tracking the kinetics with which cells retract following chemical detachment from the ECM.21 In this assay, cells are treated with trypsin to rapidly sever cell–ECM contacts and time-lapse phase-contrast imaging is used to track the projected cell area as a function of time. The idea is that as cell–ECM contacts are severed, tensile loads within the cytoskeleton suddenly become unbalanced, causing the cell to rapidly contract to a rounded morphology. We showed that the kinetics of this retraction obeys sigmoidal kinetics with characteristic time constants, and that the magnitude of these time constants closely track cellular elasticity as measured by AFM. The time constants also change in predictable ways when intracellular tension is either increased or decreased by pharmacologic inhibition or activation of cell contractility. An important unresolved issue with this assay, and with controlled-detachment assays in general, is that it is not clear to what extent cell–ECM adhesion itself contributes to the kinetics of the detachment process. In an extreme case, one might imagine that the rate of retraction could be limited by the time needed to dissipate cell–ECM contacts rather than relaxation of tensile forces within the cytoskeleton and, therefore, primarily reflect the strength of adhesion to the matrix. Distinguishing between contributions from these two sources is critical if this technology is to be explored for high-throughput screening of cell mechanical properties.
In this study, we address this open question by experimentally distinguishing contributions of cellular prestress and cell–ECM adhesion to retraction kinetics following de-adhesion. We culture cells on ECM substrates coated with varying densities of fibronectin and in the presence or in the absence of lysophosphatidic acid (LPA) to stimulate contractility. Critically, we conduct these studies in serum-free medium to minimize contributions of serum to matrix density and baseline contractility. We then perform de-adhesion assays and show that the time constants of retraction are relatively insensitive to changes in matrix density but are highly sensitive to perturbations in cell contractility. These conclusions are supported by immunofluorescence localization of key adhesive and contractile markers and by AFM nanoindentation measurements of cortical stiffness. Together, our data support the notion that cellular retraction kinetics following de-adhesion is dominated by the tensile state of the cell rather than the extent of cell–ECM adhesive engagement.
Materials and Methods
Cell Culture
U373 MG human glioblastoma cells (American Type Culture Collection, MD) were cultured at 37 °C in a humidified atmosphere of 95% air and 5% CO2 in growth medium composed of DMEM supplemented with 10% fetal calf serum, 1% sodium pyruvate (Invitrogen), 1% non-essential amino acids (Invitrogen), and 1% penicillin/streptomycin (Invitrogen). Cells were maintained in 75 cm2 cell culture flasks (Corning, NY), harvested with 0.25% trypsin-EDTA (Invitrogen), and passaged every 2–3 days. For experiments, cells were plated on glass coverslips coated with human plasma fibronectin (Milipore Corp.) at various dilutions to achieve nominal (theoretical) coating densities of 0.1, 1.0, or 10.0 μg/cm2, respectively. Experiments were performed either in 10% serum-containing medium as described above or in serum-free medium (DMEM supplemented with 1% sodium pyruvate, 1% non-essential amino acids, and 1% penicillin/streptomycin).
Immunofluorescence Labeling and Antibodies
Cultured cells were rinsed twice with phosphate-buffered saline (PBS) (Fisher Scientific), fixed with 4% para-formaldehyde solution for 10 min, permeabilized with 0.5% Triton X-100, and blocked in 5% bovine serum albumin for 1 h at room temperature. Cells were incubated for 1 h at room temperature in one of the following primary antibody solutions in PBS at the following dilutions: mouse anti-vinculin (1:200, Sigma), rabbit anti-NMMII (1:200 anti-NMMIIA and 1:200 anti-NMMIIB, Sigma), and phosphorylated MLC (pMLC) (1:500, Cell Signaling). After incubation with primary antibody, cells were rinsed twice with PBS and then incubated with secondary antibody (either Alexa 543 goat anti-mouse IgG (1:500, Invitrogen) or Alexa 543 goat anti-rabbit IgG (1:500, Invitrogen)), and Alexa 488-phalloidin (1:200, Invitrogen) for 1 h at room temperature. Samples were mounted onto slides using Cytoseal 60 (Richard-Allan Scientific).
Measurement of ECM Coating
The fibronectin coating of glass coverslips at the different theoretical densities was quantified using immunofluorescence and Western blots. For quantitative immunofluorescence analysis, camera exposure and gain settings were fixed, and images were taken at 10 random positions for each condition. Median intensities of the histograms from each image were combined to determine the intensity distributions arising from surface-attached fibronectin across the different conditions. For Western blots, the total surface-adsorbed fibronectin for each condition was extracted by incubating fibronectin-coated surfaces with RIPA buffer for 5 min at room temperature and subsequently scraping with a cell scraper. The total amounts of collected protein corresponding to the three coating densities were boiled, run on a 4–12% Bis-Tris gel (Invitrogen), transferred to a PVDF membrane (Invitrogen), and probed with mouse monoclonal fibronectin antibody (1:500, Sigma). The blot was then probed with HRP-conjugated goat-anti-mouse (Invitrogen) and developed using TMB chromogenic substrates (Invitrogen). The developed blot was scanned, and the band intensities were quantified using Image J (NIH) software.
Image Acquisition and Analysis
Live-cell and epifluorescence images were obtained with a Nikon TE2000E2 microscope equipped with an incubator chamber for controlled temperature, humidity, and CO2 (In Vivo Scientific). Images were captured with a CCD camera (Photometrics CoolSNAP HQ2) integrated with image acquisition software (Compix SimplePCI). De-adhesion measurements were acquired as described previously.21 Briefly, the medium was aspirated off, cells were washed with PBS and incubated with warm trypsin, and images were acquired every 2 s at 20× magnification until cells became rounded while still remaining attached to the substrate. De-adhesion dynamics were quantified by fitting the plot of the time-dependent normalized area to a sigmoidal curve with two time constants (τ1 and τ2).
Atomic Force Microscopy (AFM)
Force curves were obtained with an Asylum MFP3D AFM (Asylum Research, CA) mounted on a Nikon TE2000U epifluorescence microscope. Cells were cultured overnight on glass coverslips, and 50–100 cells were indented for each condition using a pyramid-tipped probe (Veeco Instruments). Tips with nominal spring constant of 60 pN/nm were calibrated using the thermal noise method, and the calibrated values were used for all calculations. The elastic modulus of the cell was determined by fitting each force curve with a modified Hertzian model of a cone indenting a semi-infinite elastic solid.20
Results
Quantification of Fibronectin Surface Adsorption
To systematically probe contributions of ECM ligand density to cellular de-adhesion and retraction kinetics, we created ECM substrates of varying fibronectin density by functionalizing glass coverslips at three theoretical coating densities, 0.1, 1.0, and 10 μg/cm2. We then characterized the degree of surface coverage in each case by immunofluorescence imaging (Fig. 1a). Quantitative analysis of total surface fluorescence revealed a tenfold increase in intensity levels from 0.1 to 1.0 μg/cm2, with no further increase in intensity from 1.0 to 10 μg/cm2, implying saturation of fibronectin adsorption or detection under these conditions (Fig. 1b). Quantification of immunofluorescence images is often complicated by sample-to-sample variations in photobleaching, illumination intensity, and other parameters. Thus, we further characterized adsorption densities by harvesting surface-adsorbed fibronectin and subjecting this lysate to SDS-PAGE followed by Western blot against fibronectin (Fig. 1c). As expected, we observed multiple bands scattered across 200–250 kDa, the expected molecular weight range for fibronectin fragments.11 Subsequent densitometric analysis of the band intensities revealed a fivefold increase in intensity between a theoretical 1 μg/cm2 surface and a 0.1 μg/cm2 surface, and a much smaller increase in intensity between 1 μg/cm2 and 10 μg/cm2.
Figure 1.

Quantification of surface adsorption of fibronectin (FN) at different densities. (a) Representative images of glass coverslips coated with FN at theoretical densities of 0.1, 1.0, and 10.0 μg/cm2, respectively, and probed with FN antibody (scale bar = 20 μm). (b) Intensity analysis of surface-adsorbed FN at different theoretical densities. Box–whisker plots of median intensities (10 random positions per condition) at different theoretical densities. (c) Quantification of surface-adsorbed FN by Western blot. FN adsorbed to glass coverslips at different densities was extracted and run on a blot to determine the amount of attached FN. Quantification of the band intensities by densitometry revealed a saturating trend with a fivefold increase in surface-adsorbed FN between 0.1 and 1.0 μg/cm2
Fibronectin Density-Dependent Adhesion Under Serum-Free Conditions
We next used these ECM substrates to investigate the relationship between surface ECM density and cell spreading and retraction kinetics following de-adhesion. To simultaneously eliminate contributions of fibronectin adsorption from the medium and minimize intrinsic levels of cell contractility, we chose to work with serum-free medium and then add lysophosphatidic acid (LPA) to specifically stimulate Rho GTPase-dependent cell contractility.13 To quantify the effect of ECM density on cell spreading area under serum-free conditions, we cultured equal numbers of U373 MG glioma cells overnight on fibronectin-coated glass coverslips at specific coating densities. Phase contrast imaging and subsequent morphometric analysis (Fig. 2a, top panel; Fig. 2b, left side) demonstrated that the spreading of these cells is sensitive to changes in ECM density. First, as expected, fewer cells adhered to 0.1 μg/cm2 substrates than to 1.0 and 10 μg/cm2 substrates. Second, although cells exhibited a spindle-like shape on all substrates, their projected spreading area increased with increasing fibronectin density. Consistent with our observation that fibronectin adsorption saturates above 1.0 μg/cm2 (Fig. 1), cell spreading increased significantly (blue bars, * p < 0.05) from ~3600 μm2 on 0.1 μg/cm2 substrates to ~5000 μm2 on 1.0 μg/cm2 and then increased only modestly to ~5500 μm2 on 10 μg/cm2 substrates (Fig. 2b). When we treated cells with a low dose of LPA (1 μM for 10 min.), we found that spreading area remained constant compared to untreated controls on the 0.1 and 1 μg/cm2 substrates and fell modestly on the 10 μg/cm2 substrates (red bar, * p < 0.05) (Fig. 2a, bottom panel; Fig. 2b, right side). Under these conditions, then, LPA stimulates Rho GTPase but not to such an extent as to produce dramatic cell contraction.
Figure 2.

FN density-dependent spreading of U373 MG glioma cells under no-serum conditions. (a) U373 MG glioma cells were cultured on fibronectin-coated coverslips at different densities for 24 h under no-serum conditions. Representative images are shown of untreated cells (−LPA) and cells exposed to 1 μM LPA for 10 min (+LPA). Scale bar = 10 μm. (b) For untreated cells, increase in fibronectin density led to increase in cell spreading in a statistically significant manner (blue bars, * p < 0.05). Contractile stimulation with LPA led to a significant reduction (red bar, * p < 0.05) in cell spreading only on 10 μg/cm2 density substrates
Fibronectin Density-Dependent De-adhesion Dynamics Under Serum-Free Conditions
Having established conditions under which we could systematically vary matrix density and cell spreading, we next measured the kinetics of cell retraction following trypsin-mediated de-adhesion as described in our earlier study.21 Briefly, trypsin de-adhesion involves severing cell–matrix adhesive contacts with trypsin and following the cellular retraction that precedes cell detachment. During this process, projected cell area reduces from an initial area A
i to a final area A
f (Fig. 3a). We then quantify retraction kinetics by tracking the average normalized cell–substrate contact area 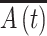 as a function of time. In the absence of LPA, retraction kinetics was largely independent of ECM coating density, with all cells fully retracting within 40 s. In the presence of LPA, retraction kinetics remained independent of coating density but was greatly accelerated over untreated controls, with cells detaching within 20 s (Fig. 3b). The detachment response of the cells under all conditions was sigmoidal in nature, enabling us to fit each of these curves to a Boltzmann equation, which yielded two characteristic time constants, τ1 and τ2 (Fig. 3c). Whereas τ1 ~ 8 s for control cells, τ1 ~ 4 s for LPA-treated cells, which represents a statistically significant (red bars, *p < 0.05) difference of 50%. Similarly, LPA treatment led to a statistically significant (red bars, *p < 0.05) 45% reduction in τ2 (~3 s and ~1.65 s for control and LPA-treated cells, respectively). Taken together, these results demonstrate that under serum-free conditions, de-adhesion time constants are independent of fibronectin density but highly sensitive to contractile stimulation.
as a function of time. In the absence of LPA, retraction kinetics was largely independent of ECM coating density, with all cells fully retracting within 40 s. In the presence of LPA, retraction kinetics remained independent of coating density but was greatly accelerated over untreated controls, with cells detaching within 20 s (Fig. 3b). The detachment response of the cells under all conditions was sigmoidal in nature, enabling us to fit each of these curves to a Boltzmann equation, which yielded two characteristic time constants, τ1 and τ2 (Fig. 3c). Whereas τ1 ~ 8 s for control cells, τ1 ~ 4 s for LPA-treated cells, which represents a statistically significant (red bars, *p < 0.05) difference of 50%. Similarly, LPA treatment led to a statistically significant (red bars, *p < 0.05) 45% reduction in τ2 (~3 s and ~1.65 s for control and LPA-treated cells, respectively). Taken together, these results demonstrate that under serum-free conditions, de-adhesion time constants are independent of fibronectin density but highly sensitive to contractile stimulation.
Figure 3.
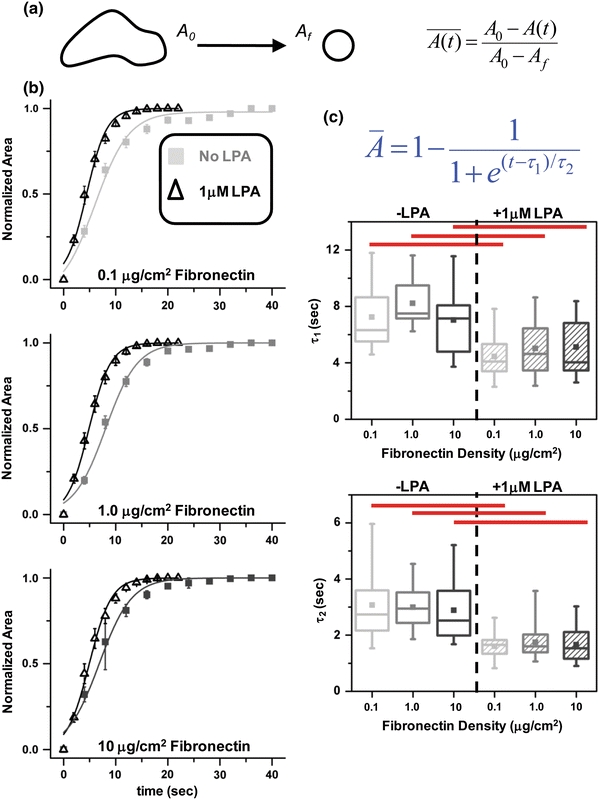
FN density-dependent de-adhesion dynamics of U373 MG glioma cells under no-serum conditions with and without LPA treatment. (a) Schematic of trypsin de-adhesion assay. Upon exposure to warm trypsin, the cell area reduces from an initial area A i to a final area A f. Cells were imaged every 2 s until cells became rounded but remained attached. (b) Quantification of cell shape changes during de-adhesion on different FN-coated substrates under no-serum conditions. Plot of normalized cell area of untreated U373 MG glioma cells (−LPA) and cells exposed to 1 μM LPA for 10 min (+LPA) cultured on different density FN-coated substrates. Irrespective of the FN density, LPA treatment caused faster de-adhesion. (c) Effect of FN density on time constants of retraction with and without LPA treatment. Under no-serum conditions, τ1 and τ2 were insensitive to changes in FN coating density. However, differences in τ1 and τ2 were statistically significant for LPA treatment (red bars, * p < 0.05) across all the densities
Retraction Dynamics Correlate with Cytoskeletal Architecture and Cell Mechanics on Fibronectin-Coated Substrates Under Serum-Free Conditions
The previous results illustrate that while spreading increases with fibronectin density under serum-free conditions, de-adhesion dynamics remain insensitive to density differences. Further, LPA treatment leads to faster de-adhesion dynamics despite only modestly altering cell shape. This suggests that cell contractility rather than adhesive ligand density is the principal determinant of cellular retraction kinetics. To address the possibility that matrix density might intrinsically contribute to cell contractility under these conditions, we investigated the effect of matrix density on cell stiffness and the assembly of the actomyosin cytoskeleton and cell–matrix adhesions. First, we used AFM nanoindentation to measure the elastic moduli of U373 MG glioma cells cultured on different density substrates with or without LPA treatment. AFM measurements confirmed that the distributions of cell stiffness for the three different densities were similar with mean stiffness of ~3 kPa (Fig. 4). LPA treatment did not alter cell stiffness for the 0.1 and 1.0 μg/cm2 densities, as evidenced by the substantial overlap in the box–whisker plots. However, on 10 μg/cm2 substrates, LPA-treated cells were significantly stiffer (red bar) than untreated controls with mean stiffness of ~4 kPa. To gain additional microscale insight into these results, we used immunofluorescence imaging to visualize the localization of key elements of the actomyosin cytoskeleton and focal adhesion machinery. For all coating densities, both untreated control cells and LPA-treated cells exhibited prominent cortical actin bundles at cell margins but relatively few well-developed central stress fibers (Fig. 5). In the absence of LPA, vinculin and NMMII were diffusely distributed throughout the cytoplasm, with poor localization of NMMII to stress fibers and the absence of mature, well-defined focal adhesions. Addition of LPA increased localization of pMLC to stress fibers and membrane ruffles, even at coating densities <10 μg/cm2 (white arrows). Overall, then, these data suggest that matrix density does not intrinsically alter contractile mechanics under these conditions and that LPA primes cell contractility through mobilization of NMMII to stress fibers and focal adhesions.
Figure 4.
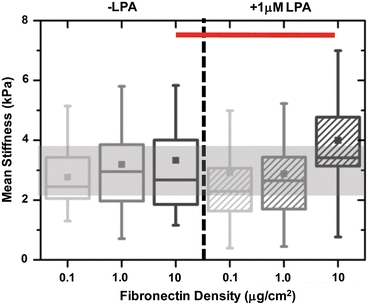
Effect of LPA on cortical stiffness of U373 MG cells cultured on different density FN substrates under no-serum conditions. Box–whisker plots of single cell stiffness of U373 MG glioma cells cultured on different density FN substrates with or without LPA treatment. Cell stiffness remained unchanged across all densities and upon LPA treatment on 0.1 and 1.0 μg/cm2 substrates. However, on 10.0 μg/cm2 substrates, LPA treatment led to a significant (red bar) increase in cell stiffness
Figure 5.
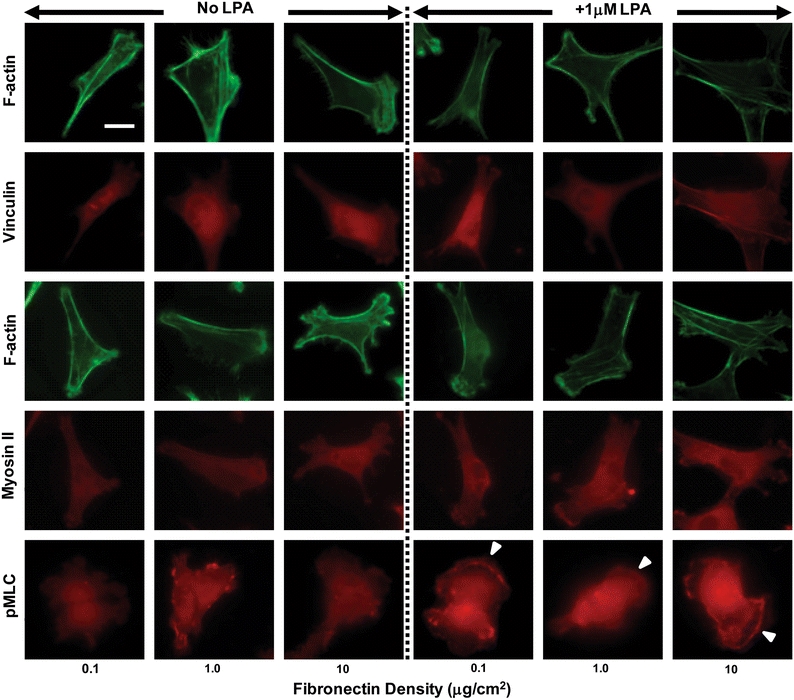
Cytoskeletal architecture, focal adhesions, NMMII, and pMLC localization. U373 MG cells cultured overnight in serum-free media on FN-coated substrates of different densities were treated with 1 μM LPA for 10 min and immunostained for F-actin, vinculin, NMMII, and pMLC, respectively. Under serum-free conditions, glioma cells exhibit a predominantly peripheral actin staining with few central stress fibers. Vinculin staining at focal adhesions and NMMII were diffused both in controls and in LPA-treated cells across all the different densities. pMLC was mostly diffusely distributed in untreated cells, but sometimes localized to peripheral membrane ruffles in LPA-treated cells (white arrows). Scale bar = 20 μm
Effect of Serum on Density-Dependent Spreading, De-adhesion Dynamics and Cortical Mechanics on Fibronectin-Coated Substrates
Our results above establish that under serum-free, LPA-free conditions de-adhesion dynamics and cortical mechanics are independent of ECM density. While these studies enable us to work with well-defined ECM coatings that minimize ECM contributions from the medium, serum contains multiple components that are expected to influence cellular structure and organization, as evidenced by the fact that serum starvation induces a spindle-like morphology with poor formation of stress fibers and focal adhesions. Moreover, media formulations for many continuous cell lines contain high quantities of serum, and it would be valuable to know if these results also hold in serum-rich medium. Therefore, we repeated all of the above experiments in culture medium containing 10% serum, standard growth conditions for U373MG glioma cells. In the presence of serum but in the absence of additional LPA, we observed consistent projected spreading areas (~5000 μm2) across all fibronectin densities (Fig. 6a). As in the serum-free case, LPA reduced cell spreading only on the 10 μg/cm2 substrates (red bar, * p < 0.05). Intriguingly, in the presence of serum, cortical stiffness was found to be mildly responsive to changes in fibronectin density. The mean cortical stiffness of glioma cells increased significantly (blue bars, * p < 0.05) from ~2.75 kPa on 1 μg/cm2 substrates to ~4 kPa on 1 μg/cm2 and 10 μg/cm2 substrates (Fig. 6b). This is presumably due to the presence of contractile agonists in serum that require threshold levels of integrin engagement for full activity. LPA treatment brought cells to a uniform stiffness value (~3.5 kPa) at all matrix densities.
Figure 6.
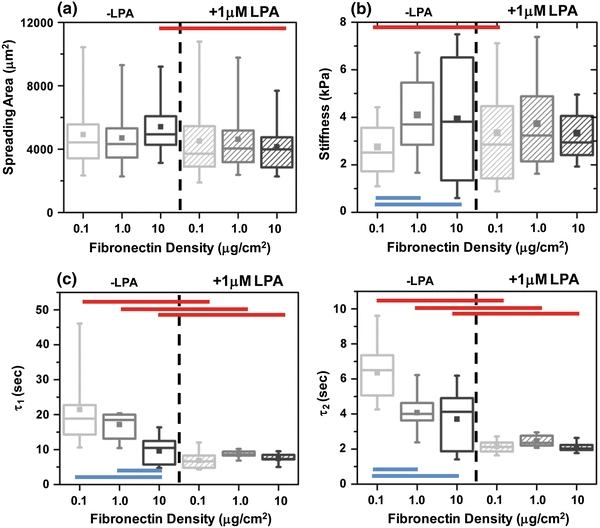
Effect of serum on the spreading, cortical stiffness, and de-adhesion dynamics of U373 MG cells cultured on different density FN substrates with and without LPA. (a) Under regular serum conditions, spreading remained unaltered on different FN-coated substrates. LPA treatment led to a reduction in spreading area with statistically significant differences observed only for the 10 μg/cm2 case (red bar, * p < 0.05). (b) Cortical stiffness of U373 MG glioma cells cultured in the presence of serum on different density FN-coated substrates in the presence or in the absence of LPA. For untreated cells, increase in FN density led to cell stiffening from ~2.85 kPa measured on 0.1 μg/cm2 density substrates to ~4 kPa measured on 1 and 10 μg/cm2 density substrates. LPA treatment eliminated these density-dependent differences in cell stiffness. (c) Effect of serum on FN density-dependent time constants of retraction with and without LPA treatment. For untreated cells cultured in serum-supplemented media, time constants exhibited an inverse density-dependent behavior with slowest retraction on 0.1 μg/cm2 substrates and fastest retraction on 10 μg/cm2 substrates. LPA treatment abolished this density-dependent behavior but led to even faster de-adhesion (red bars, * p < 0.05)
To determine whether these stiffness differences are also detectable in cellular retraction kinetics, we repeated our de-adhesion experiments in the presence of serum. Indeed, we observed density-dependent de-adhesion dynamics in the presence of serum with τ1 ~ 21 s, 17 s, and 10 s, respectively, for 0.1, 1.0, and 10 μg/cm2 substrates (Fig. 6c). The corresponding values for τ2 for the different densities were 6 s, 4.1 s, and 3.7 s, respectively. In LPA-treated cells, de-adhesion was further accelerated (τ1 ~ 7–8 s and τ2 ~ 2 s, respectively) and led to ~70% reduction in both τ1 and τ2 on 0.1 μg/cm2 substrates. Further, as in the serum-free case, LPA treatment abolished density-dependent differences in de-adhesion dynamics. Taken together, these results demonstrate that in the presence of serum, while spreading responses become insensitive to changes in fibronectin density, de-adhesion dynamics and cortical mechanics are modulated in a density-dependent manner.
Discussion
In this study we sought to separate contributions of cell–ECM adhesion and cytoskeletal prestress to cellular retraction kinetics following chemical detachment from the substrate. We have shown that in serum-free medium, increasing ECM coating density increases projected spreading area while only modestly affecting retraction kinetics. Conversely, stimulation of Rho GTPase-dependent contractility with LPA substantially accelerates retraction (i.e., reduces the time constant of retraction) regardless of the ECM coating density. When serum is re-introduced into the medium, the time constants of retraction fall with increasing ECM density; however, when LPA is added to further stimulate contractility, the time constants flatten to a constant, minimal value for all coating densities. Together, these data are consistent with a model in which cytoskeletal prestress dominates the total degree of cell–ECM adhesive engagement in determining retraction kinetics.
Importantly, these results neither demonstrate nor imply that the details of the cell–ECM adhesive interface bear no influence on retraction kinetics. Indeed, when we include serum in the medium, we find that the time constant of retraction rises significantly either in the presence or in the absence of LPA (Fig. 3c vs. Fig. 6c), with a moderate increase in cell stiffness by AFM (Fig. 4 vs. Fig 6b). Serum contains very high concentrations of fibronectin, albumin, and IgG, which rapidly adsorb to culture surfaces and radically alter their surface chemistry.2,26 Serum also contains factors that strongly stimulate cellular synthesis of fibronectin and other matrix components,16 and adsorption of these factors can in turn alter the expression and activation of integrins and other adhesion receptors on the cell surface that are critical to mechanochemical feedback. The presence of these additional factors may require substantial time to fully disassemble and may even have varying degrees of trypsin sensitivity, which would affect retraction kinetics in unpredictable ways. LPA-induced adhesion maturation, as evidenced by pMLC recruitment, might also increase affinity per adhesion without changing spreading area and, therefore, require additional time for adhesion disassembly. The notion that adhesion disassembly may contribute to retraction measurements is also apparent from our earlier report,21 in which we showed that the speed of retraction increases with increasing activity (concentration) of trypsin. It is also potentially conceivable that differences in trypsin-induced signaling across conditions could contribute to differences in measured retraction kinetics; however, we suspect this is unlikely given the extremely short time scale (tens of seconds) over which the measurements are made. All of that said, we do find that in the presence of serum, LPA stimulation of contractility still accelerates retraction kinetics, suggesting that even under “saturating” ECM ligand densities, modulations in contractility continue to measurably govern de-adhesion kinetics.
Somewhat surprisingly, we find that under serum-free conditions, AFM measurements of cortical stiffness are relatively insensitive to either ECM coating density or addition of LPA (except at the highest ECM density) and that addition of serum does not dramatically increase the range of cortical stiffnesses (Figs. 4 and 5b). While the finding that addition of serum per se does not drastically alter cell stiffness may seem somewhat counterintuitive given the well-described ability of serum to induce stress fiber formation and focal adhesion maturation,19 this result is consistent with a previous set of observations with fibroblasts.18 In that study, cell stiffness by AFM did not change significantly in the setting of serum starvation, which the authors attributed to compensatory compaction of intermediate filaments around the nucleus. In fact, Hemmer et al.9 reported that serum starvation increases the AFM-measured stiffness of vascular smooth muscle cells, which they suggested may be secondary to serum-induced changes in shape and spreading area. The equivocal effects of serum on cellular elasticity further validate our decision to establish proof-of-principle in serum-free medium and stimulate contractility with LPA, which is a chemically homogeneous stimulus that induces contractility via a single, defined intracellular target.
Remarkably, we observe that retraction dynamics measurements are frequently much more sensitive than AFM indentation measurements to LPA-induced stimulation of contractility. This is despite the fact that many studies support a close correlation between cellular contractility (or prestress) and cortical stiffness and have shown that manipulation of NMMII activity leads to predictable changes in cell stiffness.21,23,29 We hypothesize that the higher apparent sensitivity of the de-adhesion assay arises from three sources. First, de-adhesion measurements represent an integrative measure of the contractile state of the entire cell, whereas AFM measurements are localized to nano- to microscale regions of the cell surface. Second, the high throughput of the de-adhesion measurements enables significantly larger sample sizes and statistical power, which in turn may more easily elicit differences between conditions. Third, it is possible that LPA may induce stiffening of basal stress fibers, which presumably make the largest contributions to the retraction dynamics, to a much greater degree or on a faster time scale than it induces stiffening of the cortical actomyosin elements probed by AFM. An alternative explanation is that AFM may not be sufficiently sensitive to detect changes in contractile forces exerted at the cell base, and so in the future it would be valuable to repeat these experiments with direct measurements of traction forces, including traction force microscopy. Evidence that such an approach could yield results consistent with ours was recently provided by Califano et al.,1 who demonstrated that traction forces exerted by endothelial cells depend strongly on substrate stiffness, but not on ligand density.
Several open questions remain. First, whereas we performed all of our studies with fibronectin, these studies clearly require repetition with matrix components that assemble into higher-order, micron-scale structures, such as collagen I. Collagen fiber assembly and dimensions depend strongly on collagen concentration,25 and it is conceivable that as matrix features begin to approach the size of individual cells, fiber microarchitecture could influence retraction kinetics in ways that are both related to and independent of effects on cytoskeletal prestress. Second, it would be valuable to develop a more quantitative relationship between prestress and the time constant of retraction, with an eye toward creating a “standard curve” that could permit deduction of one parameter from the other. The dose–response performance of LPA may not be sufficiently high to permit this level of control, but other perturbations might. Third, our studies make no attempt to control cell geometry, which could influence both prestress and adhesive area. It would, therefore, be informative to repeat these studies with single cells under constant ECM protein density but variable geometry (e.g., by micropatterning single cells) and ask whether de-adhesion kinetics are affected by spreading area, aspect ratio, and other parameters.
In summary, our data show that the retraction kinetics of adherent cells following trypsin-mediated of detachment are more sensitive to cytoskeletal prestress than to the extent of cell–matrix adhesion. These studies support further exploration of these measurements as a low-resolution, high-throughput approach for screening mechanical properties of adherent cells, and additional effort should help assess the generalizability of these findings across cell types, matrix formulations, and media conditions.
Acknowledgments
WPN was supported by a UC Berkeley Graduate Fellowship. SK gratefully acknowledges the support of the Arnold and Mabel Beckman Young Investigator Award, the NSF (CMMI 0727420), a PECASE award from the Army Research Office (W911NF-09-1-0507), an NIH Physical Sciences Oncology Center Grant (1U54CA143836), and an NIH Director’s New Innovator Award (1DP2OD004213), a part of the NIH Roadmap for Medical Research.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- GBM
Glioblastoma Multiforme
- ECM
Extracellular matrix
- NMMII
Non-muscle myosin II
- pMLC
Phosphorylated myosin light chain
References
- 1.Califano J, Reinhart-King C. Substrate stiffness and cell area predict cellular traction stresses in single cells and cells in contact. Cel. Mol. Bioeng. 2010;3(1):68–75. doi: 10.1007/s12195-010-0102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dekker A, Reitsma K, Beugeling T, Bantjes A, Feijen J, van Aken WG. Adhesion of endothelial cells and adsorption of serum proteins on gas plasma-treated polytetrafluoroethylene. Biomaterials. 1991;12(2):130–138. doi: 10.1016/0142-9612(91)90191-C. [DOI] [PubMed] [Google Scholar]
- 3.Discher D, Dong C, Fredberg JJ, Guilak F, Ingber D, Janmey P, Kamm RD, Schmid-Schonbein GW, Weinbaum S. Biomechanics: cell research and applications for the next decade. Ann. Biomed. Eng. 2009;37(5):847–859. doi: 10.1007/s10439-009-9661-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engel A, Gaub HE. Structure and mechanics of membrane proteins. Annu. Rev. Biochem. 2008;77:127–148. doi: 10.1146/annurev.biochem.77.062706.154450. [DOI] [PubMed] [Google Scholar]
- 5.Engler AJ, Rehfeldt F, Sen S, Discher DE. Microtissue elasticity: measurements by atomic force microscopy and its influence on cell differentiation. Methods Cell Biol. 2007;83:521–545. doi: 10.1016/S0091-679X(07)83022-6. [DOI] [PubMed] [Google Scholar]
- 6.Engler AJ, Humbert PO, Wehrle-Haller B, Weaver VM. Multiscale modeling of form and function. Science. 2009;324(5924):208–212. doi: 10.1126/science.1170107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guck J, Ananthakrishnan R, Mahmood H, Moon TJ, Cunningham CC, Kas J. The optical stretcher: a novel laser tool to micromanipulate cells. Biophys. J. 2001;81(2):767–784. doi: 10.1016/S0006-3495(01)75740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hale, C. M., S. X. Sun, and D. Wirtz. Resolving the role of actoymyosin contractility in cell microrheology. PLoS ONE 4(9):e7054, 2009. [DOI] [PMC free article] [PubMed]
- 9.Hemmer JD, Dean D, Vertegel A, Langan E, III, LaBerge M. Effects of serum deprivation on the mechanical properties of adherent vascular smooth muscle cells. Proc. Inst. Mech. Eng. H. 2008;222(5):761–772. doi: 10.1243/09544119JEIM371. [DOI] [PubMed] [Google Scholar]
- 10.Hochmuth RM. Micropipette aspiration of living cells. J. Biomech. 2000;33(1):15–22. doi: 10.1016/S0021-9290(99)00175-X. [DOI] [PubMed] [Google Scholar]
- 11.Ignotz R, Massagué J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986;261(9):4337–4345. [PubMed] [Google Scholar]
- 12.Lincoln B, Wottawah F, Schinkinger S, Ebert S, Guck J. High-throughput rheological measurements with an optical stretcher. Methods Cell Biol. 2007;83:397–423. doi: 10.1016/S0091-679X(07)83017-2. [DOI] [PubMed] [Google Scholar]
- 13.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer. 2003;3(8):582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 14.Mills JP, Qie L, Dao M, Lim CT, Suresh S. Nonlinear elastic and viscoelastic deformation of the human red blood cell with optical tweezers. Mech. Chem. Biosyst. 2004;1(3):169–180. [PubMed] [Google Scholar]
- 15.Musielak M. Red blood cell-deformability measurement: review of techniques. Clin. Hemorheol. Microcirc. 2009;42(1):47–64. doi: 10.3233/CH-2009-1187. [DOI] [PubMed] [Google Scholar]
- 16.Namura T, Arai S, Okawa K, Koike A, Yamada S, Saita N, Nagae A, Itoh H, Totani M, Uemoto S, Ikemoto M. Identification of serum proteins that bind with S100A8, S100A9 and S100A8/A9: clinical significance of using proteins for monitoring the postoperative condition of liver recipients. Clin. Chim. Acta. 2010;411(21–22):1766–1773. doi: 10.1016/j.cca.2010.07.029. [DOI] [PubMed] [Google Scholar]
- 17.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8(3):241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 18.Pelling AE, Dawson DW, Carreon DM, Christiansen JJ, Shen RR, Teitell MA, Gimzewski JK. Distinct contributions of microtubule subtypes to cell membrane shape and stability. Nanomedicine. 2007;3(1):43–52. doi: 10.1016/j.nano.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 19.Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70(3):389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 20.Rotsch C, Jacobson K, Radmacher M. Dimensional and mechanical dynamics of active and stable edges in motile fibroblasts investigated by using atomic force microscopy. Proc. Natl. Acad. Sci. USA. 1999;96(3):921–926. doi: 10.1073/pnas.96.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sen S, Kumar S. Cell–matrix de-adhesion dynamics reflect contractile mechanics. Cel. Mol. Bioeng. 2009;2(2):218–230. doi: 10.1007/s12195-009-0057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sen S, Kumar S. Combining mechanical and optical approaches to dissect cellular mechanobiology. J. Biomech. 2010;43(1):45–54. doi: 10.1016/j.jbiomech.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sen, S., and S. Kumar. Isoform-specific contributions of α-actinin to glioma cell mechanobiology. PLoS ONE 4(12):e8427, 2009. [DOI] [PMC free article] [PubMed]
- 24.Suresh S. Biomechanics and biophysics of cancer cells. Acta Biomater. 2007;3(4):413–438. doi: 10.1016/j.actbio.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ulrich TA, Jain A, Tanner K, MacKay JL, Kumar S. Probing cellular mechanobiology in three-dimensional culture with collagen-agarose matrices. Biomaterials. 2010;31(7):1875–1884. doi: 10.1016/j.biomaterials.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 26.van Wachem PB, Vreriks CM, Beugeling T, Feijen J, Bantjes A, Detmers JP, van Aken WG. The influence of protein adsorption on interactions of cultured human endothelial cells with polymers. J. Biomed. Mater. Res. 1987;21(6):701–718. doi: 10.1002/jbm.820210603. [DOI] [PubMed] [Google Scholar]
- 27.Vogel V, Sheetz MP. Cell fate regulation by coupling mechanical cycles to biochemical signaling pathways. Curr. Opin. Cell Biol. 2009;21(1):38–46. doi: 10.1016/j.ceb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang JH, Lin JS. Cell traction force and measurement methods. Biomech. Model. Mechanobiol. 2007;6(6):361–371. doi: 10.1007/s10237-006-0068-4. [DOI] [PubMed] [Google Scholar]
- 29.Wang N, Tolic-Norrelykke IM, Chen J, Mijailovich SM, Butler JP, Fredberg JJ, Stamenovic D. Cell prestress. I. Stiffness and prestress are closely associated in adherent contractile cells. Am. J. Physiol. Cell Physiol. 2002;282(3):C606–C616. doi: 10.1152/ajpcell.00269.2001. [DOI] [PubMed] [Google Scholar]
- 30.Wildt B, Wirtz D, Searson PC. Programmed subcellular release for studying the dynamics of cell detachment. Nat. Methods. 2009;6(3):211–213. doi: 10.1038/nmeth.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wirtz D. Particle-tracking microrheology of living cells: principles and applications. Annu. Rev. Biophys. 2009;38:301–326. doi: 10.1146/annurev.biophys.050708.133724. [DOI] [PubMed] [Google Scholar]
- 32.Yao A, Tassieri M, Padgett M, Cooper J. Microrheology with optical tweezers. Lab Chip. 2009;9(17):2568–2575. doi: 10.1039/b907992k. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Liu KK. Optical tweezers for single cells. J. R. Soc. Interface. 2008;5(24):671–690. doi: 10.1098/rsif.2008.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]