

Abstract
We previously found that the pro-apoptotic DNA damaging agent, cisplatin, mediated the proteasome-dependent degradation of ΔNp63α associated with its increased phosphorylated status. Since ΔNp63α usually plays an opposite role to p53 and TAp63 in human cancers, we tested the notion that phosphorylation events induced by DNA damage would affect the protein degradation of ΔNp63α in HNSCC cells upon cisplatin exposure. We found that ΔNp63α is phosphorylated in the time-dependent fashion at the following positions: S385, T397 and S466, which were surrounded by recognition motifs for ATM, CDK2 and p70s6K kinases, respectively. We showed that chemical agents or siRNA inhibiting the activity of ATM, CDK2 and p70s6K kinases blocked degradation of ΔNp63α in HNSCC cells after cisplatin exposure. Site-specific mutagenesis of ΔNp63α residues targeted for phosphorylation by ATM, CDK2 or p70s6k led to dramatic modulation of ΔNp63α degradation. Finally, we demonstrated that the ΔNp63α protein is a target for direct in vitro phosphorylation by ATM, CDK2 or p70s6K. Our results implicate specific kinases, and target phosphorylation sites in the degradation of ΔNp63α following DNA damage.
Keywords: DNA damage, cisplatin, p53, squamous, stress, kinases, phosphorylation
Introduction
Although p63 displays a similar modular structure and extensive homology to that of p53, it encodes at least six different protein isotypes resulting from alternative splicing.64,76 P63 isotypes consist of two groups, including those with the transactivation (TA)-domain at the amino terminus (TA-) or those without it (ΔN-). Similarly to p53, TAp63 isotypes bind specific DNA sequences activating transcription from p53-responsive genes that induce cell cycle arrest or apoptosis.13,22,23,26,27,38,45,46,53,67,73 ΔNp63α is the most abundant p63 isotype in proliferating basal layers of many epithelial tissues and is highly expressed in various human cancers (e.g., head and neck squamous cell carcinomas, HNSCC), supporting its role in cell proliferation and neoplastic development.42,61,74,76,79,82,83 Since ΔNp63 proteins retain the core DNA-binding domain, simple competition for DNA sites might prevent p53 or TAp63 from binding target gene promoters.42,71,74,76 ΔNp63α was also shown to play a more proactive role as a transcriptional regulator by activating or downregulating several critical downstream gene targets.42,61,74 Moreover, ΔNp63α was found to induce accumulation and signaling of β-catenin supporting an oncogenic role for ΔNp63 in HNSCC cells.61
Previous studies showed that genotoxic stress agents including ultraviolet (UV)-irradiation, actinomycin D, bleomycin and etoposide led to accumulation of TAp63α and TAp63gamma proteins, and TAp63 proteins were degraded via a proteasome complex under normal cellular circumstances.40,56 In addition, TAp63 was shown to activate p53-responsive genes involved in regulation of cell cycle arrest and apoptosis in response to UV-irradiation, or actinomycin D.40,56 However, the effect of genotoxic stress on ΔNp63 isotypes in most cancers, including head and neck cancers has not been well studied. A few reports showed that UV-irradiation decreased levels of ΔNp63 in normal keratinocyte cultures.46,79,80
Apoptosis is a common feature of the cytotoxicity caused by DNA damaging anti-cancer agents.1,2,4,7,18,29,34,43,58,59,63,70 The overexpression of ΔNp63α was shown to an increase in cell proliferation, and enhanced tumor growth in vitro and in vivo and also to block UV-induced p53-dependent apoptosis in vivo.42,46 However, siRNA silencing of ΔNp63α inhibited proliferation and mediated the programmed cell death of HNSCC cells supporting the notion that ΔNp63α functions as an anti-apoptotic factor.13,45,67
We previously found that cisplatin dramatically decreased the survival of HNSCC cells and also led to a proteasome-dependent degradation of ΔNp63α25,37,85 We further found that cisplatin exposure decreased protein levels of ΔNp63α in HNSCC cells, while increasing overall phosphorylation levels of ΔNp63α based on in vivo cell labeling and treatment with a serine/threonine kinase inhibitor.25,37 This degradation of ΔNp63α was also enhanced by the physical interaction of ΔNp63α and stratifin, which preferentially binds to phosphorylated protein targets, such as ΔNp63α25,80
It is well known that p53 is phosphorylated upon DNA damage at the multiple sites, in both the N-termini and C-termini.1,2,18,72 Although stress-related phosphorylation of the p53 tumor suppressor at the N-terminus leads to its stabilization, there are a few reports showing that p53 phosphorylation occurring at the C-terminus might lead to its subsequent degradation.14 However, little is known about p63 phosphorylation events, especially after genotoxic stress. Since ΔNp63α usually plays an opposite role in human cancers to p53 and TAp63, we hypothesized that this protein might react differently to DNA damage. In this scenario, the cisplatin-induced degradation of ΔNp63α in HNSCC cells might occur through its phosphorylation rather than dephosphorylation. Earlier reports suggested that p63 is phosphorylated under unstressed conditions and after exposure of epithelial cells to cisplatin or UV-irradiation.25,37,79,80 Therefore, we examined the type and consequences of phosphorylation events induced by DNA damage on ΔNp63α levels in HNSCC cells after cisplatin exposure.
Results
To examine whether ΔNp63α, the predominant p63 isotype in HNSCC cells (reviewed in refs. 42, 60, 61 and 65) is modified after exposure to cisplatin and to track down changes in the posttranslational modification status of ΔNp63α, we employed a combination of immunoprecipitation with Ab-1 antibody against ΔNp63 and subsequent 2D-gel separation of target proteins followed by mass spectrometry analysis.25,37
We previously found that endogenous ΔNp63α produced multiple modified p63 proteins in HaCaT cells shown by subsequent immunoprecipitation (with the Ab-1 antibody) and 2D-gel isolectrofocusing/SDS-electrophoresis.37 This observation suggested that ΔNp63α is phosphorylated and degraded in HNSCC cell lines exposed to genotoxic stress agents (cisplatin, UV) supporting our previous findings and those of other groups.25,37,79 ,80 The initial bioinformatics searches performed by us (www.cbs.dtu.dk/services/NetPhosK/, see, Table 1) and others (reviewed in refs. 20, 21, 31, 41 and 57) suggested that the ΔNp63α protein might be phosphorylated at the multiple motifs by a variety of protein kinases (e.g., ATM, CDK, p70s6K, MAPK and protein C kinase).
Table 1.
Predicted phosphorylation sites in ΔNp63α
| (A) Potentially phosphorylated serine residues | |||
|---|---|---|---|
| Position | Sequence | Score | Predictions |
| 36 | QNGSSSTSP | 0.928 | *S* |
| 53 | VTAPSPYAQ | 0.994 | *S* |
| 60 | AQPSSTFDA | 0.958 | *S* |
| 73 | PAIPSNTDY | 0.911 | *S* |
| 95 | STAKSATWT | 0.981 | *S* |
| 156 | NHELSREFN | 0.987 | *S* |
| 191 | TGRQSVLVP | 0.878 | *S* |
| 264 | ADEDSIRKQ | 0.920 | *S* |
| 273 | QVSDSTKNG | 0.997 | *S* |
| 295 | IQMTSIKKR | 0.826 | *S* |
| 301 | KKRRSPDDE | 0.998 | *S* |
| 364 | QSPSSYGNS | 0.996 | *S* |
| 369 | YGNSSPPLN | 0.800 | *S* |
| 385 | NKLPSVSQL | 0.866 | *S* |
| 466 | LGCSSCLDY | 0.925 | *S* |
| 492 | DDLASLKIP | 0.871 | *S* |
| 533 | ASTVSVGSS | 0.961 | *S* |
| 536 | VSVGSSETR | 0.865 | *S* |
| 537 | SVGSSETRG | 0.979 | *S* |
| (B) Potentially phosphorylated threonine residues | |||
|---|---|---|---|
| Position | Sequence | Score | Prediction |
| 187 | VEDPITGRQ | 0.987 | *T* |
| 207 | EFTTYLYNF | 0.977 | *T* |
| 316 | RGRETYEML | 0.862 | *T* |
| 397 | RNALTPTTI | 0.927 | *T* |
| 428 | GLSPTQALP | 0.913 | *T* |
| 551 | AVRFTLRQT | 0.952 | *T* |
| (C) Potentially phosphorylated tyrosine residues | |||
|---|---|---|---|
| Position | Sequence | Score | Prediction |
| 17 | SEPQYTNLG | 0.993 | *Y* |
| 77 | SNTDYPGPH | 0.935 | *Y* |
| 100 | ATWTYSTEL | 0.977 | *Y* |
| 181 | SHAQYVEDP | 0.850 | *Y* |
| 365 | SPSSYGNSS | 0.872 | *Y* |
To further pursue these observations, we used HNSCC 028 cells (108) infected with Ad5-ΔNp63α-myc for 18 h and then treated with 10 μM cisplatin or control medium for an additional 18 h. ΔNp63α-myc was then immunoprecipitated overnight from cell lysates (500–2000 μg) using the ProFound Mammalian C-myc Tag IP/Co-IP kit and purified by fast-performance liquid chromatography. The purified ΔNp63α-myc was in-gel digested with chymotrypsin, and the peptide mixture was subjected directly to peptide profiling by MALDI-TOF-MS. Among a few phosphorylation sites in ΔNp63α that were affected by cisplatin exposure (Table 2), we found ATM, CDK2 and p70s6K kinase recognition motifs [S385, ATM motif, PSVSQL), T397, CDK2 motif, NALTPTT), and S466, p70s6K, GCSSCLD].
Table 2.
ΔNp63α phosphorylation sites identified by mass spectrometry
| Observed proteolytic | Residue | Kinase | Mascot | Xcorr |
|---|---|---|---|---|
| phosphopeptide1 | Score2 | Value3 | ||
| Chymotrypsin: VSQLINPQQRNAL (384–396) |
S385 | ATM | 41 | 2.21 |
| Chymotrypsin: LTPTTIPDGM (396–405) |
T397 | CDK2 | 43 | 2.37 |
| Plus Proteinase K (396 397 399 400 401) | ||||
| Chymotrypsin: LGCSSCL (462–468) |
S466 | p70s6K | 44 | 2.53 |
| Plus NTCB: GCS (463–466) | S466 | p70s6K | 49 | 3.23 |
To examine protein levels and phosphorylation levels of ΔNp63α possibly affected by cisplatin exposure, we used HNSCC 028 cells (without endogenous p63 expression) ectopically expressing Ad5-ΔNp63α-myc, HaCaT immortalized keratinocytes (with endogenous p63 expression) and Flip-In HaCaT stable clones that expressed wild type or mutated ΔNp63α (L514F mutation derived from Hay-Wells Syndrome), introduced into a genomic DNA (reviewed in ref. 37).
Cells were treated with 10 μM cisplatin or control media for 24 h. First, we examined levels of ΔNp63α using immunoblotting with a custom antibody generated against the phosphorylated S385 (NKLPSVSQLINPQQ, residues 379–392, designated as the anti-pATM motif). We found evidence of an increased ΔNp63α phosphorylation at the ATM motif upon exposure of HNSCC cells to cisplatin (Fig. 1A and B).
Figure 1.
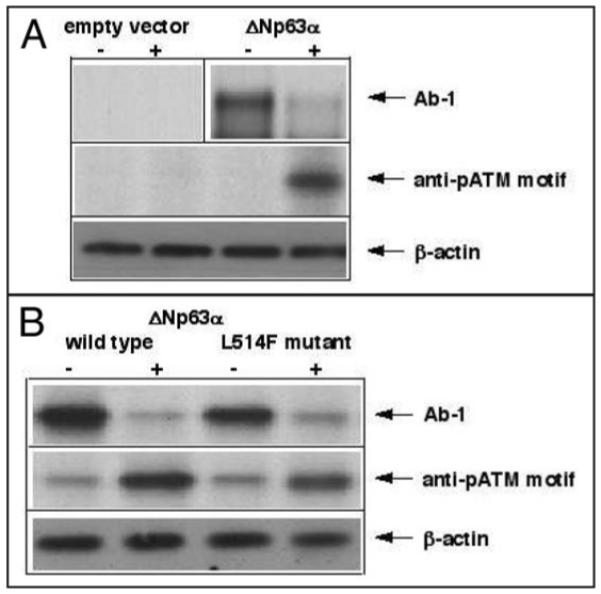
Cisplatin increased phosphorylation levels and decreased protein levels of ΔNp63α. (A). HNSCC 028 cells (105) were infected with an empty vector Ad5 or Ad5-ΔNp63α myc for 18 h and then treated with mock medium (−) or 10 μg/ml cisplatin (+) for an additional 18 h. The vertical line in the upper panel divides two independent gel experiments. (B). Flp-In HaCaT cells (105) expressing wild type or L514F mutant ΔNp63 were treated with mock medium (−) or 10 μg/ml cisplatin (+) for 18 h. Immunoblotting was performed with the indicated antibodies. Protein levels were normalized with antibody to β-actin.
We further used HNSCC 029 cells endogenously expressing an abundant amount of ΔNp63α (reviewed in ref. 32) and treated the cells with 10 μM cisplatin for 0, 12 and 24 h. Initially, we tested the effect of cisplatin exposure on cell survival using the MTT assay, as previously described.32 Using both immunoblotting and combined immunoprecipitation/in vitro protein kinase assay, we also tested whether the protein and activity levels of ATM, CDK2 or p70s6K were affected upon exposure of HNSCC 029 cells to cisplatin. We observed that cisplatin exposure decreased the survival of HNSCC 029 cells (by 2-fold in 0–48 h time frame), associated with clear induction of protein levels, and most importantly enzymatic activity, of all the kinases tested (data not shown). To evaluate the effect of cisplatin on phosphorylation levels of ΔNp63α, we employed immunoblotting of total lysates obtained from HNSCC 029 cells with custom antibodies against a phosphorylated ATM motif, CDK2 motif or p70s6K motif in the ΔNp63α polypeptide. We found that phosphorylated protein levels at the indicated positions/motifs dramatically increased upon exposure of HNSCC 029 cells to cisplatin in a time-dependent fashion relative to total ΔNp63α protein levels (Fig. 2).
Figure 2.
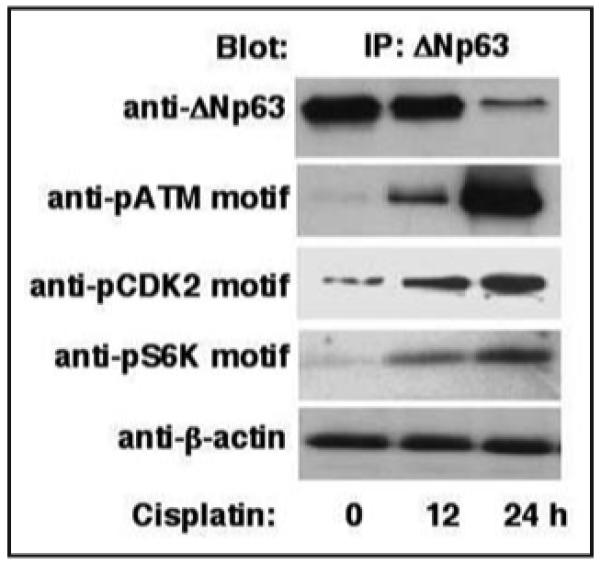
Custom antibodies to phosphorylated motifs of ATM (S385), CDK2 (T397) and p70s6K (S466) kinase recognize site-specific phosphorylation of ΔNp63α in HNSCC 029 cells upon cisplatin exposure. HNSCC 029 cells (105) were treated with 10 μg/ml cisplatin for the indicated time periods in the presence of 20 μM MG-132 (26S proteasome inhibitor). Total lysates resolved by SDS-PAGE were immunoblotted with the indicated antibodies. Protein levels were normalized with antibody to β-actin.
Next, we grew HNSCC 029 cells in the presence or absence of 10 μM cisplatin for 0, 6, 12 or 24 h and cell-permeable inhibitors for ATM (5 μM KU-55933, reviewed in refs. 34, 39, 44 and 63), CDK2 (20 μM roscovitine, reviewed in refs. 14, 17, 18, 24, 52 and 81) or p70s6K (20 nM rapamycin, reviewed in refs. 16 and 30) kinases (added for the last 60 min before cell harvesting). We first showed that these protein kinase inhibitors dramatically blocked the enzymatic activities of ATM, CDK2 and p70s6K, while increasing the cell survival compared to the inhibitory effect of cisplatin exposure (data not shown). Then, the protein levels for ΔNp63α were tested by immunoblotting with Ab-1 antibody. We observed that all protein kinase inhibitors used in this set of experiments clearly blocked degradation of ΔNp63α in HNSCC cells induced by cisplatin exposure (Fig. 3).
Figure 3.
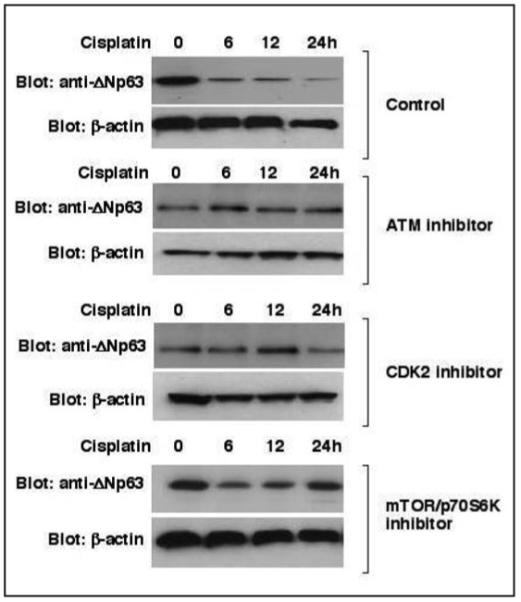
Inhibitors of protein kinases blocked the protein degradation of ΔNp63α in HNSCC 029 cells. HNSCC 029 cells (105) were treated with 10 μg/ml cisplatin for the indicated time periods in the presence or absence (control, DMSO) of inhibitors of ATM (5 μM KU-55933), Cdk2 (20 μM roscovitine) or mTOR/p70s6K (20 nM rapamycin), which were added for the last 60 min before cell harvesting. Total lysates resolved by SDS-PAGE were immunoblotted with Ab-1 antibody against ΔNp63. Protein levels were normalized with antibody to β-actin.
We then transiently introduced siRNA against ATM, CDK2 or p70s6K (300 nM each) into HNSCC 029 cells versus scrambled siRNA to examine their effectiveness in silencing expression of their respective target (Fig. 4). We observed that siRNA dramatically silenced the expression of the protein kinases tested (Fig. 4A), and in turn decreased the cisplatin-induced degradation of ΔNp63α in HNSCC 029 cells, while scrambled siRNA failed to do so (Fig. 4B).
Figure 4.
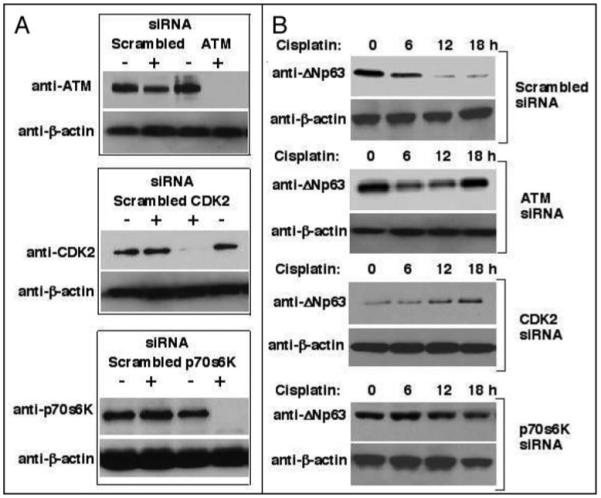
Small interference RNA (siRNA) to protein kinases inhibited the protein degradation of ΔNp63α in HNSCC 029 cells. (A). In contrast to a negative control (scrambled siRNA), siRNA against ATM, CDK2 or S6K dramatically inhibited the protein levels of ATM, CDK2 or p70S6K in 029 cells (105, 48 h post-transfection, 300 nM of siRNA), respectively, as shown by immunoblotting with the indicated antibodies. (B). HNSCC 029 cells were transiently transfected with siRNA for 48 h and then subsequently treated with 10 μg/ml cisplatin for indicated additional time periods. SiRNA against ATM, CDK2 or p70S6K dramatically inhibited the protein degradation of ΔNp63α upon cisplatin exposure. Protein levels for ΔNp63α were analyzed by immunoblotting with Ab-1 antibody, and normalized with antibody to β-actin.
We further examined whether genetic changes in putative phosphorylation sites in the ΔNp63α protein expressed in HNSCC 029 cells would affect its degradation upon cisplatin exposure. We established HNSCC 029 stable cell lines expressing the wild type ΔNp63α or mutated ΔNp63α (ATM phosphorylation site, CDK2 phosphorylation site and p70s6K phosphorylation site) using the Flp-In technology, as previously described.37 This approach allowed us to introduce p63 mutations (S385, T397 and S466) into the genomic DNA of target 029 cells resembling a knock-in technique. The cells harboring p63 mutations exclusively expressed RNA transcripts with desired mutations, as confirmed by sequencing of individual clones derived from RT-PCR of mRNA obtained from mutated cells (data not shown). Interestingly, cells expressing mutated ΔNp63α grew and proliferated at a similar rate to cells expressing the wild type ΔNp63α, while protein and activity levels for ATM, CDK2 or p70s6K were the same (data not shown). These cells were then treated with 10 mM cisplatin and protein levels of ΔNp63α were examined by immunoblotting with Ab-1 antibody against ΔNp63. We observed that the absence of serine or threonine residues necessary for kinase phosphorylation [due to mutations in Ser (S385 or S466), or Thr (T397 for Ala] completely blocked the cisplatin-induced degradation of ΔNp63α suggesting a critical role of all three of these residues in the phosphorylation-mediated degradation of ΔNp63α in HNSCC cells (Fig. 5).
Figure 5.
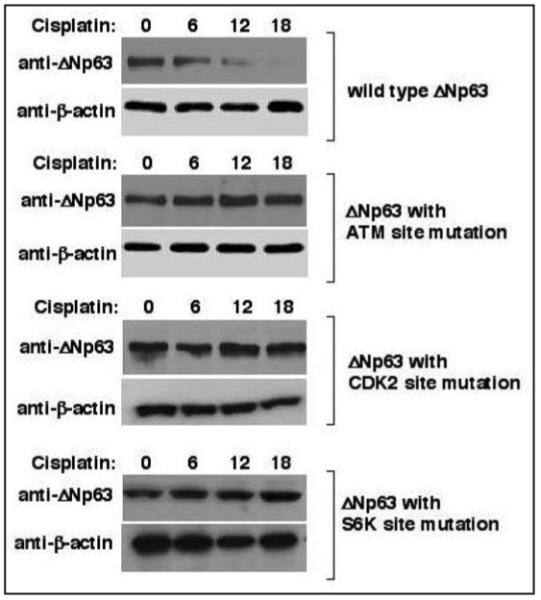
Mutations of serine (S/A-385, S/A-466) or threonine (T/A-397) in the ΔNp63α protein kinase motifs prevented the protein degradation of ΔNp63α in HNSCC 029 cells. Stable clones of HNSCC 029 cells (105) harboring plasmids with mutations in ΔNp63α for the putative phosphorylation motifs for ATM, CDK2 or p70s6K protein kinases were generated. HNSCC 029 cells were then treated with 10 μg/ml cisplatin for the indicated time periods. Protein levels for ΔNp63α were analyzed by immunoblotting with Ab-1 antibody, and normalized with antibody to β-actin.
Next, we further explored whether the cisplatin exposure of HNSCC cells induced a possible sequence of events leading to phosphorylation of ΔNp63α. Stable clones of HNSCC 029 cells harboring plasmids with wild type ΔNp63α or ΔNp63α mutated in the putative phosphorylation motifs (ATM, CDK2 or p70s6K) were grown up in the presence of 10 μg/ml cisplatin for 0, 6, 12, 18, 24 h (Fig. 6). Levels for phospho-ΔNp63α were analyzed by immunoblotting with the indicated custom antibodies against the pATM, pCDK2 or pp70s6k motifs, respectively (Fig. 6). We found that in cells expressing the wild type ΔNp63α cisplatin-inducedATM-mediated phosphorylation of ΔNp63α started to reach maximal levels at ~6 h, while CDK2 phosphorylation occurred at 12 h and p70s6K phosphorylation at 18 h (Fig. 6A). We further observed that when testing cells with ATM-mutated phosphorylation site, neither of our custom antibodies recognized the phospho-ΔNp63α protein (Fig. 6B). Moreover, the antibody to the pATM-motif (in cells harboring mutated CDK2 site) or antibodies to pATM-motif or pCDK2 motif (in cells harboring mutated p70s6K site) recognized the phospho-ΔNp63α protein (Fig. 6C and D). Therefore, in HNSCC cells treated with cisplatin, the ΔNp63α protein is phosphorylated first by ATM, then by CDK2 and finally by p70s6K kinases.
Figure 6.
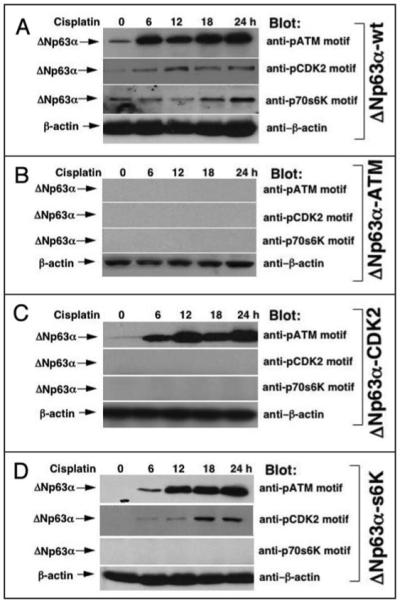
Time-course of the ΔNp63α phosphorylation by protein kinases in HNSCC cells upon cisplatin exposure. Stable clones of HNSCC 029 cells (105) harboring the wild type ΔNp63α (A. ΔNp63α-wt) or mutations in the certain ΔNp63α putative phosphorylation motifs (B, ΔNp63α-ATM), (C, ΔNp63α-CDK2) or (D, ΔNp63α-s6K) were grown-up in the presence of 10 μg/ml cisplatin for the indicated time periods (0, 6, 12, 18, 24 h). Protein levels for phospho-ΔNp63α were analyzed by immunoblotting with indicated custom antibodies against pATM motif, pCDK2 motif or p70s6k motif, and normalized with antibody to β-actin.
Finally, we tested the ability of purified protein kinases (ATM, CDK2 or p70s6K) to phosphorylate the purified ΔNp63α protein in vitro.11,25 We showed that all tested kinases phosphorylated ΔNp63α in vitro suggesting that ΔNp63α is a direct target of these protein kinases (Fig. 7).
Figure 7.
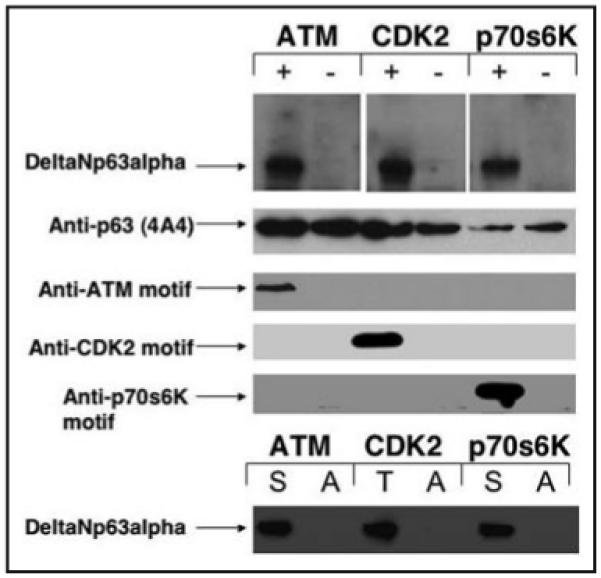
In vitro phosphorylation of ΔNp63α by ATM, CDK2 or p70s6K. Kinase reactions contained 25 mM Tris-HCl, pH 8, 50 mM KCl, 5% glycerol, 0.5 mM DTT, 5 μCi of [gamma32P]-ATP, 10 μM cold ATP, 10 mM MnCl2, 0.25–0.5 μg of PHAS-I (Stratagene) 2 ng of purified ATM (CDK2 or p70s6K or nothing) and 2 ng of ΔNp63α. ΔNp63α was precipitated with Ab-1 antibody, analyzed by 10% SDS-PAGE, and subjected to autoradiography. Blots were immunoblotted with 4A4 antibody to p63, with custom antibodies against ATM motif, CDK2 motif or p70s6K motif. The cDNA for ΔNp63α was mutated at the following positions [S-385 (S-A), T-397 (T-A), or S-466 (S-A)] for alanine residue (A) using the QuickStep site-directed mutagenesis assay. In vitro translated unlabeled products representing wild type (S or T) or mutated (A) ΔNp63α polypeptides were synthesized using the Promega TNT assay as previously described (reviewed in refs. 25, 37 and 61), and mixed with 1 ng of purified ATM, CDK2 or p70s6K for kinase reaction as described above. Resulting mixes were precipitated with Ab-1 antibody, analyzed by 10% SDS-PAGE, and subjected to autoradiography.
Discussion
We previously found that genotoxic stress agents (e.g., cisplatin, doxorubicin, etc.,) induced phosphorylation of ΔNp63α leading to a dramatic decrease of the ΔNp63α protein levels in HNSCC cells.25,85 By using MALDI-TOF-MS, we have identified phosphorylated sites in ΔNp63α in HNSCC cells upon cisplatin exposure. They included the S385, T397 and S466 sites surrounded by recognition motifs for the ATM, CDK2 and p70s6K kinases, respectively. We also showed that the cisplatin-exposed HNSCC cells harbored phosphorylated ΔNp63α at the ATM, CDK2 and p70s6K kinase recognition sites in a time-dependent fashion. We further showed that chemicals or siRNA inhibiting the activity of ATM, CDK2 and p70s6K kinases blocked degradation of ΔNp63α in HNSCC cells usually induced upon cisplatin exposure. We also found that engineered genetic changes in ΔNp63α that replace specific residues targeted for phosphorylation by ATM, CDK2 or p70s6k led to a dramatic modulation of ΔNp63α degradation. We also showed that the ΔNp63α protein in HNSCC cells exposed to cisplatin is probably phosphorylated first by ATM, then by CDK2 and finally by p70s6K kinases ultimately targeting ΔNp63α to a proteasome-dependent degradation pathway.12,25 Finally, we demonstrated that the ΔNp63α protein is a target for direct in vitro phosphorylation by ATM, CDK2 or p70s6K. Overall, we suggest that ATM kinase is a master switch for the ΔNp63α phosphorylation/degradation in human HNSCC cells upon DNA damage.
Accumulating evidence points to a role for multiple posttranslational modifications (e.g., phosphorylation) in response of cells to DNA damage induced by various extracellular stimuli mediating these events.1,2,6,7,20,21,27,43,50,72 Recent studies suggest that N-terminal phosphorylations are important for stabilizing p53 in cells exposed to genotoxic stresses.6 However, the modifications to the C-terminus of p53 inhibit its ability to negatively regulate sequence-specific DNA-binding, and also modulate protein stability, the oligomerization state, the nuclear import/export process, and the degree of p53 ubiquitination.14
From other hand, ΔNp63 isotypes were shown to manifest cellular response to DNA damage in an opposite fashion via protein degradation, because they lack the N-terminal region of TA-isotypes found in all p53 family members.25 Keeping this in mind, we now have evidence that cisplatin-induced degradation of ΔNp63α in HNSCC cells occurs after its phosphorylation at specific sites. Since ΔNp63α usually plays an opposite role in human cancers compared to p53 and TAp63, we now add additional molecular evidence that this protein reacts differently to cisplatin ands possibly to other DNA damaging agents.
Although both exogenous ΔNp63α protein expressed in transfected cells under unstressed conditions (reviewed in ref. 79) and endogenous ΔNp63α expressed in human keratinocytes and HNSCC cells under stressed conditions25,37,80 were reported to be phosphorylated, a comprehensive and direct analysis of the p63 sites phosphorylated by specific kinases was not previously performed. The relationship between p63 and key protein kinases is mutually intertwined. A few protein kinases were found to be involved in the upstream regulation of p63 transcription and several other protein kinases were shown to be p63 downstream transcription targets.5,48,54,64,78,82
Westfall and coworkers have shown that certain amino acid residues are phosphorylated in ΔNp63α in human keratinocytes upon UV-irradiation.79,80 All the modified species of ΔNp63α (12—in UV-treated cells, and 7—in untreated cells) were reduced to one by treatment of ΔNp63α precipitates with calf intestine phosphatase supporting the notion that ΔNp63α is indeed a phosphoprotein.79,80 Using antibodies to certain phosphorylated residues of ΔNp63α, Westfall and coworkers have also found an increase in phosphorylation of S66/S68 and S361 in ΔNp63α upon paclitaxel or UV exposure.80 The latter was also shown to induce the rapid phosphorylation of ΔNp63α by the p38 stress MAPK leading to inhibition of ΔNp63α transcriptional activity and its degradation.35,59 Phosphorylated ΔNp63 displayed a decreased ability to bind certain cell cycle arrest and apoptotic promoters, thus allowing rapid activation of the p53-dependent transcriptional apoptotic program.59 Interestingly, the Abl inhibitor, Gleevec was shown to reduce TAp63 expression in a dose-dependent manner in HNSCC cells thereby overriding its induction by DNA damaging agents.58,77
Since p63 proteins share a strong homology with p53, it is likely that many protein kinases implicated in phosphorylation of p53 might play a similar role in p63 (reviewed in refs. 20 and 21). Using a bioinformatics approach (GPS kinase prediction module), Finlan and Hupp uncovered several putative protein kinase motifs (Abl, PKB, ATM, S6K, CDK, MAPK and EGFR) for potential TAp63α phosphorylation (Fig. 8A and reviewed in ref. 21). A fewer motifs were predicted in the protein sequence of ΔNp63α that lacks the N-terminal domain (Fig. 8B).
Figure 8.
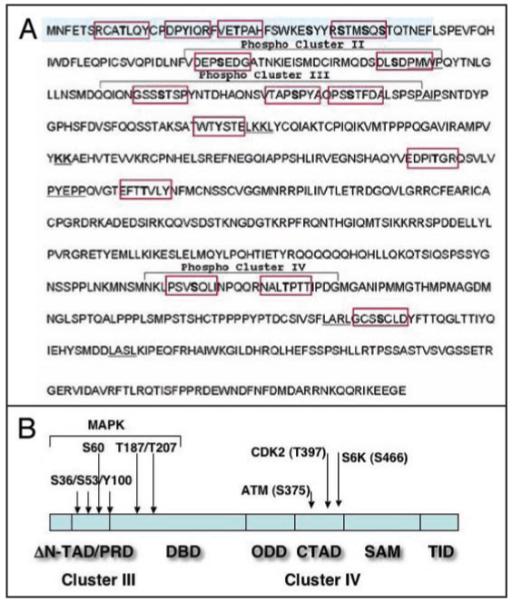
Schematic representation of phosphorylation sites in ΔNp63α. (A) “The sequence of the TAp63α protein with highlighted motifs depicting potential phosphorylation sites within TAp63α. Four clusters for potential kinases have been identified using the GPS phosphorylation prediction module. Of note, cluster I and II reside within the N-terminal transactivation domain (TAp63 isoforms, and devoid in ΔNp63 isoforms).” ((A) is courtesy of Lee Finlan and Ted Hupp. P63. The Phantom of the Tumor Suppressor. Cell Cycle 2007; 6:1062–71. Permission has been received from Dr. Ted Hupp and Cell Cycle Editorial. Copyright of Landes Biosciences Press). (B) Modular structure of ΔNp63α (reviewed in ref. 64) with putative phosphorylation sites predicted for TAp63α (reviewed in ref. 21) and adjusted according to differences between TAp63α and ΔNp63α. ΔN-transactivation domain ΔN-TAD, residues 1–26), proline-rich domain (PRD, residues 37–97), DNA-binding domain (DBD, residues 98–322), oligomerization domain (ODD, residues 323–357), C-terminal TAD (CTAD, residues 380–470), sterile α-motif (SAM, residues 472–537), and transcription inhibitory domain (TID, last 71 residue). The arrows indicate the newly identified phosphorylation sites for ATM (S385), CDK2 (T397) and p70s6K (S466) kinases found in p63 (the current study) and also reported by Westfall et al.80
Multiple signaling pathways exist to stabilize p53 in response to different forms of stress through phosphorylation of p53 by a variety of protein kinases.1,6,20,50 They include the following kinases serving as DNA damage checkpoints and sensor proteins: ATM, ATR, AURKA, DNA-PK, CHK1 and 2, CDK2 and 5, HIPK2, PKC, PKR, FACT-CK2, ERK, p38 stress MAPK, JNK, GSK3β, etc.1,6 It is not surprising that at least two of these kinases (ATM and CDK2) shown in the current study play a role in modification of ΔNp63α after DNA damage induced by cisplatin exposure.
Mammalian cells respond to DNA damage by activating signal transduction pathways that arrest cell cycle progression and initiate DNA repair.1,27,29,50,55 A key regulator of the cell response to DNA damage is the ATM protein kinase, whose activation/homodimerization leads to association of ATM with its protein targets and their phosphorylation with subsequent effects on cell cycle checkpoints, apoptosis and DNA repair.1,43,50,55 Abnormal regulation of progression from G1 to S phase of the cell cycle by altered activity of CDKs was shown to be a hallmark of cancer.18,55,87 There is also evidence implicating the mTOR/p70s6K signaling pathway in regulating cell proliferation and the resistance of cancer cells to cisplatin.47,77,84
Our study shows that the above-mentioned protein kinases phosphorylate ΔNp63α in HNSCC cells upon cisplatin exposure. Additional studies are necessary to perform a comprehensive mapping of phosphorylation events and protein kinases involved in the regulation of p63 in a variety of cell types. Moreover, it is still unclear how phosphorylation and/or dephosphorylation events affect the transcriptional function of p63. As in the case of p53 posttranslational modifications, these modifications are likely to play a critical role in regulating p63 function as a transcriptional regulator of tumorigenesis, cell proliferation/differentiation or DNA replication stress.28,55 Since ΔNp63 isotypes play an opposite role towards p53, we suggest that the long-term DNA damage induces a dramatic decrease of the ΔNp63α protein levels decrease leading to an increase in the ability of the wild type p53 to function as tumor suppressor, thereby modulating cell proliferation (by cell cycle arrest or apoptosis) and directing cells to senesce.85
Experimental Procedures
Cells, reagents and antibodies
Head and neck squamous cell carcinoma (HNSCC) cell line 029 (expressing wild type p53 and p63), and 028 (no p63 expression, while expressing wild type p53) were initially isolated from primary tissues and maintained at the Department of Otolaryngology/Head and Neck Surgery of the Johns Hopkins University School of Medicine.32 Cells were maintained in RPMI medium 1640, 10% fetal bovine serum (FBS). Cells were incubated with cisplatin (10 μg/ml, Sigma) and a 26S proteasome inhibitor (MG-132, 20 μM, American Peptide Company). We also treated cells for 60 min with the cell-permeable inhibitors for ATM kinase, 5 μM KU-55933 (2-Morpholin-4-yl-6-thianthren-1-yl-pyran-4-one (#118500, EMD Chemicals Inc.,), for CDK2, 20 μM roscovitine 2-(R)-(1-ethyl-2-hydroxyethylamino)-6-benzylamino-9-isopropylpurine (EMD Chemicals), and for mTOR/p70s6K kinase, 20 nM rapamycin (EMD Chemicals).
We used a rabbit polyclonal antibody Ab-1 directed against human ΔNp63 (EMD Chemicals), a mouse monoclonal antibody against all human p63 isotypes (4A4, Santa Cruz Biotechnology), a monoclonal antibody against human β-actin (Sigma), a mouse anti-human Cdk1/Cdk2 monoclonal antibody (clone AN21.2, ab6434, Abcam) and a rabbit anti-human s6K polyclonal antibody (ab36864, Abcam), a mouse anti-human ATM monoclonal antibody (clone 10H11.E12, ab36810, Abcam), a mouse anti-human p70s6k monoclonal antibody (clone 16, #611260, BD Biosciences/Pharmingen). Custom rabbit polyclonal antibodies against phosphorylated peptides encompassing the ΔNp63α protein sequences (ATM motif, NKLPSV-pS-QLINPQQ, residues 379-392; CDK2 motif, QQRNAL-pT-PTTIPDG, residues 391–404; p70s6K motif, LARLGC-pS-CLDYFT, residues 459–472) were prepared and purified against the phosphorylated peptide vs. non-phosphorylated peptide with the aid of Sigma Genosys. Ad-ΔNp63α-myc was prepared in our laboratory, as previously described.61
The following recombinant proteins: ATM (#HZ-2029-10), Cdk2 (HZ-2018-10), p70s6K (HZ-2053-10) were purchased from Humanzyme, Inc., Recombinant GST-ΔNp63α was purified, as previously described.25 About ~1.0–2.0 μg of fusion protein was purified from 3–4 g of bacterial cells. To purify intact ΔNp63α, GST-taq was cleaved by thrombin and separated by second round of glutathione-agarose column chromatography (Sigma) according to the manufacturer’s protocol.
Immunoblotting and immunoprecipitation
Cells were lysed in buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 0.5% Brij-50, 1 mM PMSF, 0.5 mM NaF, 0.1 mM Na3VO4, 2X complete protease inhibitor cocktail), sonicated five times for 10 sec time intervals, and clarified for 30 min at 15,000 x g.61,65 Supernatants (designated as total lysates) were resolved by 4–10% SDS-PAGE and then analyzed by immunoblotting or immunoprecipitation, as previously described.61
Generation of mutated p63 stable clones
The stable HNSCC 029 clones expressing wild type and mutated ΔNp63α were generated using the Flp-In technology (Invitrogen), as previously described.37 Flp-In HNSCC 029 host cells were generated by stable insertion of pFRT/lacZeo plasmid and selected on Zeocin.37 To establish the isogeneic HNSCC 029 clones expressing wild type and mutant ΔNp63α, we subcloned the wild type or mutated cDNA for ΔNp63α (prepared by site-directed mutagenesis using the Quick-Step mutagenesis kit (Stratagene) with the following primers:
ΔNp63S385G_F (a1153g):
5′-CAAGCTGCCTTCTGTGGGCCAGCTTATCAACCC-3′ ΔNp63S385G_R:
5′-GGGTTGATAAGCTGGCCCACAGAAGGCAGCTTG-3′; ΔNp63T397A_F (a1189g):
5′-CAGCGCAACGCCCTCGCTCCTACAACCATTCC-3′ ΔNp63T397A_R:
5′-GGAATGGTTGTAGGAGCGAGGGCGTTGCGCTG-3′; ΔNp63S466A_F (t1396g):
5′-CGAGGTTGGGCTGTTCAGCATGTCTGGACTATTTC-3′ ΔNp63S466A_R,
5′-GAAATAGTCCAGACATGCTGAACAGCCCAACCTCG-3′ and then confirmed by sequencing) into the pcDNA5/FRT/V5-His-TOPO vector.
Flp-In HNSCC 029 cells were transfected with an empty pcDNA5/FRT/V5-His-T
OPO vector or expression cassettes for ΔNp63α (wild type or mutant) along with
pOG44 plasmid, bearing the Flp recombinase.37 Resulting Flp-In HNSCC 029 clones were selected on hygromycin B and verified by RT-PCR and sequencing. Flp-In 029 cells expressing wild type or mutated p63 grew and proliferate under unstressed conditions. The resulting cells were treated with 10 μg/ml cisplatin in a time-dependent fashion.
Small-interfering RNA (siRNA)
We used the human ATM siRNA, CDK2 siRNA, p70s6K siRNA and validated negative control scrambled siRNA (SMARTpool, Dharmacon (Lafayette, CO, USA). SiRNA were combined with DharmaFECT transfection reagent (Dharmacon), and the HNSCC 029 cells were transfected according to the recommended protocol with siRNA (100 nM final concentration). After 48 h of transfection, cells were starved in RPMI medium 1640 containing 0.5% FBS before treatment. After 48 h incubation, cells were treated with cisplatin for indicated time periods and then total cell lysates were used for immunoblotting analysis, as previously described.25,37
Mass-spectrometry analysis of phosphorylation sites in ΔNp63α
028 cells were infected with the recombinant Ad5-ΔNp63α-myc for 18 h (reviewed in refs. 25, 61, 82 and 83). Resulting cells were treated with 10 μM cisplatin or with control medium for an additional 18 h, and ΔNp63α-myc was immunoprecipitated overnight from cell lysates (500–2000 μg) using the ProFound Mammalian C-myc Tag IP/Co-IP kit (Pierce) following the manufacturer’s protocol #2. The protein then was purified by fast-performance liquid chromatography allowing to achieve ~90% purity of ΔNp63α and yielding ~10 μg/108 cells. Purified ΔNp63α-myc proteins from cells treated and untreated with cisplatin were subjected to the enzyme in-gel digestion, and the peptide mixture was subjected directly to peptide profiling by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) to pinpoint naturally occurring phosphopeptides, and to sequence and identify each peptide within the mixture.3,9,19,36,49,68,86
MALDI-TOF-MS was performed using the Voyager DE ProTM mass spectrometer (Applied Biosystems). Aliquots of digested peptides (1 pmol) in 1 μl of H2O and 0.1% trifluoroacetic acid were mixed with 1 μl of cyano-4-hydroxycinnamic acid, spotted onto a sample target, dried and loaded into the mass spectrometer. The MS-digest computer programs (MS-Fit, Protein Prospector, version 3.4.1. and ProFound, version 4.10.5) were provided by the Johns Hopkins University Mass Spectrometry Core and used to calculate the average masses of all possible peptide and phosphopeptide fragments of ΔNp63α, and the m/z value of the mass spectral peaks for the corresponding MH+ ions.8,28 The peptide masses were entered into the MASCOT Search Engine-2 and the National Center for Biotechnology Information database was searched to match the tryptic peptide fingerprint with a parent polypeptide.49,62
In vitro protein kinase assay
Kinase reactions contained 25 mM Tris-HCl, pH 8, 50 mM KCl, 5% glycerol, 0.5 mM DTT, 5 μCi of [gamma32P]-ATP, 10 μM cold ATP, 10 mM MnCl2, 0.25–0.5 μg of PHAS-I (Stratagene), 2–4 ng of purified recombinant ATM (CDK2 or p70s6K or nothing, all purified kinases were purchased from Humanzyme) and 2–4 ng of purified ΔNp63α as previously described.11,25 The reactions were incubated at 30°C for 30 min. ΔNp63α was precipitated with Ab-1 antibody, and analyzed by 10% SDS-PAGE. Gels were stained with Coomassie Blue, destained, dried and exposed to X-ray film with intensifying screen at 80°C.
References
- 1.Appella E, Anderson C. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem. 2001;268:2764–72. doi: 10.1046/j.1432-1327.2001.02225.x. [DOI] [PubMed] [Google Scholar]
- 2.Ashcroft M, Taya Y, Vousden K. Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol. 2000;20:3224–33. doi: 10.1128/mcb.20.9.3224-3233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakhtiar R, Nelson R. Mass Spectrometry of the Proteome. Mol Pharmacol. 2001;60:405–15. [PubMed] [Google Scholar]
- 4.Banin S, Moyal L, Shieh S, Taya Y, Anderson C, Chessa L, Smorodinsky N, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1677–9. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 5.Barbieri CE, Barton CE, Pietenpol JA. DeltaNp63alpha expression is regulated by the phosphoinositide 3-kinase pathway. J Biol Chem. 2003;278:51408–14. doi: 10.1074/jbc.M309943200. [DOI] [PubMed] [Google Scholar]
- 6.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 7.Borst P, Rottenberg S, Jonkers J. How do real tumors become resistant to cisplatin? Cell Cycle. 2008;7:1353–9. doi: 10.4161/cc.7.10.5930. [DOI] [PubMed] [Google Scholar]
- 8.Candi E, Dinsdale D, Rufini A, Salomoni P, Knight RA, Mueller M, Krammer PH, Melino G. TAp63 and ΔNp63 in Cancer and Epidermal Development. Cell Cycle. 2007;6:274–84. doi: 10.4161/cc.6.3.3797. [DOI] [PubMed] [Google Scholar]
- 9.Carr SA, Annan RS, Huddleston MJ. Mapping posttranslational modifications of proteins by MS-based selective detection: application to phosphoproteomics. Meth Enzymol. 2005;405:82–115. doi: 10.1016/S0076-6879(05)05005-6. [DOI] [PubMed] [Google Scholar]
- 10.Casaday RJ, Bailey JR, Kalb SR, Brignole EJ, Loveland AN, Cotter RJ, Gibson W. Assembly protein precursor (pUL80.5 homolog) of simian cytomegalovirus is phosphorylated at a glycogen synthase kinase 3 site and its downstream “priming” site: phosphorylation affects interactions of protein with itself and with major capsid protein. J Virol. 2004;78:13501–11. doi: 10.1128/JVI.78.24.13501-13511.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan D, Son SC, Block W, Ye R, Khanna K, Wold M, Douglas P, Goodarzi A, Pelley J, Taya Y, Lavin M, Lees-Miller S. Purification and characterization of ATM from human placenta. A Manganese-dependent, wortmannin-sensitive serine/threonine protein kinase. J Biol Chem. 2000;275:7803–10. doi: 10.1074/jbc.275.11.7803. [DOI] [PubMed] [Google Scholar]
- 12.Chatterjee A, Upadhyay S, Chang X, Nagpal J, Trink B, Sidransky D. U-box-type ubiquitin E4 ligase attenuates cisplatin-mediated degradation of DeltaNp63alpha. Cell Cycle. 2008;7:1231–7. doi: 10.4161/cc.7.9.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng W, Jacobs WB, Zhang JJ, Moro A, Park JH, Kushida M, Qiu W, Mills AA, Kim PC. DeltaNp63 plays an anti-apoptotic role in ventral bladder development. Development. 2006;133:4783–92. doi: 10.1242/dev.02621. [DOI] [PubMed] [Google Scholar]
- 14.Chernov M, Bean L, Lerner N, Stark G. Regulation of ubiquitination and degradation of p53 in unstressed cells through C-terminal phosphorylation. J Biol Chem. 2001;276:31819–24. doi: 10.1074/jbc.M103170200. [DOI] [PubMed] [Google Scholar]
- 15.Chiang GG, Abraham RT. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70s6 kinase. J Biol Chem. 2005;280:25485–90. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]
- 16.De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M, Kim SH. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur J Biochem. 1997;243:518–26. doi: 10.1111/j.1432-1033.1997.0518a.x. [DOI] [PubMed] [Google Scholar]
- 17.Deb-Basu D, Aleem E, Kaldis P, Felsher DW. CDK2 Is Required By MYC to Induce Apoptosis. Cell Cycle. 2006;5:1342–7. doi: 10.4161/cc.5.12.2859. [DOI] [PubMed] [Google Scholar]
- 18.Ferguson M, Luciani MG, Finlan L, Rankin EM, Ibbotson S, Fersht A, Hupp TR. The development of a CDK2-docking site peptide that inhibits p53 and sensitizes cells to death. Cell Cycle. 2004;3:80–9. [PubMed] [Google Scholar]
- 19.Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol. 2002;20:301–5. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 20.Finlan LE, Nenutil R, Ibbotson SH, Vojtesek B, Hupp TR. CK2-site Phosphorylation of p53 is Induced in DeltaNp63 Expressing Basal Stem Cells in UVB Irradiated Human Skin. Cell Cycle. 2006;5:2489–94. doi: 10.4161/cc.5.21.3393. [DOI] [PubMed] [Google Scholar]
- 21.Finlan LE, Hupp TR. p63: the phantom of the tumor suppressor. Cell Cycle. 2007;6:1062–71. doi: 10.4161/cc.6.9.4162. [DOI] [PubMed] [Google Scholar]
- 22.Flores E, Tsai K, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T. P63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–4. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 23.Flores ER. The Roles of p63 in Cancer. Cell Cycle. 2007;6:300–4. doi: 10.4161/cc.6.3.3793. [DOI] [PubMed] [Google Scholar]
- 24.Fischer PM, Endicott J, Meijer L. Cyclin-dependent kinase inhibitors. Prog Cell Cycle Res. 2003;5:235–48. [PubMed] [Google Scholar]
- 25.Fomenkov A, Zangen R, Huang Y, Osada M, Guo X, Fomenkov T, Trink B, Sidransky D, Ratovitski E. RACK1 and stratifin target DeltaNp63alpha for a proteasome degradation in head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle. 2004;3:1285–95. doi: 10.4161/cc.3.10.1155. [DOI] [PubMed] [Google Scholar]
- 26.Gressner O, Schilling T, Lorenz K, Schulze Schleithoff E, Koch A, Schulze-Bergkamen H, Lena AM, Candi E, Terrinoni A, Catani MV, Oren M, Melino G, Krammer PH, Stremmel W, Muller M. TAp63alpha induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J. 2005;24:2458–71. doi: 10.1038/sj.emboj.7600708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo X, Mills AA. p63, Cellular Senescence and Tumor Development. Cell Cycle. 2007;6:305–11. doi: 10.4161/cc.6.3.3794. [DOI] [PubMed] [Google Scholar]
- 28.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 29.Hammond EM, Dorie MJ, Giaccia AJ. ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J Biol Chem. 2003;278:12207–13. doi: 10.1074/jbc.M212360200. [DOI] [PubMed] [Google Scholar]
- 30.Han JW, Pearson R, Dennis P, Thomas G. Rapamycin, wortmannin, and the methylxan-thine SQ20006 inactivate p70 by Inducing dephosphorylation of the same subset of sites. J Biol Chem. 1995;270:21396–403. doi: 10.1074/jbc.270.36.21396. [DOI] [PubMed] [Google Scholar]
- 31.Hunter T. Protein kinase classification. Meth. Enzymol. 1991;200:3–37. doi: 10.1016/0076-6879(91)00125-g. [DOI] [PubMed] [Google Scholar]
- 32.Hibi K, Trink B, Patturajan M, Westra W, Caballero O, Hill D, Ratovitski E, Jen J, Sidransky D. AIS is an oncogene amplified in squamous cell carcinoma. Proc Natl Acad Sci USA. 2000;97:5462–7. doi: 10.1073/pnas.97.10.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 34.Higginbottom K, Jahnke U, Newland AC, Cotter FE, Allen PD. New alternative phosphorylation sites on the cyclin dependent kinase 1/cyclin a complex in p53-deficient human cells treated with etoposide: possible association with etoposide-induced apoptosis. Apoptosis. 2007;12:1847–55. doi: 10.1007/s10495-007-0104-6. [DOI] [PubMed] [Google Scholar]
- 35.Hildesheim J, Belova GI, Tyner SD, Zhou X, Vardanian L, Fornace AJ., Jr. Gadd45a regulates matrix metalloproteinases by suppressing DeltaNp63alpha and beta-catenin via p38 MAP kinase and APC complex activation. Oncogene. 2004;23:1829–33. doi: 10.1038/sj.onc.1207301. [DOI] [PubMed] [Google Scholar]
- 36.Hoffman E, Stroobant V. Principles and Applications. John Wiley and Sons Ltd Press; Chichester, West Sussex, England: 2007. Mass Spectrometry; pp. 189–217. [Google Scholar]
- 37.Huang YP, Kim Y, Li Z, Fomenkov T, Fomenkov A, Ratovitski EA. AEC-associated p63 mutations lead to alternative splicing/protein stabilization of p63 and modulation of Notch signaling. Cell Cycle. 2005;4:1440–7. doi: 10.4161/cc.4.10.2086. [DOI] [PubMed] [Google Scholar]
- 38.Jacobs WB, Govoni G, Ho D, Atwal JK, Barnabe-Heider F, Keyes WM, Mills AA, Miller FD, Kaplan DR. P63 is an essential proapoptotic protein during neural development. Neuron. 2005;48:743–56. doi: 10.1016/j.neuron.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 39.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 40.Katoh I, Aisaki KI, Kurata SI, Ikawa S, Ikawa Y. P51A (TAp63gamma), a p53 homolog, accumulates in response to DNA damage for cell regulation. Oncogene. 2000;19:3126–30. doi: 10.1038/sj.onc.1203644. [DOI] [PubMed] [Google Scholar]
- 41.Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274:37538–43. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- 42.King KE, Ponnamperuma RM, Yamashita T, Tokino T, Lee LA, Young MF, Weinberg WC. DeltaNp63alpha functions as both positive and a negative transcriptional regulator and blocks in vitro differentiation of murine keratinocytes. Oncogene. 2003;22:3635–44. doi: 10.1038/sj.onc.1206536. [DOI] [PubMed] [Google Scholar]
- 43.Lavin M, Kozlov S. ATM Activation and DNA Damage Response. Cell Cycle. 2007;6:931–42. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- 44.Lau A, Swinbank KM, Ahmed PS, Taylor DL, Jackson SP, Smith GC, O’Connor MJ. Suppression of HIV-1 infection by a small molecule inhibitor of the ATM kinase. Nat Cell Biol. 2005;7:493–500. doi: 10.1038/ncb1250. [DOI] [PubMed] [Google Scholar]
- 45.Leong CO, Vidnovic N, DeYoung MP, Sgroi D, Ellisen LW. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J Clin Invest. 2007;117:1370–80. doi: 10.1172/JCI30866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liefer K, Koster M, Wang X, Yang A, McKeon F, Roop D. Downregulation of p63 is required for epidermal UV-B-induced apoptosis. Cancer Res. 2000;60:4016–20. [PubMed] [Google Scholar]
- 47.Liu LZ, Zhou XD, Qian G, Shi X, Fang J, Jiang BH. AKT1 amplification regulates cisplatin resistance in human lung cancer cells through the mammalian target of rapamycin/p70s6K1 pathway. Cancer Res. 2007;67:6325–32. doi: 10.1158/0008-5472.CAN-06-4261. [DOI] [PubMed] [Google Scholar]
- 48.Matheny KE, Barbieri CE, Sniezek JC, Arteaga CL, Pietenpol JA. Inhibition of epidermal growth factor receptor signaling decreases p63 expression in head and neck squamous carcinoma cells. Laryngoscope. 2003;113:936–9. doi: 10.1097/00005537-200306000-00004. [DOI] [PubMed] [Google Scholar]
- 49.MacCoss MJ, Wu CC, Yates JR., 3rd. Probability-based validation of protein identifications using a modified SEQUEST algorithm. Anal Chem. 2002;74:5593–9. doi: 10.1021/ac025826t. [DOI] [PubMed] [Google Scholar]
- 50.Mallette F, Ferbeyre G. The DNA Damage Signaling Pathway Connects Oncogenic Stress to Cellular Senescence. Cell Cycle. 2007;6:1831–6. doi: 10.4161/cc.6.15.4516. [DOI] [PubMed] [Google Scholar]
- 51.McLachlin DT, Chait BT. Analysis of phosphorylated proteins and peptides by mass spectrometry. Curr Opin Chem Biol. 2001;5:591–602. doi: 10.1016/s1367-5931(00)00250-7. [DOI] [PubMed] [Google Scholar]
- 52.Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–36. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 53.Mills AA. P63: oncogene or tumor suppressor? Curr Opin Genet Dev. 2006;16:38–44. doi: 10.1016/j.gde.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 54.Nishi H, Senoo M, Nishi KH, Murphy B, Rikiyama T, Matsumura Y, Habu S, Johnson AC. p53 Homologue p63 represses epidermal growth factor receptor expression. J Biol Chem. 2001;276:41717–24. doi: 10.1074/jbc.M101241200. [DOI] [PubMed] [Google Scholar]
- 55.Nussenzweig A. Causes and Consequences of the DNA Damage Response. Cell Cycle. 2007;6:2339–40. doi: 10.4161/cc.6.19.4995. [DOI] [PubMed] [Google Scholar]
- 56.Okada Y, Osada M, Kurata S, Sato S, Aisaki K, Kageyama Y, Kihara K, Ikawa Y, Katoh I. p53 gene family p51 (p63)-encoded, secondary transactivator p51B (TAp63alpha) occurs without forming an immunoprecipitable complex with MDM2, but responds to genotoxic stress by accumulation. Exp Cell Res. 2002;276:194–200. doi: 10.1006/excr.2002.5535. [DOI] [PubMed] [Google Scholar]
- 57.O’Neill T, Dwyer AJ, Ziv Y, Chan DW, Lees-Miller SP, Abraham RH, Lai JH, Hill D, Shiloh Y, Cantley LC, Rathbun GA. Utilization of oriented peptide libraries to identify substrate motifs selected by ATM. J Biol Chem. 2000;275:22719–27. doi: 10.1074/jbc.M001002200. [DOI] [PubMed] [Google Scholar]
- 58.Ongkeko WM, An Y, Chu TS, Aguilera J, Dang CL, Wang-Rodriguez J. Gleevec suppresses p63 expression in head and neck squamous cell carcinoma despite p63 activation by DNA-damaging agents. Laryngoscope. 2006;116:1390–6. doi: 10.1097/01.mlg.0000225941.60901.0f. [DOI] [PubMed] [Google Scholar]
- 59.Papoutsaki M, Moretti F, Lanza M, Marinari B, Sartorelli V, Guerrini L, Chimenti S, Levrero M, Costanzo A. A p38-dependent pathway regulates DeltaNp63 DNA binding to p53-dependent promoters in UV-induced apoptosis of keratinocytes. Oncogene. 2005;24:6970–5. doi: 10.1038/sj.onc.1208835. [DOI] [PubMed] [Google Scholar]
- 60.Parsa R, Yang A, McKeon F, Green H. Association of p63 with proliferative potential in normal and neoplastic human keratinocytes. J Invest Dermatol. 1999;113:1099–105. doi: 10.1046/j.1523-1747.1999.00780.x. [DOI] [PubMed] [Google Scholar]
- 61.Patturajan M, Nomoto S, Sommer M, Fomenkov A, Hibi K, Zangen R, Poliak N, Califano J, Trink B, Ratovitski E, Sidransky D. DeltaNp63 induces beta-catenin nuclear accumulation and signaling. Cancer Cell. 2002;1:369–79. doi: 10.1016/s1535-6108(02)00057-0. [DOI] [PubMed] [Google Scholar]
- 62.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–67. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 63.Pereg Y, Shkedy D, de Graaf P, Meulmeester E, Edelson-Averbukh M, Salek M, Biton S, Teunisse AF, Lehmann WD, Jochemsen AG, Shiloh Y. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc Natl Acad Sci USA. 2005;102:5056–61. doi: 10.1073/pnas.0408595102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perez C, Pietenpol J. Transcriptional programs regulated by p63 in normal epithelium and tumors. Cell Cycle. 2007;6:246–54. doi: 10.4161/cc.6.3.3801. [DOI] [PubMed] [Google Scholar]
- 65.Ratovitski E, Patturajan M, Hibi K, Trink B, Yamaguchi K, Sidransky D. P53 associates with and targets DeltaNp63 into a protein degradation pathway. Proc Natl Acad Sci USA. 2001;98:1817–22. doi: 10.1073/pnas.98.4.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ratovitski E, Trink B, Sidransky D. P63 and p73: teammates or adversaries? Cancer Cell. 2006;9:1–2. doi: 10.1016/j.ccr.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 67.Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. P63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9:45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 68.Rosenberg I. Benchtop Techniques. Vol. 230. Birkhauser Press; Boston, Basel, New York: 2005. Protein Analysis and Purification; pp. 278–87. [Google Scholar]
- 69.Samuel T, Weber OH, Funk JO. Linking DNA Damage to Cell Cycle Checkpoints. Cell Cycle. 2002;1:162–8. [PubMed] [Google Scholar]
- 70.Sancar A, Lindsey-Boltz LA, Unsal-Kaccmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 71.Shimada A, Kato S, Enjo K, Osada M, Ikawa Y, Kohno K, Obinata M, Kanamaru R, Ikawa S, Ishioka C. The transcriptional activities of p53 and its homologue p51/p63: similarities and differences. Cancer Res. 1999;59:2781–6. [PubMed] [Google Scholar]
- 72.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–15. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 73.Thurfjell N, Coates PJ, Vojtesek B, Benham-Motlagh P, Eisold M, Nylander K. Endogenous p63 acts as a survival factor for tumour cells of SCCHN origin. Int J Mol Med. 2005;16:1065–70. [PubMed] [Google Scholar]
- 74.Trink B, Osada M, Ratovitski E, Sidransky D. p63 Transcriptional Regulation of Epithelial Integrity and Cancer. Cell Cycle. 2007;6:240–5. doi: 10.4161/cc.6.3.3803. [DOI] [PubMed] [Google Scholar]
- 75.Upadhyay S, Liu C, Chatterjee A, Hoque M, Kim M, Engles J, Westra W, Trink B, Ratovitski E, Sidransky D. LKB1/STK11 suppresses COX-2 induction and cellular invasion through PEA3 in lung cancer. Cancer Res. 2006;66:7870–9. doi: 10.1158/0008-5472.CAN-05-2902. [DOI] [PubMed] [Google Scholar]
- 76.Yang A, Kaghad M, Wang Y, Gillett E, Fleming M, Dotsch V, Andrews N, Caput D, McKeon F. P63, a p53 homologue at 3q27-29, encodes multiple products with transactivating, death-inducing and dominant-negative activities. Mol Cell. 1998;2:305–16. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 77.Wan X, Helman LJ. Effect of insulin-like growth factor II on protecting myoblast cells against cisplatin-induced apoptosis through p70s6 kinase pathway. Neoplasia. 2002;4:400–8. doi: 10.1038/sj.neo.7900242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang-Rodriguez J, Lopez JP, Altuna X, Chu TS, Weisman RA, Ongkeko WM. STI-571 (Gleevec) potentiates the effect of cisplatin in inhibiting the proliferation of head and neck squamous cell carcinoma in vitro. Laryngoscope. 2006;116:1409–16. doi: 10.1097/01.mlg.0000225895.40732.52. [DOI] [PubMed] [Google Scholar]
- 79.Westfall MD, Mays DJ, Sniezek JC, Pietenpol JA. The DeltaNp63alpha phosphoprotein binds the p21 and 14-3-3sigma promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol Cell Biol. 2003;23:2264–76. doi: 10.1128/MCB.23.7.2264-2276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Westfall MD, Joyner AS, Barbieri CE, Livingstone M, Pietenpol JA. Ultraviolet radiation induces phosphorylation and ubiquitin-mediated degradation of DeltaNp63alpha. Cell Cycle. 2005;4:710–6. doi: 10.4161/cc.4.5.1685. [DOI] [PubMed] [Google Scholar]
- 81.Whittaker S, te Poele R, Chan F, Linardopoulos S, Walton M, Garrett M, Workman P. The Cyclin-Dependent Kinase Inhibitor Seliciclib (R-roscovitine; CYC202) Decreases the Expression of Mitotic Control Genes and Prevents Entry into Mitosis. Cell Cycle. 2007;6:3114–31. doi: 10.4161/cc.6.24.5142. [DOI] [PubMed] [Google Scholar]
- 82.Wu G, Nomoto S, Hoque MO, Dracheva T, Osada M, Lee CC, Dong SM, Guo Z, Benoit N, Cohen Y, et al. DeltaNp63alpha and TAp63alpha regulate transcription of genes with distinct biological functions in cancer and development. Cancer Res. 2003;63:2351–7. [PubMed] [Google Scholar]
- 83.Wu G, Osada M, Fomenkov A, Begum S, Nomoto S, Zhao M, Upadhyay S, Xing M, Mantovani R, Moon C, Westra W, Koch W, Califano J, Ratovitski E, Sidransky D, Trink B. DeltaNp63alpha upregulates Hsp70 expression in human head and neck cancers. Cancer Res. 2005;65:758–66. [PubMed] [Google Scholar]
- 84.Wu C, Wangpaichitr M, Feun L, Kuo MT, Robles C, Lampidis T, Savaraj N. Overcoming cisplatin resistance by mTOR inhibitor in lung cancer. Mol Cancer. 2005;4:25–31. doi: 10.1186/1476-4598-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zangen R, Ratovitski E, Sidransky D. DeltaNp63alpha levels correlate with clinical tumor response to cisplatin. Cell Cycle. 2005;4:1313–5. doi: 10.4161/cc.4.10.2066. [DOI] [PubMed] [Google Scholar]
- 86.Zhou H, Watts JD, Aebersold R. A systematic approach to the analysis of protein phosphorylation. Nat Biotechnol. 2001;19:375–8. doi: 10.1038/86777. [DOI] [PubMed] [Google Scholar]
- 87.Zhu Y. A Model for CDK2 in Maintaining Genomic Stability. Cell Cycle. 2004;3:1358–62. doi: 10.4161/cc.3.11.1226. [DOI] [PubMed] [Google Scholar]