

Abstract
The human T-lymphotropic virus type I (HTLV-I) causes a chronic inflammatory disorder of the central nervous system termed HTLV-I–associated myelopathy/tropical spastic paraparesis (HAM/TSP). HTLV-I encodes a protein known to activate several host-signaling pathways involved in inflammation, such as the nuclear factor-κB (NF-κB). The contribution of the NF-κB pathway to the pathogenesis of HAM/TSP, however, has not been fully defined. We show evidence of canonical NF-κB activation in short-term cultures of peripheral blood mononuclear cells (PBMCs) from subjects with HAM/TSP. NF-κB activation was closely linked to HTLV-I viral protein expression. The NF-κB activation in HAM/TSP PBMCs was reversed by a novel small-molecule inhibitor that demonstrates potent and selective NF-κB antagonist activity. Inhibition of NF-κB activation led to a reduction in the expression of lymphocyte activation markers and resulted in reduced cytokine signaling in HAM/TSP PBMCs. Furthermore, NF-κB inhibition led to a reduction in spontaneous lymphoproliferation, a key ex vivo correlate of the immune activation associated with HAM/TSP. These results indicate that NF-κB activation plays a critical upstream role in the immune activation of HAM/TSP, and identify the NF-κB pathway as a potential target for immunomodulation in HAM/TSP.
Introduction
Infection with the retrovirus human T-lymphotropic virus type I (HTLV-I) is associated with the development of HTLV-I–associated myelopathy/tropical spastic paraparesis (HAM/TSP) and adult T-cell leukemia/lymphoma (ATLL). HAM/TSP is an immune-mediated inflammatory disorder of the central nervous system that leads to progressive neurologic disability in affected individuals.1 A key mechanism in the pathogenesis of HAM/TSP is considered to be the HTLV-I–induced immune activation that supports the establishment of central nervous system inflammation.2 Immune activation is a hallmark of HAM/TSP, as evidenced by the increased expression of lymphocyte activation markers, the induction of pro-inflammatory cytokines, and spontaneous lymphoproliferation.3–5 The HTLV-I–encoded transactivating protein Tax is thought to play a role in the immune activation associated with HAM/TSP by activating host-signaling molecules such as the cyclic AMP-responsive element-binding protein, the serum response factor, and the nuclear factor-κB (NF-κB), thereby up-regulating the expression of pro-inflammatory cytokines and/or their receptors.6 The activation of the NF-κB pathway is considered a key event in the HTLV-I–induced leukemogenesis leading to ATLL,7 but the contribution of the NF-κB pathway to the pathogenesis of HAM/TSP has not been fully defined.
The NF-κB proteins, which include the RelA (p65), c-Rel, RelB, NF-κB1 (p105/p50), and NF-κB2 (100/p52) subunits, comprise a family of Rel-homology domain–containing transcription factors that play a key role in regulating inflammation.8 NF-κB signaling occurs by activation of either the canonical or the noncanonical pathways, leading to nuclear translocation of the RelA/p50 or RelB/p52 heterodimers, respectively.9 Key signaling events involve the release of NF-κB subunits from the cytoplasmic sequestration by the inhibitor of NF-κB (IκB), the subsequent nuclear translocation, and the binding of NF-κB heterodimers to NF-κB response elements that ultimately lead to gene transcription. The HTLV-I protein Tax is capable of activating both the canonical and the noncanonical NF-κB pathways by interacting with the IκB kinase subunits, leading to the release of NF-κB from cytoplasmic sequestration.10,11 The NF-κB–dependent induction of pro-inflammatory cytokines such as IL-6,12 IL-9,13 and IL-15,14 and the induction of IL-2 receptorα (IL-2Rα)15 in HTLV-I–infected cells suggests that NF-κB activation may play a critical role in the development of diseases associated with HTLV-I infection.
To further define the contribution of NF-κB activation to the pathogenesis of HAM/TSP, we compared NF-κB activation in peripheral blood mononuclear cells (PBMCs) from subjects with HAM/TSP against that of healthy donors, and examined the relationship of HTLV-I viral protein expression and NF-κB activation. We developed several series of novel inhibitor of NF-κB targeting the DNA-binding Rel transcription factors.16–18 To define the contribution of NF-κB activation to immune activation in HAM/TSP, we tested the impact of NF-κB inhibition on key ex vivo correlates of immune activation in HAM/TSP, such as the expression of lymphocyte activation markers,3 the induction of cytokine production and signaling,4 and spontaneous lymphoproliferation.5
Methods
Samples
Peripheral blood was obtained from subjects with HAM/TSP diagnosed according to published criteria19 and from healthy donors. PBMCs were obtained by density centrifugation and cryopreserved before use. Written, informed consent was obtained from each subject in compliance with the Declaration of Helsinki. The study was reviewed and approved by a National Institute of Neurologic Disorders and Stroke institutional review board.
Cell culture
PBMCs were resuspended in RPMI supplemented with 1% penicillin/streptomycin, 1% l-glutamine, and 5% FCS (CRPMI) and placed in 96-well round-bottom plates at 4 × 105 cells/well with or without the NF-κB inhibitors dehydroxymethylepoxyquinomicin (DHMEQ) or PBS-1086 in the doses indicated. PBMCs treated with either dimethylsulfoxide (DMSO; vehicle) or the control compound PBS-1143, a regioisomer of PBS-1086 without anti–NF-κB activity, were used as controls. The antibodies anti-Tac and Mik-β1 used in some experiments were kind gifts from Dr Thomas A. Waldmann.
NF-κB DNA-binding ELISA
NF-κB activation was measured using a DNA-binding enzyme-linked immunosorbent assay (ELISA) (TransAM NF-κB Family Transcription Factor Assay Kit; Active Motif) according to manufacturer's protocol. NF-κB proteins bound to the target sequence were detected with primary antibodies specific for each NF-κB family member (RelA, c-Rel, or RelB) and a horseradish peroxidase (HRP)–conjugated secondary antibody. A colorimetric HRP substrate was added and, after stopping the reaction, the absorbance at 450 nm was recorded as a relative measure of NF-κB protein bound. To test compounds for NF-κB–inhibitory activity, stock solutions (5 mg/mL or approximately 15mM) and dilutions (150× final concentration) of the compounds were prepared in DMSO. Immediately before the assay, the compounds were further diluted to the final concentration in the assay buffer without dithiothreitol. Compounds were typically tested at 5μM and 50μM or at a dilution series from 0.2-500μM. Next, the compounds were incubated with recombinant NF-κB proteins (RelA or p50, 1 μg/mL) or nuclear extracts (Raji or tumor necrosis factor-α [TNF-α]–treated HeLa, 150-250 μg/mL) for 30 minutes to 2 hours at room temperature. Percent inhibition (% inhibition = 1 − (value/maximum value) × 100) was determined from the quantity of NF-κB protein bound in the compound treated wells relative to the maximum quantity bound in the DMSO-treated control wells. If a dilution series was tested, the EC50 for the compounds was calculated using a SigmaPlot macro that fits a sigmoidal dose-response curve to the log10 (μM) concentration versus percent inhibition. To test NF-κB activation in PBMCs, nuclear extracts were obtained using the Nuclear Extract Kit (Active Motif) according to manufacturer's protocol. After the total protein concentration was determined by Bradford assay, 1.25 μg of nuclear extract per well was added to microplates coated with the NF-κB consensus oligonucleotide. NF-κB proteins bound to the oligonucleotide were detected using RelA-, RelB-, or c-Rel–specific antibodies, and an HRP-conjugated secondary antibody. The Rel-binding activities were determined by chemiluminescence according to the manufacturer's instructions. Nuclear extract prepared from stimulated Jurkat cells served as a positive control in each experiment. Specificity was confirmed in each experiment by the use of wild-type and mutant NF-κB oligonucleotide competitors.
NF-κB–driven luciferase expression assay
293/NF-κB-luciferase cells (Panomics) were plated into 96-well white/clear bottom tissue-culture plates at a concentration of 5 × 105 cells/mL (0.1 mL/well) in Dulbecco modified Eagle medium supplemented with 10% FCS, 100 U/mL of penicillin, 100 μg/mL of streptomycin, 2mM l-glutamine, and 100 μg/mL of hygromycin B. Plated cells were incubated overnight at 37°C in a humidified 5% CO2 atmosphere, then 0.5 μL of 200× compounds or 100% DMSO (vehicle control) were added to each well in triplicate and incubated with the cells for 2 hours. After the incubation period, TNF-α (PeproTech) was added to each well at a final concentration of 20 ng/mL. Cells were then incubated at 37°C for 14-18 hours. Luciferase activity was detected by adding 100 μL of Bright-Glo reagent (Promega) to each well, mixing with a pipette, and incubating at room temperature for 2 minutes. Luminescence was read with a DTX 880 Multimode Detector (Beckman Coulter) using Multimode Analysis Software Version 3.1.0.1 (no filter, 0.1-second integration time).
Immunofluorescence analysis
PBMCs from subjects with HAM/TSP were cultured in RPMI supplemented with 10% FCS 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 2mM l-glutamine for 20 hours. Cells were fixed in 4% paraformaldehyde for 15 minutes at 4°C, then permeabilized by washing twice in 0.1% Triton X-100 in phosphate-buffered saline (PBS). Antibodies to HTLV-I Tax (clone LT-4) and/or RelA (clone C22B4; Cell Signaling Technology) were added at 1:500 and 1:50 dilutions, respectively, for 30 minutes at room temperature. After washing with 0.1% Triton X-100 in PBS, Alexa Fluor 488 goat anti–mouse immunoglobulin G3 (IgG3) and/or Alexa Fluor 594 goat anti–rabbit IgG (Invitrogen) were added at 1:200 dilution as secondary antibodies for 30 minutes at room temperature and protected from light. After washing with 0.1% Triton X-100 in PBS and subsequently in PBS, the cell suspension was placed on a glass slide and allowed to dry. The cells were mounted with Prolong Gold with 4,6-diamidino-2-phenylindole, dihydrochloride (DAPI; Invitrogen) and observed with an Axiovert 200M equipped with mercury lamp house HBO-100 and appropriate filters (all from (Carl Zeiss MicroImaging) for detecting DAPI and the 2 secondary antibodies. Images were deconvoluted using the imaging software Volocity (Improvision).
Flow cytometry
All flow cytometric data were acquired on an LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software Version 8 (TreeStar). For fluorescence-assisted cell sorting (FACS) analysis of surface markers, cells were washed and then stained with CD3-PacificBlue, CD4-FITC, CD8-PerCP-Cy5.5, CD25-APC, and CD69-APC-Cy7 (BD Biosciences) for 20 minutes before washing and acquisition. For phosphorylated STAT5 detection, PBMCs from subjects with HAM/TSP were resuspended in CRPMI and placed in 96-well round-bottom plates at 4 × 105 cells/well with NF-κB inhibitors or controls. After overnight (20 hours) culture, cells were fixed in 1.5% paraformaldehyde for 10 minutes at 37°C, then incubated in 90% methanol for 20 minutes at 4°C. After 3 washings in FACS buffer (PBS, 1% FCS, 0.1% NaN3), cells were stained with fluorochrome-conjugated antibodies against pSTAT5 and CD3 (BD Biosciences) for 30 minutes at room temperature, and then washed before acquisition.
Proliferation assay
PBMCs from subjects with HAM/TSP and healthy volunteers were resuspended in complete RPMI, and placed in triplicate in 96-well round-bottom plates at 4 × 105 cells/well with NF-κB inhibitors or controls. Proliferation was measured by tritiated thymidine incorporation on day 4. Phytohemagglutinin-treated PBMCs and healthy donor PBMCs were used as positive and negative controls, respectively. Alternatively, PBMCs from subjects with HAM/TSP were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen) according to the manufacturer's instructions, resuspended in complete RPMI with NF-κB inhibitors or controls, and the data acquired on an LSRII flow cytometer at day 5.
HTLV-I proviral load
HTLV-I proviral load was determined by real-time polymerase chain reaction, as described previously.20 The HTLV-1 proviral DNA load was calculated by the following formula: copy number of HTLV-1 (pX) per 100 cells = (copy number of pX)/(copy number of β-actin/2) × 100.
Statistical analysis
Kolmogorov-Smirnov was used as a test of normality, and either parametric or nonparametric tests were used accordingly. Unpaired t test or Mann-Whitney was used for comparisons between groups. Paired t test or Wilcoxon signed rank test was used for within-group comparisons. For experiments with multiple groups, analysis of variance (ANOVA) with Dunnett posttest or Kruskal-Wallis with Dunn posttest was used. All statistical analyses were carried out using Prism software Version 5.02 (GraphPad).
Results
PBMCs from subjects with HAM/TSP demonstrate increased NF-κB activation in association with viral protein expression
NF-κB activation was examined in PBMCs from healthy donors and subjects with HAM/TSP by measuring nuclear translocation of the NF-κB subunits RelA, RelB, and c-Rel. When assayed directly ex vivo, PBMCs from subjects with HAM/TSP and healthy donors showed comparable levels of nuclear RelA. After short-term (20 hours) culture, PBMCs from subjects with HAM/TSP, but not healthy donors, showed increased nuclear translocation of RelA (Figure 1A). Compared with healthy donors, PBMCs from subjects with HAM/TSP showed higher levels of nuclear RelA (P < .05), indicating activation of the canonical NF-κB pathway (Figure 1B). RelB and c-Rel nuclear translocation were also higher in HAM/TSP, but did not reach statistical significance. To more precisely define the role of the HTLV-I virus in NF-κB activation, we examined the relationship between HTLV-I viral protein expression and the subcellular location of the NF-κB subunit RelA in PBMCs from subjects with HAM/TSP. Immunofluorescence analysis showed that whereas the majority of the cells demonstrated intracytoplasmic sequestration of RelA, cells expressing HTLV-I Tax protein showed nuclear translocation of RelA (Figure 1C), indicating that viral protein expression in HLTV-I–infected cells is associated with NF-κB activation in HAM/TSP.
Figure 1.

PBMCs from subjects with HAM/TSP demonstrate increased NF-κB activation in association with viral protein expression. NF-κB activation was measured by an NF-κB DNA-binding ELISA (TransAM NF-κB transcription factor ELISA; Active Motif) on nuclear extracts from PBMCs of subjects with HAM/TSP and healthy donors to detect nuclear translocation of NF-κB subunits. (A) Nuclear RelA (p65) levels in PBMCs from subjects with HAM/TSP compared with healthy donors (HD), measured directly ex vivo and after 20 hours (20h) of culture. RLU, relative LIGHT units. (B) RelA, RelB, and c-Rel levels in nuclear extracts from healthy donor (HD) PBMCs (n = 6) compared with HAM/TSP PBMCs (n = 6). *P < .05, unpaired t test. (C) Immunofluorescence analysis of PBMCs from a subject with HAM/TSP showing HTLV-I Tax protein expression (left; Alexa Fluor 488), RelA expression (center; Alexa Fluor 594), and merged image (right), with DAPI nuclear counterstain. One representative image from 2 subjects is shown. Note the nuclear translocation of RelA in cells expressing Tax (arrow) compared with the intracytoplasmic sequestration of RelA in cells not expressing Tax. Observation was performed with a Zeiss Axiovert 200M equipped with mercury lamp house HBO-100 and appropriate filters for detecting DAPI and the 2 secondary antibodies conjugated to Alexa Fluor 488 and Alexa Fluor 594, using a 63× oil objective.
NF-κB activation in HAM/TSP is inhibited by PBS-1086, a novel dual inhibitor of the canonical and noncanonical pathways of NF-κB
We have synthesized several families of NF-κB inhibitors.16–18 The potency and selectivity of the compounds in inhibiting NF-κB DNA binding was assessed. We found PBS-1086 to be more potent than either parthenolide or DHMEQ in inhibiting recombinant RelA binding to the NF-κB site (Figure 2A). In addition, PBS-1086 inhibited RelB binding to the NF-κB site, whereas parthenolide and DHMEQ were ineffective (Figure 2B). Thus, PBS-1086 demonstrated dual activity against NF-κB, inhibiting both the canonical and noncanonical pathways. The inhibitory effect of PBS-1086 on NF-κB activation was further demonstrated in a cell-based assay. PBS-1086 inhibited the TNF-α–induced expression of luciferase in a 293/NF-κB-luciferase reporter cell line, while a regioisomer, PBS-1143, was ineffective (Figure 2C). PBS-1086 had no direct inhibitory effect on STAT3 and STAT5-binding activities (data not shown).
Figure 2.

NF-κB activation in HAM/TSP is inhibited by PBS-1086, a novel inhibitor of NF-κB. The potency and selectivity of several small-molecule inhibitors of NF-κB were assessed using the NF-κB DNA-binding ELISA (A-B) and a 293/NF-κB–luciferase reporter cell line (C). (A) Percent inhibition of RelA binding as a function of inhibitor concentration (μM) for parthenolide, DHMEQ, and PBS-1086. (B) Percent inhibition of RelB binding as a function of inhibitor concentration (μM) for parthenolide, DHMEQ, and PBS-1086. (C) Percent inhibition of luciferase activity in TNF-α–treated 293/NF–κB-luciferase reporter cell line as a function of inhibitor concentration (μM) for PBS-1086 and its regioisomer PBS-1143. The efficacy of DHMEQ and PBS-1086 at inhibiting NF-κB activation in HAM/TSP PBMCs was tested using the NF-κB DNA-binding ELISA. (D) RelA-, RelB-, and c-Rel–binding activity in nuclear extracts from untreated, DMSO (vehicle), DHMEQ (10μM)–, or PBS-1086 (10μM)–treated HAM/TSP PBMCs (n = 6). **P < .01, ANOVA and Dunnett posttest.
We examined the efficacy of DHMEQ and PBS-1086 in inhibiting NF-κB activation in PBMCs from subjects with HAM/TSP (Figure 2D). The addition of 10μM DHMEQ to short-term (20 hours) cultures of HAM/TSP PBMCs resulted in a 19% reduction in nuclear RelA-binding activity. The addition of 10μM PBS-1086 resulted in an 83% reduction in nuclear RelA-binding activity (P < .01; ANOVA and Dunnett posttest). The addition of PBS-1086 also resulted in a 70% and 81% reduction in nuclear RelB- and c-Rel–binding activity, respectively. These results indicate that the NF-κB activation in PBMCs of subjects with HAM/TSP can be effectively inhibited by the use of the novel small-molecule inhibitor PBS-1086.
Inhibition of NF-κB leads to modulation of immune activation in HAM/TSP
To test the contribution of the NF-κB pathway to immune activation in HAM/TSP, we examined the effect of NF-κB inhibition on the ex vivo correlates of HAM/TSP activation. The addition of NF-κB inhibitors resulted in reduced expression of the lymphocyte activation markers CD25 and CD69 in cultured PBMCs from subjects with HAM/TSP. Figure 3A shows the increased expression of CD25 and CD69 on PBMCs from subjects with HAM/TSP compared with healthy donors after short-term (24 hours) culture, and the inhibition of CD25 and CD69 expression in the presence of PBS-1086. PBS-1086 treatment yielded significant reduction in the mean fluorescent intensity of CD25 expression (P < .01; ANOVA and Dunnett posttest; Figure 3B). The frequency of CD69-expressing cells was also significantly reduced in the presence of NF-κB inhibitors (P < .01; ANOVA with Dunnett posttest; Figure 3B).
Figure 3.

Inhibition of NF-κB leads to the modulation of immune activation in HAM/TSP. PBMCs from subjects with HAM/TSP were placed in short-term (20 hours) culture and analyzed for the expression of CD25, CD69, and phosphorylated STAT5 (pSTAT5) by FACS analysis. (A) Representative FACS histograms (left panels) showing CD25 and CD69 expression (blue lines) in healthy donor (HD) PBMCs after 20 hours of culture. Shaded histograms are isotype control staining. Representative FACS histograms (right panels) showing CD25 and CD69 expression (blue lines) in untreated PBMCs from a HAM/TSP subject after 20 hours of culture. Green lines indicate CD25 and CD69 expression in HAM/TSP PBMCs treated with PBS-1086 (10μM). Shaded histograms are isotype control staining. (B) The mean (± SD) expression of CD25 and CD69 in untreated, DMSO, regioisomer PBS-1143 (10μM)–, DHMEQ (10μM)–, or PBS-1086 (10μM)–treated PBMCs from HAM/TSP subjects (n = 5). **P < .01, ANOVA and Dunnett posttest. (C) Representative FACS histograms showing STAT5 activation (pSTAT5) in cultured PBMCs from a healthy donor (HD, left), in untreated PBMCs from a subject with HAM/TSP (center), and in HAM/TSP PBMCs treated with the NF-κB inhibitor PBS-1086 (10μM, right). (D) representative FACS histogram showing STAT5 activation (pSTAT5) in HAM/TSP PBMCs in the presence of antibodies against the IL-2Rα (anti-Tac) and the IL-2/IL-15Rβ (Mik-β1): (1) anti-Tac (5 μg/mL) + Mik-β1 (5 μg/mL), (2) Mik-β1 (10 μg/mL), (3) anti-Tac (10 μg/mL), or (4) untreated and (5) isotype control antibody staining. (E) The mean (±SD) T-cell STAT5 activation (pSTAT5 + CD3+) in untreated, msIgG2a (10 μg/mL)–, anti-Tac (10 μg/mL)–, Mik-β1 (10 μg/mL)–, and anti-Tac (5 μg/mL) + Mik-β1 (5 μg/mL)–treated HAM/TSP PBMCs (n = 4). *P < .05, Kruskal-Wallis, Dunn posttest. (F) The mean (±SD) frequency of pSTAT5+ cells in cultured (20 hours) HAM/TSP PBMCs (n = 3) as a function of inhibitor concentration for PBS-1086 and control regioisomer PBS-1143.
The induction of inflammatory cytokines in HTLV-I infection has been implicated in the pathogenesis of HAM/TSP.21 STAT proteins function to mediate cytokine signal transduction, such that the detection of STAT activation can be used as an indicator of cytokine production and signaling.22 We analyzed the activation of STAT5 protein to examine the induction of cytokines in PBMCs from subjects with HAM/TSP. As shown in Figure 3C, STAT5 phosphorylation was detected in short-term (20 hours) cultures of PBMCs from subjects with HAM/TSP, but not those from healthy donors, indicating induction of cytokine production and signaling in the absence of exogenous stimulation in HAM/TSP. To confirm the identity of STAT5-signaling cytokines in HAM/TSP, we used 2 antibodies against the IL-2Rα and IL-2/IL-15Rβ subunits to test whether STAT5 activation could be attributed to signaling by IL-2 and/or IL-15, 2 cytokines implicated in the pathogenesis of HAM/TSP.23 As shown in Figure 3D-E, the addition of either anti-Tac or Mik-β1 to block IL-2Rα and IL-2/IL-15Rβ, respectively, partially inhibited STAT5 activation in HAM/TSP PBMCs. The combination of anti-Tac and Mik-β1 abolished T-cell STAT5 activation in HAM/TSP, indicating that signaling via the IL-2Rα and IL-2/IL-15Rβ subunits accounts for nearly all of the T-cell STAT5 activation in HAM/TSP. To test the contribution of the NF-κB pathway to cytokine induction in HAM/TSP, we examined the effect of NF-κB inhibition on STAT5 activation. The addition of the NF-κB inhibitor PBS-1086 resulted in a dose-dependent reduction in STAT5 activation in cultures of HAM/TSP PBMCs, with a nearly complete absence of pSTAT5 at the 10μM concentration and above (Figure 3F). In contrast, the control regioisomer PBS-1143 had no effect on STAT5 activation.
NF-κB inhibitors suppress spontaneous lymphoproliferation in HAM/TSP PBMCs
PBMCs from subjects with HAM/TSP undergo spontaneous lymphoproliferation in culture, without requirement for exogenous stimulation.5 We tested the effect of NF-κB inhibition on spontaneous lymphoproliferation of PBMCs from subjects with HAM/TSP. As shown in Figure 4A, the addition of NF-κB inhibitors to cultures of HAM/TSP PBMCs resulted in the inhibition of spontaneous lymphoproliferation. Reduction in proliferation, measured by tritiated thymidine incorporation, was observed in a dose-dependent manner beginning at the 5μM inhibitor concentration. At 10μM concentrations, DHMEQ and PBS-1086 demonstrated 34% and 71% reduction in spontaneous lymphoproliferation, respectively, compared with untreated HAM/TSP PBMCs. The control regioisomer PBS-1143 showed no inhibition of spontaneous lymphoproliferation. The effect of NF-κB inhibition on spontaneous lymphoproliferation was further confirmed by CFSE dilution. As shown in Figure 4B, 10μM PBS-1086 abolished cell division in HAM/TSP PBMCs as detected by CFSE dilution, whereas cell division was virtually unchanged with the control regioisomer PBS-1143 in PBMCs from HAM/TSP subjects.
Figure 4.

NF-κB inhibitors suppress spontaneous lymphoproliferation in HAM/TSP PBMCs. Proliferation was measured by either tritiated thymidine incorporation or CFSE dilution in PBMCs from subjects with HAM/TSP. (A) Percent inhibition of tritiated thymidine incorporation (day 4) in HAM/TSP PBMCs (n = 3) treated with increasing concentrations of the regioisomer PBS-1143 (control), or the NF-κB inhibitors DHMEQ and PBS-1086 compared with untreated PBMCs. Error bars indicate standard deviation. (B) Representative FACS plots showing CFSE dilution in untreated, control regioisomer PBS-1143 (10μM)– or PBS-1086 (10μM)–treated HAM/TSP PBMCs after 5 days of culture.
The effect of NF-κB inhibition on HTLV-I proviral load
Finally, we asked whether inhibition of NF-κB activation alters the HTLV-I proviral load in PBMCs from subjects with HAM/TSP. The HTLV-I proviral load increased by approximately 80% over a period of 72 hours of culture in untreated PBMCs from subjects with HAM/TSP (P = .0174; Friedman test; Figure 5A), suggesting preferential survival and/or proliferation of HTLV-I–infected cells. The addition of either DHMEQ (10μM) or PBS-1086 (10μM) had only a modest effect on HTLV-I proviral load, resulting in approximately 20% reduction compared with the untreated PBMCs after 72 hours of culture (Figure 5B). NF-κB inhibition had no significant impact on viral protein (HTLV-I Tax) expression (data not shown). Thus, NF-κB activation appeared to have only a modest impact on the HTLV-I proviral load in subjects with HAM/TSP.
Figure 5.
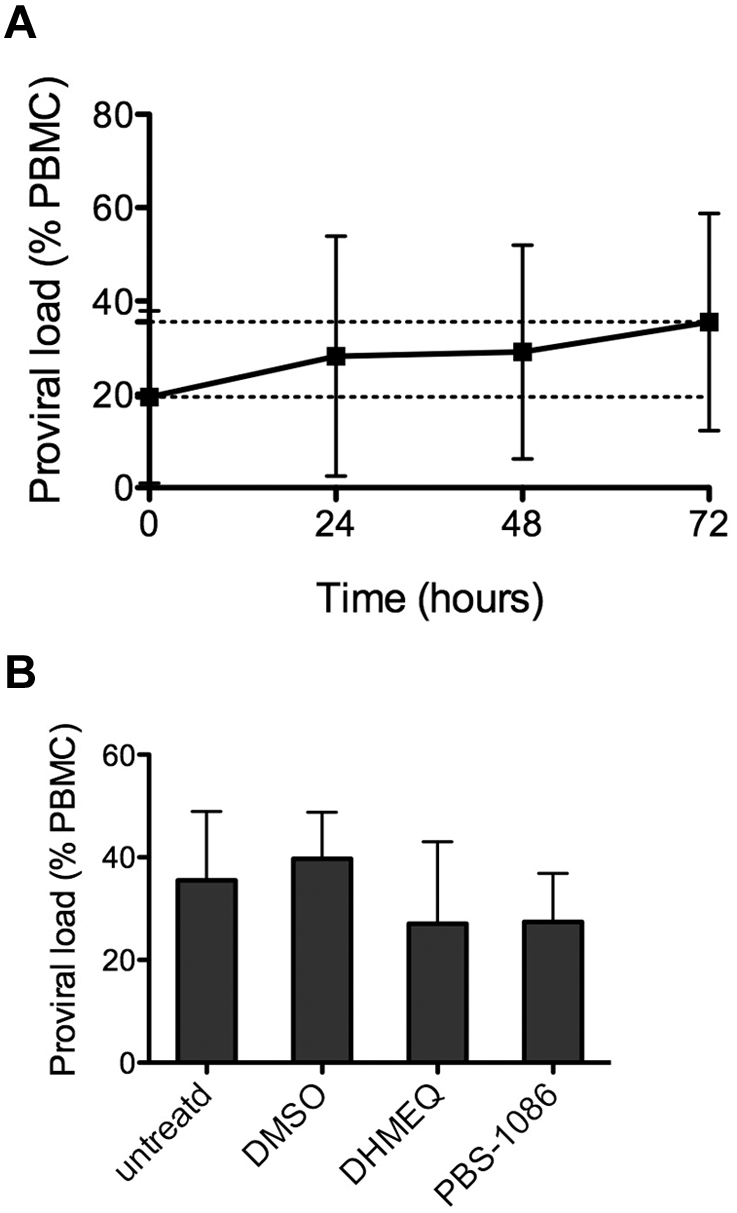
The effect of NF-κB inhibition on HTLV-I proviral load. PBMCs from subjects with HAM/TSP were placed in culture, and serial HTLV-I proviral loads were measured by real-time polymerase chain reaction. (A) The mean (±standard deviation [SD]) HTLV-I proviral load in untreated PBMCs from subjects with HAM/TSP measured at the indicated time points in culture (n = 3; P = .0174, Friedman test). (B) The mean (±SD) HTLV-I proviral load in untreated, DMSO (vehicle)–, DHMEQ (10μM)–, or PBS-1086 (10μM)–treated HAM/TSP PBMCs (n = 3).
Discussion
Subjects with HAM/TSP showed increased NF-κB activation compared with healthy donors. Unlike leukemic cells of ATLL, which show constitutive NF-κB activation,24 PBMCs from subjects with HAM/TSP showed increased NF-κB activation after short-term ex vivo culture. The NF-κB activation in HAM/TSP PBMCs involved the canonical pathway and was reversed by the use of NF-κB–specific small-molecule inhibitors. Increased RelB nuclear translocation, suggesting noncanonical NF-κB activation, was also observed, although this finding was not statistically significant, perhaps because of the small sample size. To our knowledge, this is the first report to directly demonstrate and inhibit increased NF-κB activation in primary cells from subjects with HAM/TSP.
The NF-κB activation in HAM/TSP was closely linked to HTLV-I viral protein expression. Whereas the NF-κB activation in ATLL is Tax independent,7 we found an association between Tax expression and NF-κB activation in HAM/TSP PBMCs. Previous studies have shown that Tax can activate both the canonical and the noncanonical NF-κB–signaling pathways by interacting with the IκB kinase subunits.10,11 The finding from previous studies that in situ Tax expression occurs in the spinal cord of subjects with HAM/TSP25 suggests the likelihood that NF-κB activation may contribute to central nervous system inflammation in these patients. The time course of NF-κB activation in short-term cultures of PBMCs from subjects with HAM/TSP is likely explained by the time course of Tax expression, which is typically marginal directly ex vivo and peaks at 16 to 24 hours in cultured PBMCs from subjects with HAM/TSP.26 Although the mechanism whereby Tax is up-regulated in short-term cultures of HAM/TSP PBMCs has yet to be fully characterized, the activation of stress kinases such as p38 mitogen-activated protein kinase after cytokine withdrawal, and the subsequent cAMP response element-binding protein activation, may explain HTLV-I viral gene expression in culture.27 Thus, local environmental cues may contribute to induction of Tax expression and ultimately to NF-κB activation in the HTLV-I–infected cells of HAM/TSP.
NF-κB activation was a causal influence on lymphocyte activation in HAM/TSP. We showed that the induction of the lymphocyte activation markers CD25 and CD69 was inhibited by antagonizing NF-κB activation. Induction of CD25 expression is a characteristic feature of HTLV-I–infected lymphocytes,28 and is known to be mediated in an NF-κB–dependent manner by the HTLV-I–transactivating protein Tax.15 CD69 is an early lymphocyte activation marker whose expression can be induced by IL-15,29 a pro-inflammatory cytokine that is induced by HTLV-I infection in an NF-κB–dependent manner.14 Consistent with their NF-κB dependence, the induction of the activation markers CD25 and CD69 was significantly reduced by the use of NF-κB inhibitors in HAM/TSP.
The establishment of aberrant cytokine production and signaling is considered to play a key role in the immunopathogenesis of HAM/TSP and has been a target for therapeutic intervention.23 We showed that STAT5 activation occurs in PBMCs from subjects with HAM/TSP in the absence of exogenous stimulation, and showed by the use of 2 antibodies (anti-Tac and Mik-β1) that preferentially block IL-2 and IL-15, respectively,30 that the cytokines IL-2 and IL-15 account for nearly all of the T-cell STAT5 activation in short-term cultures of HAM/TSP PBMCs. The NF-κB inhibitor PBS-1086 reduced STAT5 activation in a dose-dependent manner, indicating that the establishment of IL-2/IL-15 cytokine signaling in HAM/TSP is NF-κB dependent and can be targeted by inhibiting NF-κB activation. A functional consequence of cytokine induction in HAM/TSP is the spontaneous lymphoproliferation of PBMCs from subjects with HAM/TSP.5 PBMCs from subjects with HAM/TSP proliferate in culture in the absence exogenous stimulation, a process largely dependent on the actions of IL-2 and IL-15.31 The inhibition of spontaneous lymphoproliferation by NF-κB inhibitors in this study provides functional confirmation that the NF-κB pathway can be targeted to modulate immune activation in HAM/TSP.
The HTLV-I proviral load in PBMCs from HAM/TSP subjects nearly doubled during 72 hours of culture. The increased HTLV-I proviral load suggests preferential survival and/or increased proliferation of infected cells. NF-κB inhibition resulted in approximately 20% relative reduction in proviral load in PBS-1086–treated PBMCs compared with untreated PBMCs from HAM/TSP subjects. One possible interpretation of the modest impact of NF-κB inhibition on the proviral load is that although immune activation and proliferation are effectively suppressed by NF-κB inhibition, the survival of HTLV-I–infected cells may be less dependent on the NF-κB pathway in HAM/TSP. If so, the pathways that influence the survival of HTLV-I–infected cells in HAM/TSP likely differ from that of ATLL, which are highly susceptible to apoptosis by NF-κB inhibition.32
Although the findings of this study are highly suggestive that NF-κB activation contributes to the pathogenesis of HAM/TSP, we have not yet excluded the possibility that asymptomatic carriers exhibiting comparably high HTLV-I proviral loads might also exhibit levels of NF-κB activation above that of healthy donors. Further validation of the role of the NF-κB pathway in the pathogenesis of HAM/TSP will require testing asymptomatic carriers, identifying correlations with disease activity, and performing clinical trials targeting the NF-κB pathway in HAM/TSP.
In summary, PBMCs from subjects with HAM/TSP demonstrate increased NF-κB activation compared with healthy donors. Activation of the canonical NF-κB pathway was shown in PBMCs from subjects with HAM/TSP and was linked to viral protein expression. Small-molecule inhibitors such as PBS-1086 effectively inhibit NF-κB activation in HAM/TSP, leading to the modulation of key ex vivo correlates of immune activation. These results indicate that NF-κB activation is a critical upstream mediator of immune activation pathways involved in HAM/TSP and establish NF-κB as a potential target for intervention.
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke (NINDS), and in part by Profectus Biosciences Inc.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: U.O. designed and performed research, collected, analyzed, and interpreted data, performed statistical analysis, and wrote the manuscript; M.J.M and D.D. performed research and collected, analyzed, and interpreted data; R.V.T., K.B., and D.D.M. performed research, collected, analyzed, and interpreted data, and wrote the manuscript; D.R.S and Y.T. contributed vital new reagents or analytical tools; J.Z. and S.J. designed research, analyzed and interpreted data, and wrote the manuscript; and J.M. designed research and analyzed and interpreted data.
Conflict-of-interest disclosure: K.B., D.D.M., J.Z., and J.M. are employees of Profectus BioSciences Inc, which is developing NF-κB inhibitors as novel therapeutics. D.R.S. is a consultant for Profectus BioSciences Inc. The remaining authors declare no competing financial interests.
Correspondence: Steven Jacobson, PhD, 10 Center Dr, Bldg 10, Rm 5C103, Bethesda, MD 20892; e-mail: jacobsons@ninds.nih.gov.
References
- 1.Oh U, Jacobson S. Treatment of HTLV-I-associated myelopathy/tropical spastic paraparesis: toward rational targeted therapy. Neurol Clin. 2008;26(3):781–797. doi: 10.1016/j.ncl.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobson S. Immunopathogenesis of human T cell lymphotropic virus type I-associated neurologic disease. J Infect Dis. 2002;186(Suppl 2):S187–192. doi: 10.1086/344269. [DOI] [PubMed] [Google Scholar]
- 3.Al-Fahim A, Cabre P, Kastrukoff L, Dorovini-Zis K, Oger J. Blood mononuclear cells in patients with HTLV-I-associated myelopathy: lymphocytes are highly activated and adhesion to endothelial cells is increased. Cell Immunol. 1999;198(1):1–10. doi: 10.1006/cimm.1999.1580. [DOI] [PubMed] [Google Scholar]
- 4.Tendler CL, Greenberg SJ, Blattner WA, et al. Transactivation of interleukin 2 and its receptor induces immune activation in human T-cell lymphotropic virus type I-associated myelopathy: pathogenic implications and a rationale for immunotherapy. Proc Natl Acad Sci U S A. 1990;87(13):5218–5222. doi: 10.1073/pnas.87.13.5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itoyama Y, Minato S, Kira J, et al. Spontaneous proliferation of peripheral blood lymphocytes increased in patients with HTLV-I-associated myelopathy. Neurology. 1988;38(8):1302–1307. doi: 10.1212/wnl.38.8.1302. [DOI] [PubMed] [Google Scholar]
- 6.Peloponese JM, Yeung ML, Jeang KT. Modulation of nuclear factor-kappaB by human T cell leukemia virus type 1 Tax protein: implications for oncogenesis and inflammation. Immunol Res. 2006;34(1):1–12. [PubMed] [Google Scholar]
- 7.Sun SC, Yamaoka S. Activation of NF-kappaB by HTLV-I and implications for cell transformation. Oncogene. 2005;24(39):5952–5964. doi: 10.1038/sj.onc.1208969. [DOI] [PubMed] [Google Scholar]
- 8.Sha WC. Regulation of immune responses by NF-kappa B/Rel transcription factor. J Exp Med. 1998;187(2):143–146. doi: 10.1084/jem.187.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senftleben U, Cao Y, Xiao G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293(5534):1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 10.Chu ZL, Shin YA, Yang JM, DiDonato JA, Ballard DW. IKKgamma mediates the interaction of cellular IkappaB kinases with the tax transforming protein of human T cell leukemia virus type 1. J Biol Chem. 1999;274(22):15297–15300. doi: 10.1074/jbc.274.22.15297. [DOI] [PubMed] [Google Scholar]
- 11.Xiao G, Cvijic ME, Fong A, et al. Retroviral oncoprotein Tax induces processing of NF-kappaB2/p100 in T cells: evidence for the involvement of IKKalpha. EMBO J. 2001;20(23):6805–6815. doi: 10.1093/emboj/20.23.6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muraoka O, Kaisho T, Tanabe M, Hirano T. Transcriptional activation of the interleukin-6 gene by HTLV-1 p40tax through an NF-kappa B-like binding site. Immunol Lett. 1993;37(2-3):159–165. doi: 10.1016/0165-2478(93)90026-x. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Petrus M, Bryant BR, et al. Induction of the IL-9 gene by HTLV-I Tax stimulates the spontaneous proliferation of primary adult T-cell leukemia cells by a paracrine mechanism. Blood. 2008;111(10):5163–5172. doi: 10.1182/blood-2007-09-113654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Azimi N, Brown K, Bamford RN, Tagaya Y, Siebenlist U, Waldmann TA. Human T cell lymphotropic virus type I Tax protein trans-activates interleukin 15 gene transcription through an NF-kappaB site. Proc Natl Acad Sci U S A. 1998;95(5):2452–2457. doi: 10.1073/pnas.95.5.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ballard DW, Bohnlein E, Lowenthal JW, Wano Y, Franza BR, Greene WC. HTLV-I tax induces cellular proteins that activate the kappa B element in the IL-2 receptor alpha gene. Science. 1988;241(4873):1652–1655. doi: 10.1126/science.241.4873.1652. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Sliskovic DR, Ducker CE. Inhibitors of NF-kB. WO 2010/111460. 2010 Sep 30; ( www.wipo.int) [Google Scholar]
- 17.Zhang J, Sliskovic DR, Ducker CE. Benzamide and naphthamide derivatives inhibiting nuclear factor-kappa(B) (NF-kB). WO 2010/127058. 2010 Nov 4; ( www.wipo.int) [Google Scholar]
- 18.Zhang j, Sliskovic DR, Ducker CE. Oxabicyclo [4.1.0]Hept-B-en-S-ylcarbamoyl derivatives inhibiting the nuclear factor-kappa(B) (NF-kB). WO 2010/148042. 2010 Dec 23; ( www.wipo.int) [Google Scholar]
- 19.De Castro-Costa CM, Araujo AQ, Barreto MM, et al. Proposal for diagnostic criteria of tropical spastic paraparesis/HTLV-I-associated myelopathy (TSP/HAM). AIDS Res Hum Retroviruses. 2006;22(10):931–935. doi: 10.1089/aid.2006.22.931. [DOI] [PubMed] [Google Scholar]
- 20.Nagai M, Kubota R, Greten TF, Schneck JP, Leist TP, Jacobson S. Increased activated human T cell lymphotropic virus type I (HTLV-I) Tax11-19-specific memory and effector CD8+ cells in patients with HTLV-I-associated myelopathy/tropical spastic paraparesis: correlation with HTLV-I provirus load. J Infect Dis. 2001;183(2):197–205. doi: 10.1086/317932. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe H, Nakamura T, Nagasato K, et al. Exaggerated messenger RNA expression of inflammatory cytokines in human T-cell lymphotropic virus type I-associated myelopathy. Arch Neurol. 1995;52(3):276–280. doi: 10.1001/archneur.1995.00540270068021. [DOI] [PubMed] [Google Scholar]
- 22.Krutzik PO, Nolan GP. Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A. 2003;55(2):61–70. doi: 10.1002/cyto.a.10072. [DOI] [PubMed] [Google Scholar]
- 23.Waldmann TA. The IL-2/IL-15 receptor systems: targets for immunotherapy. J Clin Immunol. 2002;22(2):51–56. doi: 10.1023/a:1014416616687. [DOI] [PubMed] [Google Scholar]
- 24.Mori N, Fujii M, Ikeda S, et al. Constitutive activation of NF-kappaB in primary adult T-cell leukemia cells. Blood. 1999;93(7):2360–2368. [PubMed] [Google Scholar]
- 25.Lehky TJ, Fox CH, Koenig S, et al. Detection of human T-lymphotropic virus type I (HTLV-I) tax RNA in the central nervous system of HTLV-I-associated myelopathy/tropical spastic paraparesis patients by in situ hybridization. Ann Neurol. 1995;37(2):167–175. doi: 10.1002/ana.410370206. [DOI] [PubMed] [Google Scholar]
- 26.Yamano Y, Nagai M, Brennan M, et al. Correlation of human T-cell lymphotropic virus type 1 (HTLV-1) mRNA with proviral DNA load, virus-specific CD8(+) T cells, and disease severity in HTLV-1-associated myelopathy (HAM/TSP). Blood. 2002;99(1):88–94. doi: 10.1182/blood.v99.1.88. [DOI] [PubMed] [Google Scholar]
- 27.Washiyama M, Nishigaki K, Ahmed N, et al. IL-2 withdrawal induces HTLV-1 expression through p38 activation in ATL cell lines. FEBS Lett. 2007;581(27):5207–5212. doi: 10.1016/j.febslet.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 28.Yamano Y, Cohen CJ, Takenouchi N, et al. Increased expression of human T lymphocyte virus type I (HTLV-I) Tax11-19 peptide-human histocompatibility leukocyte antigen A*201 complexes on CD4+ CD25+ T cells detected by peptide-specific, major histocompatibility complex-restricted antibodies in patients with HTLV-I-associated neurologic disease. J Exp Med. 2004;199(10):1367–1377. doi: 10.1084/jem.20032042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sancho D, Yanez-Mo M, Tejedor R, Sanchez-Madrid F. Activation of peripheral blood T cells by interaction and migration through endothelium: role of lymphocyte function antigen-1/intercellular adhesion molecule-1 and interleukin-15. Blood. 1999;93(3):886–896. [PubMed] [Google Scholar]
- 30.Guex-Crosier Y, Raber J, Chan CC, et al. Humanized antibodies against the alpha-chain of the IL-2 receptor and against the beta-chain shared by the IL-2 and IL-15 receptors in a monkey uveitis model of autoimmune diseases. J Immunol. 1997;158(1):452–458. [PubMed] [Google Scholar]
- 31.Azimi N, Jacobson S, Leist T, Waldmann TA. Involvement of IL-15 in the pathogenesis of human T lymphotropic virus type I-associated myelopathy/tropical spastic paraparesis: implications for therapy with a monoclonal antibody directed to the IL-2/15R beta receptor. J Immunol. 1999;163(7):4064–4072. [PubMed] [Google Scholar]
- 32.Mori N, Yamada Y, Ikeda S, et al. Bay 11-7082 inhibits transcription factor NF-kappaB and induces apoptosis of HTLV-I–infected T-cell lines and primary adult T-cell leukemia cells. Blood. 2002;100(5):1828–1834. doi: 10.1182/blood-2002-01-0151. [DOI] [PubMed] [Google Scholar]