

Abstract
Limb-girdle muscular dystrophy 2I (LGMD2I) is caused by mutations in the fukutin-related protein (FKRP) gene. Unlike its severe allelic forms, LGMD2I usually involves slower onset and milder course without defects in the central nervous system. The lack of viable animal models that closely recapitulate LGMD2I clinical phenotypes led us to use RNA interference technology to knock down FKRP expression via postnatal gene delivery so as to circumvent embryonic lethality. Specifically, an adeno-associated viral vector was used to deliver short hairpin (shRNA) genes to healthy ICR mice. Adeno-associated viral vectors expressing a single shRNA or two different shRNAs were injected one time into the hind limb muscles. We showed that FKRP expression at 10 months postinjection was reduced by about 50% with a single shRNA and by 75% with the dual shRNA cassette. Dual-cassette injection also reduced a-dystroglycan glycosylation and its affinity to laminin by up to 70% and induced α-dystrophic pathology, including fibrosis and central nucleation, in more than 50% of the myofibers at 10 months after injection. These results suggest that the reduction of approximately or more than 75% of the normal level of FKRP expression induces chronic dystrophic phenotypes in skeletal muscles. Furthermore, the restoration of about 25% of the normal FKRP level could be sufficient for LGMD2I therapy to correct the genetic deficiency effectively and prevent dystrophic pathology.
Limb-girdle muscular dystrophies (LGMD) are a group of clinically and genetically heterogeneous muscular diseases that have both autosomal dominant (type 1) and autosomal recessive (type 2) inheritance. The disorders are generally characterized by progressive muscle wasting and weakness of the shoulder and pelvic girdles and often are associated with a wide range of clinical severity.1–5 To date, at least 13 subtypes (A–M) of LGMD type 2 have been reported, and the causative genes for each subtype have also been identified; LGMD2I (OMIM_607155) is one of the subsets and is caused by mutations in the gene encoding fukutin-related protein (FKRP). The disease is also one of the more common types of LGMD in Denmark,6 the United Kingdom,7 Brazil,8 and the United States.9,10 The onset of LGMD2I can occur from early childhood to adulthood. In addition, cardiac involvement has been frequently reported in patients with LGMD2I.6,7,11–13 By far, the most common mutation in the FKRP gene is the point mutation C826A in the coding sequence, which results in an amino acid change from leucine to isoleucine (L276I) at position 276.6–10 Several studies have reported that homozygous L276I mutation is generally associated with a mild phenotype, whereas compound heterozygous mutation tends to produce a more severe course.6,14–16 Currently, the diagnosis for LGMD2I is based mainly on clinical evaluations and immunohistochemical analyses of muscle biopsies followed by genetic screening for the FKRP gene,17 muscle magnetic resonance imaging, 18 and cardiovascular magnetic resonance imaging.11
The human FKRP gene is mapped to chromosome 19q13.3 and consists of four exons,19 with exon 4 being the single coding exon. The FKRP transcript is expressed predominantly in the skeletal muscle, placenta, and heart.20 The FKRP protein has been shown to localize to the Golgi apparatus,21–24 but other studies have reported its localization to the endoplasmic reticulum25 and sarcolemma.26 Although the precise function of FKRP is not clearly understood, evidence strongly suggests that the protein is involved in post-translational modification of Eα-dystroglycan,16,26–28 a critical component of the dystrophin-glycoprotein complex at the sarcolemma.29 For example, mutations in the FKRP gene are often associated with secondary abnormal glycosylation of α-dystroglycan (hypoglycosylation)20,30–33 and can cause more severe types of muscular dystrophies, including Walker-Warburg syndrome, muscle-eye-brain disease,34 and congenital muscular dystrophy type 1C.20,35
Although recent clinical studies have made rapid progress in understanding LGMD2I, the lack of a viable animal model for LGMD2I has impeded research into its pathobiology and the development of therapeutics. Targeted deletion of the mouse FKRP gene was embryonically lethal, indicating that FKRP is required for embryo development (unpublished results, Q.L.L.). In humans, no patient has ever been reported to carry homozygous null mutations of the FKRP gene until recently. Dr. van Reeuwijk and colleagues reported that two siblings carrying a homozygous mutation (c.1 A>G, Met1Val) in the start codon of FKRP resulted in Walker-Warburg syndrome, the most severe disorder in the disease spectrum of dystroglycanopathies.36 This is highly likely to be a homozygous null FKRP mutation. On the other hand, experiments in our own laboratory and others showed that mice engineered homozygous for the mild L276I missense mutation in the FKRP gene exhibited no appreciable phenotypes (unpublished observations). Recently, Ackroyd and colleagues37 reported that knockin mice carrying a disease-causing Y307N mutation in the FKRP gene also failed to generate discernible phenotypes. It is interesting to note that if the neomycin-resistant cassette was kept in the mutant FKRP allele, the homozygous Y307N neo mice developed very severe phenotypes, including abnormalities in muscle, eye, and brain, and died immediately after birth. In addition, the levels of FKRP transcripts and α-dystroglycan glycosylation (α-DG) were reduced. These results suggest that a knockdown strategy might be a good alternative for investigating FKRP functions. In support of this idea, it has been demonstrated that down-regulation of FKRP has led to developmental abnormalities in zebrafish morphant embryos.38,39 There is still a need for the generation of a viable LGMD2I mouse model that will recapitulate the clinical phenotypes seen in the majority of LGMD2I patients without brain involvement.
In an effort to circumvent the problem of embryonic and neonatal lethality, we adopted RNA interference (RNAi) technology to postnatally knock down FKRP gene expression. Specifically, we used adeno-associated virus (AAV) as an efficient and long-term gene delivery vehicle40–48 to deliver short-hairpin RNA (shRNA) to down-regulate FKRP expression in the skeletal muscles of wild-type ICR mice. We initially screened for small interference RNA (siRNA) oligonucleotides and shRNA capable of effectively knocking down FKRP expression in cultured cells. Subsequently, the selected shRNA sequences were cloned and packaged in AAV vectors and then delivered to the limb muscles of the animals by intramuscular injection. Our results showed that specific targeting of FKRP mRNA by RNAi reduced the level of FKRP transcripts in the skeletal muscle. Furthermore, long-term down-regulation of FKRP expression also resulted in hypoglycosylation of α-dystroglycan. AAV-shFKRP-vector–treated mice developed signs of chronic pathological changes associated with muscular dystrophy in limb muscles. Thus, postnatal knockdown of FKRP gene expression by means of AAV-mediated long-term transfer of shRNA is a useful approach for generating an LGMD2I mouse model and investigating the function of FKRP.
Materials and Methods
Cell Culture and Transfection
Both Hepa 1–6 cells (a C57L mouse hepatoma cell line) and the 293 cells (a human embryonic kidney cell line) were grown in Dulbecco's modified Eagle's medium (Invitrogen, San Diego, CA) with antibiotics (penicillin and streptomycin; Invitrogen) and 10% heat-inactivated fetal bovine serum (HyClone, Thermo Scientific, Waltham, MA). For 293 cells, before the transfection, the density of cells was allowed to reach 80% to 85% in six-well culture plates. In each well, the transfection was followed by Lipofectamine with plus reagent method (Invitrogen). A total of 2 μg of plasmid DNA were applied to each well. After a 48-hour incubation, the protein was extracted and kept at -20°C for future testing.
For Hepa 1–6 cells, the transfection was preceded by the DharmaFECT+ 1 method (Thermo Scientific). Briefly, the Hepa 1–6 cells were cultured to 80% confluence and trypsinized with trypsin-EDTA (Invitrogen) and counted; 5 × 105 cells were remixed with the mixture of siRNA (Integrated DNA Technologies) and the DharmaFECT 1 reagent. The final siRNA concentration per well was 100 nmol/L for 2.5 × 105 cells (24-well plate). It was incubated in a 37°C/5% CO2 incubator for 48 hours. After the first 24 hours of incubation, the medium was aspirated from the cells and replaced with 500 μL of fresh growth medium. The total RNA was extracted and kept at -80°C for further assay. All samples were prepared at least in triplicate, and the results were confirmed by independent experiments.
Construction of Plasmids Expressing Short-Hairpin RNAs
All of the FKRP inhibitory 19-mer siRNA (total, 10 candidates) were designed as shRNA forms and cloned into an AAV vector driven by mouse RNA polymerase III promoters-U6 promoter, which was adapted from the commercial plasmid-pSilencer 1.0. The sequence TTCAAGAGA was used as the loop of shFKRP.49 For the FKRP2 shRNA construct, the sense (5′-GATCCCCGCACTTCTGTCCCGCTTCATTCAAGAGATGAAGCGGGACAGAAGTGCTTTTTGGA-3′) and antisense (5′-AGCTTCCAAAAAGCACTTCTGTCCCGCTTCATCTCTTGAATGAAGCGGGACAGAAGTGCGGG-3′) strands of shFKRP2 were designed with BamH I and HindIII restriction site linkers flanking the 5′ and 3′ termini of shRNA, respectively. Two oligos of 62 nucleotides, sense and antisense strands of the shRNA, were synthesized by Integrated DNA Technologies. The annealed shFKRP2 was cloned into the BamH I and HindIII sites on the double-stranded AAV vector plasmid containing the U6 promoter. Similarly for the FKRP5 shRNA construct, sense (5′-GATCCCCGCGACTTCTTCCGAGTACATTCAAGAGATGTACTCGGAAGAAGTCGCTTTTTGGA-3′) and antisense (5′-AGCTTCCAAAAAGCGACTTCTTCCGAGTACATCTCTTGAATGTACTCGGAAGAAGTCGCGGG-3′) strands of shFKRP5 were also cloned to the AAV vector. To test the additional effects of shFKRP2 and shFKRP5, a head-to tail dual-cassette vector (pAAV-U6-shFKRP2-U6-shFKRP5) was also constructed. A control vector pAAV-U6-shGFP has been described previously.48 The 19-mer shRNA target sequence for GFP was 5′-CGGCCACAAGTTCAGCGTG-3′. Another vector containing a randomly scrambled shFKRP sequence, 5-TTCGGCTACTCACCGAGTA-3′, was used as an in vivo control vector.
AAV Vectors Production
The recombinant viral vector stocks were produced according to the three-plasmid cotransfection method.50 Briefly, the viral particles were purified twice through a CsCl density gradient ultracentrifugation using the previously published protocol.51 The vector titers of viral particle numbers were determined by the viral DNA dot blot method. The concentration of viral vectors was kept in the range of 2 × 1012 to 5 × 1012 vector genomes/ml. storage at -80°C for future use. The final vectors were named AAV6-shFKRP2, AAV6-shFKRP5 (single cassette), and AAV6-shFKRP2 + 5 (dual cassette).
Mice and Vector Administration
All protocols involving animal experiments were approved by the University of North Carolina Animal Care and Use Committee. ICR mice were purchased from the Jackson Laboratory (Bar Harbor, ME). The AAV serotype 6 shFKRP vectors were injected into two sites, the tibialis anterior (TA) and the gastrocnemius (GAS) muscles of both hind legs of anesthetized 6- to 8-week-old ICR mice (10 mice/group) with 30 and 100 μL (total, 3 × 1010 and 1 × 1011 vg) at each site, respectively.42 The shRNA of the single-cassette groups (FKRP2, 5) and the dual-cassette group (shFKRP2 + 5) vectors were all driven by RNA polymerase III promoter U6.
Western Blotting
Western analysis was carried out according to a previously published method.43,52 Briefly, frozen GAS muscle sections were homogenized with 500 μL of ice-cold 1% TX-100 buffer (50 mmol/L Tris, pH 8.0, 150 mmol/L NaCl, 1% Triton X-100, and 0.1% SDS) supplemented by protease inhibitor cocktail (Roche, Indianapolis, IN). The mixtures were centrifuged at 18,000 × g at 4°C for 15 minutes to remove the cell debris. The supernatants were then cleaned using Micro Bio-Spin 6 columns (Bio-Rad Microscience, Hercules, CA), and the protein concentration was determined by Bio-Rad DC Protein assay. Of the total muscle protein, 85 mg were loaded on precast 4% to 20% Tris-glycine gel (Invitrogen) and transferred to polyvinylidene (PVDF) membranes. The membranes were blocked with Pierce Protein-Free T20 Blocking Buffer (Thermo Scientific) at room temperature for 1 hour. Western blots were performed using the following primary antibodies at predetermined dilutions: α-DG mouse monoclonal IIH6 (Millipore, Billerica, MA; 1:1000)29; β-DG rabbit polyclonal DAG1 (Sigma-Aldrich, St. Louis, MO, D1945, 1:4000); actin: rabbit polyclonal anti-actin (Sigma A2066, 1:1000). Primary antibodies were diluted in 1× TBST (20 mmol/L Tris, pH7.4, 150 mmol/L NaCl, 0.1% Tween20) with 0.5% gelatin. Blots were incubated with the antibodies overnight at 4°C. After washing 6 × 10 minutes with buffer, blots were incubated with goat anti-mouse IgM (Millipore) or anti-rabbit IgG secondary antibodies (BioRad, Hercules, CA) at room temperature for 1 hour. Membranes were then washed 6 × 10 minutes with buffer before developing with ECL kit (Amersham Corp., Arlington Heights, IL). Images were taken and processed using the Fuji LAS-3000 Imaging System.
Real-Time Polymerase Chain Reaction
The RNA from the tissues of interest as isolated by the Trizol method (Invitrogen) and about 2 μg of the total RNA were reverse transcripted using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). Quantitative real-time polymerase chain reaction (RT-PCR) was performed on the 7300 RT-PCR system (Applied Biosystems) by using the TaqMan gene expression kit (Applied Biosystems). For each reaction, 2 μL of cDNA were used as a template in the PCR reaction mix, which contained 12.5 μL Universal PCR Master Mix (Applied Biosystems); 9.25 μL RNase-free H2O; and the 1.25 μL primer/probe mixture. The primer and probe for the FKRP (Mm00557870_m1) gene expression assay were purchased from Applied Biosystems, and β-glucuronidase (Gusb, Mm00446953_m1) was chosen as the endogenous control.
Immunofluorescent Staining
The immunofluorescent staining was performed according to the previous published protocol.43,53 Cryostatic sectioning of the GAS muscle tissue was performed at 8-μm thickness (Leica, Bannockburn, IL). For immunofluorescence staining, the unfixed muscle cryosections were immediately blocked in 10% horse serum and phosphate-buffered saline (PBS) with mouse-on-mouse blocking kit MKB-2213 (Vector Laboratories, Burlingame, CA) at room temperature for 1 hour, respectively. Monoclonal antibodies against α-dystroglycan (IIH6, adapted from Dr. Kevin P. Campbell) was diluted 1:50 in 2% horse serum–PBS and incubated with the cryosections for 2 hours at room temperature. After three washes with PBS (10 minutes/time), the sections were incubated with Cy-3-labeled anti-mouse secondary antibodies at 1:500 dilutions in 2% horse serum-PBS. After three washes, the samples were mounted in gel-mount aqueous antifading medium (Fisher, Pittsburgh, PA). Photographs were taken with a Nikon Eclipse TE300 microscope, using a Spot RT Slider digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI).
Laminin Ligand Overlay Assay
On a basis of a paper published by Michele and colleagues,54 ligand overlay assays were performed on PVDF membranes using mouse Engelbreth-Holm-Swarm laminin (Sigma). Briefly, PVDF membranes were blocked in laminin-binding buffer (LBB) (10 mmol/L triethanolamine, 140 mmol/L NaCl, 1 mmol/L MgCl2, 1 mmol/L CaCl2, pH 7.6) containing 5% nonfat dry milk followed by incubation of each ligand overnight at 4°C in LBB. Membranes were washed and incubated with antilaminin (Sigma) followed by anti-rabbit IgG-HRP. Blots were developed by enhanced chemiluminescence.
Solid-Phase Assay
Wheat germ agglutinin (WGA) eluates were diluted 1:50 in TBS and coated on polystyrene ELISA microplates (Corning Costar, Lowell, MA) for 16 hours at 4°C. Plates were washed in LBB and blocked for 2 hours in 3% bovine serum albumin in LBB. Mouse Engelbreth-Holm-Swarm laminin was diluted in LBB and applied for 2 hours. Wells were washed with 3% bovine serum albumin in LBB, incubated for 30 minutes with 1:10,000 anti-laminin (Sigma) followed by anti-rabbit horseradish peroxidase. Plates were developed with o-phenylenediamine dihydrochloride and H2O2; reactions were stopped with 2N H2SO4; and values were obtained on a microplate reader. The data were analyzed by the equation A = Bmax × (Kd + ×), where Kd is the dissociation constant, A is absorbance, and Bmax is maximal binding.
Myofiber Size Measurement
The measurement of the myofiber size in our study was done according our previously published protocol.55 We subjected 8-μm cryo-thin sections of the GAS muscles to immunofluorescence (IF) staining against the laminin-α2 to display the circumferences of the myofibers. Pictures were taken and the radius of the myofiber was analyzed by using Metamorph software with a minimum of 300 myofibers from each mouse and at least three mice in each group.
Results
In Vitro Screening of FKRP siRNA Oligonucleotides
Small-interference RNA design requires optimization of candidate sequences. We selected 10 potential target sequences against the mouse FKRP mRNA. The mouse FKRP gene (GenBank NM 173430) contains two noncoding exons (exon 1 and 2) and a 2.7-kb coding exon (exon 3). The exon 3 consists of 5′ untranslated region (5′-UTR), the open reading frame, and the 3′-UTR. All 10 siRNAs (FKRP1-10) were designed to target exon 3, with FKRP1 through FKRP4 targeting the 3′-UTR, whereas FKRP5 through FKRP10 targeted the coding sequence (Table 1). To perform a pilot screening on the efficiencies of RNAi-mediated FKRP knockdown, we synthesized four pairs of siRNA oligos against 4 of the 10 target sequences. The four duplex siRNA oligos (siFKRP1, siFKRP2, siFKRP3, and siFKRP5) were transfected into mouse Hepa 1–6 cell line that expresses FKRP. Quantitative RT-PCR on RNA isolated at 2 days post-transfection of siFKRP1 and siFKRP3 demonstrated approximately a 60% and a 37% reduction of FKRP mRNA, respectively, compared to the controls (see Supplemental Figure 1 at http://ajp.amjpathol.org). Meanwhile, siFKRP2 and siFKRP5 each achieved about 70% knockdown efficiency, whereas co-transfection of both siRNA oligos into Hepa 1–6 cells led to a nearly 80% reduction of FKRP mRNA. These results demonstrated that the use of two effective siRNA oligos together enhanced FKRP knockdown efficiency, which would make a significant difference in our subsequent experiments in vivo.
Table 1.
Target Sequences of FKRP siRNAs
| siRNA | Sequence | Target position in FKRP exon 3 |
|---|---|---|
| FKRP1 | 5′-GTTACCCAGTCTGTTTCAT-3′ | 2021-2040, 3′ UTR |
| FKRP2 | 5′-GCACTTCTGTCCCGCTTCA-3′ | 2118-2137, 3′ UTR |
| FKRP3 | 5′-GGCACTTTCCTTTGAGAGT-3′ | 2250-2269, 3′ UTR |
| FKRP4 | 5′-GTTTCATTTGAGATACCGA-3′ | 2033-2052, 3′ UTR |
| FKPR5 | 5′-GCGACTTCTTCCGAGTACA-3′ | 1336-1355, ORF |
| FKPR6 | 5′-GTGCCTAGCTCTGAACGTC-3′ | 0638-0657, ORF |
| FKPR7 | 5′-GCATCTACCTGGAGGACGT-3′ | 1234-1253, ORF |
| FKPR8 | 5′-GCCCTTTGCGGGTTTCATG-3′ | 1493-1512, ORF |
| FKPR9 | 5′-GGCTAGAGTGGTTTGGCTG-3′ | 0985-1004, ORF |
| FKPR10 | 5′-GTGAGAACAACCACCTGCA-3′ | 1360-1379, ORF |
Construction of shRNA Vectors and Testing Their Efficiency
Because the synthetic siRNA oligos FKRP2 and FKRP5 showed the highest knockdown efficiency, we separately subcloned each sequence in the form of an shRNA gene into an AAV vector expression plasmid. The resulting plasmid constructs were referred to as pAAV-U6-shFKRP2 and pAAV-U6-shFKRP5 (Figure 1A). The expression cassette was under the control of a mouse U6 promoter and therefore had an advantage over the synthetic siRNA in long-term expression.48 Because of the positive effect of co-transfecting both siRNA oligos, a dual-cassette vector pAAV-U6-shFKRP2 + 5 containing both shFKRP2 and shFKRP5 expression cassettes was also constructed (Figure 1A). We then examined whether plasmid transfection of our designed plasmid AAV FKRP shRNA vectors would yield similar knockdown efficiencies of the FKRP gene comparable to the synthetic siRNA oligos in Hepa 1–6 cells. Quantitative RT-PCR was performed using the RNA samples isolated from the transfected Hepa 1–6 cells at 48 hours post-transfection. As shown in Supplemental Figure S2A (http://ajp.amjpathol.org), pAAV-U6-shFKRP2 achieved a knockdown efficiency of approximately 60% (42.6% ± 12% FKRP remaining) comparable to that of FKRP2 siRNA oligo. In contrast, pAAV-U6-shFKRP5 led to only a 52.7% reduction of FKRP transcripts (47.3% ± 4.2% remaining), which was lower than the nearly 70% knockdown efficiency obtained by the FKRP5 siRNA oligo. The dual cassette pAAV-U6-shFKRP2 + 5 resulted in 66% knockdown of FKRP gene (34.4 ± 13.1% remaining). Cotransfection of pAAV-U6-shFKRP2 and pAAV-U6-shFKRP5 also resulted in similar knockdown efficiency at roughly 65% (35.6% ± 14.5% remaining). Altogether, the effects of the plasmid vectors of AAV-shRNA, especially the dual cassette plasmid on FKRP transcripts, were largely consistent with those of synthetic siRNA oligos (compare Supplemental Figure S2A with Supplemental Figure S1 at http://ajp.amjpathol.org). Subsequently, we also cloned the remaining six shRNAs (FKRP4, 6–10; see Table 1) into the pAAV-U6 backbone for shRNA expression and examined all 10 single-cassette shRNA plasmids side-by-side for their FKRP mRNA knockdown activities after transfection and quantitative RT-PCR. Among the six new constructs, only pAAV-U6-shFKRP9 achieved significant knockdown (59.2% ± 7.1% remaining), which was still less effective than pAAV-U6-shFKRP2 or pAAV-U6-shFKRP5 (data not shown).
Figure 1.
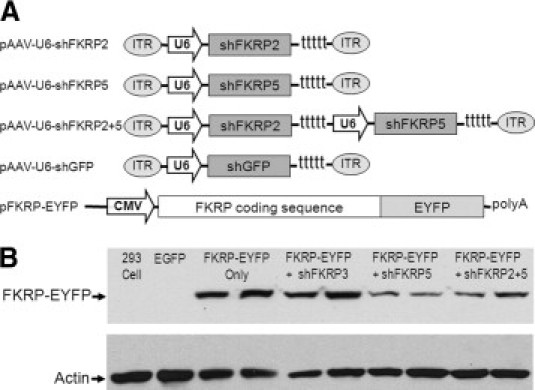
Design and testing of the constructs of recombinant adeno-associated viral vectors expressing the shRNA against FKRP. A: Schematic of pAAV-U6-shFKRP vectors. The FKRP2 and FKRP5 expression cassettes in the dual cassette lie in a head-to-tail orientation. The EYFP-tagged mouse FRKP plasmid was driven by the CMV promoter. B: Knockdown of FKRP-EYFP protein synthesis shown by Western analysis of 293 cells cotransfected with pFKRP-EYFP and pAAV-U6-shFKRP plasmids. FKRP-EYFP fusion protein, with a molecular weight of about 100 kDa, was detected by anti-GFP antibody, which cross-reacts with EYFP. Actin was stained as a sample loading control. Samples were prepared in duplicate.
Next we investigated the effects of FKRP knockdown on FKRP protein synthesis. Given that endogenous FKRP was difficult to detect,21,56 we had previously engineered a construct pFKRP-EYFP encoding mouse FKRP and a C-terminal fusion of enhanced yellow fluorescent protein (EYFP) (Figure 1A). The FKRP-EYFP fusion was used as an indirect indicator of FKRP protein after knockdown by plasmid vectors of AAV-shRNA. Cotransfection of pFKRP-EYFP expression plasmid and pAAV-U6-FKRP vectors was carried out in the 293 cell line. The FKRP-EYFP protein was monitored by fluorescent microscopy and Western blot. Our results showed that the intensity of EYFP fluorescence in the cotransfected cells was significantly reduced by pAAV-U6-shFKRP5 and the dual-cassette pAAV-U6-shFKRP2 + 5 compared to cells transfected with pFKRP-EYFP only (see Supplemental Figure S2B at http://ajp.amjpathol.org). Similar results were observed on Western blots of the cotransfected cells using an anti-GFP antibody that cross-reacts with EYFP (Figure 1B). On the other hand, pAAV-U6-shFKRP3 had no effects (Figure 1B, and see Supplemental Figure S2B at http://ajp.amjpathol.org). It is important to mention that the pFKRP-EYFP reporter did not contain the 3′-UTR of FKRP mRNA and thus can be targeted only by AAV vectors containing the shRNA5 sequence. The apparently more effective inhibition by shRNA5 on the reporter FKRP-EYFP fusion protein (Figure 1B) than on the endogenous FKRP mRNA (see Supplemental Figure S2A at http://ajp.amjpathol.org) is likely to be due to the co-entry of the shRNA5 plasmid and its target FKRP-EYFG plasmid into the same cotransfected cells. On the other hand, inhibition of the endogenous FKRP mRNA in Hepa 6-1 cell line is heavily dependent on the percentage of cells transfected by the shRNA plasmid. In summary, our findings showed that pAAV-U6-shFKRP2 + 5 was able to suppress FKRP expression effectively and to reduce its mRNA and the protein.
Effective FKRP Knockdown in Adult Mouse Muscle by AAV shRNA Vectors
To examine the effects of the shFKRP in vivo, we packaged the U6-shFKRP2, the U6-shFKRP5, and the dual-expression cassette U6-shFKRP2 + 5 into the AAV serotype 6 (AAV6) vector for effective gene transfer in the skeletal muscles by local intramuscular injection.57–60 Lower hind-leg muscles (GAS and tibalis anterior [TA]) of 2-month-old ICR mice (n = 10) were injected intramuscularly with the respective AAV vector or control PBS saline. Both the AAV- and PBS-injected contralateral control muscles were collected at 2 months and 10 months later (n = 5 in each time point) for analyses of FKRP expression as well as of dystrophic pathology. Quantitative RT-PCR was performed using RNA samples extracted from the GAS muscles. At 2 months and 10 months after injection, the single-cassette shFKRP2 reduced FKRP expression by 10.6% ± 10.8% and 46.5% ± 4.1%, respectively, when compared to the PBS-treated GAS muscles (Figure 2). Meanwhile, the single-cassette shFKRP5 attained a knockdown efficiency of 48.3% ± 10.1% (2 months) and 45.0% ± 7.4% (10 months). In contrast, the dual-cassette shFKRP2 + 5 achieved 71.6% ± 2% and 72.3% ± 7.8% knockdown of FKRP expression at the two time points (Figure 2). The observations that the dual cassette was more effective than the single cassette in knocking down FKRP gene expression was in agreement with our previous results of screening siRNA and shRNA in cell cultures. It remains unclear why shFKRP2 at 2 months after injection attained only a 10% knockdown.
Figure 2.

Analysis of FKRP mRNA knockdown in GAS muscles after intramuscular injection of AAV6 shFKRP vectors. At the 2-month time point, quantitative RT-PCR analyses of FKRP mRNA in the muscle samples showed significant knockdown in groups treated with shFKRP5 (48.3 ± 10.1%) and shFKRP2 + 5 (71.6 ± 1.2%) but not in the group treated with shFKRP2 (10.6 ± 10.8%). However, at the 10-month time point, FKRP mRNA was knocked down in all AAV-treated groups, shFKRP2 (46.4 ± 4.1%), shFKRP5 (45.0 ± 7.4%), and dual shFKRP2 + 5 (72.3% ± 7.8%) (n = 5, mean ± SD; * P < 0.05).
FKRP Knockdown in Adult Mouse Muscle Caused Dystrophic Pathology
When we examined the AAV-treated mice, we noticed that muscle pathology was essentially absent at 2 months after AAV U6-shFKRP treatment and was indistinguishable from both of the PBS-treated controls (Figure 3A, left panels) and from the scrambled control muscles (see Supplemental Figure S3, upper row, at http://ajp.amjpathol.org). Although quantitative RT-PCR of the same muscle samples showed the highest reduction of FKRP expression by the dual-cassette shFKRP2 + 5 (Figure 2), the mice apparently did not develop dystrophic phenotypes. It is striking that at 10 months after AAV treatment, both the TA and GAS muscles of these mice were found to contain large number of myofibers with centrally localized nuclei (CN), a hallmark of muscle degeneration and regeneration (Figure 3A, right panels). However, muscles treated with the single-cassette AAV vector, either shFKRP2 or shFKRP5, showed only minimal signs of pathology, displaying much fewer centrally nucleated myofibers (Figure 3, A, right panels). Masson trichrome staining was also performed on GAS muscles from each group 10 months after injection to detect signs of fibrosis. Our findings demonstrated that deposition of connective tissues was more prominent in the GAS muscles of the shFKRP2 + 5–treated mice than the other two single-cassette–treated mice or the control group treated with PBS (Figure 3B). Altogether, these results suggest that a reduction of FKRP gene expression by approximately 75% in the normal muscle is sufficient for the mice to develop progressive dystrophic pathology between 2 and 10 months. The phenotypes of our FKRP knockdown mice are consistent with the mild pathology and late-onset progression commonly observed in patients with LGMD2I.
Figure 3.

Analysis of the muscle fiber pathology and morphology. GAS and tibialis anterior muscles transduced with AAV6-shFKRP were collected 2 months and 10 months postinjection. A: Cryo-thin sections of the muscle samples were analyzed using H&E staining. Similar pathological signs in the GAS and TA muscles were observed in both AAV-treated and PBS-injected groups at 2 months postinjection. Note that more centrally nucleated myofibers were found in the shFKRP2 + 5–treated muscle at 10 months postinjection (arrows). Scale bar, 100 μm. B: Increase of connective tissue deposition in the shFKRP-treated muscle. Masson trichrome staining was performed on the cryo-thin section of GAS muscles from each treatment group at 10 months postinjection. Note that connective tissues were frequently found in the intermediate of myofibers (arrows) in both single- and dual-cassette shFKRP-treated groups. Scale bar, 100 μm.
To further explore this issue, quantitative analysis of the myofibers showed a CN rate of 51.0% ± 8.5% in the dual-cassette shFKRP2 + 5–treated group, which was much higher than the PBS-treated control group of 1.0% ± 0.5% (Figure 4A, t-test P < 0.001). Although the U6-shFKRP2–treated muscles showed a CN rate of 12.1% ± 3.3%, which was statistically higher than the PBS-treated group, the overall rate of CN in the single-cassette shFKRP-treated muscles was still significantly lower than that observed in the shFKRP2 + 5–treated group (Figure 4A). Subsequently, quantitative analysis of the myofiber radius revealed a much broader distribution of fiber size (more smaller and larger myofibers) in shFKRP2 + 5–treated muscles (Figure 4B), suggesting chronic muscle degeneration and regeneration. Finally, we also monitored serum creatine kinase (CK) levels because the vector-injected dystrophic limb muscles could release CK systemically. Indeed, we found that the serum CK activities in mice that were treated with the shFKRP5 (1480.4 ± 214.4 IU/L) and the dual shFKRP2 + 5 (2104 ± 675.7 UI/L) were significantly higher than those of the control mice (884.1 ± 214.5 IU/L) (Figure 4C). The elevated serum CK levels are in agreement with the dystrophic pathology of the vector-treated limb muscles.
Figure 4.
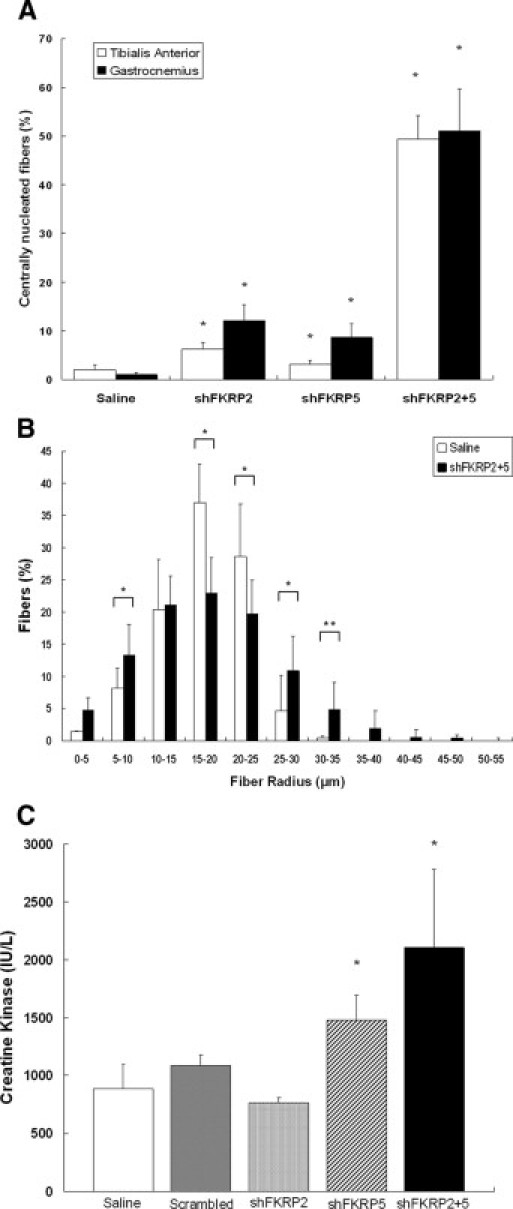
Quantification of the muscle fibers' pathology and morphology. A:. The numbers of centrally nucleated fibers in AAV-shFKRP-treated TA and GAS muscles were counted in five random fields of view under the microscope. In both TA and GAS muscles, not only the single-cassette groups treated with shFKRP2 (6.3% ± 1.4% and 12.1% ± 3.3%) and shFKRP5 (3.1% ± 0.9% and 8.7% ± 2.8%), but also the dual-cassette group treated with shFKRP2 + 5 (49.4% ± 4.9% and 51.1% ± 8.5%) showed centrally nuclear myopathy that was significantly different from that of the control group (2.0% ± 1.05% and 1.01% ± 0.48%, n = 5, mean ± SD, *P < 0.001). B: The radius of individual GAS myofibers in each treatment group was measured and evaluated by MetaMorph software. Note that the shFKRP2 + 5–treated GAS muscles (black column) displayed more variable and uneven distribution of myofiber radius than did the PBS-treated muscle (white column) (n = 5, mean ± SD, Student's t-test, *P < 0.001; **P < 0.05). C: The level of creatine kinase (CK) in each treatment group was analyzed. Note that compared with the control (884.1 ± 214.5 IU/L; white column), the levels of CK in shFKRP2 + 5–treated mice (2104 ± 675.7 UI/L; black column) and in shFKRP5 (1480.4 ± 214.4 IU/L; diagonal column) were significantly increased (n = 5, mean ± SD, *P < 0.001).
FKRP Knockdown in Adult Mouse Muscle Resulted in Hypoglycosylation of α-Dystroglycan
It has been reported that FKRP mutations often lead to secondary defects in α-dystroglycan glycosylation, that is, hypoglycosylation.20,30–33 The monoclonal antibody IIH6 specifically recognizes the glycosylated epitopes on α-DG61–63 and was often used to detect abnormal glycosylation of α-DG in the muscle samples of patients having LGMD2I.13,31–33 We therefore used this antibody to examine whether hypoglycosylation of α-DG occurred in AAV U6-shRNA–treated muscles as a result of FKRP knockdown. First, immunofluorescent staining was performed on cryo-thin sections of the GAS muscles that were collected 10 months after treatment. The GAS muscle from mdx mice was used as a negative control because it lacks α-DG at the sarcolemma.64 When muscle sections were labeled with the IIH6 antibody, the immunofluorescent signal of AAV-U6-shRNA–treated muscles was noticeably reduced in comparison to the PBS-treated muscles, suggesting aberrant modification of α-DG as a result of FKRP knockdown (Figure 5A). The reduction of signal was especially apparent in the dual-cassette shFKRP2 + 5–treated muscle. Furthermore, Western blots were performed using proteins extracted from the GAS muscle of each treatment group (Figure 5B). Consistent with the immunofluorescent results, reduced IIH6 immunoreactivity at the expected molecule weight for α-DG was observed in all AAV-shFKRP–treated muscles (Figure 5B, lanes 3–6). The other component of the dystroglycan, β-dystroglycan (β-DG), was also monitored by Western blot. As expected,65 there was no reduction of β-DG in all AAV-treated muscle when compared to saline control muscle except muscle from the mdx control, which has been well documented to have much reduced dystroglycans due to dystrophin deficiency. These results suggest that the shKFRPs did not inhibit dystroglycan gene expression, that is, precursor mRNA and protein synthesis; but the shFKRPs did significantly affect and reduce the level of the fully glycosylated α-DG.
Figure 5.

Abnormal glycosylation of α-dystroglycan in shFKRP-treated muscles after 10 months. A: Immunofluorescent staining of the AAV-shFKRP-treated GAS muscles with α-dystroglycan–specific antibody IIH6 at 10 months postinjection. Note the reduction of staining in the shFKRP2 + 5–treated muscles. B: Western blot of proteins extracted from GAS muscles of each treated group were done using anti-α-dystroglycan (IIH6) and β-dystroglycan (DAG-1) antibodies to evaluate the glycosylation of α-dystroglycan and the expression of β-dystroglycan, respectively. The GAS muscles from mdx mice and PBS-treated mice were used as the negative and positive controls, respectively. C: A ligand overlay assay using laminin on a blot of WGA-enriched shFKPR knockdown muscles was performed. Note that the affinity of the α-dystroglycan to laminin in shFKRP2 + 5–treated muscles (lanes 6 and 7) was weaker than in the control group (lane 2). D: Solid-phase assays were performed to quantitate the maximum laminin binding ability to α-DG in the shFKRP-treated muscles. Note that the binding ability of α-DG in shFKRP2 (53.7% ± 5.0%); -5(40.6% ± 4.4%); and -2 + 5 (31.0% ± 3.0%) treated groups was decreased and weaker than in the control group (n = 5, mean ± SD, *P < 0.001.)
Furthermore, we performed the ligand overlay assay and solid-phase assay using laminin on a blot of WGA-enriched shFKPR knockdown muscles (Figure 5, C and D). In muscles from the shFKRP2 + 5 group, the ligand binding activities were markedly reduced, similar to those of the dystrophin-deficient mdx mouse group, and much lower than those of the saline-treated and scrambled-vector–treated control groups (Figure 5C). To quantify the total ligand-binding activity of intact (nondenatured) α-DG, we performed solid-phase laminin-binding assays of WGA-enriched protein fractions from the same muscle samples (Figure 5D). The total high-affinity laminin-binding activity in the shFKRP2, shFKRP5, and shFKRP2 + 5 group was reduced by approximately 50%, 60%, and 70%, consistent with the data from the laminin overlay assay (Figure 5C). These results suggest that the reduced laminin-binding activity in the FKRP knockdown muscles are likely to be caused by the reduced glycosylation of α-DG.
Discussion
LGMD2I was originally reported in a Tunisian family.19 Patients who have LGMD2I with mutations in the FKRP gene usually manifest mild muscular dystrophy with late onset and no central nervous system abnormalities. However, FKRP mutations can also cause more severe and early-onset congenital muscular dystrophy, muscular dystrophy type 1C, and muscle-eye-brain disease. Despite the recent gains in knowledge about LGMD2I and FKRP, there is still a lack of a representative animal model that recapitulates LGMD2I phenotypes. The classical gene knockout approach resulted in embryonic lethality in mice when the FKRP gene was completely ablated. To overcome this problem, genetic knockin of the mutant human FKRP gene has been investigated in other studies. Disease-causing missense mutations, for example L276I and Y307N, were engineered in mice, anticipating that these mutations would cause phenotypes characteristic of human patients. However, these mice often did not generate phenotypes unless the neomycin selection cassette was present in the mutant FKRP allele,37 which reduced FKRP transcripts and also caused a severe muscle-brain-eye phenotype. A recent study by our group showed that homozygous knockin of a missense P448L mutation also manifested severe muscle-brain-eye phenotypes, but some homozygous mice were viable to adult age.66 In order to avoid disrupting embryonic development and complications in the central nervous system, we used RNAi technology to effectively knockdown FKRP gene expression in normal adult mouse muscle via AAV-mediated postnatal gene transfer. Intramuscular injection of AAV shFKRP to the hind leg muscles of adult mice was shown to sustain efficient and long-term FKRP gene knockdown, resulting in a progressive and mild dystrophic pathology similar to that observed in patients having LGMD2I.
In the first part of our study, we selected 10 potential target sites (Table 1) on the mouse FKRP gene to screen for the most effective siRNA sequences. Four randomly selected siRNAs in duplex forms were synthesized and tested in an FKRP-expressing cell line Hepa 1–6. Two of the four synthetic siRNAs (FKRP2 and FKRP5) were shown to have the best knockdown efficiency, approximately 70% to 80% (see Supplemental Figure S1 at http://ajp.amjpathol.org). To take advantages of the long-term effects of AAV-mediated gene transduction in vivo,40–46,67 we cloned the two most effective siRNA sequences into an expression vector driven by the U6 promoter in either single- or dual-expression cassettes. It is worth mentioning that the shFKRP expression constructs seemed to be less effective in the transfection experiments when compared to the synthetic siRNAs (compare Supplemental Figure S1 and Supplemental Figure S2A at http://ajp.amjpathol.org). In addition to the possibility of transfection-efficiency differences between the oligos and plasmids, their structural differences might also contribute to their biological activities. The synthetic siRNA forms an open-ended duplex, whereas the AAV-shRNA takes the shape of a short hairpin. The latter requires additional enzymatic processing in the cells and therefore might not behave in the same way as the synthetic counterpart in some cases (for example, siFKRP1 vs. shFKRP1, in Supplemental Figure S1 and Supplemental Figure S2A at http://ajp.amjpathol.org).
Adeno-associated virus-based vectors are well documented for their ability to provide efficient and long-term gene delivery to the muscles.43,48,68,69 AAV vector-mediated gene delivery of shRNA has been used to successfully knockdown dystrophin expression in mouse skeletal muscle.60 Suppression of dystrophin expression was maintained for up to 1 year. It is interesting that no dystrophic pathology was observed in these mice. Dickson and colleagues60 have discussed a number of possible reasons for this finding, including the long half-life of dystrophin protein. Another reason that might account for the failure to induce dystrophic pathology in normal mice might be directly related to the residual amount of dystrophin that escaped shRNA-mediated knockdown. Previous transgenic mouse studies showed that 10% of the normal dystrophin expression would be sufficient to prevent dystrophic pathology in mdx mice,70 suggesting that more than a 90% knockdown of the normal level of dystrophin expression is required to cause dystrophic phenotypes. In another study, we reported that AAV-mediated shRNA knockdown in TA muscle caused approximately 70% down-regulation of A-type lamin at 2 weeks postinjection and generated phenotypes reminiscent of neuromuscular junction defect.48 These studies indicated that there might be a threshold for the minimal amount of the knockdown gene required for function in each disease model.
Our study might provide clues about how much reduction of FKRP expression would be necessary to generate dystrophic phenotypes. When the single-expression cassette shFKRP (shFKRP2 or shFKRP5) was delivered to the muscle via AAV6, nearly 50% reduction of FKRP gene expression was observed. However, a near 75% reduction in FKRP gene expression was reached by the dual-cassette shFKRP2 + 5 (Figure 2). In fact, the dual cassette has consistently achieved the highest knockdown efficiency in both cell cultures and muscle, suggesting an additive effect. The 75% knockdown of the FKRP gene expression was apparently sufficient to induce overt dystrophic pathology in the treated muscles 10 months after injection (Figure 3A). Approximately one half of the myofibers showed centrally localized nuclei (Figure 4A); the morphology of the myofibers was smaller and more uneven (Figure 4B); and the serum CK was 2.5-fold of normal controls (Figure 4C). On the other hand, very limited pathology and much fewer centrally localized nuclei were observed in the single-cassette shRNA-treated muscles, despite a nearly 50% knockdown of FKRP gene expression by the single cassette shFKRP for the same period. It should be noted that the percentage of knockdown determined by quantitative RT-PCR represented an average of all of the myofibers in the tissue that was used to extract the RNA. We suspect that the small number of myofibers with central nucleation in the single-cassette–treated muscle was the result of extra-high uptake of the vector by a small number of myofibers near the needle track of intramuscular injection. The extra-high vector uptake would lead to more effective knockdown, sufficient to induce limited pathology even in the cases of the single shFKRP cassette. However, the majority of the myofibers a distance from the needle track might not have the extra-high expression needed to achieve sufficient FKRP knockdown, given that the single shFKRP was not as potent as the dual shFKRP2 + 5. This could offer a plausible explanation for the low-level central nucleation in the single-cassette shFKRP2- and shFKRP5-treated muscles. By the same token, the uneven nature of vector in intramuscular injections should be taken into consideration when interpreting data.
These observations suggest that a reduction of approximately 75%, but not 50%, of the normal level of FKRP expression induces overt dystrophic pathology. This could provide useful guidance and help to narrow the range or threshold of effectively therapeutic restoration of the FKRP, which we believe is in the neighborhood of 25% of the wild-type levels. It is clearly important to take this value into consideration when designing gene therapy and small-molecule drug therapy such as stop codon read through. However, because of the lack of an antibody to detect endogenous FKRP, either by immunofluorescent staining or by Western blot, we were unable to determine the level of FKRP protein in the knockdown muscles. As a result, our data concerning FKRP expression were indirectly based on quantitative RT-PCR of mRNA.
Another important finding in our FKRP knockdown mice was the development of progressive and relatively mild pathology, despite the fact that FKRP gene expression was strongly suppressed at young adulthood. When we examined the hind-leg muscles (GAS and TA) 2 months after AAV-shFKRP vector treatment, quantitative RT-PCR showed that FKRP gene expression was significantly reduced as much as 70%. However, we did not detect apparent muscle pathology in the mice until 10 months of age. This observation is in line with the mild phenotypes and late onset seen in patients with LGMD2I. It will be of interest to investigate whether other genes in the knockdown muscle are compensating for FKRP deficiency during the course of disease development.
The function of FKRP is not clearly understood, but it is thought to play an important role in the post-translational modification of α-DG. Consistent with this hypothesis, glycosylation of α-DG was indeed much reduced in the FKRP knockdown muscles, as revealed by the presence of IIH6 antibody, specific for the polysaccharide side chain (Figure 5B). Three lines of evidence unequivocally point to the reduction of α-DG glycosylation in knockdown of FKRP expression. First, the protein quantity of β-DG, which shares the same mRNA and protein precursor with α-DG, was not reduced other than slightly increased as shown by Western blot (Figure 5B). Second, quantitative PCR of dystroglycan mRNA transcripts did not reveal appreciable alteration in any of the muscles (data not shown). Third, and most important, the laminin-binding activity of the α-DG in the shFKRP-treated muscles as reduced in both the ligand binding assay (Figure 5C) and solid-phase assay (Figure 5D). Thus, the reduced detection of α-DG by IIH6 antibody was not caused by reduction of dystroglycan gene expression. It is interesting that the molecular weight of the α-DG in the FKRP knockdown muscles was unchanged, unlike the evident reduction on the molecular weight of α-DG seen in some patients with FKRP missense mutations. A possible explanation is that certain mutant FKRP proteins may exhibit altered enzymatic properties, which incorrectly glycosylate the α-DG. On the other hand, shRNA knockdown of FKRP reduced the quantity but not the property of the enzyme. Thus, reduced amounts of wildtype FKRP should still be able to correctly and fully glycosylate α-DG, although only a fraction of all α-DG, resulting in reduced intensity but normal molecular weight of α-DG on Western blot as detected by the IIH6 antibody. It has been well documented that α-DG interacts with laminin and plays a critical role in linking the cytoskeleton and the extracellular matrix.71,72 In several clinical reports, the expression of the laminin α2 chain was found to be lower in the biopsies of patients with LGMD2I.16,33,73 In our knockdown mice, however, the level of laminin α2 chain, as judged by immunofluorescent staining, had not changed in the treated GAS muscle (data not shown); this could explain the relatively mild pathology.
This study demonstrated that the expression of FKRP can be down-regulated by RNAi technology and that using the AAV vector as a delivery system can achieve effective and long-term suppression of FKRP gene expression in vivo. Based on the pathology and progression of the muscle phenotypes, our mice could be a useful model for the study of the mild form of LGMD2I.
Acknowledgments
We thank the McColl Foundation and the Carolinas Muscular Dystrophy Research Endowment at the Carolinas HealthCare Foundation (Charlotte, NC) for the generous support of this work. We thank Dr. Kevin P. Campbell (University of Iowa) for kindly providing the α-dystroglycan antibody (IIH6) and Dr. Heather Hui Zhou for her helpful scientific advice and discussion of this work.
Footnotes
Supported by the McColl Foundation and the Carolinas Muscular Dystrophy Research Endowment at the Carolinas HealthCare Foundation, Charlotte, NC.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.11.020.
Supplementary data
Supplemental Figure 1.
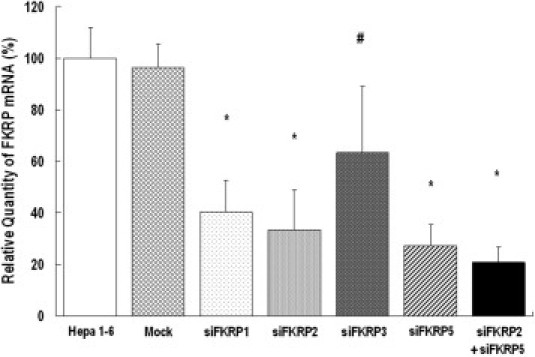
FKRP expression knockdown in Hepa 1-6 cell line. The given FKRP siRNA synthetic oligos in duplex form were transfected into Hepa 1-6 cells. mRNAs were harvested at 48 hours post-transfection, which was followed by quantitative RT-PCR to determine the level of FKRP transcripts. The transfection of the two most effective, siFKRP2 and siFKRP5, resulted in a 60% to 70% reduction of endogenous FKRP mRNA expression. Furthermore, knockdown efficiency was improved to approximately 80% with co-transfection of both siFKRP2 and siFKRP5 (n = 12, mean ± SD, *P < 0.001, #P < 0.005).
Supplemental Figure 2.
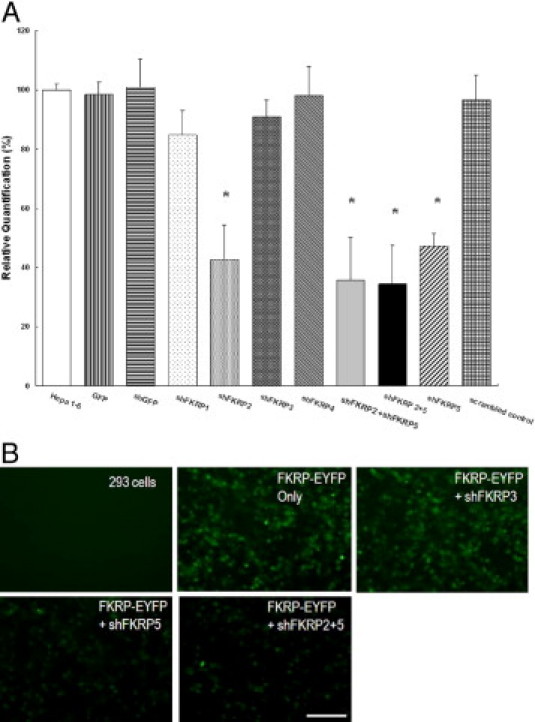
Testing of the constructs of recombinant adeno-associated viral vectors expressing the shRNA against FKRP. A: Reduction of the FKRP mRNA transcript by transfected pAAV-U6-shFKRP (2 μg) in the Hepa 1-6 cell line (n = 9, mean ± SD, *P < 0.001, #P < 0.005). B: Overexpressed mouse FKRP-EYFP fusion protein in 293 cells was partially knocked down by pAAV-U6-shFKRP2 + 5. Transfected cells were observed under fluorescent microscope. Scale bar, 100 μm.
Supplemental Figure 3.

Muscle biopsy of the scrambled vector-treated muscle. H&E staining was performed in the cryo-thin sections of the scrambled vector-control-treated muscles (GAS and TA) at 2 months and 10 months after injection. Note that no significant pathological signs like centrally nucleated myofibers were found at either time points (n = 5).
References
- 1.Bushby K.M. The limb-girdle muscular dystrophies: multiple genes, multiple mechanisms. Hum Mol Genet. 1999;8:1875–1882. doi: 10.1093/hmg/8.10.1875. [DOI] [PubMed] [Google Scholar]
- 2.Moreira E.S., Vainzof M., Marie S.K., Sertié A.L., Zatz M., Passos-Bueno M.R. The seventh form of autosomal recessive limb-girdle muscular dystrophy is mapped to 17q11-12. Am J Hum Genet. 1997;61:151–159. doi: 10.1086/513889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bushby K.M. Making sense of the limb girdle muscular dystrophies. Brain. 1999;122(Pt 8):1403–1420. doi: 10.1093/brain/122.8.1403. [DOI] [PubMed] [Google Scholar]
- 4.Guglieri M., Straub V., Bushby K., Lochmuller H. Limb-girdle muscular dystrophies. Curr Opin Neurol. 2008;21:576–584. doi: 10.1097/WCO.0b013e32830efdc2. [DOI] [PubMed] [Google Scholar]
- 5.Laval S.H., Bushby K.M. Limb-girdle muscular dystrophies: from genetics to molecular pathology. Neuropathol Appl Neurobiol. 2004;30:91–105. doi: 10.1111/j.1365-2990.2004.00555.x. [DOI] [PubMed] [Google Scholar]
- 6.Sveen M.L., Schwartz M., Vissing J. High prevalence and phenotype-genotype correlations of limb girdle muscular dystrophy type 2I in Denmark. Ann Neurol. 2006;59:808–815. doi: 10.1002/ana.20824. [DOI] [PubMed] [Google Scholar]
- 7.Poppe M., Cree L., Bourke J., Eagle M., Anderson L.V., Birchall D., Brockington M., Buddles M., Busby M., Muntoni F., Wills A., Bushby K. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology. 2003;60:1246–1251. doi: 10.1212/01.wnl.0000058902.88181.3d. [DOI] [PubMed] [Google Scholar]
- 8.Frosk P., Greenberg C.R., Tennese A.A., Lamont R., Nylen E., Hirst C., Frappier D., Roslin N.M., Zaik M., Bushby K., Straub V., Zatz M., de Paula F., Morgan K., Fujiwara T.M., Wrogemann K. The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populations. Hum Mutat. 2005;25:38–44. doi: 10.1002/humu.20110. [DOI] [PubMed] [Google Scholar]
- 9.Moore S.A., Shilling C.J., Westra S., Wall C., Wicklund M.P., Stolle C., Brown C.A., Michele D.E., Piccolo F., Winder T.L., Stence A., Barresi R., King N., King W., Florence J., Campbell K.P., Fenichel G.M., Stedman H.H., Kissel J.T., Griggs R.C., Pandya S., Mathews K.D., Pestronk A., Serrano C., Darvish D., Mendell J.R. Limb-girdle muscular dystrophy in the United States. J Neuropathol Exp Neurol. 2006;65:995–1003. doi: 10.1097/01.jnen.0000235854.77716.6c. [DOI] [PubMed] [Google Scholar]
- 10.Kang P.B., Feener C.A., Estrella E., Thorne M., White A.J., Darras B.T., Amato A.A., Kunkel L.M. LGMD2I in a North American population. BMC Musculoskelet Disord. 2007;8:115. doi: 10.1186/1471-2474-8-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaul C., Deschauer M., Tempelmann C., Vielhaber S., Klein H.U., Heinze H.J., Zierz S., Grothues F. Cardiac involvement in limb-girdle muscular dystrophy 2I: conventional cardiac diagnostic and cardiovascular magnetic resonance. J Neurol. 2006;253:1317–1322. doi: 10.1007/s00415-006-0213-0. [DOI] [PubMed] [Google Scholar]
- 12.Poppe M., Bourke J., Eagle M., Frosk P., Wrogemann K., Greenberg C., Muntoni F., Voit T., Straub V., Hilton-Jones D., Shirodaria C., Bushby K. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann Neurol. 2004;56:738–741. doi: 10.1002/ana.20283. [DOI] [PubMed] [Google Scholar]
- 13.Boito C.A., Melacini P., Vianello A., Prandini P., Gavassini B.F., Bagattin A., Siciliano G., Angelini C., Pegoraro E. Clinical and molecular characterization of patients with limb-girdle muscular dystrophy type 2I. Arch Neurol. 2005;62:1894–1899. doi: 10.1001/archneur.62.12.1894. [DOI] [PubMed] [Google Scholar]
- 14.Mercuri E., Brockington M., Straub V., Quijano-Roy S., Yuva Y., Herrmann R., Brown S.C., Torelli S., Dubowitz V., Blake D.J., Romero N.B., Estournet B., Sewry C.A., Guicheney P., Voit T., Muntoni F. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol. 2003;53:537–542. doi: 10.1002/ana.10559. [DOI] [PubMed] [Google Scholar]
- 15.Walter M.C., Petersen J.A., Stucka R., Fischer D., Schroder R., Vorgerd M., Schroers A., Schreiber H., Hanemann C.O., Knirsch U., Rosenbohm A., Huebner A., Barisic N., Horvath R., Komoly S., Reilich P., Muller-Felber W., Pongratz D., Muller J.S., Auerswald E.A., Lochmuller H. FKRP (826C>A) frequently causes limb-girdle muscular dystrophy in German patients. J Med Genet. 2004;41:e50. doi: 10.1136/jmg.2003.013953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brockington M., Yuva Y., Prandini P., Brown S.C., Torelli S., Benson M.A., Herrmann R., Anderson L.V., Bashir R., Burgunder J.M., Fallet S., Romero N., Fardeau M., Straub V., Storey G., Pollitt C., Richard I., Sewry C.A., Bushby K., Voit T., Blake D.J., Muntoni F. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet. 2001;10:2851–2859. doi: 10.1093/hmg/10.25.2851. [DOI] [PubMed] [Google Scholar]
- 17.Evans V., Foster H., Graham I.R., Foster K., Athanasopoulos T., Simons J.P., Dickson G., Owen J.S. Human apolipoprotein E expression from mouse skeletal muscle by electrotransfer of nonviral DNA (plasmid) and pseudotyped recombinant adeno-associated virus (AAV2/7) Hum Gene Ther. 2008;19:569–578. doi: 10.1089/hum.2007.169. [DOI] [PubMed] [Google Scholar]
- 18.Fischer D., Walter M.C., Kesper K., Petersen J.A., Aurino S., Nigro V., Kubisch C., Meindl T., Lochmuller H., Wilhelm K., Urbach H., Schroder R. Diagnostic value of muscle MRI in differentiating LGMD2I from other LGMDs. J Neurol. 2005;252:538–547. doi: 10.1007/s00415-005-0684-4. [DOI] [PubMed] [Google Scholar]
- 19.Driss A., Amouri R., Ben Hamida C., Souilem S., Gouider-Khouja N., Ben Hamida M., Hentati F. A new locus for autosomal recessive limb-girdle muscular dystrophy in a large consanguineous Tunisian family maps to chromosome 19q13.3. Neuromuscul Disord. 2000;10:240–246. doi: 10.1016/s0960-8966(00)00099-7. [DOI] [PubMed] [Google Scholar]
- 20.Brockington M., Blake D.J., Prandini P., Brown S.C., Torelli S., Benson M.A., Ponting C.P., Estournet B., Romero N.B., Mercuri E., Voit T., Sewry C.A., Guicheney P., Muntoni F. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet. 2001;69:1198–1209. doi: 10.1086/324412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Esapa C.T., Benson M.A., Schroder J.E., Martin-Rendon E., Brockington M., Brown S.C., Muntoni F., Kroger S., Blake D.J. Functional requirements for fukutin-related protein in the Golgi apparatus. Hum Mol Genet. 2002;11:3319–3331. doi: 10.1093/hmg/11.26.3319. [DOI] [PubMed] [Google Scholar]
- 22.Dolatshad N.F., Brockington M., Torelli S., Skordis L., Wever U., Wells D.J., Muntoni F., Brown S.C. Mutated fukutin-related protein (FKRP) localizes as wild type in differentiated muscle cells. Exp Cell Res. 2005;309:370–378. doi: 10.1016/j.yexcr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 23.Keramaris-Vrantsis E., Lu P.J., Doran T., Zillmer A., Ashar J., Esapa C.T., Benson M.A., Blake D.J., Rosenfeld J., Lu Q.L. Fukutin-related protein localizes to the Golgi apparatus and mutations lead to mislocalization in muscle in vivo. Muscle Nerve. 2007;36:455–465. doi: 10.1002/mus.20833. [DOI] [PubMed] [Google Scholar]
- 24.Torelli S., Brown S.C., Brockington M., Dolatshad N.F., Jimenez C., Skordis L., Feng L.H., Merlini L., Jones D.H., Romero N., Wewer U., Voit T., Sewry C.A., Noguchi S., Nishino I., Muntoni F. Sub-cellular localization of fukutin-related protein in different cell lines and in the muscle of patients with MDC1C and LGMD2I. Neuromuscul Disord. 2005;15:836–843. doi: 10.1016/j.nmd.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto H., Noguchi S., Sugie K., Ogawa M., Murayama K., Hayashi Y.K., Nishino I. Subcellular localization of fukutin and fukutin-related protein in muscle cells. J Biochem. 2004;135:709–712. doi: 10.1093/jb/mvh086. [DOI] [PubMed] [Google Scholar]
- 26.Beedle A.M., Nienaber P.M., Campbell K.P. Fukutin-related protein associates with the sarcolemmal dystrophin-glycoprotein complex. J Biol Chem. 2007;282:16713–16717. doi: 10.1074/jbc.C700061200. [DOI] [PubMed] [Google Scholar]
- 27.Esapa C.T., McIlhinney R.A., Blake D.J. Fukutin-related protein mutations that cause congenital muscular dystrophy result in ER-retention of the mutant protein in cultured cells. Hum Mol Genet. 2005;14:295–305. doi: 10.1093/hmg/ddi026. [DOI] [PubMed] [Google Scholar]
- 28.Aravind L., Koonin E.V. The fukutin protein family; predicted enzymes modifying cell-surface molecules. Curr Biol. 1999;9:R836–R837. doi: 10.1016/s0960-9822(00)80039-1. [DOI] [PubMed] [Google Scholar]
- 29.Ervasti J.M., Campbell K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 30.Mercuri E., Messina S., Bruno C., Mora M., Pegoraro E., Comi G.P., D'Amico A., Aiello C., Biancheri R., Berardinelli A., Boffi P., Cassandrini D., Laverda A., Moggio M., Morandi L., Moroni I., Pane M., Pezzani R., Pichiecchio A., Pini A., Minetti C., Mongini T., Mottarelli E., Ricci E., Ruggieri A., Saredi S., Scuderi C., Tessa A., Toscano A., Tortorella G., Trevisan C.P., Uggetti C., Vasco G., Santorelli F.M., Bertini E. Congenital muscular dystrophies with defective glycosylation of dystroglycan: a population study. Neurology. 2009;72:1802–1809. doi: 10.1212/01.wnl.0000346518.68110.60. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto L.U., Velloso F.J., Lima B.L., Fogaca L.L., de Paula F., Vieira N.M., Zatz M., Vainzof M. Muscle protein alterations in LGMD2I patients with different mutations in the fukutin-related protein gene. J Histochem Cytochem. 2008;56:995–1001. doi: 10.1369/jhc.2008.951772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jimenez-Mallebrera C., Torelli S., Feng L., Kim J., Godfrey C., Clement E., Mein R., Abbs S., Brown S.C., Campbell K.P., Kroger S., Talim B., Topaloglu H., Quinlivan R., Roper H., Childs A.M., Kinali M., Sewry C.A., Muntoni F. A comparative study of alpha-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of alpha-dystroglycan does not consistently correlate with clinical severity. Brain Pathol. 2009;19:596–611. doi: 10.1111/j.1750-3639.2008.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown S.C., Torelli S., Brockington M., Yuva Y., Jimenez C., Feng L., Anderson L., Ugo I., Kroger S., Bushby K., Voit T., Sewry C., Muntoni F. Abnormalities in alpha-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am J Pathol. 2004;164:727–737. doi: 10.1016/s0002-9440(10)63160-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beltran-Valero de Bernabe D., Voit T., Longman C., Steinbrecher A., Straub V., Yuva Y., Herrmann R., Sperner J., Korenke C., Diesen C., Dobyns W.B., Brunner H.G., van Bokhoven H., Brockington M., Muntoni F. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. 2004;41:e61. doi: 10.1136/jmg.2003.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Louhichi N., Triki C., Quijano-Roy S., Richard P., Makri S., Meziou M., Estournet B., Mrad S., Romero N.B., Ayadi H., Guicheney P., Fakhfakh F. New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities: identification of a founder mutation in Tunisian families. Neurogenetics. 2004;5:27–34. doi: 10.1007/s10048-003-0165-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Reeuwijk J., Olderode-Berends M.J., van den Elzen C., Brouwer O.F., Roscioli T., van Pampus M.G., Scheffer H., Brunner H.G., van Bokhoven H., Hol F.A. A homozygous FKRP start codon mutation is associated with Walker-Warburg syndrome, the severe end of the clinical spectrum. Clin Genet. 2010;78:275–281. doi: 10.1111/j.1399-0004.2010.01384.x. [DOI] [PubMed] [Google Scholar]
- 37.Ackroyd M.R., Skordis L., Kaluarachchi M., Godwin J., Prior S., Fidanboylu M., Piercy R.J., Muntoni F., Brown S.C. Reduced expression of fukutin-related protein in mice results in a model for fukutin-related protein associated muscular dystrophies. Brain. 2009;132(2):439–451. doi: 10.1093/brain/awn335. [DOI] [PubMed] [Google Scholar]
- 38.Thornhill P., Bassett D., Lochmuller H., Bushby K., Straub V. Developmental defects in a zebrafish model for muscular dystrophies associated with the loss of fukutin-related protein (FKRP) Brain. 2008;131:1551–1561. doi: 10.1093/brain/awn078. [DOI] [PubMed] [Google Scholar]
- 39.Kawahara G., Guyon J.R., Nakamura Y., Kunkel L.M. Zebrafish models for human FKRP muscular dystrophies. Hum Mol Genet. 2010;19:623–633. doi: 10.1093/hmg/ddp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao X., Li J., Samulski R.J. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol. 1996;70:8098–8108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kessler P.D., Podsakoff G.M., Chen X., McQuiston S.A., Colosi P.C., Matelis L.A., Kurtzman G.J., Byrne B.J. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc Natl Acad Sci U S A. 1996;93:14082–14087. doi: 10.1073/pnas.93.24.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang B., Li J., Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci USA. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao X., Li J., Tsao Y.P., Dressman D., Hoffman E.P., Watchko JF Full functional rescue of a complete muscle (TA) in dystrophic hamsters by adeno-associated virus vector-directed gene therapy. J Virol. 2000;74:1436–1442. doi: 10.1128/jvi.74.3.1436-1442.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greelish J.P., Su L.T., Lankford E.B., Burkman J.M., Chen H., Konig S.K., Mercier I.M., Desjardins P.R., Mitchell M.A., Zheng X.G., Leferovich J., Gao G.P., Balice-Gordon R.J., Wilson J.M., Stedman H.H. Stable restoration of the sarcoglycan complex in dystrophic muscle perfused with histamine and a recombinant adeno-associated viral vector. Nat Med. 1999;5:439–443. doi: 10.1038/7439. [DOI] [PubMed] [Google Scholar]
- 45.Cordier L., Hack A.A., Scott M.O., Barton-Davis E.R., Gao G., Wilson J.M., McNally E.M., Sweeney H.L. Rescue of skeletal muscles of gamma-sarcoglycan-deficient mice with adeno-associated virus-mediated gene transfer. Mol Ther. 2000;1:119–129. doi: 10.1006/mthe.1999.0019. [DOI] [PubMed] [Google Scholar]
- 46.Dressman D., Araishi K., Imamura M., Sasaoka T., Liu L.A., Engvall E., Hoffman E.P. Delivery of alpha- and beta-sarcoglycan by recombinant adeno-associated virus: efficient rescue of muscle, but differential toxicity. Hum Gene Ther. 2002;13:1631–1646. doi: 10.1089/10430340260201725. [DOI] [PubMed] [Google Scholar]
- 47.High K.A. Clinical gene transfer studies for hemophilia B. Semin Thromb Hemost. 2004;30:257–267. doi: 10.1055/s-2004-825639. [DOI] [PubMed] [Google Scholar]
- 48.Mejat A., Decostre V., Li J., Renou L., Kesari A., Hantai D., Stewart C.L., Xiao X., Hoffman E., Bonne G., Misteli T. Lamin A/C-mediated neuromuscular junction defects in Emery-Dreifuss muscular dystrophy. J Cell Biol. 2009;184:31–44. doi: 10.1083/jcb.200811035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brummelkamp T.R., Bernards R., Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 50.Xiao X., Li J., Samulski R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Snyder R., Xiao X., Samulski R.J. Edited by N Dracopoli. John Wiley & Sons Ltd; New York: 1996. Production of recombinant adeno-associated viral vectors: Current Protocols in Human Genetics; pp. 12.11.11–12.12.23. [Google Scholar]
- 52.Chan Y.M., Bonnemann C.G., Lidov H.G., Kunkel L.M. Molecular organization of sarcoglycan complex in mouse myotubes in culture. J Cell Biol. 1998;143:2033–2044. doi: 10.1083/jcb.143.7.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watchko J., O'Day T., Wang B., Zhou L., Tang Y., Li J., Xiao X. Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum Gene Ther. 2002;13:1451–1460. doi: 10.1089/10430340260185085. [DOI] [PubMed] [Google Scholar]
- 54.Michele D.E., Barresi R., Kanagawa M., Saito F., Cohn R.D., Satz J.S., Dollar J., Nishino I., Kelley R.I., Somer H., Straub V., Mathews K.D., Moore S.A., Campbell K.P. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- 55.Qiao C., Li J., Zhu T., Draviam R., Watkins S., Ye X., Chen C., Xiao X. Amelioration of laminin-alpha2-deficient congenital muscular dystrophy by somatic gene transfer of miniagrin. Proc Natl Acad Sci U S A. 2005;102:11999–12004. doi: 10.1073/pnas.0502137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu P.J., Zillmer A., Wu X., Lochmuller H., Vachris J., Blake D., Chan Y.M., Lu Q.L. Mutations alter secretion of fukutin-related protein. Biochim Biophys Acta. 2010;1802:253–258. doi: 10.1016/j.bbadis.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 57.Wang Z., Allen J.M., Riddell S.R., Gregorevic P., Storb R., Tapscott S.J., Chamberlain J.S., Kuhr C.S. Immunity to adeno-associated virus-mediated gene transfer in a random-bred canine model of Duchenne muscular dystrophy. Hum Gene Ther. 2007;18:18–26. doi: 10.1089/hum.2006.093. [DOI] [PubMed] [Google Scholar]
- 58.Wang Z., Kuhr C.S., Allen J.M., Blankinship M., Gregorevic P., Chamberlain J.S., Tapscott S.J., Storb R. Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol Ther. 2007;15:1160–1166. doi: 10.1038/sj.mt.6300161. [DOI] [PubMed] [Google Scholar]
- 59.Odom G.L., Gregorevic P., Allen J.M., Finn E., Chamberlain J.S. Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol Ther. 2008;16:1539–1545. doi: 10.1038/mt.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghahramani Seno M.M., Graham I.R., Athanasopoulos T., Trollet C., Pohlschmidt M., Crompton M.R., Dickson G. RNAi-mediated knockdown of dystrophin expression in adult mice does not lead to overt muscular dystrophy pathology. Hum Mol Genet. 2008;17:2622–2632. doi: 10.1093/hmg/ddn162. [DOI] [PubMed] [Google Scholar]
- 61.Matsumura K., Chiba A., Yamada H., Fukuta-Ohi H., Fujita S., Endo T., Kobata A., Anderson L.V., Kanazawa I., Campbell K.P., Shimizu T. A role of dystroglycan in schwannoma cell adhesion to laminin. J Biol Chem. 1997;272:13904–13910. doi: 10.1074/jbc.272.21.13904. [DOI] [PubMed] [Google Scholar]
- 62.Leschziner A., Moukhles H., Lindenbaum M., Gee S.H., Butterworth J., Campbell K.P., Carbonetto S. Neural regulation of alpha-dystroglycan biosynthesis and glycosylation in skeletal muscle. J Neurochem. 2000;74:70–80. doi: 10.1046/j.1471-4159.2000.0740070.x. [DOI] [PubMed] [Google Scholar]
- 63.Ervasti J.M., Ohlendieck K., Kahl S.D., Gaver M.G., Campbell K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 64.Ohlendieck K., Campbell K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol. 1991;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matsumura K., Campbell K.P. Dystrophin-glycoprotein complex: its role in the molecular pathogenesis of muscular dystrophies. Muscle Nerve. 1994;17:2–15. doi: 10.1002/mus.880170103. [DOI] [PubMed] [Google Scholar]
- 66.Chan Y.M., Keramaris-Vrantsis E., Lidov H., Norton J.H., Zinchenko N., Gruber H.E., Thresher R., Blake D.J., Ashar J., Rosenfeld J., Lu Q.L. Fukutin-related protein is essential for mouse muscle, brain and eye development, and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum Mol Genet. 2010;19:3995–4006. doi: 10.1093/hmg/ddq314. [DOI] [PubMed] [Google Scholar]
- 67.Adriaansen J., Tas S.W., Klarenbeek P.L., Bakker A.C., Apparailly F., Firestein G.S., Jorgensen C., Vervoordeldonk M.J., Tak P.P. Enhanced gene transfer to arthritic joints using adeno-associated virus type 5: implications for intra-articular gene therapy. Ann Rheum Dis. 2005;64:1677–1684. doi: 10.1136/ard.2004.035063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Monahan P.E., Samulski R.J., Tazelaar J., Xiao X., Nichols T.C., Bellinger D.A., Read M.S., Walsh C.E. Direct intramuscular injection with recombinant AAV vectors results in sustained expression in a dog model of hemophilia. Gene Ther. 1998;5:40–49. doi: 10.1038/sj.gt.3300548. [DOI] [PubMed] [Google Scholar]
- 69.Li J., Dressman D., Tsao Y.P., Sakamoto A., Hoffman E.P., Xiao X. rAAV vector-mediated sarcogylcan gene transfer in a hamster model for limb girdle muscular dystrophy. Gene Ther. 1999;6:74–82. doi: 10.1038/sj.gt.3300830. [DOI] [PubMed] [Google Scholar]
- 70.Corrado K., Rafael J.A., Mills P.L., Cole N.M., Faulkner J.A., Wang K., Chamberlain J.S. Transgenic mdx mice expressing dystrophin with a deletion in the actin-binding domain display a “mild Becker” phenotype. J Cell Biol. 1996;134:873–884. doi: 10.1083/jcb.134.4.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ibraghimov-Beskrovnaya O., Ervasti J.M., Leveille C.J., Slaughter C.A., Sernett S.W., Campbell K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 72.Ervasti J.M., Campbell K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin Y.C., Murakami T., Hayashi Y.K., Nishino I., Nonaka I., Yuo C.Y., Jong Y.J. A novel FKRP gene mutation in a Taiwanese patient with limb-girdle muscular dystrophy 2I. Brain Dev. 2007;29:234–238. doi: 10.1016/j.braindev.2006.09.006. [DOI] [PubMed] [Google Scholar]