

Abstract
Integrins are heterodimeric receptors that regulate cell adhesion, migration, and apoptosis. Integrin αvβ8 is most abundantly expressed in kidney and brain, and its major ligand is latent transforming growth factor-β (TGF-β). Kidney αvβ8 localizes to mesangial cells, which appose glomerular endothelial cells and maintain glomerular capillary structure by mechanical and poorly understood paracrine mechanisms. To establish kidney αvβ8 function, mice with homozygous Itgb8 deletion (Itgb8−/−) were generated on outbred and C57BL/6 congenic backgrounds. Most Itgb8−/− mice died in utero, and surviving Itgb8−/− mice failed to gain weight, and rarely survived beyond 6 weeks. A renal glomerular phenotype included azotemia and albuminuria, as well as increased platelet endothelial cell adhesion molecule-1 (PECAM-1) expression, which was surprisingly not associated with conventional functions, such as endothelial cell hyperplasia, hypertrophy, or perivascular inflammation. Itgb8−/− mesangial cells demonstrated reduced latent TGF-β binding, resulting in bioactive TGF-β release, which stimulated glomerular endothelial cell apoptosis. Using PECAM-1 gain and loss of function strategies, we show that PECAM-1 provides endothelial cytoprotection against mesangial cell TGF-β. These results clarify a singular mechanism of mesangial-to-endothelial cell cross-talk, whereby mesangial cell αvβ8 homeostatically arbitrates glomerular microvascular integrity by sequestering TGF-β in its latent conformation. Under pathological conditions associated with decreased mesangial cell αvβ8 expression and TGF-β secretion, compensatory PECAM-1 modulation facilitates glomerular endothelial cell survival.
See related Commentary on page 485
Integrins are heterodimeric transmembrane receptors that bidirectionally transduce signals that are vital for cell adhesion, migration, proliferation, and survival during development and tissue homeostasis. Eighteen α and eight β subunits assemble to form 24 heterodimers, and the integrin β8 polypeptide subunit partners exclusively with αv.1 αvβ8 is expressed most abundantly in kidney, brain, and placenta,1 although expression in other tissues has been documented.2–5
β8 integrin gene (Itgb8) deletion caused embryonic lethality because of vascular defects in the yolk sac and placenta, as well as perinatal death from brain capillary malformations.6 A similar phenotype was observed in Itgαv−/− mice.7,8 Integrin β8 is not expressed in brain endothelial cells,6,9 however, Itgb8 or Itgαv deletion from glial precursor (nestin-positive) neuroepithelial cells resulted in intracerebral hemorrhage, as well as capillary and astroglial disorganization,9,10 indicating that communication between endothelial cells and αvβ8-expressing parenchymal cells is required for cerebral vascular development. Glial αvβ8-endothelial cross-talk is further substantiated by brain endothelial cell hyperplasia in Itgb8−/− mice,6 which is dependent on genetic background and age.11 Studies involving conditional Itgαv or Itgb8 deletion from dendritic cells have also demonstrated an αvβ8 role in innate immunity.4,5
Unlike extensive studies in the brain, kidney β8 integrin function is not as well characterized. We have previously shown that αvβ8 localizes to glomerular mesangial cells (MC) and integrin β8 expression is reduced in sclerosing glomerular diseases.12 MC share neuroepithelial features; both cell types abut with capillaries and express nestin during organogenesis and recovery from injury.6,10,13,14 MC are modified pericytes that contact the glomerular basement membrane (GBM) and glomerular endothelial cells (GEC) to mechanically counteract glomerular capillary distention.15–17 Deletion of GEC, GBM or podocyte genes reveals MC dysfunction and defective glomerular capillary development and maintenance,15,18–20 thereby supporting primacy of the MC as a receiver of cues from other glomerular cells. However, reciprocal communication involving MC-derived molecules affecting GEC or podocytes has not been described.17
The major ligands for αvβ8 are the latency-associated peptide (LAP) component of latent transforming growth factor (TGF)-β1 (LAP TGF-β1), LAP TGF-β3, and vitronectin.21,22 Latent TGF-β bound to αvβ8 is coordinately activated through LAP cleavage by the transmembrane matrix metalloproteinase-14 [MMP-14, also known as membrane type 1 metalloproteinase (MT1-MMP)], which releases bioactive TGF-β.22,23 LAP binding to αvβ8 is a high-affinity interaction that generally leads to efficient latent TGF-β activation in MMP-14-expressing cells.22,23 However, MMP-14 is not expressed in MC, but can be induced in pathological states,24 suggesting that MC αvβ8 normally sequesters latent TGF-β to prevent activation. The large latent complex TGF-β [noncovalently attached to LAP, which is bound to latent TGF-β binding protein (LTBP)] may also be secreted, enabling LTBP to target latent TGF-β to extracellular matrix and proteases for activation in an integrin-independent fashion.25,26
To interrogate β8 integrin function in kidney, Itgb8−/− mice were generated on outbred and C57BL/6 congenic backgrounds that harbor modifier genes27,28 that permitted embryonic survival and evaluation of renal phenotypes. Both strains showed glomerular dysfunction, including azotemia, albuminuria and enhanced platelet/endothelial cell adhesion molecule-1 (PECAM-1, also known as CD31) expression, which facilitated GEC cytoprotection against TGF-β-stimulated apoptosis. The data indicate that MC αvβ8 controls glomerular capillary integrity through cross-talk with GEC, by regulating TGF-β release and PECAM-1 expression.
Materials and Methods
Mouse Models
Methods for generation and characterization of Itgb8−/− mice on a C57BL/6J-129Sv background have been described.6 C57BL/6 and CD-1 mice were purchased from Jackson Laboratories (Bar Harbor, ME). Itgb8−/− mice on a congenic C57BL/6 genetic background were developed following more than12 generations of backcrosses. Outbred Itgb8−/− mice were initially generated by pairing C57BL/6J-129Sv-Itgb8+/− male mice with CD-1-Itgb8+/+ female mice. Itgb8+/− progeny were then mated and mice from F2 intercrosses were evaluated. All protocols were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University.
Antibodies
Rabbit polyclonal anti-β8 integrin antibodies were generated against a cytosolic domain peptide as previously described.9 Other antibodies were purchased from the following sources: Wilm's tumor protein (WT-1), latent TGF-β1 binding protein (LTBP-1), β-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA); desmin (Chemicon, Temecula, CA); PECAM-1, TGF-β1, LAP-β1 (R&D Systems, Minneapolis, MN); Tie2 (eBioscience, San Diego, CA); Ki-67 (Novacastra, Newcastle-On-Tyne, UK); F4/80 (Cedarlane, Hornby, Ontario); proliferating cell nuclear antigen (GeneTex, San Antonio, TX); Phospho-Smad2/3 (Cell Signaling, Beverly, MA); MMP-14 (Millipore, Billerica, MA).
Immunohistochemistry
For the experiments in which we assessed β8 integrin expression in kidney, freshly sectioned unfixed frozen mouse kidney tissue was air dried for 20 to 30 minutes, then permeabilized with 0.2% Triton X-100 (Sigma, St. Louis, MO) in PBS for 10 minutes. Sections were blocked with goat serum (5% in PBS, 1 hour, at room temperature), then incubated with anti-β8 integrin antibodies (10 μg/ml, overnight at 4°C), followed by Alexa 488-conjugated goat anti-rabbit IgG (Invitrogen; 1:200, 1 hour, at room temperature). Nuclei were counterstained with TOTO-3 (1:1000 in PBS, 30 minutes, at room temperature; Molecular Probes, Eugene, OR), and sections were mounted in Aqueous Mounting Media (Vector, Burlingame, CA).
All other immunohistochemistry experiments were conducted according to previously described protocols.29 Primary antibody incubation conditions were as follows: WT1 (1:200, overnight, 4°C), desmin (1:200, overnight, 4°C), PECAM-1 (1:200, overnight, 4°C), Tie2 (1:25, overnight, 4°C), Ki-67 (1:1000, overnight, 4°C), F4/80 (1:200, overnight, 4°C), proliferating cell nuclear antigen (1:200, overnight, 4°C), LAP (1:50, overnight, 4°C), LTBP-1 (1:50, 2 hours, at room temperature). Fluorochrome-conjugated secondary antibodies were then routinely added (1:300, 1 hour, at room temperature), followed by mounting media. All cells were examined with a Leica TCS SP2 Confocal system (Leica Microsystems, Wetzlar, Germany), and digital images were processed with Molecular Devices deconvolution software v9.1 (Molecular Devices, Sunnyvale, CA) and Adobe Photoshop v9.0 (Adobe Acrobat, San Jose, CA).
Histology
For light microscopy, kidneys were fixed in formaldehyde, embedded in paraffin, then sectioned, de-paraffinized and stained with PAS reagent or Masson trichrome stain. For transmission electron microscopy, kidneys were fixed in 2.5% glutaraldehyde and processed by the Department of Pathology at the Metro Health Medical Center, Case Western Reserve University. Glomerular and tubulointerstitial ultrastructure was analyzed with the assistance of Dr. Moonja Chung-Park (Department of Pathology, Case Western Reserve University).
Renal Function Assays
Mice were phenotyped for renal function by measuring albuminuria using the Mouse Albumin ELISA Quantitation Set (Bethyl Lab, Montgomery, TX) as previously described,30 and by assaying for blood urea nitrogen with a Hitachi 911 autoanalyzer (Roche Diagnostics, Basel, Switzerland). Hematuria was screened by Multistix 7 dipstick analysis (Bayer, Elkhart, IN).
Glomerular Microdissection
Mouse kidneys were rapidly removed. The renal cortex was dissected and minced through a 180-μm wire mesh sieve (Newark Wire Cloth Company, Clifton, NJ) onto a Petri dish containing Dulbecco's phosphate buffered saline (Hyclone, 137 mmol/L NaCl, 8.1 mmol/L Na2HPO4, 2.7 mmol/L KCl, 1.5 mmol/L KH2PO4, 0.9 mmol/L CaCl2, 0.5 mmol/L MgCl2). Renal tissue was then passed through a 100-μm nylon filter (Falcon), and washed in Dulbecco's phosphate buffered saline. The flow was passed through a 70 μm nylon filter, washed again, and the glomeruli-enriched fraction, which does not flow through, was collected and resuspended in Dulbecco's phosphate buffered saline. Aliquots of the kidney suspension were spread on 10-cm cell culture dishes and individual glomeruli were visualized with an inverted light microscope (10X magnification) and separated from other elements with a tool fashioned from an eyelash, which was glued to a toothpick handle. A minimum of 10 to 20 glomeruli was required for RT-PCR experiments.
Quantitative RT-PCR
Real-time quantitative RT-PCR analysis was performed using LightCycler and SYBR Green technology (Roche, Mannheim, Germany) according to previously described methods.12 Each analysis included mouse kidney samples of unknown mRNA concentration, and to confirm amplification specificity, random-primed RNA in the absence of reverse transcriptase (RT) or RNA template as negative controls. Total RNA (3 μg) was first treated with DNase I, and then reverse transcribed using the Thermoscript RT-PCR System (Invitrogen, Carlsbad, CA), in 20 μL. Two-microliter cDNA products were PCR-amplified in buffer containing 2 μL LightCycler – FastStart DNA Master SYBR Green I mix (Roche Molecular Systems, Alameda, CA), 18 μL hybridization buffer, 5 μmol/L gene-specific primers, and 3 mmol/L MgCl2. PECAM-1 (NM_001032378.1) isoforms were amplified with primers 5′-AGGAAAGCCAAGGCCAAA-3′ (nt 2072-2089) and 5′-TTGACTGTCTTAAGTTCC-3′ (nt 2325-2342). β-actin mRNA (NM_007393.3) internal standard was detected using 5′-ATCTGGCACCACACCTTCTACAATGAGCTGCG-3′ (nt 333-364) and 5′-CGTCATACTCCTGCTTGCTGATCCACATCTGC-3′ (nt 1139-1169) primers (BD Biosciences Clontech, Palo Alto, CA). Nephrin (NM_019459.2) was amplified with primers 5′- AGGTACAGCCTGGAAGGAGACA-3′ (nt 372-393) and 5′-CGCACTTACTCCCTAGTCTCCT-3′ (nt 685-706). Glyceraldehyde-3-phosphate dehydrogenase mRNA standard (XR_032630.1) was detected using 5′-GGAGCCAAACGGGTCATC-3′ (nt 204-221) and 5′-TACTTATGCCGATGTCGTTGT-3′ (nt 808-828) primers. Transcript quantification was determined by the comparative CT (ΔΔCT) method.12 Data are expressed as PECAM-1 normalized to β-actin, or nephrin normalized to GAPDH transcript content within the same sample.
Cell Culture
Mouse MC from neonatal Itgb8+/+ and Itgb8−/− kidneys were obtained from microdissected glomeruli as described,12 and distinguished from other glomerular cells by immunocytochemical analysis of desmin expression. Third-passage MC in primary culture were immortalized by infection with temperature-sensitive SV40. Immortalized mouse GEC were obtained from Dr. M. Madaio (Medical College of Georgia).31 Methods for generation and maintenance of wild type and PECAM-1−/− kidney endothelial cells have previously been described.32 Minor deviations from this protocol include cell propagation at 31°C in media containing 10% fetal bovine serum. To generate MC conditioned media, cells were first grown to near confluence in complete media. Wells were then washed with Hanks' solution, and media was changed to serum-free Dulbecco's modified Eagle's medium (DMEM). After 48 to 72 hours, media was harvested and stored at 4°C until it was needed for experiments.
Apoptosis Assays
Apoptosis was assessed in GEC exposed to Itgb8+/+ or Itgb8−/− MC-conditioned media supplemented with TGF-β1 (R&D Systems, 2.5 ng/ml) or neutralizing anti-TGF-β1 antibodies (10 μg/ml) by simultaneously labeling externalized phosphatidylserine with annexin V in unfixed cells, then nuclear morphology was determined using DAPI in cells postfixed in paraformaldehyde, according to previously described methods.33 In some experiments, GEC were transfected with a PECAM-1 adenoviral vector, which has been described.32
Immunoblot Analysis
Methods have previously been described.33 Briefly, cell monolayers were lysed and denatured in boiling buffer (125 mmol/L Tris, pH 6.8, 2% SDS, 5% glycerol, 1% β-mercaptoethanol, 0.003% bromphenol blue) for 5 minutes. Samples (20 μg protein/lane) were resolved by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to polyvinylidine difluoride membranes. Blots were blocked in 5% dried milk and 2% bovine serum albumin, probed with primary antibody (1 μg/ml, 1 hour, at room temperature), and peroxidase conjugated IgG (1:5000, 1 hour, at room temperature). Band intensity was detected by enhanced chemiluminescence.
TGF-β Bioactivity Assays
TGF-β activity within Itgb8+/+ or Itgb8−/− MC-conditioned media was determined using mink lung epithelial cells (MLEC) stably expressing a truncated promoter of plasminogen activator inhibitor-1 fused to the firefly luciferase reporter gene,34 which were provided by Dr. Daniel Rifkin (New York University). MLEC were plated for 24 hours in 6-well tissue culture dishes, washed twice with PBS, and maintained for 4 hours in DME-H21 medium without fetal bovine serum. Conditioned medium collected from the indicated cells was maintained for 18 hours in DMEM that was supplemented with 0.2% fetal bovine serum and was directly applied to MLEC to determine the active TGF-β. In parallel cultures, MLEC cells were incubated in DMEM supplemented with recombinant human TGF-β1 (0.1 to 5000 ng/ml; PeproTech, Rocky Hill, NJ) to generate a standard curve. After 24 hours, MLEC cell extracts were prepared and assayed for luciferase activity using the Luciferase Assay System (Promega) according to the manufacturer's instructions, and luminescence was measured as described above. Luciferase units obtained from conditioned medium samples were normalized to cell number, and secreted TGF-β was quantified relative to luciferase units from recombinant human TGF-β1 standard curves.
Statistics
All data are representative of a minimum of three experiments per condition. Quantitative results are presented as mean ± SEM. Comparisons between multiple groups were made by one-way analysis of variance with the Student-Newman-Keuls or Kruskal-Wallis tests for parametric and nonparametric data, respectively. Comparison between two groups was made by paired t-test. A χ2 test was used to determine statistical significance of observed versus expected genotype distributions in Table 1. Statistical significance is defined as P ≤ 0.05.
Table.
Genotypes of Progeny From Heterozygous Intercrosses
| β8 integrin genotypes |
|||||
|---|---|---|---|---|---|
| Intercrosses | +/+ | +/− | −/− | Expected viable −/− | P value |
| Outbred embryos | |||||
| E14.5-16.5 | 5 | 13 | 1 | 6 | |
| E17.5 | 12 | 22 | 7 | 11 | |
| E18.5 | 8 | 21 | 4 | 10 | |
| Total | 25 (27) | 56 (60) | 12 (13) | 27 | 0.09 |
| P14-P21 | |||||
| Outbred | 196 (35) | 353 (63) | 14 (2) | 183 | <0.0001 |
| Congenic | 168 (38) | 267 (61) | 6 (1) | 145 | <0.0001 |
Data are presented as total number of progeny with percentages in parentheses. Expected viable is calculated with the assumption that the sum of (+/+) and (+/−) equals 75%. A Chi-square test was used to determine statistical significance.
E, embryonic day; P, postnatal day.
Results
Integrin β8 Expressed in Mesangial Cells
We previously determined that integrin β8 mRNA localized to glomerular MC in vivo, and immunoblots from cell culture lysates confirmed integrin β8 protein expression in MC, but not in GEC or podocytes.12 Figure 1 shows integrin β8 protein labeling in a predominant mesangial pattern in vivo.
Figure 1.
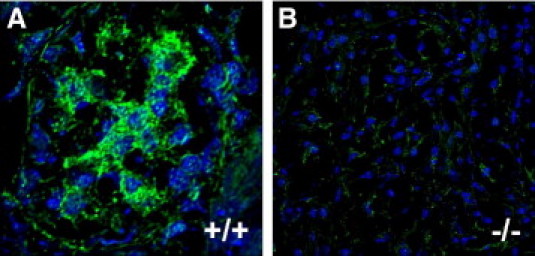
Integrin β8 is expressed in a mesangial distribution. Unfixed frozen sections from Itgb8+/+ (A, +/+) and Itgb8−/− (B, −/−) mouse kidneys were permeabilized in 0.2% Triton X-100, blocked with goat serum, followed by incubation with rabbit anti-human β8 integrin IgG, then Alexa 488-conjugated goat anti-rabbit IgG (green), and TOTO-3 nuclear counterstain (blue). Slides were viewed by confocal microscopy at 1000X magnification. Digital images were processed with Molecular Devices deconvolution software v9.1 and Adobe Photoshop v7.0.
Itgb8−/− Extrarenal Phenotypes
Previous evaluation of Itgb8−/− mice showed developmental defects that resulted in embryonic lethality.6 No gross kidney abnormalities were noted. To investigate more subtle phenotypes in older animals, two strains were created on backgrounds enriched for genetic modifiers27,28: Itgb8−/− mice with the C57BL/6J-129/Sv background were intercrossed with CD-1 mice to create the outbred F2 progeny, and backcrossed with C57BL/6 mice to create a congenic strain.
Examination of E14.5-E18.5 embryos revealed decreased numbers of Itgb8−/− mice, and the proportion surviving to birth decreased even further (Table 1). However, surviving Itgb8−/− mice lived long enough to be evaluated for postnatal phenotypes. The most striking extrarenal feature among Itgb8−/− mice was smaller body size compared to Itgb8+/+ littermates (Figure 2B–D), which became more marked over time, consistent with a runting syndrome. Kidney weights were also smaller in Itgb8−/− mice (Figure 2E), although kidney:body weight ratios were not significantly different between genotypes (Figure 2F).
Figure 2.

Itgb8−/− mice have extrarenal phenotypes. A:Itgb8 genotypes were established by PCR analysis. Representative Itgb8+/+ (+/+) and Itgb8−/− (−/−) mice on outbred (B) and C57BL/6 congenic (C) backgrounds. Body weights (D), kidney weights (E) and kidney:body weight ratios (F) are shown from 4-week-old outbred male mice with Itgb8+/+ (+/+), Itgb8+/− (+/−), and Itgb8−/− (−/−) genotypes (n = 4 to 8 per group). *P < 0.05 compared to +/+ group.
Few Itgb8−/− mice on the outbred background survived beyond 12 weeks, whereas no C57BL/6 Itgb8−/− mice lived more than 5 weeks. Otherwise, no differences could be discerned between strains. By 8 to 12 weeks, gait abnormalities (hind limb dragging and ataxia) were observed. Similar defects have been noted in conditional Itgαv−/− mice, and attributed to cerebellar and axonal degeneration.9 Rarely, Itgb8−/− mice had hydrocephalus, which was also observed in Itgαv−/− mice.7 Gross examination of brains from Itgb8−/− mice revealed no intracerebral hemorrhage. The longest surviving mice were euthanized because they developed hemorrhagic rectal prolapse.
Itgb8−/− Glomerular Phenotype
Renal function studies in Itgb8+/+ and Itgb8−/− mice revealed albuminuria at 4 weeks, which was significantly greater in Itgb8−/− compared to Itgb8+/+ mice, and more pronounced at 12 weeks (Figure 3A). Azotemia was also greater in Itgb8−/− compared to Itgb8+/+ or Itgb8+/− mice (Figure 3B). Dipstick tests for hematuria were negative in all mice. Figure 3, C and D, show glomerular histology from Itgb8+/+ and Itgb8−/− mice, with mildly increased mesangial PAS staining in Itgb8−/− glomeruli and equivalent glomerular cellularity between genotypes. However, Masson trichrome staining revealed no differences between Itgb8+/+ and Itgb8−/− kidneys (not shown), indicating absence of scar matrix protein deposition. With the exception of rare interstitial infiltrates and eosinophilic casts in Itgb8−/− kidneys, the tubulointerstitium was histologically normal (not shown). Transmission electron micrographs revealed focal podocyte foot process fusion in Itgb8−/− mice (Figure 3, F and H), consistent with albuminuria. Mesangial and GEC morphology appeared normal (Figure 3, E–H). The number of slit diaphragms per micrometer GBM was 2.31 ± 0.28 in wild type glomeruli, in agreement with previous reports for rodent glomeruli.35,36 Slit diaphragm density per GBM length was slightly less in Itgb8−/− glomeruli (1.68 ± 0.17/μm, P = 0.11, Figure 3I). Nephrin expression, normalized to GAPDH, was assessed by quantitative PCR, and revealed a 7% decrease in transcript levels in Itgb8−/− versus Itgb8−/− kidneys. These results reflect mild foot process effacement and proteinuria. Glomerulogenesis at E17.5 to E19.5 was similar between Itgb8+/+ and Itgb8−/− kidneys (not shown).
Figure 3.
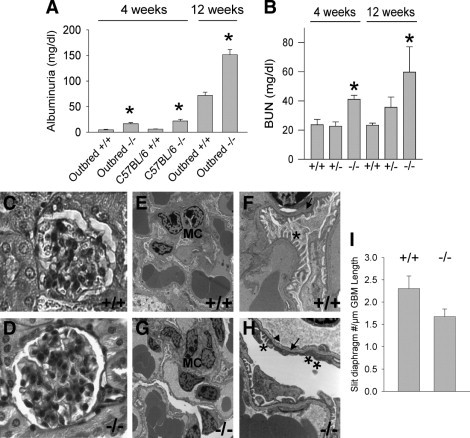
Itgb8−/− mice have glomerular dysfunction. A: Spot urine samples were assayed for albumin from 4-week-old and 12-week-old Itgb8+/+ (+/+) and Itgb8−/− (−/−) outbred and C57BL/6 congenic mice by enzyme-linked immunosorbent assay methods. N = 6 mice per group. B: Blood urea nitrogen (BUN) was measured in 4-week-old Itgb8+/+ (+/+), Itgb8+/− (+/−), and Itgb8−/− (−/−); and 12-week-old Itgb8+/+ (+/+), Itgb8+/− (+/−), and Itgb8−/− (−/−) mice with a Beckman-Coulter LX20 analyzer. N = 3 to 9 mice per group. *P < 0.01 compared to age-matched +/+ mice. Paraffin sections from 12-week-old outbred Itgb8+/+ (C) and Itgb8−/− (D) mice were stained with PAS. Images are viewed using light microscopy, original magnifications, 1000. E–H: Transmission electron micrographs of glomeruli from 12-week-old Itgb8+/+ and Itgb8−/− kidneys. E: Glomerulus from wild-type (+/+) kidney, original magnification, 4,400. F: Glomerulus from wild-type (+/+) kidney, original magnification, 15,000. G: Glomerulus from Itgb8−/− (−/−) kidney, original magnification, 4,400. H: Glomerulus from Itgb8−/− (−/−) kidney, original magnification, 15,000. I: Slit diaphragms, defined as discrete areas of separation between podocyte foot processes, were tabulated from electron micrographs of Itgb8+/+ and Itgb8−/− glomeruli. Data are expressed as number of slit diaphragms per μm glomerular basement membrane length. MC, mesangial cells; Arrows, glomerular basement membrane; Arrowhead, normal-appearing, fenestrated glomerular endothelial cells; asterisks indicate normal podocyte foot processes; double asterisks indicate, areas of foot process fusion.
Increased Glomerular PECAM-1 Expression in Itgb8−/− Mice
To further explore effects of Itgb8 deletion, resident glomerular cell populations were characterized by immunohistochemical techniques. No difference in podocyte number, as defined by WT1 labeling [Itgb8−/− = 16.2 ± 3.1 (mean ± SD per glomerulus); Itgb8+/+ = 16.0 ± 2.9] or desmin staining of MC (not shown) was detected between Itgb8−/− and Itgb8+/+ animals. However, PECAM-1 staining of GEC was significantly more intense in Itgb8−/− compared to Itgb8+/+ glomeruli (Figure 4). To verify these results, PECAM-1 mRNA expression from isolated glomeruli was measured using quantitative PCR, and revealed a four-fold increase in Itgb8−/− mice (Figure 4C), consistent with the immunohistochemistry findings.
Figure 4.
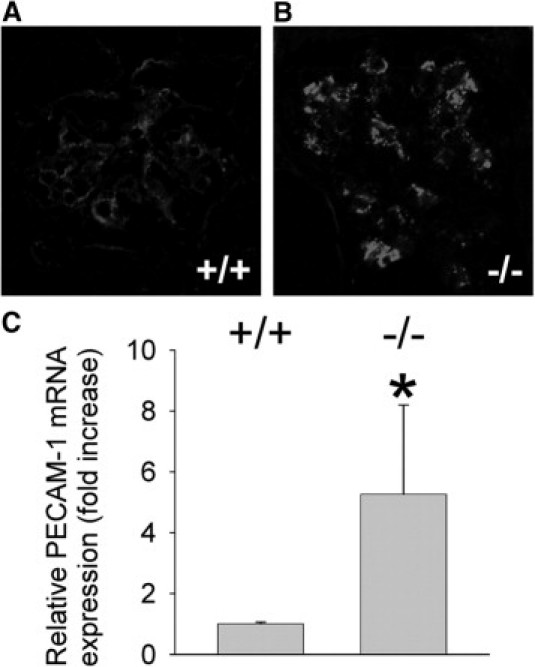
Itgb8−/− mice display increased glomerular PECAM-1 expression. Frozen sections from 12-week-old Itgb8+/+ (+/+) (A) and Itgb8−/− (−/−) (B) kidneys were labeled for endothelial cells by blocking with MOM mouse blocking reagent (Vector, 1 hour), then incubating with rat anti-mouse PECAM-1 (1:200, 1 hour), followed by Texas red-conjugated goat anti-rat IgG (1:300, 1 hour). Slides were viewed by confocal microscopy, original magnification, 1000. (C) PECAM-1 expression was determined by quantitative RT-PCR in isolated glomeruli from Itgb8+/+ (+/+) and Itgb8−/− (−/−) kidneys. Data are expressed as relative PECAM-1 mRNA expression (PECAM-1 normalized to β-actin transcript content within the same sample). *P < 0.05 compared to +/+ group.
PECAM-1 Regulates GEC Cytoprotection
PECAM-1 forms homophilic interactions between adjacent endothelial cells37,38 and is often used as a marker of hyperplasia or hypertrophy. To test for GEC proliferation, kidneys were examined for co-localization of glomerular PECAM-1 with Ki-67 nucleolar antigen39 (Figure 5A) or proliferating cell nuclear antigen40 (not shown). As seen in Figure 5B, the number of Ki-67-positive GEC cells was equivalent between Itgb8−/− and Itgb8+/+ mice. To verify that increased PECAM-1 in Itgb8−/− glomeruli is not caused by proliferation, kidneys were labeled for a different endothelial cell marker, Tie2. Figure S1 (see Supplemental Figure S1 at http://ajp.amjpathol.org) reveals similar Tie2 staining between GEC from Itgb8−/− and Itgb8+/+ mice. No difference in GEC size was noted between Itgb8−/− and Itgb8+/+ glomeruli by electron microscopy, suggesting that enhanced PECAM-1 expression was also not caused by GEC hypertrophy.
Figure 5.
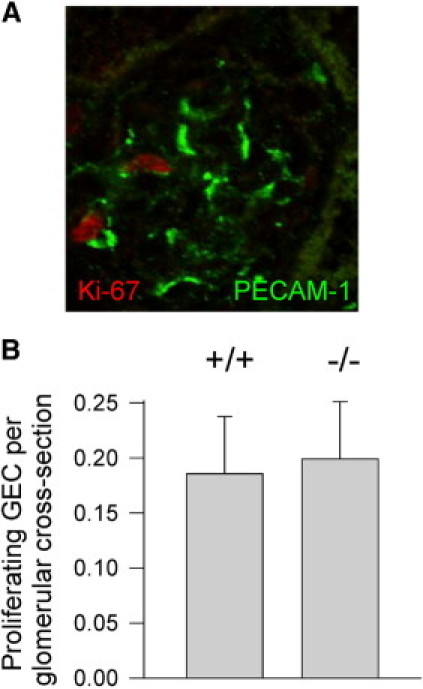
Increased PECAM-1 expression is not associated with GEC hyperplasia in Itgb8−/− kidneys. A: GEC proliferation was assessed by co-localization of Ki-67-positive cells (red) within PECAM-1-stained capillaries (green) in a glomerulus from a 4-week-old Itgb8−/− kidney. B: Quantitation of GEC from 60 glomeruli in three 4-week-old Itgb8+/+ (+/+) and Itgb8−/− (−/−) kidneys.
PECAM-1 regulates pleiotropic endothelial cell functions,37,38,41–46 including inflammatory cell diapedesis38,41 and cytoprotection.37,42,43 Therefore, we determined whether increased PECAM-1 expression is associated with glomerular inflammation using F4/80 labeling of macrophages and dendritic cells.47,48 Although F4/80-positive cells could occasionally be detected within the tubulointerstitium, they were seldom observed in Itgb8−/− or Itgb8+/+ glomeruli (not shown).
Apoptotic glomerular cells were not detected in Itgb8−/− or Itgb8+/+ kidneys (not shown), indicating that PECAM-1 upregulation does not represent compensation for GEC loss. However, lack of GEC death in Itgb8−/− mice could be caused by PECAM-1-mediated cytoprotection.37,42,43 To address this possibility, apoptosis was assayed in PECAM-1-null versus PECAM-1 overexpressing GEC in response to conditioned media from Itgb8−/− and Itgb8+/+ MC, to simulate in situ paracrine effects of MC on GEC. At baseline, PECAM-1 expression was undetectable (Figure 6A), and neither exogenous TGF-β (0.1–10 ng/ml), Itgb8−/− nor Itgb8+/+ MC media induced GEC PECAM-1 (not shown), suggesting that the in vitro co-culture system did not fully model the in vivo scenario. As a result, it was necessary to transfect GEC with adenovirus vector to investigate the effects of PECAM-1 upregulation. Figures 6B and S2 (see Supplemental Figure S2 at http://ajp.amjpathol.org) show that Itgb8−/− MC-conditioned media caused significant apoptosis of PECAM-1-deficient GEC, which was completely blunted by PECAM-1 overexpression. To assess whether there is a PECAM-1 dose/response, apoptosis induced by Itgb8−/− MC conditioned media was compared in wild type and PECAM-1−/− kidney endothelial cells. Figure 6C shows significant susceptibility of PECAM-1−/− cells to apoptosis, which is incompletely resolved in PECAM-1+/+ cells. These data support a paradigm whereby an Itgb8−/− MC-secreted factor induces GEC apoptosis unless PECAM-1 is upregulated to provide cytoprotection.
Figure 6.
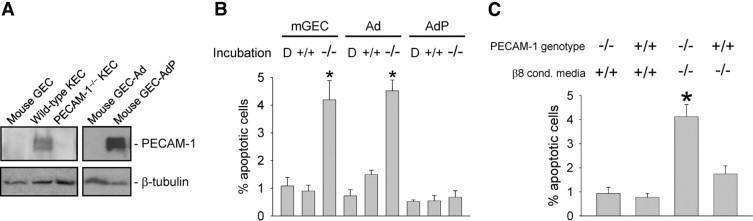
MC integrin β8 and GEC PECAM-1 are synergistically cytoprotective. A: Immunoblots from whole cell lysates (20 μg total protein) from mouse glomerular endothelial cells (GEC), kidney endothelial cells (KEC) from wild-type and PECAM-1−/− mice, and GEC transfected with adenovirus (Ad) or PECAM-1 adenovirus (AdP). The PECAM-1 band appears as a doublet due either to the presence of more than one isoform76 or glycosylated and unglycosylated proteins.77 Blots were stripped and re-probed with anti-tubulin antibodies as a loading control (lower panel). B: Untransfected (mGEC), adenoviral vector-transfected (Ad), and PECAM-1 adenoviral vector-transfected (AdP) mouse GEC were incubated for 24 hours with serum-free DMEM, followed by conditioned media obtained from quiescent wild-type MC (+/+), Itgb8−/− (−/−) MC or DMEM (D) only (16 hours, 37°C), and then evaluated for apoptosis by annexin V labeling. *P < 0.01 compared to D- and +/+ treated groups. C: Conditionally immortalized endothelial cells derived from PECAM-1−/− or PECAM-1+/+ mouse kidneys were grown to near confluence in complete media under permissive 31°C conditions, and then placed in serum-free DMEM (72 hours, 37°C). Cells were then washed with Hanks' solution, and incubated with conditioned media from wild-type (+/+) or Itgb8−/− (−/−) MC (8 hours, 37°C) and then evaluated for apoptotic nuclear morphology by DAPI labeling by two observers blinded to experimental conditions. *P < 0.01 compared to other groups.
Itgb8−/− MC Release Bioactive TGF-β
Latent TGF-β is the major ligand for αvβ8. Unlike other latent TGF-β-binding integrins that activate TGF-β through non-enzymatic, mechanical strain, causing a conformational change in the LAP-TGF-β interaction,49,50 αvβ8-bound latent TGF-β activation requires LAP cleavage by MMP-14.22 However, MMP-14 expression and activity are undetectable in MC that are rendered quiescent by prolonged incubation in serum-free media24 (see Supplemental Figure S3 at http://ajp.amjpathol.org), which mimics the normal in vivo state,51 suggesting that MC αvβ8 normally sequesters TGF-β in the latent conformation.
To test whether Itgb8 deletion leads to reduced TGF-β binding in vivo, mouse glomeruli were analyzed for integrin β8 and latent TGF-β co-localization with immunohistochemical techniques. Figure 7A again shows β8 integrin expression in a glomerular mesangial pattern. LAP also labeled glomeruli in a predominant mesangial distribution (Figure 7B), in agreement with LTBP-1 expression.52,53 Importantly, integrin β8 and LAP co-localized in glomeruli (Figure 7C), consistent with LAP as the cognate ligand.22
Figure 7.
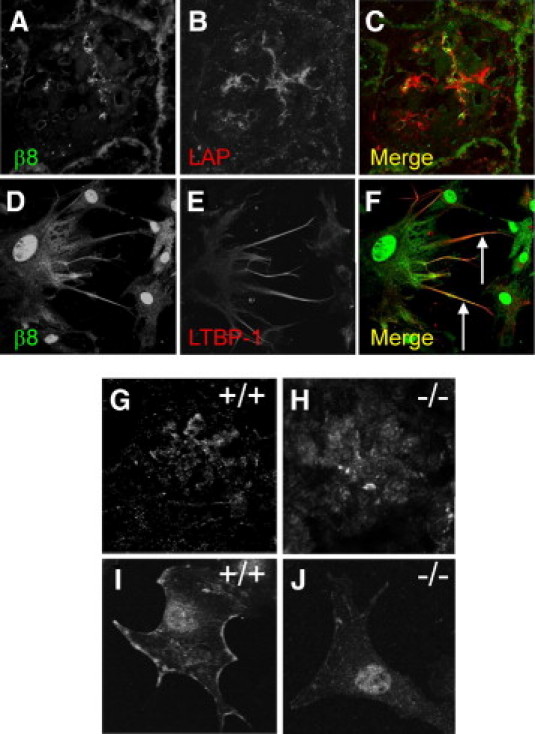
Latent TGF-β binds to MC integrin β8. A–C: Unfixed cryosections from wild-type C57BL/6 mouse kidney were blocked with donkey serum, then incubated with goat anti-human LAP-β1 IgG and rabbit anti-β8 integrin IgG. Sections were washed with PBS, then incubated with Texas red-conjugated donkey anti-goat IgG and Alexa 488-conjugated donkey anti-rabbit IgG. Slides were mounted and viewed with a Leica confocal microscope, original magnification, 630. D–F: Human MC were grown to subconfluence on coverslips. Unfixed cells were labeled for latent TGF-β expression by initially incubating with goat anti-LTBP-1 antibodies (E). To determine integrin β8 expression, slides then were washed in serum-free DMEM, followed by fixation in paraformaldehyde, blocking with donkey serum, and incubation with rabbit anti-β8 integrin antibodies (D). Slides were then incubated with Texas red-conjugated donkey anti-goat and Alexa 488-conjugated donkey anti-rabbit antibodies, and viewed by confocal microscopy, original magnification, 1000. All digital images were analyzed with deconvolution software. Arrows denote regions of colocalization. G–H: Frozen sections from formaldehyde-fixed Itgb8+/+ (G, +/+) or Itgb8−/− (H, −/−) kidneys were probed for LAP-β1, as in B. MC derived from Itgb8+/+ (I, +/+) or Itgb8−/− (J, −/−) mice were labeled for LTBP-1 as in E.
To more extensively assess latent TFG-β as the ligand for MC integrin β8, cultured human MC were colabeled with LTBP-1 and β8 integrin antibodies. As seen in Figure 7D, integrin β8 was expressed primarily within fine, filopodia-like membrane extensions, similar to the integrin β8 cellular distribution observed in other cell types.22,54 Co-localization of LAP with integrin β8 was restricted to the membrane extensions (Figure 7F).
We next analyzed whether deletion of MC integrin β8 results in diminished latent TGF-β binding to MC. Figure 7G shows the discrete mesangial latent TGF-β distribution in a wild type glomerulus, whereas the pattern is more diffuse in Itgb8−/− glomeruli (Figure 7H). In cultured MC, Figure 7I again shows plasma membrane latent TGF-β binding in Itgb8+/+ cells. However, in MC derived from Itgb8−/− mice, latent TGF-β surface staining is nil (Figure 7J), which is consistent with absence of integrin β8-null cell adhesion to LAP,22 and suggests that integrin β8 deletion results in latent TGF-β release from MC plasma membrane binding sites.
To determine whether β8 integrin deletion and decreased latent TFG-β binding results in altered MC release of bioactive TGF-β, conditioned media from Itgb8+/+ and Itgb8−/− MC was assayed for TGF-β activity using an MLEC reporter system. Itgb8−/− media contained a significantly greater quantity of bioactive TGF-β compared to media derived from Itgb8+/+ MC (Figure 8A). To address whether released TGF-β is biologically active toward GEC, conditioned media from Itgb8−/− and Itgb8+/+ MC was applied to GEC and then assayed for Phospho-Smad translocation to nuclei. Figures 8B and S4 (see Supplemental Figure 4 at http://ajp.amjpathol.org) show that Itgb8−/− media contained significantly greater TGF-β activity compared to Itgb8+/+ media.
Figure 8.
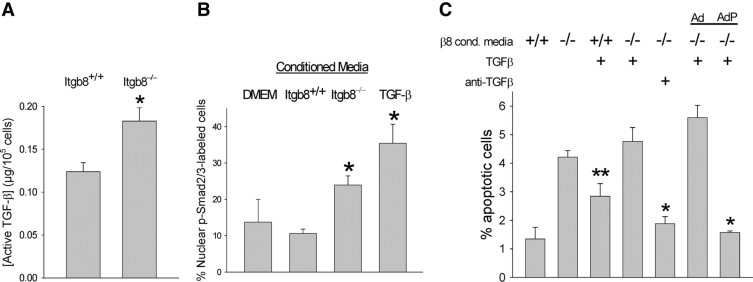
PECAM-1-deficient GEC are susceptible to apoptosis from TGF-β released by Itgb8−/− MC. A: Aliquots of conditioned media from Itgb8+/+ or Itgb8−/− MC were obtained over 48 hours and then incubated with plasminogen activator inhibitor-1 promoter/luciferase-expressing MLEC for 6 hours. TGF-β bioactivity was determined by luciferase luminescence as described in Methods. *P = 0.05 compared to the Itgb8+/+ conditioned media group. B: GEC cultured on glass coverslips were incubated for 24 hours with serum-free DMEM in all groups, followed by conditioned media harvested from quiescent Itgb8+/+ (+/+) or Itgb8−/− (−/−) MC as in A, or DMEM supplemented with TGF-β1 (2.5 ng/ml) or DMEM only (16 hours, 37°C). GEC were then washed in PBS, fixed in paraformaldehyde, and incubated with rabbit monoclonal anti-phospho-Smad2/3 IgG (1 hour, at room temperature), followed by FITC-conjugated goat anti-rabbit IgG (1 hour,at room temperature). Cells were examined from six different fields and counted for nuclear staining of phospho-Smad2/3 as an index of TGF-β bioactivity (original magnification, 400). *P < 0.05 compared to Itgb8+/+ conditioned media alone. C: PECAM-1-deficient GEC, adenoviral vector-transfected GEC (Ad), and PECAM-1 adenoviral vector-transfected GEC (AdP) were incubated for 24 hours with serum-free DMEM at 37°C, followed by conditioned media harvested as in A from Itgb8+/+ (+/+) or Itgb8−/− (−/−) MC (16 hours, 37°C). In some groups, conditioned media was supplemented with TGF-β1 (2.5 ng/ml) or neutralizing anti-TGF-β1 antibodies (10 μg/ml). GEC were evaluated for apoptosis by nuclear morphology of DAPI-stained cells by two observers blinded to experimental conditions. *P < 0.01 compared to other groups incubated with Itgb8−/− conditioned media. **P < 0.05 compared to Itgb8+/+ conditioned media alone.
Release of TGF-β Causes Paracrine GEC Apoptosis
Because TGF-β stimulates apoptosis in GEC,55,56 the next set of studies tested whether the pro-apoptotic factor in Itgb8−/− MC conditioned media is indeed TGF-β. This was accomplished by exposing PECAM-1-deficient GEC to conditioned media from Itgb8+/+ or Itgb8−/− MC, which was supplemented with TGF-β1 or neutralizing anti-TGF-β1 antibodies. Co-incubation of TGF-β1 with Itgb8−/− MC conditioned media caused minimal additive apoptosis (Figure 8C), implying that TGF-β is a major pro-apoptotic component in the Itgb8−/− MC media. This was established more definitively in experiments with neutralizing TGF-β1 antibody co-incubation, which decreased GEC apoptosis to near basal levels. Finally, the effect of TGF-β1 was tested in GEC transfected with PECAM-1 adenovirus. Figure 8C (last 2 bars) reveals that PECAM-1 overexpression completely abolished TGF-β1-induced apoptosis. These results expose cross-talk pathways between MC and GEC, whereby MC β8 integrin normally sequesters TGF-β, which is capable of stimulating paracrine GEC apoptosis unless PECAM-1 is upregulated to provide cytoprotection.
Discussion
Our interest in integrin αvβ8 was initially stimulated by the observation that integrin β8 mRNA expression is restricted to few tissues, most prominently in the kidney.1 A comprehensive human glomerular transcriptome showed that β8 is the most prevalent β-integrin subunit,57 consistent with our previous findings that integrin β8 mRNA expression is localized to the glomerular MC.12 In this report we confirmed, using immunohistochemical techniques, that kidney β8 integrin protein is expressed predominantly in MC.
An impediment to elucidating kidney αvβ8 function had been that Itgb8 disruption resulted in lethal vascular morphogenesis defects,6 which prevented evaluation of renal function beyond embryogenesis. Because genetic background influences phenotype, we established congenic C57BL/6 Itgb8−/− mice, with anticipation of greater longevity. Because the C57BL/6 background may also suppress disease genes27,28 that could minimize the kidney phenotype, we also generated outbred mice on a less permissive CD-1 background.27,28 These outbred Itgb8−/− mice had similar renal function and histology compared to age-matched congenic C57BL/6 Itgb8−/− mice, indicating that kidney phenotype is caused by Itgb8 deletion per se.
In contrast to two recent reports of Itgb8−/− mice on outbred genetic backgrounds,11,58 we observed significant embryonic lethality in Itgb8−/− mice and a shortened lifespan among survivors. Otherwise, examined (extrarenal) phenotypes were similar between studies. An explanation for decreased body size and failure to gain weight is not obvious, although we speculate that residual compromised placental vascularization is a plausible reason for decreased birth weight. We also considered that cleft palate, which was noted in the original Itgb8−/− mice,6 could contribute by prohibiting suckling, and account for poor growth; however, this was not observed in Itgb8−/− mice on congenic C57BL/6 or outbred backgrounds.11,58
A major finding is that Itgb8 deletion resulted in renal insufficiency, consistent with decreased kidney integrin β8 expression in progressive renal disease.12 In addition, GEC PECAM-1 was upregulated in Itgb8−/− kidneys. Glomerular PECAM-1 expression is decreased in human renal diseases and in animals with disordered glomerular architecture.59,60 In reversible models, such as anti-Thy1 treatment in rats, PECAM-1 expression increases during the recovery phase.61,62 Because integrin β8 localizes to MC and the Thy1 model is characterized by mesangiolysis, we speculate that PECAM-1 is induced as compensation for MC integrin β8 loss.
Increased PECAM-1 expression in Itgb8−/− glomeruli was not surprising because it was also observed in the brain cells of both the Itgb8−/− and Itgαv−/−.6,8 The mechanism of PECAM-1 induction is unclear, although TGF-β1 has been shown to induce PECAM-1 expression is some systems,63 suggesting complex interplay between pro-aptopic and anti-apoptotic effects of TGF-β on endothelial cells.55,56,64 The fact that apoptosis, rather than PECAM-1 induction (data not shown), predominated following in vitro application of conditioned media suggests that apoptosis is a default pathway regulated by TGF-β concentration and sustained stimulation, whereas additional TGF-β-independent factors may be required for PECAM-1 regulation.
Unlike studies in Itgb8−/− brain, in which increased PECAM-1 expression was attributed to endothelial hyperplasia,6 we found that PECAM-1 directed GEC cytoprotection against MC-secreted factors, most notably TGF-β. This is relevant because GEC apoptosis is a component of chronic kidney disease pathophysiology,65 which has previously been ascribed to loss of podocyte-dependent vascular endothelial growth factor secretion.17,20 Regarding other possible PECAM-1-dependent mechanisms that could regulate the Itgb8−/− renal phenotype, we observed no glomerular inflammation in Itgb8−/− mice, but we cannot exclude roles for mechanotransduction in response to shear stress44,45 or inhibition of thrombosis.46 Because MC are modified glomerular pericytes,66 we conclude that integrin β8 plays a homeostatic role through endothelial stabilization.
To establish the mechanism of endothelial stabilization, we found that TGF-β within Itgb8−/− MC-conditioned media stimulated apoptosis of PECAM-1-deficient GEC, whereas ectopic PECAM-1 expression in GEC abrogated TGF-β-stimulated apoptosis. TGF-β is known to be pro-apoptotic toward human umbilical vein endothelial cells and GEC,55,56 but here we describe a unique mode of TGF-β presentation to the GEC. Latent TGF-β is an αvβ8 ligand, and in other systems, binding results in constitutive TGF-β activation through a MMP-14-dependent mechanism.22,23 However, we show that MC maintained in serum-free conditions to mimic the normal in vivo state did not express MMP-14, whereas sustained exposure to serum, which induces myofibroblast differentiation,24,51 stimulated MMP-14 expression. Our interpretation of these data is that TGF-β is normally maintained in its latent conformation through binding to MC integrin β8, whereas glomerular diseases associated with MC MMP-14 expression permit TGF-β activation.
Our data support an additional mechanism of MC TGF-β activation whereby Itgb8 deletion results in reduced binding and release of latent TGF-β. We speculate that the liberated latent TGF-β is targeted to extracellular matrix binding sites, resulting in both activation by extracellular proteases or thrombospondin-125,26,67 and enhanced presentation to GEC receptors. The glomerular effects of TGF-β are protean, including permeability to albumin68 as was observed in Itgb8−/− mice, and glomerulosclerosis.69 The importance of TGF-β in glomerular disease pathophysiology is exemplified by a recently completed phase I clinical trial that examines safety of pan-TGF-β antibodies in focal and segmental glomerulosclerosis (http://www.clinicaltrials.gov #NCT00464321). Our data highlight an additional consequence of unbridled glomerular TGF-β activity - GEC apoptosis, if not for compensatory PECAM-1 upregulation (summarized in Figure 9).
Figure 9.
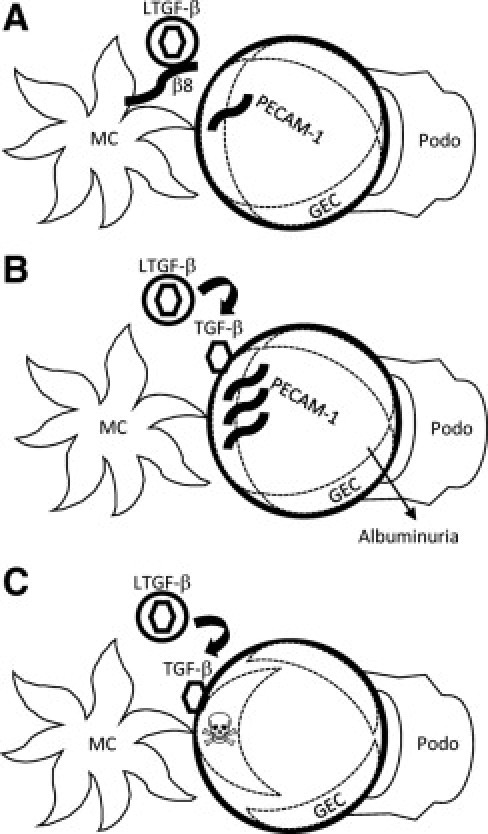
Schematic model of MC integrin β8 effects on GEC. A: Baseline conditions, whereby integrin β8 serves as a reservoir for TGF-β by maintaining the cytokine in its latent conformation through binding of an RGD tri-peptide recognition sequence within LAP to integrin β8. B: Pathological conditions, such as chronic glomerular diseases, in which MC β8 expression is decreased or MMP-14 activity is stimulated. In the former situation, which is modeled with Itgb8−/− mice, TGF-β is no longer secluded in its latent form, and TGF-β could then be released and activated by glomerular matrix proteases or GEC-secreted factors. Alternatively, TGF-β could be enzymatically activated by MMP-14, which is upregulated in MC undergoing myofibroblast differentiation. A glomerular capillary phenotype is minimized by GEC PECAM-1 induction, perhaps in response to TGF-β stimulation.63C: Based on in vitro data with PECAM-1-deficient endothelial cells and conditioned media from MC, TGF-β released by Itgb8−/− MC causes apoptosis in PECAM-1-null GEC. By extrapolation, a parallel in vivo scenario (diminished integrin β8 and GEC with insufficiently upregulated PECAM-1 expression) would result in a more severe renal phenotype characterized by GEC apoptosis. β8, integrin β8; GEC, glomerular endothelial cell; LTGF-β, latent TGF-β; MC, mesangial cell; Podo, podocyte; skull and crossbones represents an apoptotic GEC.
The mechanism by which TGF-β stimulates GEC apoptosis was not investigated. TGF-β-mediated apoptosis regulation is cell-specific, but generally conforms to either a Smad2/3-dependent pathway, which suppresses survivin and Akt, and stimulates the pro-apoptotic Bcl-2 member Bim, or a Smad-independent pathway that activates a TRAF6-TAK1-MKK3/6-JNK/p38 MAP kinase cascade.70 Because Itgb8−/− MC conditioned media stimulated Smad 2/3 phosphorylation, we speculate that the Smad-dependent pathway is more likely. The phosphatase Shp2 is a critical effector for PECAM-1-regulated cytoprotection42 and acts in some circumstances by activating Akt71,72 and destabilizing Bim.73 However, we cannot exclude the possibility that PECAM-1 inhibits TGF-β-directed apoptosis by enhancing parallel cell survival signaling pathways.
Prior studies evoking glomerular cell cross-talk emphasize that GEC- and podocyte-derived growth factors influence MC.20,74 However, to our knowledge this report represents the first example of MC-derived factors affecting GEC function. Although we demonstrated paracrine communication between MC and GEC, a juxtacrine mechanism, whereby MC αvβ8 might directly interact with GEC or GBM ligands, is possible, particularly because MC directly contact GEC16 and genetic deletion of GBM components results in developmental MC defects.15,75
In conclusion, we found that kidney αvβ8 integrin is expressed in MC, and Itgb8 deletion in mice with two different genetic backgrounds developed glomerular dysfunction. In vitro experiments revealed communication between MC and GEC. Effects of resident glomerular cells on MC function have previously been proposed, but our results highlight a unique mechanism in the opposite direction, whereby MC integrin β8 arbitrates GEC viability through TGF-β sequestration and PECAM-1 induction.
Acknowledgments
We appreciate the assistance of Vincent Li with animal husbandry and mouse genotyping and Dr. Moonja Chung-Park for preparation of samples and evaluation of the transmission electron microscopy data.
Footnotes
Supported by Public Health Service grants DK067528, DK072348, and DK064719 from the National Institutes of Health.
CME Disclosure: The authors did not disclose any relevant financial relationships.
Supplemental material for this article can be found at http://ajp.amjpathol.org and at doi:10.1016/j.ajpath.2010.10.031.
Supplementary data
References
- 1.Moyle M., Napier M.A., McLean J.W. Cloning and expression of a divergent integrin subunit β8. J Biol Chem. 1991;266:19650–19658. [PubMed] [Google Scholar]
- 2.Cambier S., Mu D.Z., O'Connell D., Boylen K., Travis W., Liu W.H., Broaddus V.C., Nishimura S.L. A role for the integrin αvβ8 in the negative regulation of epithelial cell growth. Cancer Res. 2000;60:7084–7093. [PubMed] [Google Scholar]
- 3.Stepp M.A. α9 and β8 integrin expression correlates with the merger of the developing mouse eyelids. Dev Dyn. 1999;214:216–228. doi: 10.1002/(SICI)1097-0177(199903)214:3<216::AID-AJA5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 4.Travis M.A., Reizis B., Melton A.C., Masteller E., Tang Q., Proctor J.M., Wang Y., Bernstein X., Huang X., Reichardt L.F., Bluestone J.A., Sheppard D. Loss of integrin αvβ8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007;449:361–365. doi: 10.1038/nature06110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lacy-Hulbert A., Smith A.M., Tissire H., Barry M., Crowley D., Bronson R.T., Roes J.T., Savill J.S., Hynes R.O. Ulcerative colitis and autoimmunity induced by loss of myeloid αv integrins. Proc Natl Acad Sci USA. 2007;104:15823–15828. doi: 10.1073/pnas.0707421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu J.W., Motejlek K., Wang D.N., Zang K.L., Schmidt A., Reichardt L.F. β8 integrins are required for vascular morphogenesis in mouse embryos. Development. 2002;129:2891–2903. doi: 10.1242/dev.129.12.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bader B.L., Rayburn H., Crowley D., Hynes R.O. Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all αv integrins. Cell. 1998;95:507–519. doi: 10.1016/s0092-8674(00)81618-9. [DOI] [PubMed] [Google Scholar]
- 8.McCarty J.H., Monahan-Earley R.A., Brown L.F., Keller M., Gerhardt H., Rubin K., Shani M., Dvorak H.F., Wolburg H., Bader B.L., Dvorak A.M., Hynes R.O. Defective associations between blood vessels and brain parenchyma lead to cerebral hemorrhage in mice lacking αv integrins. Mol Cell Biol. 2002;22:7667–7677. doi: 10.1128/MCB.22.21.7667-7677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCarty J.H., Lacy-Hulbert A., Charest A., Bronson R.T., Crowley D., Housman D., Savill J., Roes J., Hynes R.O. Selective ablation of αv integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development. 2005;132:165–176. doi: 10.1242/dev.01551. [DOI] [PubMed] [Google Scholar]
- 10.Proctor J.M., Zang K., Wang D., Wang R., Reichardt L.F. Vascular development of the brain requires β8 integrin expression in the neuroepithelium. J Neurosci. 2005;25:9940–9948. doi: 10.1523/JNEUROSCI.3467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mobley A.K., Tchaicha J.H., Shin J., Hossain M.G., McCarty J.H. β8 integrin regulates neurogenesis and neurovascular homeostasis in the adult brain. J Cell Sci. 2009;122:1842–1851. doi: 10.1242/jcs.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lakhe-Reddy S., Khan S., Konieczkowski M., Jarad G., Wu K.L., Reichardt L.F., Takai Y., Bruggeman L.A., Wang B., Sedor J.R., Schelling J.R. β8 integrin binds Rho GDP dissociation inhibitor-1 and activates Rac1 to inhibit mesangial cell myofibroblast differentiation. J Biol Chem. 2006;281:19688–19699. doi: 10.1074/jbc.M601110200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertelli E., Regoli M., Fonzi L., Occhini R., Mannucci S., Ermini L., Toti P. Nestin expression in adult and developing human kidney. J Histochem Cytochem. 2007;55:411–421. doi: 10.1369/jhc.6A7058.2007. [DOI] [PubMed] [Google Scholar]
- 14.Daniel C., Albrecht H., Ludke A., Hugo C. Nestin expression in repopulating mesangial cells promotes their proliferation. Lab Invest. 2008;88:387–397. doi: 10.1038/labinvest.2008.5. [DOI] [PubMed] [Google Scholar]
- 15.Kikkawa Y., Virtanen I., Miner J.H. Mesangial cells organize the glomerular capillaries by adhering to the G domain of laminin α5 in the glomerular basement membrane. J Cell Biol. 2003;161:187–196. doi: 10.1083/jcb.200211121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potter E.L. Development of the human glomerulus. Arch Pathol. 1965;80:241–255. [PubMed] [Google Scholar]
- 17.Schlondorff D., Banas B. The mesangial cell revisited: no cell is an island. J Am Soc Nephrol. 2009;20:1179–1188. doi: 10.1681/ASN.2008050549. [DOI] [PubMed] [Google Scholar]
- 18.Leveen P., Pekny M., Gebre-Medhin S., Swolin B., Larsson E., Betsholtz C. Mice deficient for PDGF-B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8:1875–1887. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]
- 19.Takemoto M., He L., Norlin J., Patrakka J., Xiao Z., Petrova T., Bondjers C., Asp J., Wallgard E., Sun Y., Samuelsson T., Mostad P., Lundin S., Miura N., Sado Y., Alitalo K., Quaggin S.E., Tryggvason K., Betsholtz C. Large-scale identification of genes implicated in kidney glomerulus development and function. EMBO J. 2006;25:1160–1174. doi: 10.1038/sj.emboj.7601014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eremina V., Sood M., Haigh J., Nagy A., Lajoie G., Ferrara N., Gerber H.P., Kikkawa Y., Miner J.H., Quaggin S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishimura S.L., Sheppard D., Pytela R. Integrin αvβ8: Interaction with vitronectin and functional divergence of the β8 cytoplasmic domain. J Biol Chem. 1994;269:28708–28715. [PubMed] [Google Scholar]
- 22.Mu D., Cambier S., Fjellbirkeland L., Baron J.L., Munger J.S., Kawakatsu H., Sheppard D., Broaddus V.C., Nishimura S.L. The integrin αvβ8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-β1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishimura S.L. Integrin-mediated transforming growth factor-β activation, a potential therapeutic target in fibrogenic disorders. Am J Pathol. 2009;175:1362–1370. doi: 10.2353/ajpath.2009.090393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turck J., Pollock A.S., Lee L.K., Marti H.P., Lovett D.H. Matrix metalloproteinase 2 (gelatinase A) regulates glomerular mesangial cell proliferation and differentiation. J Biol Chem. 1996;271:15074–15083. doi: 10.1074/jbc.271.25.15074. [DOI] [PubMed] [Google Scholar]
- 25.Annes J.P., Munger J.S., Rifkin D.B. Making sense of latent TGFβ activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 26.Hynes R.O. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gharavi A.G., Bruggeman L.A., Oller S., Klotman P.E., Lifton R.P. Genetic background modifies the development of renal disease in HIV-1 transgenic mice. J Am Soc Nephrol. 1999;10:404A. [Google Scholar]
- 28.George M., Ayuso E., Casellas A., Costa C., Devedjian J.C., Bosch F. Beta cell expression of IGF-I leads to recovery from type 1 diabetes. J Clin Invest. 2002;109:1153–1163. doi: 10.1172/JCI12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schelling J.R., Nkemere N., Kopp J.B., Cleveland R.P. Fas-dependent fratricidal apoptosis is a mechanism of tubular epithelial cell deletion in chronic renal failure. Lab Invest. 1998;78:813–824. [PubMed] [Google Scholar]
- 30.Khan S., Wu K.L., Sedor J.R., Abu Jawdeh B.G., Schelling J.R. The NHE1 Na+/H+ exchanger regulates cell survival by activating and targeting ezrin to specific plasma membrane domains. Cell Mol Biol. 2006;52:115–121. [PubMed] [Google Scholar]
- 31.Akis N., Madaio M.P. Isolation, culture, and characterization of endothelial cells from mouse glomeruli. Kidney Int. 2004;65:2223–2227. doi: 10.1111/j.1523-1755.2004.00634.x. [DOI] [PubMed] [Google Scholar]
- 32.Kondo S., Scheef E.A., Sheibani N., Sorenson C.M. PECAM-1 isoform-specific regulation of kidney endothelial cell migration and capillary morphogenesis. Am J Physiol Cell Physiol. 2007;292:C2070–C2083. doi: 10.1152/ajpcell.00489.2006. [DOI] [PubMed] [Google Scholar]
- 33.Khan S., Koepke A., Jarad G., Schlessman K., Cleveland R.P., Wang B.C., Konieczkowski M., Schelling J.R. Apoptosis and JNK activation are differentially regulated by Fas expression level in renal tubular epithelial cells. Kidney Int. 2001;60:65–76. doi: 10.1046/j.1523-1755.2001.00771.x. [DOI] [PubMed] [Google Scholar]
- 34.Abe M., Harpel J.G., Metz C.N., Nunes I., Loskutoff D.J., Rifkin D.B. An assay for transforming growth factor-β using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- 35.Kerjaschki D., Miettinen A., Farquhar M.G. Initial events in the formation of immune deposits in passive Heymann nephritis: gp330-anti-gp330 immune complexes form in epithelial coated pits and rapidly become attached to the glomerular basement membrane. J Exp Med. 1987;166:109–128. doi: 10.1084/jem.166.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baleato R.M., Guthrie P.L., Gubler M.C., Ashman L.K., Roselli S. Deletion of Cd151 results in a strain-dependent glomerular disease due to severe alterations of the glomerular basement membrane. Am J Pathol. 2008;173:927–937. doi: 10.2353/ajpath.2008.071149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bird I.N., Taylor V., Newton J.P., Spragg J.H., Simmons D.L., Salmon M., Buckley C.D. Homophilic PECAM-1(CD31) interactions prevent endothelial cell apoptosis but do not support cell spreading or migration. J Cell Sci. 1999;112:1989–1997. doi: 10.1242/jcs.112.12.1989. [DOI] [PubMed] [Google Scholar]
- 38.Mamdouh Z., Chen X., Pierini L.M., Maxfield F.R., Muller W.A. Targeted recycling of PECAM from endothelial surface-connected compartments during diapedesis. Nature. 2003;421:748–753. doi: 10.1038/nature01300. [DOI] [PubMed] [Google Scholar]
- 39.Scholzen T., Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 40.Hohenstein B., Renk S., Lang K., Daniel C., Freund M., Leon C., Amann K.U., Gachet C., Hugo C.P. P2Y1 gene deficiency protects from renal disease progression and capillary rarefaction during passive crescentic glomerulonephritis. J Am Soc Nephrol. 2007;18:494–505. doi: 10.1681/ASN.2006050439. [DOI] [PubMed] [Google Scholar]
- 41.Duncan G.S., Andrew D.P., Takimoto H., Kaufman S.A., Yoshida H., Spellberg J., Elia A., Wakeham A., Karan-Tamir B., Muller W.A., Senaldi G., Zukowski M.M., Mak T.W. Genetic evidence for functional redundancy of platelet/endothelial cell adhesion molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J Immunol. 1999;162:3022–3030. Luis dlP. [PubMed] [Google Scholar]
- 42.Gao C., Sun W., Christofidou-Solomidou M., Sawada M., Newman D.K., Bergom C., Albelda S.M., Matsuyama S., Newman P.J. PECAM-1 functions as a specific and potent inhibitor of mitochondrial-dependent apoptosis. Blood. 2003;102:169–179. doi: 10.1182/blood-2003-01-0003. [DOI] [PubMed] [Google Scholar]
- 43.Dimaio T.A., Wang S., Huang Q., Scheef E.A., Sorenson C.M., Sheibani N. Attenuation of retinal vascular development and neovascularization in PECAM-1-deficient mice. Dev Biol. 2008;315:72–88. doi: 10.1016/j.ydbio.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osawa M., Masuda M., Kusano K., Fujiwara K. Evidence for a role of platelet endothelial cell adhesion molecule-1 in endothelial cell mechanosignal transduction: is it a mechanoresponsive molecule. J Cell Biol. 2002;158:773–785. doi: 10.1083/jcb.200205049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tzima E., Irani-Tehrani M., Kiosses W.B., Dejana E., Schultz D.A., Engelhardt B., Cao G., DeLisser H., Schwartz M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 46.Zhang J.J., Kelm R.J., Biswas P., Kashgarian M., Madri J.A. PECAM-1 modulates thrombin-induced tissue factor expression on endothelial cells. J Cell Physiol. 2007;210:527–537. doi: 10.1002/jcp.20908. [DOI] [PubMed] [Google Scholar]
- 47.Kruger T., Benke D., Eitner F., Lang A., Wirtz M., Hamilton-Williams E.E., Engel D., Giese B., Muller-Newen G., Floege J., Kurts C. Identification and functional characterization of dendritic cells in the healthy murine kidney and in experimental glomerulonephritis. J Am Soc Nephrol. 2004;15:613–621. doi: 10.1097/01.asn.0000114553.36258.91. [DOI] [PubMed] [Google Scholar]
- 48.Soos T.J., Sims T.N., Barisoni L., Lin K., Littman D.R., Dustin M.L., Nelson P.J. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 2006;70:591–596. doi: 10.1038/sj.ki.5001567. [DOI] [PubMed] [Google Scholar]
- 49.Munger J.S., Huang X., Kawakatsu H., Griffiths M.J., Dalton S.L., Wu J., Pittet J.F., Kaminski N., Garat C., Matthay M.A., Rifkin D.B., Sheppard D. The integrin αvβ6 binds and activates latent TGFβ1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 50.Annes J.P., Chen Y., Munger J.S., Rifkin D.B. Integrin αvβ6-mediated activation of latent TGF-β requires the latent TGF-β binding protein-1. J Cell Biol. 2004;165:723–734. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schelling J.R., Sinha S., Konieczkowski M., Sedor J.R. Myofibroblast differentiation: plasma membrane microdomains and cell phenotype. Exp Nephrol. 2002;10:313–319. doi: 10.1159/000065309. [DOI] [PubMed] [Google Scholar]
- 52.Tamaki K., Okuda S., Nakayama M., Yanagida T., Fujishima M. Transforming growth factor-β1 in hypertensive renal injury in Dahl salt-sensitive rats. J Am Soc Nephrol. 1996;7:2578–2589. doi: 10.1681/ASN.V7122578. [DOI] [PubMed] [Google Scholar]
- 53.Sterzel R.B., Hartner A., Schlotzer-Schrehardt U., Voit S., Hausknecht B., Doliana R., Colombatti A., Gibson M.A., Braghetta P., Bressan G.M. Elastic fiber proteins in the glomerular mesangium in vivo and in cell culture. Kidney Int. 2000;58:1588–1602. doi: 10.1046/j.1523-1755.2000.00320.x. [DOI] [PubMed] [Google Scholar]
- 54.McCarty J.H., Cook A.A., Hynes R.O. An interaction between αvβ8 integrin and Band 4.1B via a highly conserved region of the Band 4.1 C-terminal domain. Proc Natl Acad Sci USA. 2005;102:13479–13483. doi: 10.1073/pnas.0506068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pollman M.J., Naumovski L., Gibbons G.H. Vascular cell apoptosis: cell type-specific modulation by TGF-β1 in endothelial cells versus smooth muscle cells. Circulation. 1999;99:2019–2026. doi: 10.1161/01.cir.99.15.2019. [DOI] [PubMed] [Google Scholar]
- 56.Fierlbeck W., Liu A., Coyle R., Ballermann B.J. Endothelial cell apoptosis during glomerular capillary lumen formation in vivo. J Am Soc Nephrol. 2003;14:1349–1354. doi: 10.1097/01.asn.0000061779.70530.06. [DOI] [PubMed] [Google Scholar]
- 57.Nystrom J., Fierlbeck W., Granqvist A., Kulak S.C., Ballermann B.J. A human glomerular SAGE transcriptome database. BMC Nephrol. 2009;10:13. doi: 10.1186/1471-2369-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aluwihare P., Mu Z., Zhao Z., Yu D., Weinreb P.H., Horan G.S., Violette S.M., Munger J.S. Mice that lack activity of αvβ6- and αvβ8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci. 2009;122:227–232. doi: 10.1242/jcs.035246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hauser I.A., Riess R., Hausknecht B., Thuringer H., Sterzel R.B. Expression of cell adhesion molecules in primary renal disease and renal allograft rejection. Nephrol Dial Transplant. 1997;12:1122–1131. doi: 10.1093/ndt/12.6.1122. [DOI] [PubMed] [Google Scholar]
- 60.Sivridis E., Giatromanolaki A., Touloupidis S., Pasadakis P., Vargemezis V. Platelet endothelial cell adhesion molecule-1 and angiogenic factor expression in idiopathic membranous nephropathy. Am J Kidney Dis. 2003;41:360–365. doi: 10.1053/ajkd.2003.50044. [DOI] [PubMed] [Google Scholar]
- 61.Iruela-Arispe L., Gordon K., Hugo C., Duijvestijn A.M., Claffey K.P., Reilly M., Couser W.G., Alpers C.E., Johnson R.J. Participation of glomerular endothelial cells in the capillary repair of glomerulonephritis. Am J Pathol. 1995;147:1715–1727. [PMC free article] [PubMed] [Google Scholar]
- 62.Isome M., Fujinaka H., Yaoita E., Feng L., Adhikary L.P., Abe A., Tsuchida S., Kawasaki K., Suzuki H., Kihara I., Wilson C.B., Yamamoto T. Involvement of endothelial cell adhesion molecules in the development of anti-Thy-1 nephritis. Exp Nephrol. 2002;10:338–347. doi: 10.1159/000065298. [DOI] [PubMed] [Google Scholar]
- 63.Lastres P., Almendro N., Bellon T., Lopez-Guerrero J.A., Eritja R., Bernabeu C. Functional regulation of platelet/endothelial cell adhesion molecule-1 by TGF-β1 in promonocytic U-937 cells. J Immunol. 1994;153:4206–4218. [PubMed] [Google Scholar]
- 64.Cambier S., Gline S., Mu D., Collins R., Araya J., Dolganov G., Einheber S., Boudreau N., Nishimura S.L. Integrin αvβ8-mediated activation of transforming growth factor-β by perivascular astrocytes: an angiogenic control switch. Am J Pathol. 2005;166:1883–1894. doi: 10.1016/s0002-9440(10)62497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Isermann B., Vinnikov I.A., Madhusudhan T., Herzog S., Kashif M., Blautzik J., Corat M.A., Zeier M., Blessing E., Oh J., Gerlitz B., Berg D.T., Grinnell B.W., Chavakis T., Esmon C.T., Weiler H., Bierhaus A., Nawroth P.P. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–1358. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]
- 66.Armulik A., Abramsson A., Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 67.Shi Y.G., Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 68.Sharma R., Khanna A., Sharma M., Savin V.J. Transforming growth factor-β1 increases albumin permeability of isolated rat glomeruli via hydroxyl radicals. Kidney Int. 2000;58:131–136. doi: 10.1046/j.1523-1755.2000.00148.x. [DOI] [PubMed] [Google Scholar]
- 69.Yamamoto T., Noble N.A., Miller D.E., Border W.A. Sustained expression of TGF-β1 underlies development of progressive kidney fibrosis. Kidney Int. 1994;45:916–927. doi: 10.1038/ki.1994.122. [DOI] [PubMed] [Google Scholar]
- 70.Heldin C.H., Landstrom M., Moustakas A. Mechanism of TGF-β signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009;21:166–176. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 71.Wu C.J., Chen Z., Ullrich A., Greene M.I., O'Rourke D.M. Inhibition of EGFR-mediated phosphoinositide-3-OH kinase (PI3-K) signaling and glioblastoma phenotype by signal-regulatory proteins (SIRPs) Oncogene. 2000;19:3999–4010. doi: 10.1038/sj.onc.1203748. [DOI] [PubMed] [Google Scholar]
- 72.Tai L.K., Zheng Q., Pan S., Jin Z.G., Berk B.C. Flow activates ERK1/2 and endothelial nitric oxide synthase via a pathway involving PECAM1: SHP2, and Tie2. J Biol Chem. 2005;280:29620–29624. doi: 10.1074/jbc.M501243200. [DOI] [PubMed] [Google Scholar]
- 73.Yang W., Klaman L.D., Chen B., Araki T., Harada H., Thomas S.M., George E.L., Neel B.G. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell. 2006;10:317–327. doi: 10.1016/j.devcel.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 74.Bjarnegård M., Enge M., Norlin J., Gustafsdottir S., Fredriksson S., Abramsson A., Takemoto M., Gustafsson E., Fässler R., Betsholtz C. Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development. 2004;131:1847–1857. doi: 10.1242/dev.01080. [DOI] [PubMed] [Google Scholar]
- 75.Abrass C.K., Berfield A.K., Ryan M.C., Carter W.G., Hansen K.M. Abnormal development of glomerular endothelial and mesangial cells in mice with targeted disruption of the lama3 gene. Kidney Int. 2006;70:1062–1071. doi: 10.1038/sj.ki.5001706. [DOI] [PubMed] [Google Scholar]
- 76.Sheibani N., Sorenson C.M., Frazier W.A. Tissue specific expression of alternatively spliced murine PECAM-1 isoforms. Dev Dyn. 1999;214:44–54. doi: 10.1002/(SICI)1097-0177(199901)214:1<44::AID-DVDY5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 77.Xie Y., Muller W.A. Molecular cloning and adhesive properties of murine platelet/endothelial cell adhesion molecule 1. Proc Natl Acad Sci USA. 1993;90:5569–5573. doi: 10.1073/pnas.90.12.5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.