

Abstract
Recent analyses of complete genomes have revealed that alternative splicing became more prevalent and important during eukaryotic evolution. Alternative splicing augments the protein repertoire—particularly that of the human genome—and plays an important role in the development and function of differentiated cell types. However, splicing is also extremely vulnerable, and defects in the proper recognition of splicing signals can give rise to a variety of diseases. In this review, we discuss splicing correction therapies, by using the inherited disease Spinal Muscular Atrophy (SMA) as an example. This lethal early childhood disorder is caused by deletions or other severe mutations of SMN1, a gene coding for the essential survival of motoneurons protein. A second gene copy present in humans and few non-human primates, SMN2, can only partly compensate for the defect because of a single nucleotide change in exon 7 that causes this exon to be skipped in the majority of mRNAs. Thus SMN2 is a prime therapeutic target for SMA. In recent years, several strategies based on small molecule drugs, antisense oligonucleotides or in vivo expressed RNAs have been developed that allow a correction of SMN2 splicing. For some of these, a therapeutic benefit has been demonstrated in mouse models for SMA. This means that clinical trials of such splicing therapies for SMA may become possible in the near future.
Key words: antisense oligonucleotides, bifunctional RNA, exon definition, gene therapy, motoneurons, splicing enhancer, U7snRNA
Introduction
In higher eukaryotes, most transcripts of protein coding-genes are subject to splicing. Moreover, alternative splicing provides variant transcripts that may encode different proteins or that may be alternatively productive or non-productive with regard to a given protein product. The human genome is estimated to contain ∼25,000 protein-coding transcription units, whereas the number of proteins could be considerably higher.1,2 Although other mechanisms (such as RNA editing, post-translational modifications) contribute to this increase in protein diversity, alternative splicing is clearly its main source. Alternative splicing is particularly important for the development and differentiation of complex tissues such as the nervous system. Large scale studies reveal that ∼95% of human genes undergo alternative splicing.3–5 Considering the importance of constitutive and alternative splicing, it is not surprising that defects in these processes can generate aberrant transcripts with diverse pathologic consequences. In this review, we will briefly describe causes and effects of errors in constitutive and alternative splicing. Then, we will describe the therapeutic strategies currently being developed to circumvent these effects, with a special focus on the human inherited disease Spinal Muscular Atrophy (SMA).
The Importance of Exon Definition
In higher eukaryotes and especially in humans, introns are much longer than exons (∼3,500 bp compared to ∼150 bp, respectively).6 Moreover, the signals recognized by the splicing machinery, the 5′ and 3′ splice sites (SS) and the branch points (BP), are highly degenerate (Fig. 1A).7 Many sequences within a transcription unit have the potential to act as ss, but only a minority of them are true ones in the sense that they are used in constitutive or alternative splicing. All sequences that resemble the 5′ or 3′ SS consensus but that are not used in splicing are defined as cryptic SS.
Figure 1.

Cis acting sequence elements and trans-acting factors determining exon definition. (A) Conserved sequences flanking metazoan and yeast exons. The exon is symbolized by a yellow rectangle. The flanking introns are symbolized by lines in which the branch point (BP) sequence and polypyrimidine tract (PP tract) are highlighted as light blue boxes. The conserved nucleotides of the BP, PP tract, 3′ splice site (3′ SS) and 5′ splice site (5′ SS) are shown below with numbers indicating the percent prevalence of the most frequent nucleotides at each position. Shown in red are the branch point adenosine as well as the virtually invariant last two and first two nucleotides of the introns. (B) Role of splicing activators and repressors in splicing modulation. SR proteins bind to exonic splicing enhancers (ESE; dark blue) via their RNA recognition motifs and favour the recruitment of the splicing machinery (+) at the 5′ and 3′ SS, mainly by stabilizing the interaction between snRNPs and the pre-mRNA. SR proteins also act by direct protein-protein interactions with U1 snRNPs at the 5′ SS, and with the U2 snRNP and U2AF co-factors at the 3′ SS. In contrast, hnRNP proteins most frequently bind to intronic splicing silencers (ISS; red) and are involved in repression (−) of splicing, by exerting negative effects on U snRNPs or SR proteins. These different interactions are represented by arrows.
This degenerate nature of SS and BP sequences and the frequent occurrence of cryptic signals raises the question what distinguishes true signals from cryptic ones. The answer is that additional informations encoded in the sequence of a primary transcript help to define the exons of a transcription unit.7 These informations can be found in exons or in introns, and are correspondingly called exonic splicing enhancers (ESEs), exonic splicing silencers (ESSs), intronic splicing enhancers (ISEs) and intronic splicing silencers (ISSs). The enhancers promote exon inclusion, whereas the silencers repress it. They do so by recruiting RNA-binding proteins that act as splicing regulators (Fig. 1B).
The predominant proteins acting as promoters by binding to ESEs, the so-called SR and SR-related proteins (SRp), contain one or more RNA recognition motifs (RRM) and a region of alternating serines and arginines. The RRM allows them to bind to target sequences in pre-mRNAs, whereas the SR repeats act as protein interaction domains with which these proteins can help to recruit parts of the spliceosomal machinery to the nearest intron-exon boundary, thereby improving exon definition.8–10 In contrast, the hnRNPs (heterogenous nuclear ribonucleoproteins) rather act as silencers by antagonising the recognition of splice sites directly or by disturbing the link between enhancers and their interactors.11,12 In general, binding sites for SR proteins are more frequent in true exons, and hnRNP binding sites are more often found in introns.
Nevertheless, the positive or negative roles of these factors in splicing are not so simply defined, since in certain exons, they may also act in an opposite way. Moreover additional proteins not belonging to these two groups, such as proteins of the CELF/Bruno-like family,11,13 can act both as enhancers or modulators of splicing. In the end, the definition of an exon and hence its inclusion in the mature mRNA is the composite result of the sequences and relative positions of splicing signals (SS and BP), enhancers and silencers, the amounts of enhancing and repressing transacting factors and their interaction with each other as well as with components of the basic splicing machinery. Even the secondary structure of the RNA to be spliced may sometimes influence exon definition.14
Alternative splicing results when the interplay of all these sequences, splicing regulators and the general splicing factors that are directly involved in the spliceosome cycle can be interpreted in different ways. For many transcritpion units, multiple splicing isoforms can persist as mature mRNAs in a given cell. However, SS and exon choices can also vary within a given cell under different physiological conditions. Most important are changes in splicing patterns that occur during development while cells differentiate. Some of these splicing differences may persist and establish long-lasting tissue- or cell type-specific patterns of splicing.
The differential expression of mRNA isoforms by alternative splicing can depend on several parameters. Splicing regulatory proteins can be controlled in their abundance or activity. Of particular importance is the tissue-specific distribution of RNA-binding proteins acting as splicing regulators. In the central nervous system (CNS), alternative splicing is particularly important, as it allows for the differentiation of neurons in several ways.15–18 An interesting switch in the abundance of the splicing regulator polypyrimidine tract binding protein (PTB) to its neuronal counterpart nPTB represents an important trigger in neuronal differentiation. The downregulation of PTB is initiated by a micro-RNA, miR-124 and is then maintained by nPTB which negatively affects the inclusion of exon 11 in the PTB mRNA.19–21 This PTB/nPTB switch leads to differential splicing of a few hundred mRNAs and thus plays a major role in reprogramming the cells that will become neurons. Another set of splicing regulators, the Nova proteins, are specifically expressed in neurons and appear to induce a similar switch which leads to synaptic maturation.22–24 These are only a few examples which demonstrate the importance of alternative splicing in developmental processes, as a single splicing regulatory protein can affect the expression of dozens or hundreds of other genes.
Splicing and Disease
Viewing the importance and complexity of splicing, the fact that it plays an important role in many diseases is not surprising. Early studies indicating that ∼15% of point mutations leading to genetic disease were due to RNA splicing defects25 were probably underestimates for several reasons. The early genetic analyses relied exclusively on DNA samples and concentrated on the sequences of exons and their flanking SS. Only with recent progress in DNA sequencing, intronic sequences have really come under scrutiny. Moreover, some missense mutations may act through their effects on splicing rather than on the function of the protein.26 Additionally, silent mutations which may have been classified as polymorphisms may in fact alter binding sites for splicing regulators.27,28 Bioinformatic programs to predict regulatory binding sites (ESE, ESS, ISE, ISS) are still not very reliable. Finally, the organization of human genetic sampling is not geared to analyze RNA; this is even more of a problem when it is difficult or even impossible to obtain biopsies from the affected tissues.29
This slow development of splicing analyses as part of the genetic testing process is reflected by the fact that most splicing mutations reported so far are ones that destroy a SS, or create one by mutating a related sequence.27,28 However, it is clear that splicing defects may also arise by mutations that cause steric block of SS,30 modulate SS communication31,32 or change RNA folding.33,34 It is interesting that only very few mutations have been found in the core spliceosomal components and the splicing regulatory proteins, certainly due to lethal effects during early development.35 An increasing number of mutations that is being recognized are those that affect regulatory sites in RNA (binding sites for SR and hnRNP proteins and other splicing regulators).27,28 A particularly interesting example is Myotonic Dystrophy which is caused by a CUG trinucleotide expansion. The expanded repeats sequester the muscleblind protein that acts as a splicing regulator.36 The resulting pleiotropic splicing defects are the immediate cause of the Myotonic Dystrophy.
SMA, Not a Splicing Disease by Itself, Calls for a Splicing Therapy
The neuromuscular disorder Spinal Muscular Atrophy (SMA) occurs with a carrier frequency between 1/35 and 1/80 leading to an affected individual every 1 in 6,000 to 1 in 10,000 live births.37–39 It is the second most common autosomal recessive disease in Caucasians after Cystic Fibrosis and a frequent cause of early infant mortality. Ailment progression is characterized by a degeneration of α-motoneurons in the anterior horn of the spinal cord resulting in symmetrical muscle weakness and paralysis of both, proximal and distal muscles. Depending on the time of onset, severity of phenotypes and age at death, SMA has been divided into several types:
Type I (Werdnig-Hoffmann disease), the most severe form, characterized by appearance of muscular weakness during the first months of life and an average lifespan of 8 months.
Type II (intermediate form), with clinical manifestation starting 6–18 months after birth and life expectancy of 2–30 years.
Type III (Kugelberg-Welander disease); first impacts are typically observed after the second year of life; patients often get wheelchair-bound within or after adolescence.
Two additional types are described in the literature, namely the very severe type 0, with prenatal onset and early neonatal death and type VI, a genetically heterogeneous appearance with comparatively mild consequences emerging only during adulthood.40
More than 95% of all SMA cases are due to mutations or deletions in the gene SMN1 Survival of Motor Neuron 1, located in chromosome region 5q12–5q13.38 This region contains a 500-kb inverted duplication. The telomeric SMN1 and centromeric SMN2 genes differ in only a few nucleotides (none of which affect the encoded protein sequence). The most relevant of these is a C to T transition in exon 7 (see below). Due to gene conversion and duplication events, the number of SMN2 copies in patients can vary, and this was shown to modulate the severity of the disease phenotype.41 The SMN protein is essential for normal development and cell survival, as homozygous deletions of the single Smn gene in mice have been shown to be lethal in early embryonic stages.42,43 Human survival is only possible due to the second gene copy, and all SMA patients therefore carry at least one SMN2 gene.
The SMN protein is composed of 249 amino acids and migrates at 38 kDa on SDS gels.44 Although it is ubiquitously expressed, its levels vary between different tissues. In healthy adults, particularly high amounts of the protein are found in spinal cord, kidney, liver and brain tissue, whereas skeletal and cardiac muscle show intermediate amounts, and even lower quantities occur in fibroblasts and lymphocytes.45 Its expression is also developmentally regulated, displaying the highest abundance in prenatal phases.46
That SMN is an essential protein for all cells can be explained by its well characterized biochemical function. Together with a small number of other proteins, called Gemins 2–8, it forms the SMN complex which mediates the cytoplasmic assembly of the spliceosomal small nuclear ribonucleoproteins (snRNPs) U1, U2, U4 and U5, their counterparts from the minor spliceosome (U11, U12, U4atac) and the U7 snRNP involved in histone RNA 3′ end processing.47–52 Besides this cytoplasmic function, the SMN complex is also found in nuclear foci, called gems (for gemini of Cajal bodies), which often colocalize with Cajal bodies. In these locations, the SMN complex is proposed to play a role in snRNP maturation, recycling or turnover.53,54 Moreover, SMN has been proposed to play a role in nuclear import of snRNPs,55 together with snurportin which is an import adaptor recognizing the trimethyl guanosine cap structure of snRNAs that is generated after SMN-mediated assembly has taken place.56
Besides this well-characterized function in snRNP maturation, SMN has also been proposed to play a direct role in splicing48 or to participate in the assembly of other ribonucleoprotein particles.57
It is presently still not clear if the motoneuron defects seen in SMA patients are related to SMN's role in snRNP metabolism. Although partial and unequal deficiencies in snRNP concentrations have been observed,58,59 it is still unclear whether the primary defect is a disturbance of splicing patterns that would be most pronounced or whose effects would be most damaging for motoneurons.60 An alternative explanation is that some additional but less well characterized interactions61 and functions of SMN could be vital to the maintenance of motoneuron functionality and viability.62,63 This latter view is supported by studies showing that the SMN protein is present in transport granules of cultured motoneurons travelling down the axon.64,65 Some of SMN's interaction partners such as hnRNP R and Q,66 profilins (proteins associated with microfilaments)65,67 or the Fragile X Mental Retardation protein FMRP68 support such a peripheral function. Thus it has been proposed that SMN could be involved in the transport or translational regulation of actin mRNA or other so far not characterized mRNAs and thereby control the organisation of the axon terminal.62,65 Interestingly, the phenotypes observed in SMA mouse models all show first perturbations of the neuromuscular junctions, before an actual cell death can be confirmed in the spinal cord.69–76 This strongly favours a dying back mechanism whereby the functionality of the synapse in the periphery gets primarily disturbed, suggesting initial mechanisms being disrupted at this place.77 It has to be mentioned that the the two hypotheses—splicing defect versus motoneuron-specific role—do not exclude each other and can actually be combined.
The Architecture of SMN Genes with Regard to Exon 7 Definition
Although the two SMN gene copies are nearly identical, SMN2 is not able to completely substitute for the loss of function due to destroyed SMN1. The problem is that one of the previously mentioned nucleotide differences, the C → U transition at position 6 of exon 7, changes the splicing pattern such that this exon is mostly excluded from the native mRNA, thereby leading to the predominant production of a truncated protein during translation.78 This truncated protein (SMNΔ7) retains some functionality, as evidenced by slightly longer survival times when it is expressed in SMA mice (on a background of deleted mouse Smn genes and two copies of human SMN2).79 However, SMNΔ7 is unable to oligomerize80 and therefore very unstable.81
The question how an exchange of a single nucleotide can have such a detrimental effect on pre-mRNA splicing can partly be explained by the structure and sequence of this exon and its environment. Of three initially characterized ESEs,81 two, SE1 and SE2, seem to be functional (Fig. 2). SE2 can be bound by the SR-like splicing factor hTra2-β1, which increases the inclusion of this exon in a dose-dependent manner82 by forming stable complexes with two other splicing factors, namely SRp30c83 and hnRNP G.84 The SE1 element acts as an SF2/ASF-dependent splicing enhancer which is destroyed by the critical C6U mutation.85,86 However, this mutation additionally appears to create a new exonic splicing silencer signal (ESS) that is bound by hnRNP A1.87,88
Figure 2.

Splicing architecture of exon 7 of the human SMN1 and SMN2 genes. The diagram represents exon 7 (yellow box) and its flanking intronic regions (lines). Elements inhibiting exon 7 inclusion are shown in red, whereas the positive elements are represented in dark blue. The suboptimal branch point (BP) and polypyrimidine tract (PP tract) are indicated in light blue. SF2/ASF and Tra2/β1 bind to the exonic splicing enhancers SE1 and SE2, respectively. The recognition of SE1 by SF2/ASF is prevented in SMN2, due to the C → U transition. This sequence alteration also creates a hnRNP A1-dependent splicing silencer (see main text for refs.).
Obviously, the exon 7 seems to be heavily dependent on regulatory splicing signals, all the more as it is flanked by two intronic splicing silencer sequences (ISSs), one lying within intron 6 (termed element 1; Fig. 2)89 and the other in intron 7 (termed ISS-N1).90 The latter was recently shown to contain two binding sites for hnRNP A1/A2.91 Finally, two additional intronic splicing enhancer sequences were discovered in intron 7 that strongly influence the exon 7 inclusion ratio.92
Compared to the average length of human exons, which lies around 129 base pairs,93 this crucial exon 7 appears to be extremely short, consisting of only 54 bp. An in vivo SELEX study performed with a randomized SMN1 exon 7 sequence came to the conclusion that both, the 5′ and the 3′ endings seem to predispose it for exclusion.94 On the 5′ end, positions 5–15 create an overall negative surface for splicing. The 3′ end shows an element with negative splicing impacts as well, namely at position 45–52. This region also seems to be involved in a secondary structure that affects exon 7 inclusion.95 Finally, the last exonic nucleotide represents an A instead of the canonical G found in more than 80% of all human exons.7 As a consequence, the binding affinity of the U1 snRNP is reduced.94,96 In addition to these exonic features, the polypyrimidine tract at the 3′ SS81 and the BP97 preceding exon 7 are suboptimal.
In summary, the SMN exon 7 is rather poorly defined, and positive and negative elements create a very sensitive equilibrium towards exon inclusion. The vast majority of transcripts from the SMN1 gene (approximately 90%) contains all 9 exons (full-length transcripts) and the remaining 10% are mostly missing exon 5.45 In SMN2, however, this balance is disturbed by converting an enhancer motif into a silencer, and the vantage therefore tips to predominant exclusion. The SMN2 gene in fact produces different mRNA isoforms, some lacking exon 5, exon 7, or even both exons as well as full-length mRNA. The full-length mRNA accounts for only 20–30% of the total,98,99 even though this percentage strongly varies between different patients and tissues. Thus, the production of full-length SMN protein is severely reduced in SMA patients, since a functional SMN1 gene copy is absent.
Promising Approaches to Improve SMN2 Exon 7 Inclusion
Strategies aiming to repair splicing defects are being developed for several diseases and by many different laboratories. SMA is only one example, albeit an important one for which multiple strategies have been tested.
However, before we delve into a discussion of these approaches, it should be mentioned that strategies which are not directed at correcting SMN2 splicing are also being tried with some success. For example, it is possible to enhance the promoter activity of the SMN2 gene. Valproate, a histone deacetylase (HDAC) inhibitor has been shown to increase the production of full-length SMN and to extend the survival of transgenic SMA mice deleted for mouse Smn and expressing human SMN2.79,100,101 However, a study performed with SMA patients showed no increase of SMN.102 Another HDAC inhibitor, phenylbutyrate, yielded promising results in vitro,103 but a test performed on a large cohort of SMA type II patients did not show an improvement.104 Hydroxyurea (HU), a ribonucleotide reductase inhibitor, is also able to increase the expression of SMN in SMA cells in vitro,105,106 and a test performed on 33 patients of SMA types II and III, showed a slight increase in muscle strength and full-length SMN mRNA in some patients.106 A recent drug screen revealed that certain 2,4-diaminoquinazoline derivatives also stimulate the SMN promoter and can increase the lifespan of SMNΔ7 SMA mice by approximately 21–30% when given prior to motoneuron loss.107 Overall, these studies need further investigation and cohort selection to really evaluate the gain obtained with these treatments. In addition to such efforts to increase SMN2 transcription, a forced translational read-through of the premature termination codon of SMNΔ7 transcripts by aminoglycoside compounds was also shown to increase functional SMN protein levels in fibroblasts derived from SMA patients.108 Finally, it should be mentioned that a gene replacement therapy is also a valid option, despite the size of the SMN gene. Two recent reports have demonstrated that different adeno-associated virus (AAV) vectors expressing SMN cDNA can reach motoneurons and partly rescue newborn mice that would otherwise show a severe SMA phenotype.109,110 Moreover, neural stem cell therapy may also be an option for treating SMA.111
Small molecule drugs improving SMN2 exon 7 inclusion.
Focusing on splicing correction strategies for SMA, certain drugs have been shown to modulate alternative splicing decisions,112,113 but in general their specificity is low and side effects are therefore a major concern. Concerning SMA, beneficial splicing effects were reported for aclarubicine,114 but the drug proved to be quite toxic. Another drug found to improve SMN2 exon 7 inclusion is a tetracycline-like compound, termed PTK-SMA1.115 However, this compound does not appear to cross the blood-brain barrier, so that further chemical derivatives or sophisticated modes of application such as cerebrospinal fluid instillation may be required.
Oligonucleotide-based approaches.
Very specific splicing correction can be obtained by the use of antisense oligonucleotides (AONs). AONs are ideally suited to hybridize to specific premRNA sequences involved in the splicing of a particular gene.116 They can contain different types of modifications that stabilize them and/or allow for more efficient target sequence hybridisation.117 Most often this approach is used to block a particular splice site or splicing enhancer and thereby to induce the skipping of target exon.116,118 However, in the case of SMA the problem is not to skip an exon but to enhance its utilisation by the splicing machinery which asks for more sophisticated approaches. Another problem specific for using AONs to treat SMA may again be how to cross the blood-brain barrier and to reach the spinal cord motoneurons.
One of the AON strategies used in the SMA context (Fig. 3A) is based on the idea that exon 7 skipping at least partly results from a competition between the 3′ SS of exons 7 and 8. Thus, to change the balance of 3′ SS use in favour of exon 7, AONs targeting the 3′ SS and BP region of exon 8 were tested. Indeed, several of these AONs increased the proportion of final transcripts with inclusion of exon 7 in tissue culture experiments.119 Other AON strategies aimed at masking inhibitory sequences that limit exon 7 inclusion (Fig. 3B). In fact AONs have been instrumental at defining several of these inhibitory elements. In particular, AONs masking sequences just downstream of the exon 7 5′ SS contributed to the discovery of the potent ISS-N1 and caused reinclusion of exon 7 in SMA patient fibroblasts.90 In a recent study, even shorter AONs were tested, and an 8-mer was shown to induce specific exon 7 inclusion and to restore SMN protein levels in SMA patient cells even when applied in nanomolar concentrations.120 Finally, the periodic intracerebroventricular instillation into SMA mice of a longer AON targeting this ISS-N1 restored SMN expression in brain and spinal cord up to 50% of the level found in control mice and led to a slight amelioration of disease symptoms.121
Figure 3.

Schematic representation of the different AON-based splicing correction strategies for SMA. Red and blue arrows indicate negative and positive splicing effects, respectively. Small blue dots signify unspecified splicing factors necessary for exon definition and splicing. (A) In a concept of 3′ SS competition where exon 8 prevails over exon 7, an AON masking the 3′ SS of exon 8 will partly shift splicing factor recruitment to exon 7. (B) Masking any of the flanking ISS by an AON can stimulate exon 7 inclusion in the mRNA. (C) The bifunctional strategy allows to tether binding sequences for different SR proteins to either ISS element 1 or the altered SE1 sequence and thereby to enhance the recruitment of the splicing machinery to exon 7. As the mutated SE1 element is also a splicing silencer, both approaches have dual effects by masking a negatively acting element and by recruiting positively acting SR proteins. An AON targeting the exon 8 3′ SS that bears a tail with a binding sequence for hnRNP A1 will also more efficiently shift splicing factor recruitment to exon 7 than the corresponding tail-less AON (see main text for refs.).
A systematic screen of a large number of AONs hybridizing to different positions of exon 7 revealed several AONs that led to efficient exon 7 reinclusion.122 These oligonucleotides may either have interfered with crucial binding sites for splicing regulatory proteins or have disrupted inhibitory RNA secondary structures. The same principle of oligo walk was also performed in the intronic regions flanking exon 7, leading to the discovery of a weak ISS in intron 6 and of two tandem hnRNP A1/A2 motifs91 in the previously described strong intron 7 ISS-N1.90 Certain AONs targeting this region also increased hSMN2 exon 7 inclusion in the liver and kidney of SMA mice.
An interesting expansion of the antisense approach was introduced almost simultaneously by two groups in 2003. In this socalled bifunctional strategy, a functional moiety is added to an AON (Fig. 3C). Both groups targeted the region of SMN2 exon 7 that carries the C → T transition and used a functional moiety that should replace the missing splicing enhancer protein. In one case, the functional moiety used was a repeating SR peptide coupled to the AON.123 In the other study an RNA sequence containing a repeated binding sequence for a SR protein (ASF/SF2) was added.124 Both of these approaches indeed led to considerable exon 7 reinclusion and a partial restoration of SMN protein levels in SMA patient fibroblasts. Two further developments of this bifunctional concept were recently published by Lorson and colleagues. In one case, an AON targeting the 3′ SS region of exon 8 was combined with a functional sequence able to attract hnRNP A1 and thereby to increase the inhibitory effect on exon 8.125 In the other case, the antisense moiety targeted the element 1 ISS located in intron 6 of SMN2, whereas the functional moiety was an ESE tail recruiting positive splicing factors.126 Both of these approaches led to improved exon 7 reinclusion compared to the corresponding simple AONs lacking the functional moiety. They were also tested by intraventricular injections into the brains of SMA mice. In both studies, increases in brain SMN protein levels were observed. In the second study, SMN levels were also increased in various sections of the spinal cord and a small positive effect on weight development and survival of the mice could be demonstrated.126 This, so far, has been the only study that has demonstrated a therapeutic effect of an oligonucleotide in a mouse SMA model.
Based on all these studies, multiple oligonucleotides—simple AONs or bifunctional ones—are now available that potently improve SMN2 exon 7 inclusion. The main problem currently preventing their use in clinical studies is the fact that they do not cross the blood-brain barrier. It is not clear if cerebrospinal fluid instillations or other types of introduction into the central nervous system will be a valid option for human patient therapy, especially if the treatments have to be applied on a life-long basis. Another point to consider is whether a repeated deposition into the central nervous system of oligonucleotides that are chemically designed to be undegradable or slowly degraded will not have unexpected long-term toxic consequences.
Approaches based on in vivo expressed RNAs.
To circumvent the problems of oligonucleotide delivery and to allow for a more permanent therapy, antisense sequences designed for splicing modulation can also be introduced into RNA expression cassettes designed to be transcribed in vivo within the target cells. As splicing occurs in the nucleus, expression cassettes based on small nuclear RNAs are particularly suited for this purpose, and the most frequently used expression system is based on a modified gene for U7 snRNA.127,128 The U7 snRNP is an essential factor involved in 3′ end processing of animal replication-dependent histone mRNAs, that lack the typical poly(A) tail present on all other eukaryotic mRNAs.129,130 A set of three point mutations introduced in the non-canonical Sm binding site of U7 snRNA (AAU UUG UCU AG) that render it similar to the consensus Sm binding site of the spliceosomal snRNAs (AAU UUU UGG AG) leads to an increased nuclear accumulation of the snRNP particle (termed U7 Sm OPT), which however is non-functional in histone RNA processing.131,132 The reason for this lack of function is the replacement of the U7-specific Sm core structure, composed of Lsm10 and Lsm11 and five Sm proteins, by a standard Sm core composed of all seven Sm proteins found in spliceosomal snRNPs.52,130,133 These observations fostered the idea that this “generic,”, but function-less U7 Sm OPT snRNP could serve as a platform for antisense sequences designed to modulate specific splicing events (Fig. 4A and inset). As was the case for AONs, such modified U7 snRNAs were initially almost exclusively designed to induce exon skipping in genes involved in human diseases such as β-thalassemia,134–137 Duchenne muscular dystrophy,138–140 or HIV/AIDS.141–143
Figure 4.
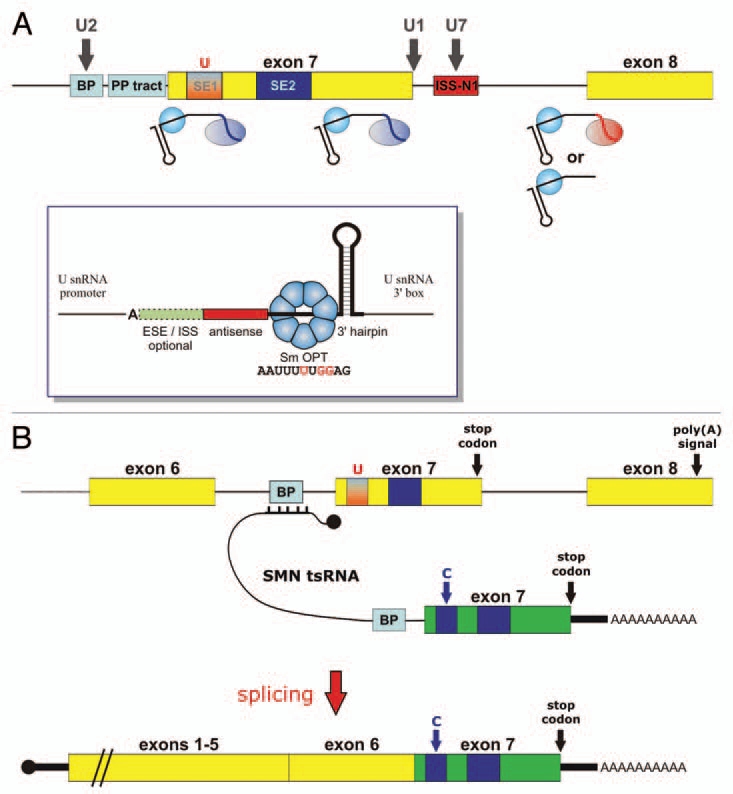
Scheme depicting splicing correction strategies for SMA based on in vivo expressed RNAs (A) SnRNA-based strategies. Bifunctional U7 Sm OPT derivatives have been designed to tether SR proteins to various positions in exon 7. Alternatively, U7 Sm OPT derivatives can mask the BP and 3′ SS of exon 8, either with or without tethering hnRNP A1. Shown with dark grey vertical arrows are snRNA-based strategies that were either not successful (U2 snRNA fully complementary to BP upstream of exon 7), inhibitory (U7 Sm OPT targeting ISS-N1) or toxic to cells (U1 snRNA fully complementary to the exon 7 5′ SS). Inset: Basic structure of a U7 Sm OPT derivative. The important elements are (from 5′ to 3′): the antisense sequence, the Sm OPT site capable of assembling with a heptameric Sm core of the standard Sm proteins (nucleotide changes respective to wild-type U7 snRNA are shown in red), and a 3′-terminal hairpin which stabilises the RNA. Splicing enhancer or silencer sequences can be added at the 5′ end to generate bifunctional U7 snRNAs. Note that transcription from a U snRNA promoter is important to allow efficient 3′ end formation at the U snRNA 3′ box and assembly into a snRNP particle. Moreover, mammalian U7 snRNAs start with an adenosine residue. (B) Trans-splicing strategy. A SMN-specific trans-splicing RNA (tsRNA) will bind to the BP/3′ SS region upstream of SMN2 exon 7 (for simplicity only the BP is shown) and will contain a strong BP/3′ SS leading into a SMN1-specific version of exon 7 (shown in green) and ending in a poly(A) tail downstream of the stop codon. After splicing, this SMN1-specific exon 7 will be fused to the body of the endogenous SMN2 mRNAs containing exons 1–6. Black knobs indicate the cap structures at the 5′ ends of the RNAs involved (see main text for refs.).
Focusing on SMA, a number of different U snRNA-based approaches have been used with variable success (Fig. 4A). Several U7 Sm OPT derivatives targeting the 3′ SS and BP region of SMN2 exon 8 were found to induce exon 7 reinclusion,144 similar to what had been observed with AONs targeting the same sequences.119 In our laboratory, we have been able to reproduce these findings, but have found this strategy to be less efficient than the bifunctional approach (see below).96 Such U7 constructs were also stably introduced into SMA patient fibroblasts by means of lentiviral96 or adenoviral145 vectors leading to a stimulation of full length SMN2 mRNA and protein.
In our laboratory, we have tested multiple U snRNA-based approaches to enhance SMN2 exon 7 inclusion. Based on the observation that the BP preceding exon 7 is suboptimal, we tried to express a U2 snRNA derivative with increased complementarity to this BP region.97 However, no improvement of SMN2 splicing could be observed, despite the fact that a BP mutation increasing the complementarity to U2 snRNA stimulated exon 7 inclusion. It is possible that the U2 modification interfered with U2 snRNP assembly or that the limiting step in SMN2 exon 7 definition is not the binding of the U2 snRNP but the prior recognition of the BP region by the splicing factor SF1.146,147 Another strategy was to design a modified U1 snRNA that would bind more efficiently to the suboptimal exon 7 5′ SS that features an adenosine in the last exon position. Although this approach led to a strong exon 7 inclusion, the stable expression of this U1 snRNA was toxic to cells, most likely because splicing efficiencies of other exons containing an A in this position were altered or because of the activation of cryptic splice sites (Marquis J, Meyer K and Schümperli D, unpublished observations). We also tried to target the intron 7 ISS-N1 with U7 Sm OPT derivatives, but this caused an even stronger skipping of exon 7, possibly due to steric interference of the U7 snRNP moiety with spliceosome assembly at the 5′ SS.96
The most efficient and viable approach in our hand, however, has been an adaptation of the bifunctional AON approach to a U7 Sm OPT construct.96 Two different regions in exon 7 were targeted, and the U7 snRNA derivatives additionally contained different sequence repeats able to bind to various SR proteins. One of these RNAs targeting the 3′ region of exon 7 with an ESE tail induced 90% of full-length SMN2 mRNA both in a HeLa cell minigene test system and in SMA patient fibroblasts. In the latter cells, the level of SMN protein was increased to about 50% of the level seen in wild-type human fibroblasts which contain two copies each of SMN1 and SMN2. Considering that human SMA carriers with a single SMN1 copy are fully viable, this increase was highly significant. More important, however, transgenic SMA mice carrying this U7 construct showed an extended lifespan (median of 124 days, compared to 5–7 days for untreated SMA animals), a greatly improved muscle performance and normal motoneuron counts at 1 month of age.148 Early ultrastructural alterations in neuromuscular junctions of the diaphragm of SMA mice were also prevented.76 These studies proved for the first time that the idea of improving SMN2 splicing to cure SMA is not a mere hypothesis but, in fact a realistic strategy. It should also be mentioned that some success in cell culture models was reported for a similar bifunctional RNA driven by the U6 promoter, but lacking a snRNA backbone.149 However, using an almost identical construct, we have not been able to reproduce this result in our hands.96 The bifunctional RNA was almost undetectable and presumably also not concentrated in the nucleus, underlining the importance of a snRNA backbone for RNA stability and nucleoplasmic accumulation.
An entirely different approach to repair the splicing defect in SMN2 is the use of a method known as spliceosome-mediated RNA trans-splicing (SMaRT).150 It consists of repressing the 3′ SS of an exon by an antisense RNA sequence that is followed by an exon to be trans-spliced to the body of the message (Fig. 4B). In particular, Coady and coworkers have developed a therapeutic trans-splicing RNA base-pairing with the intron 6 of SMN2 and containing the SMN1 exon 7 sequence.151 The final product after splicing is a chimeric mRNA containing SMN2-exons 1–6, followed by SMN1 exon 7. The transduction of SMA fibroblast cells with an AAV vector carrying this construct increased SMN protein levels.
For a somatic gene therapy in experimental animals or in human patients, these therapeutic RNA genes will have to be introduced into spinal cord motoneurons. This will require appropriate transfer vectors. Some initial success was obtained by Azzouz, Mazarakis and coworkers by using lentiviral vectors,152,153 but recent results indicate that self-complementary AAV9 vectors can efficiently transduce motoneurons, when injected into newborn mice, cats or non-human primates.109,154,155 In particular, two groups have recently reported a significant clinical amelioration in SMA mice after scAAV9- or scAAV8-mediated introduction of a SMN cDNA.109,110 Future work will have to reveal which type of gene therapy is most efficient in a human clinical setting, a gene replacement approach as used in these two studies or one of the splicing correction strategies described above.
Acknowledgements
We are thankful for financial support by the Kanton Bern as well as by grants of the AFM (Association Française contre les Myopathies), EURASNET (European Network of Excellence on Alternative Splicing), Swiss Foundation for Research on Muscle Diseases (to K.M.) and Swiss National Science Foundation (grant 3100A0-120064).
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/12206
References
- 1.Human Genome Sequencing Consortium, author. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 2.O'Donovan C, Apweiler R, Bairoch A. The human proteomics initiative (HPI) Trends Biotechnol. 2001;19:178–181. doi: 10.1016/s0167-7799(01)01598-0. [DOI] [PubMed] [Google Scholar]
- 3.Matlin AJ, Clark F, Smith CWJ. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 4.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 5.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deutsch M, Long M. Intron-exon structures of eukaryotic model organisms. Nucl Acids Res. 1999;27:3219–3228. doi: 10.1093/nar/27.15.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burge CB, Tuschl T, Sharp PA. Splicing of precursors to mRNAs by the spliceosome. In: Gesteland RF, Cech TR, Atkins JF, editors. The RNA World. Cold Spring Harbor NY: CSHL Press; 1999. pp. 525–560. [Google Scholar]
- 8.Graveley BR, Maniatis T. Arginine/serine-rich domains of SR proteins can function as activators of pre-mRNA splicing. Mol Cell. 1998;1:765–771. doi: 10.1016/s1097-2765(00)80076-3. [DOI] [PubMed] [Google Scholar]
- 9.Blencowe BJ. Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem Sci. 2000;25:106–110. doi: 10.1016/s0968-0004(00)01549-8. [DOI] [PubMed] [Google Scholar]
- 10.Shepard P, Hertel K. The SR protein family. Genome Biol. 2009;10:242. doi: 10.1186/gb-2009-10-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pozzoli U, Sironi M. Silencers regulate both constitutive and alternative splicing events in mammals. Cellular and Molecular Life Sciences. 2005;62:1579–1604. doi: 10.1007/s00018-005-5030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez-Contreras R, Cloutier P, Shkreta L, Fisette JF, Revil T, Chabot B. hnRNP proteins and splicing control. Adv Exp Med Biol. 2007;623:123–147. doi: 10.1007/978-0-387-77374-2_8. [DOI] [PubMed] [Google Scholar]
- 13.Caceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends in Genetics. 2002;18:186–193. doi: 10.1016/s0168-9525(01)02626-9. [DOI] [PubMed] [Google Scholar]
- 14.Buratti E, Baralle FE. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol Cell Biol. 2004;24:10505–10514. doi: 10.1128/MCB.24.24.10505-10514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calarco JA, Superina S, O'Hanlon D, Gabut M, Raj B, Pan Q, et al. Regulation of vertebrate nervous system alternative splicing and development by an SR-related protein. Cell. 2009;138:898–910. doi: 10.1016/j.cell.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 16.Jin Y, Suzuki H, Maegawa S, Endo H, Sugano S, Hashimoto K, et al. A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J. 2003;22:905–912. doi: 10.1093/emboj/cdg089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Underwood JG, Boutz PL, Dougherty JD, Stoilov P, Black DL. Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol Cell Biol. 2005;25:10005–1016. doi: 10.1128/MCB.25.22.10005-10016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuroyanagi H. Fox-1 family of RNA-binding proteins. Cell Mol Life Sci. 2009;66:3895–3907. doi: 10.1007/s00018-009-0120-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makeyev EV, Zhang J, Carrasco MA, Maniatis T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell. 2007;27:435–448. doi: 10.1016/j.molcel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, et al. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21:1636–1652. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coutinho-Mansfield GC, Xue Y, Zhang Y, Fu XD. PTB/nPTB switch: a post-transcriptional mechanism for programming neuronal differentiation. Genes Dev. 2007;21:1573–1577. doi: 10.1101/gad.1575607. [DOI] [PubMed] [Google Scholar]
- 22.Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, et al. Nova regulates brain-specific splicing to shape the synapse. Nat Genet. 2005;37:844–852. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- 23.Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, et al. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 24.Ule J, Darnell RB. RNA binding proteins and the regulation of neuronal synaptic plasticity. Curr Opin Neurobiol. 2006;16:102–110. doi: 10.1016/j.conb.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 26.Krawczak M, Thomas NS, Hundrieser B, Mort M, Wittig M, Hampe J, et al. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution and consequences for mRNA splicing. Hum Mutat. 2007;28:150–158. doi: 10.1002/humu.20400. [DOI] [PubMed] [Google Scholar]
- 27.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 28.Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 29.Baralle D, Baralle M. Splicing in action: assessing disease causing sequence changes. J Med Genet. 2005;42:737–748. doi: 10.1136/jmg.2004.029538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valcarcel J, Singh R, Zamore PD, Green MR. The protein Sex-lethal antagonizes the splicing factor U2AF to regulate alternative splicing of transformer pre-mRNA. Nature. 1993;362:171–175. doi: 10.1038/362171a0. [DOI] [PubMed] [Google Scholar]
- 31.Dominski Z, Kole R. Cooperation of pre-mRNA sequence elements in splice site selection. Mol Cell Biol. 1992;12:2108–2114. doi: 10.1128/mcb.12.5.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Penalva LOF, Lallena MJ, Valcarcel J. Switch in 3′ splice site recognition between exon definition and splicing catalysis Is Important for sex-lethal autoregulation. Mol Cell Biol. 2001;21:1986–1996. doi: 10.1128/MCB.21.6.1986-1996.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buratti E, Muro AF, Giombi M, Gherbassi D, Iaconcig A, Baralle FE. RNA folding affects the recruitment of SR proteins by mouse and human polypurinic enhancer elements in the fibronectin EDA exon. Mol Cell Biol. 2004;24:1387–1400. doi: 10.1128/MCB.24.3.1387-1400.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warf MB, Diegel JV, von Hippel PH, Berglund JA. The protein factors MBNL1 and U2AF65 bind alternative RNA structures to regulate splicing. PNAS. 2009;106:9203–9208. doi: 10.1073/pnas.0900342106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ward AJ, Cooper TA. The pathobiology of splicing. J Pathol. 2010;220:152–163. doi: 10.1002/path.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 37.Pearn J. Classification of spinal muscular atrophies. Lancet. 1980;1:919–922. doi: 10.1016/s0140-6736(80)90847-8. [DOI] [PubMed] [Google Scholar]
- 38.Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) Hum Mutat. 2000;15:228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 39.Talbot K, Davies KE. Spinal muscular atrophy. Semin Neurol. 2001;21:189–197. doi: 10.1055/s-2001-15264. [DOI] [PubMed] [Google Scholar]
- 40.Briese M, Esmaeili B, Sattelle DB. Is spinal muscular atrophy the result of defects in motor neuron processes? Bioessays. 2005;27:946–957. doi: 10.1002/bies.20283. [DOI] [PubMed] [Google Scholar]
- 41.Lefebvre S, Burglen L, Frezal J, Munnich A, Melki J. The role of the SMN gene in proximal spinal muscular atrophy. Hum Mol Genet. 1998;7:1531–1536. doi: 10.1093/hmg/7.10.1531. [DOI] [PubMed] [Google Scholar]
- 42.Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, et al. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 43.Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 44.Monani UR. Spinal muscular atrophy: A deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48:885–895. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 45.Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, et al. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 46.Briese M, Richter DU, Sattelle DB, Ulfig N. SMN, the product of the spinal muscular atrophy-determining gene, is expressed widely but selectively in the developing human forebrain. J Comp Neurol. 2006;497:808–816. doi: 10.1002/cne.21010. [DOI] [PubMed] [Google Scholar]
- 47.Liu Q, Fischer U, Wang F, Dreyfuss G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell. 1997;90:1013–1021. doi: 10.1016/s0092-8674(00)80367-0. [DOI] [PubMed] [Google Scholar]
- 48.Pellizzoni L, Kataoka N, Charroux B, Dreyfuss G. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell. 1998;95:615–624. doi: 10.1016/s0092-8674(00)81632-3. [DOI] [PubMed] [Google Scholar]
- 49.Meister G, Bühler D, Pillai R, Lottspeich F, Fischer U. A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat Cell Biol. 2001;3:945–949. doi: 10.1038/ncb1101-945. [DOI] [PubMed] [Google Scholar]
- 50.Meister G, Eggert C, Fischer U. SMN-mediated assembly of RNPs: a complex story. Trends Cell Biol. 2002;12:472–478. doi: 10.1016/s0962-8924(02)02371-1. [DOI] [PubMed] [Google Scholar]
- 51.Paushkin S, Gubitz AK, Massenet S, Dreyfuss G. The SMN complex, an assemblyosome of ribonucleoproteins. Curr Opin Cell Biol. 2002;14:305–312. doi: 10.1016/s0955-0674(02)00332-0. [DOI] [PubMed] [Google Scholar]
- 52.Pillai RS, Grimmler M, Meister G, Will CL, Lührmann R, Fischer U, Schümperli D. Unique Sm core structure of U7 snRNPs: assembly by a specialized SMN complex and the role of a new component, Lsm11, in histone RNA processing. Genes Dev. 2003;17:2321–2333. doi: 10.1101/gad.274403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hebert MD, Szymczyk PW, Shpargel KB, Matera AG. Coilin forms the bridge between Cajal bodies and SMN, the spinal muscular atrophy protein. Genes Dev. 2001;15:2720–2729. doi: 10.1101/gad.908401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matera AG, Shpargel KB. Pumping RNA: nuclear bodybuilding along the RNP pipeline. Current Opinion in Cell Biology. 2006;18:317–324. doi: 10.1016/j.ceb.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 55.Narayanan U, Ospina JK, Frey MR, Hebert MD, Matera AG. SMN, the spinal muscular atrophy protein, forms a pre-import snRNP complex with snurportin1 and importin beta. Hum Mol Genet. 2002;11:1785–1795. doi: 10.1093/hmg/11.15.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huber J, Cronshagen U, Kadokura M, Marshallsay C, Wada T, Sekine M, et al. Snurportin1, an m3G-capspecific nuclear import receptor with a novel domain structure. EMBO J. 1998;17:4114–4126. doi: 10.1093/emboj/17.14.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Terns MP, Terns RM. Macromolecular complexes: SMN—the master assembler. Curr Biol. 2001;11:862–864. doi: 10.1016/s0960-9822(01)00517-6. [DOI] [PubMed] [Google Scholar]
- 58.Gabanella F, Butchbach MER, Saieva L, Carissimi C, Burghes AHM, Pellizzoni L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE. 2007;2:921. doi: 10.1371/journal.pone.0000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and wide-spread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chari A, Paknia E, Fischer U. The role of RNP biogenesis in spinal muscular atrophy. Curr Opin Cell Biol. 2009;21:387–393. doi: 10.1016/j.ceb.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 61.Fuller HR, Man NT, Lam LT, Thanh LT, Keough RA, Asperger A, et al. The SMN interactome includes Myb-binding protein 1a. J Proteome Res. 2009;9:556–563. doi: 10.1021/pr900884g. [DOI] [PubMed] [Google Scholar]
- 62.Rossoll W, Jablonka S, Andreassi C, Kroning AK, Karle K, Monani UR, et al. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carrel TL, McWhorter ML, Workman E, Zhang H, Wolstencroft EC, Lorson C, et al. Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J Neurosci. 2006;26:11014–11022. doi: 10.1523/JNEUROSCI.1637-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fan L, Simard LR. Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum Mol Genet. 2002;11:1605–1614. doi: 10.1093/hmg/11.14.1605. [DOI] [PubMed] [Google Scholar]
- 65.Sharma A, Lambrechts A, Hao Lt, Le TT, Sewry CA, Ampe C, et al. A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp Cell Res. 2005;309:185–197. doi: 10.1016/j.yexcr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 66.Rossoll W, Kroning AK, Ohndorf UM, Steegborn C, Jablonka S, Sendtner M. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum Mol Genet. 2002;11:93–105. doi: 10.1093/hmg/11.1.93. [DOI] [PubMed] [Google Scholar]
- 67.Giesemann T, Rathke-Hartlieb S, Rothkegel M, Bartsch JW, Buchmeier S, Jockusch BM, et al. A Role for Polyproline Motifs in the Spinal Muscular Atrophy Protein SMN. J Biol Chem. 1999;274:37908–37914. doi: 10.1074/jbc.274.53.37908. [DOI] [PubMed] [Google Scholar]
- 68.Piazzon N, Rage F, Schlotter F, Moine H, Branlant C, Massenet S. In vitro and in cellulo evidences for association of the survival of motor neuron complex with the fragile X mental retardation protein. J Biol Chem. 2008;283:5598–5610. doi: 10.1074/jbc.M707304200. [DOI] [PubMed] [Google Scholar]
- 69.Cifuentes-Diaz C, Nicole S, Velasco ME, Borra-Cebrian C, Panozzo C, Frugier T, et al. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum Mol Genet. 2002;11:1439–1447. doi: 10.1093/hmg/11.12.1439. [DOI] [PubMed] [Google Scholar]
- 70.Jablonka S, Wiese S, Sendtner M. Axonal defects in mouse models of motoneuron disease. J Neurobiol. 2004;58:272–286. doi: 10.1002/neu.10313. [DOI] [PubMed] [Google Scholar]
- 71.Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, Gillingwater TH. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- 72.Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, et al. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McGovern VL, Gavrilina TO, Beattie CE, Burghes AHM. Embryonic motor axon development in the severe SMA mouse. Hum Mol Genet. 2008;17:2900–2909. doi: 10.1093/hmg/ddn189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kong LL, Wang XY, Choe DW, Polley M, Burnett BG, Bosch-Marce M, et al. Impaired Synaptic Vesicle Release and Immaturity of Neuromuscular Junctions in Spinal Muscular Atrophy Mice. J Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murray LM, Lee S, Baumer D, Parson SH, Talbot K, Gillingwater TH. Pre-symptomatic development of lower motor neuron connectivity in a mouse model of severe spinal muscular atrophy. Hum Mol Genet. 2009:506. doi: 10.1093/hmg/ddp506. [DOI] [PubMed] [Google Scholar]
- 76.Voigt T, Meyer K, Baum O, Schumperli D. Ultrastructural changes in diaphragm neuromuscular junctions in a severe mouse model for spinal muscular atrophy: Phenotypic rescue by bifunctional U7 snRNA correcting SMN2 splicing. Neuromuscul Disord. 2010 doi: 10.1016/j.nmd.2010.06.010. In press. [DOI] [PubMed] [Google Scholar]
- 77.Burghes AHM, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, et al. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 80.Lorson CL, Strasswimmer J, Yao JM, Baleja JD, Hahnen E, Wirth B, et al. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 81.Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 82.Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B. Htra2-beta1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2) Proc Natl Acad Sci USA. 2000;97:9618–9623. doi: 10.1073/pnas.160181697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Young PJ, DiDonato CJ, Hu D, Kothary R, Androphy EJ, Lorson CL. SRp30c-dependent stimulation of survival motor neuron (SMN) exon 7 inclusion is facilitated by a direct interaction with hTra2{beta}1. Hum Mol Genet. 2002;11:577–587. doi: 10.1093/hmg/11.5.577. [DOI] [PubMed] [Google Scholar]
- 84.Hofmann Y, Wirth B. hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-beta1. Hum Mol Genet. 2002;11:2037–2049. doi: 10.1093/hmg/11.17.2037. [DOI] [PubMed] [Google Scholar]
- 85.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 86.Cartegni L, Hastings ML, Calarco JA, de Stanchina E, Krainer AR. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet. 2006;78:63–77. doi: 10.1086/498853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 88.Kashima T, Rao N, David CJ, Manley JL. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum Mol Genet. 2007;16:3149–3159. doi: 10.1093/hmg/ddm276. [DOI] [PubMed] [Google Scholar]
- 89.Miyajima H, Miyaso H, Okumura M, Kurisu J, Imaizumi K. Identification of a cis-acting element for the regulation of SMN exon 7 splicing. J Biol Chem. 2002;277:23271–23277. doi: 10.1074/jbc.M200851200. [DOI] [PubMed] [Google Scholar]
- 90.Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26:1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gladman J, Chandler D. Intron 7 conserved sequence elements regulate the splicing of the SMN genes. Human Genetics. 2009;126:833–841. doi: 10.1007/s00439-009-0733-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Goren A, Ram O, Amit M, Keren H, Lev-Maor G, Vig I, et al. Comparative analysis identifies exonic splicing regulatory sequences—The complex definition of enhancers and silencers. Molecular Cell. 2006;22:769–781. doi: 10.1016/j.molcel.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 94.Singh NN, Androphy EJ, Singh RN. In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. RNA. 2004;10:1291–1305. doi: 10.1261/rna.7580704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Singh NN, Singh RN, Androphy EJ. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 2007;35:371–389. doi: 10.1093/nar/gkl1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marquis J, Meyer K, Angehrn L, Kämpfer SS, Rothen-Rutishauser B, Schümperli D. Spinal muscular atrophy: SMN2 pre-mRNA splicing corrected by a U7 snRNA derivative carrying a splicing enhancer sequence. Mol Ther. 2007;15:1479–1486. doi: 10.1038/sj.mt.6300200. [DOI] [PubMed] [Google Scholar]
- 97.Scholl R, Marquis J, Meyer K, Schümperli D. Spinal Muscular Atrophy: position and functional importance of the branch site preceding SMN exon 7. RNA Biol. 2007;4:34–37. doi: 10.4161/rna.4.1.4534. [DOI] [PubMed] [Google Scholar]
- 98.Gennarelli M, Lucarelli M, Capon F, Pizzuti A, Merlini L, Angelini C, et al. Survival motor-neuron gene transcript analysis in muscles from spinal muscular-atrophy patients. Biochem Biophys Res Commun. 1995;213:342–348. doi: 10.1006/bbrc.1995.2135. [DOI] [PubMed] [Google Scholar]
- 99.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 100.Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, et al. Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003;12:2481–2489. doi: 10.1093/hmg/ddg256. [DOI] [PubMed] [Google Scholar]
- 101.Sumner CJ, Huynh TN, Markowitz JA, Perhac JS, Hill B, Coovert DD, et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol. 2003;54:647–654. doi: 10.1002/ana.10743. [DOI] [PubMed] [Google Scholar]
- 102.Swoboda KJ, Scott CB, Reyna SP, Prior TW, LaSalle B, Sorenson SL, et al. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS ONE. 2009;4:5268. doi: 10.1371/journal.pone.0005268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Andreassi C, Angelozzi C, Tiziano FD, Vitali T, De Vincenzi E, Boninsegna A, et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. 2003;12:59–65. doi: 10.1038/sj.ejhg.5201102. [DOI] [PubMed] [Google Scholar]
- 104.Mercuri E, Bertini E, Messina S, Solari A, D'Amico A, Angelozzi C, et al. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology. 2007;68:51–55. doi: 10.1212/01.wnl.0000249142.82285.d6. [DOI] [PubMed] [Google Scholar]
- 105.Grzeschik SM, Ganta M, Prior TW, Heavlin WD, Wang CH. Hydroxyurea enhances SMN2 gene expression in spinal muscular atrophy cells. Ann Neurol. 2005;58:194–202. doi: 10.1002/ana.20548. [DOI] [PubMed] [Google Scholar]
- 106.Liang WC, Yuo CY, Chang JG, Chen YC, Chang YF, Wang HY, et al. The effect of hydroxyurea in spinal muscular atrophy cells and patients. J Neurol Sci. 2008;268:87–94. doi: 10.1016/j.jns.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 107.Butchbach MER, Singh J, Thorsteinsdottir M, Saieva L, Slominski E, Thurmond J, et al. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum Mol Genet. 2010;19:454–467. doi: 10.1093/hmg/ddp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mattis V, Rai R, Wang J, Chang CW, Coady T, Lorson C. Novel aminoglycosides increase SMN levels in spinal muscular atrophy fibroblasts. Hum Genet. 2006;120:589–601. doi: 10.1007/s00439-006-0245-7. [DOI] [PubMed] [Google Scholar]
- 109.Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotech. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 110.Passini MA, Bu J, Roskelley EM, Richards AM, Sardi SP, et al. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J Clin Invest. 2010;120:1253–1264. doi: 10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Corti S, Nizzardo M, Nardini M, Donadoni C, Salani S, Ronchi D, et al. Neural stem cell transplantation can ameliorate the phenotype of a mouse model of spinal muscular atrophy. J Clin Invest. 2008;118:3316–3330. doi: 10.1172/JCI35432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Soret J, Bakkour N, Maire S, Durand S, Zekri L, Gabut M, et al. Selective modification of alternative splicing by indole derivatives that target serine-arginine-rich protein splicing factors. PNAS. 2005;102:8764–8769. doi: 10.1073/pnas.0409829102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tazi J, Bakkour N, Soret J, Zekri L, Hazra B, Laine W, et al. Selective inhibition of topoisomerase I and various steps of spliceosome assembly by diospyrin derivatives. Mol Pharmacol. 2005;67:1186–1194. doi: 10.1124/mol.104.007633. [DOI] [PubMed] [Google Scholar]
- 114.Andreassi C, Jarecki J, Zhou J, Coovert DD, Monani UR, Chen X, et al. Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients. Hum Mol Genet. 2001;10:2841–2849. doi: 10.1093/hmg/10.24.2841. [DOI] [PubMed] [Google Scholar]
- 115.Hastings ML, Berniac J, Liu YH, Abato P, Jodelka FM, Barthel L, et al. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci Transl Med. 2009;1:5–12. doi: 10.1126/scitranslmed.3000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vacek M, Sazani P, Kole R. Antisense-mediated redirection of mRNA splicing. Cell Mol Life Sci. 2003;60:825–833. doi: 10.1007/s00018-003-3042-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kurreck J. Antisense technologies. Improvement through novel chemical modifications. Eur J Biochem. 2003;270:1628–1644. doi: 10.1046/j.1432-1033.2003.03555.x. [DOI] [PubMed] [Google Scholar]
- 118.Aartsma-Rus A, van Ommen GJ. Antisense-mediated exon skipping: A versatile tool with therapeutic and research applications. RNA. 2007;13:1609–1624. doi: 10.1261/rna.653607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lim SR, Hertel KJ. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3′ splice site pairing. J Biol Chem. 2001;276:45476–45483. doi: 10.1074/jbc.M107632200. [DOI] [PubMed] [Google Scholar]
- 120.Singh NN, Shishimorova M, Cao LC, Gangwani L, Singh RN. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009;6:341–350. doi: 10.4161/rna.6.3.8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Williams JH, Schray RC, Patterson CA, Ayitey SO, Tallent MK, Lutz GJ. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J Neurosci. 2009;29:7633–7638. doi: 10.1523/JNEUROSCI.0950-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides Targeting the Exon. PLoS Biol. 2007;5:73. doi: 10.1371/journal.pbio.0050073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol. 2003;10:120–125. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- 124.Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci USA. 2003;100:4114–4119. doi: 10.1073/pnas.0633863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dickson A, Osman E, Lorson C. A negatively-acting bifunctional RNA increases survival motor neuron in vitro and in vivo. Hum Gene Ther. 2008 doi: 10.1089/hum.2008.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Baughan TD, Dickson A, Osman EY, Lorson CL. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum Mol Genet. 2009;18:1600–1611. doi: 10.1093/hmg/ddp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gorman L, Schümperli D, Kole R. Alteration of pre-mRNA splicing patterns by modified small nuclear RNAs. In: Bertolotti R, Parvez SH, Nagatsu T, editors. Progress in Gene Therapy—basic and clinical frontiers. Utrecht, The Netherlands: VSP BV; 2000. pp. 455–473. [Google Scholar]
- 128.Asparuhova M, Kole R, Schümperli D. Antisense derivatives of U7 and other small nuclear RNAs as tools to modify pre-mRNA splicing patterns. Gene Therapy Regul. 2004;2:321–349. [Google Scholar]
- 129.Müller B, Schümperli D. The U7 snRNP and the hairpin binding protein: Key players in histone mRNA metabolism. Semin Cell Dev Biol. 1997;8:567–576. doi: 10.1006/scdb.1997.0182. [DOI] [PubMed] [Google Scholar]
- 130.Schümperli D, Pillai RS. The special Sm core structure of the U7 snRNP: far-reaching significance of a small nuclear ribonucleoprotein. Cell Mol Life Sci. 2004;60:2560–2570. doi: 10.1007/s00018-004-4190-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Grimm C, Stefanovic B, Schümperli D. The low abundance of U7 snRNA is partly determined by its Sm binding site. EMBO J. 1993;12:1229–1238. doi: 10.1002/j.1460-2075.1993.tb05764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Stefanovic B, Hackl W, Lührmann R, Schümperli D. Assembly, nuclear import and function of U7 snRNPs studied by microinjection of synthetic U7 RNA into Xenopus oocytes. Nucl Acids Res. 1995;23:3141–3151. doi: 10.1093/nar/23.16.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pillai RS, Will CL, Lührmann R, Schümperli D, Müller B. Purified U7 snRNPs lack the Sm proteins D1 and D2 but contain Lsm10, a new 14 kDa Sm D1-like protein. EMBO J. 2001;20:5470–5479. doi: 10.1093/emboj/20.19.5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gorman L, Suter D, Emerick V, Schümperli D, Kole R. Stable alteration of pre-mRNA splicing patterns by modified U7 small nuclear RNAs. Proc Natl Acad Sci USA. 1998;95:4929–4934. doi: 10.1073/pnas.95.9.4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Suter D, Tomasini R, Reber U, Gorman L, Kole R, Schümperli D. Double-target antisense U7 snRNAs promote efficient skipping of an aberrant exon in three human beta-thalassemic mutations. Hum Mol Genet. 1999;8:2415–2423. doi: 10.1093/hmg/8.13.2415. [DOI] [PubMed] [Google Scholar]
- 136.Lacerra G, Sierakowska H, Carestia C, Fucharoen S, Summerton J, Weller D, et al. Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients. Proc Natl Acad Sci USA. 2000;97:9591–9596. doi: 10.1073/pnas.97.17.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vacek MM, Ma H, Gemignani F, Lacerra G, Kafri T, Kole R. High-level expression of hemoglobin A in human thalassemic erythroid progenitor cells following lentiviral vector delivery of an antisense snRNA. Blood. 2003;101:104–111. doi: 10.1182/blood-2002-06-1869. [DOI] [PubMed] [Google Scholar]
- 138.Brun C, Suter D, Pauli C, Dunant P, Lochmüller H, Burgunder JM, et al. U7 snRNAs induce correction of mutated dystrophin pre-mRNA by exon skipping. Cell Mol Life Sci. 2003;60:557–566. doi: 10.1007/s000180300047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.De Angelis FG, Sthandier O, Berarducci B, Toso S, Galluzzi G, Ricci E, et al. Chimeric snRNA molecules carrying antisense sequences against the splice junctions of exon 51 of the dystrophin pre-mRNA induce exon skipping and restoration of a dystrophin synthesis in Delta 48-50 DMD cells. Proc Natl Acad Sci USA. 2002;99:9456–9461. doi: 10.1073/pnas.142302299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Goyenvalle A, Vulin A, Fougerousse F, Leturcq F, Kaplan JC, Garcia L, et al. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science. 2004;306:1796–1799. doi: 10.1126/science.1104297. [DOI] [PubMed] [Google Scholar]
- 141.Liu S, Asparuhova M, Brondani V, Ziekau I, Klimkait T, Schümperli D. Inhibition of HIV-1 multiplication by antisense U7 snRNAs and siRNAs targeting cyclophilin A. Nucl Acids Res. 2004;32:3752–3759. doi: 10.1093/nar/gkh715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Asparuhova MB, Marti G, Liu S, Serhan F, Trono D, Schümperli D. Inhibition of HIV-1 multiplication by a modified U7 snRNA inducing Tat and Rev exon skipping. J Gene Med. 2007;9:323–334. doi: 10.1002/jgm.1027. [DOI] [PubMed] [Google Scholar]
- 143.Asparuhova MB, Barde I, Trono D, Schranz K, Schümperli D. Development and characterization of a triple combination gene therapy vector inhibiting HIV-1 multiplication. J Gene Med. 2008;10:1059–1070. doi: 10.1002/jgm.1238. [DOI] [PubMed] [Google Scholar]
- 144.Madocsai C, Lim SR, Geib T, Lam BJ, Hertel KJ. Correction of SMN2 pre-mRNA splicing by antisense U7 small nuclear RNAs. Mol Ther. 2005;12:1013–1022. doi: 10.1016/j.ymthe.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 145.Geib T, Hertel KJ. Restoration of full-length SMN promoted by adenoviral vectors expressing RNA antisense oligonucleotides embedded in U7 snRNAs. PLoS ONE. 2009;4:8204. doi: 10.1371/journal.pone.0008204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Arning S, Grüter P, Bilbe G, Krämer A. Mammalian splicing factor SF1 is encoded by variant cDNAs and binds to RNA. RNA. 1996;2:794–810. [PMC free article] [PubMed] [Google Scholar]
- 147.Krämer A. The structure and function of proteins involved in mammalian pre-mRNA splicing. Ann Rev Biochem. 1996;65:367–409. doi: 10.1146/annurev.bi.65.070196.002055. [DOI] [PubMed] [Google Scholar]
- 148.Meyer K, Marquis J, Trub J, Nlend Nlend R, Verp S, Ruepp MD, et al. Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA-mediated splicing modulation. Hum Mol Genet. 2009;18:546–555. doi: 10.1093/hmg/ddn382. [DOI] [PubMed] [Google Scholar]
- 149.Baughan T, Shababi M, Coady TH, Dickson AM, Tullis GE, Lorson CL. Stimulating full-length SMN2 expression by delivering bifunctional RNAs via a viral vector. Mol Ther. 2006;14:54–62. doi: 10.1016/j.ymthe.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 150.Puttaraju M, Jamison SF, Mansfield SG, Garcia-Blanco MA, Mitchell LG. Spliceosome-mediated RNA transsplicing as a tool for gene therapy. Nat Biotechnol. 1999;17:246–252. doi: 10.1038/6986. [DOI] [PubMed] [Google Scholar]
- 151.Coady TH, Shababi M, Tullis GE, Lorson CL. Restoration of SMN Function: Delivery of a Transsplicing RNA Re-directs SMN2 Pre-mRNA Splicing. Mol Ther. 2007;15:1471–1478. doi: 10.1038/sj.mt.6300222. [DOI] [PubMed] [Google Scholar]
- 152.Mazarakis ND, Azzouz M, Rohll JB, Ellard FM, Wilkes FJ, Olsen AL, et al. Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum Mol Genet. 2001;10:2109–2121. doi: 10.1093/hmg/10.19.2109. [DOI] [PubMed] [Google Scholar]
- 153.Azzouz M, Le T, Ralph GS, Walmsley L, Monani UR, Lee DC, et al. Lentivector-mediated SMN replacement in a mouse model of spinal muscular atrophy. J Clin Invest. 2004;114:1726–1731. doi: 10.1172/JCI22922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Duque S, Joussemet B, Riviere C, Marais T, Dubreil L, Douar AM, et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]