

Abstract
PURPOSE
Microarray studies indicate medulloblastoma comprises distinct molecular disease subgroups, which offer potential for improved clinical management.
EXPERIMENTAL DESIGN
Minimal mRNA expression signatures diagnostic for the Wnt/Wingless (WNT) and Sonic Hedgehog (SHH) subgroups were developed, validated and used to assign subgroup affiliation in 173 tumours from four independent cohorts, alongside a systematic investigation of subgroup clinical and molecular characteristics.
RESULTS
WNT tumours (12% (21/173)) were diagnosed >5 years of age (peak, 10 years), displayed classic histology, CTNNB1 mutation (19/20), associated chromosome 6 loss and have previously been associated with favourable prognosis. SHH cases (24% (42/173)) predominated in infants (<3 years) and showed an age-dependent relationship to desmoplastic/nodular pathology; all infant desmoplastic/nodular cases (previously associated with a good outcome) were SHH-positive, but these relationships broke down in non-infants. PTCH1 mutations were common (34%; 11/32), but PTCH1 exon1c hypermethylation, chromosome 9q and REN (KCTD11) genetic loss were not SHH-associated, and SMO or SUFU mutation, PTCH1 exon1a or SUFU hypermethylation did not play a role, indicating novel activating mechanisms in the majority of SHH cases. SHH tumours were associated with an absence of COL1A2 methylation. WNT/SHH-independent medulloblastomas (64% (110/173)) showed all histologies, peaked at 3-6 years, and were exclusively associated with chromosome 17p loss.
CONCLUSIONS
Medulloblastoma subgroups are characterised by distinct genomic, epigenomic and clinico-pathological features, and clinical outcomes. Validated array-independent gene expression assays for the rapid assessment of subgroup affiliation in small biopsies, provide a basis for their routine clinical application, in strategies including molecular disease-risk stratification and delivery of targeted therapeutics.
Keywords: medulloblastoma, subgroups, diagnosis, biomarkers, molecular mechanisms
Introduction
Medulloblastoma, a primitive neuro-ectodermal tumour of the cerebellum, is the most common malignant brain tumour of childhood. Five-year overall survival rates have increased over recent years to 70 to 80% for standard-risk patients. However for high-risk patients (infants <3yrs, cases with metastatic disease at diagnosis or incomplete surgical resection), current treatments only cure around 40 to 60% of cases. In addition, many survivors exhibit long term therapy-associated late-effects. The development of novel targeted treatments, alongside refined patient stratification, will be essential to increase survival rates and reduce adverse sequelae(1). Improvements in understanding of the molecular basis of medulloblastoma will be fundamental to such advances.
The constitutive activation of developmental signalling pathways plays a key role in medulloblastoma pathogenesis, and pathway components represent the major mutational targets identified in the disease to date. The sonic hedgehog (SHH) pathway plays an essential role in normal cerebellar development, is activated by PTCH1 mutation in around 10% of human primary medulloblastomas, and promotes medulloblastoma development in mouse models of the disease(2-4). Likewise, mutations in components of the canonical Wnt/wingless (WNT) signalling pathway have been described in up to 20% of cases (5-7). Importantly, these pathways appear to have therapeutic significance; WNT-active cases are associated with a favourable prognosis (>90% overall survival(8, 9)), while small molecule inhibitors of the SHH pathway show pre-clinical and early-clinical activity against the disease(10, 11).
Recent array-based genome-wide genomic and transcriptomic investigations in medulloblastoma have identified distinct molecular disease subgroups, which are distinguished by their gene expression profiles, and display related clinical disease features. Two disease groupings, characterised respectively by activation and mutation of the WNT and SHH signalling pathways, are consistently supported by these studies(12, 13). The WNT subgroup is best documented, and is distinguished by nuclear β-catenin immunostaining, CTNNB1 mutations and chromosome 6 loss (5, 13-15), alongside its associated favourable prognosis(8, 9). The SHH subgroup is however less well characterised; PTCH1 mutations are only identified in a subset of SHH cases, indicating a role for other activating mechanisms and correlates. A series of putative mechanisms of SHH activation (e.g. PTCH1 hypermethylation, SUFU/SMO mutation, REN (KCDT11) genetic loss) have been reported in medulloblastoma(16-19), but any relationships to the SHH subgroup remain to be established. Moreover, SHH subgroup clinical features require clarification; SHH activation has been associated with the desmoplastic / nodular (DN) disease histological variant in some studies, but not others, and associations with infant cases have also been postulated (12, 17, 20, 21).
Identification of these medulloblastoma molecular subgroups has significant potential to (i) improve clinical management, through molecular disease-risk stratification strategies and the identification of patients who could benefit from SHH and WNT targeted molecular therapeutics, and (ii) provide a basis for biological investigations to further understand disease molecular pathogenesis and its therapeutic applications. However, subgroup identification has so far relied on advanced genomics (i.e. microarray) technologies, which are relatively expensive and have used different gene expression data collection and analysis methodologies. The development of robust biomarkers and assays for subgroup detection, which can be routinely tested in clinical material across multiple treatment centres, and are informative in small tumour biopsies, will therefore be essential for their future study and any clinical application. Moreover, investigations in modestly-sized cohorts have limited comprehensive investigation of subgroup clinical characteristics and their molecular basis.
In this study, we report the development and validation of minimal mRNA expression signatures, which are robust for the routine diagnosis of the SHH and WNT subgroups in clinical material, and can be applied rapidly and cost-effectively using array-independent methodologies. Using these signatures, we show that equivalent disease subgroups are detected in four independent medulloblastoma cohorts, independent of gene expression assay used. We then use these signatures to assign subgroup affiliation in this large combined cohort of 173 medulloblastomas, and use these data as the basis for comprehensive investigations to define the clinical and molecular characteristics of each subgroup, and their utility for improved disease management.
Methods
Tissue samples
A representative cohort of 55 medulloblastoma samples was analysed, comprising 39 classic, 5 large cell/anaplastic (LCA) and 11 tumours of the DN histopathological subtype(22), 21 female and 34 male cases, 11 infants (≤3 years), 41 children (>3-15 years) and 3 adults (≥16 years). DNA (n=55) and RNA (n=39) were extracted from these snap-frozen tissues using standard methods. A panel of constitutional DNA samples from 100 normal individuals was obtained from the North Cumbria Community Genetics Project. Research Ethics Committee approval has been obtained for the collection, storage and biological study of all material.
Mutation Screening
All coding exons of the PTCH1 and SMO genes were PCR amplified using the primers and conditions shown in Supplementary Table 1. Mutation screening methods for the SUFU gene have been described previously(23). Mutation screening was performed by analysis of PCR products for heteroduplex formation, before and after ‘spiking’ with equal amounts of control wild-type DNA using denaturing high performance liquid chromatography (Wave DNA Fragment Analysis System, Transgenomic, Elancourt, France), according to the manufacturer’s instructions. Products detected as containing a heteroduplex were sequenced directly on an ABI 377 sequencer (Applied Biosystems, Foster City, CA, USA). In reported studies including our own, DHPLC has been reported to identify >90% of sequence variants (23). Mutation analysis of the CTNNB1 and APC genes was performed as previously described(8).
Analysis of promoter methylation status
Two promoter-associated CpG islands of the PTCH1 gene, spanning exons 1a and 1c(16), were identified and characterised using the Emboss CpGPlot website (http://www.ebi.ac.uk/emboss/cpgplot/): 1a methylation status was determined by methylation specific PCR (MSP), and 1c by bisulphite sequencing using previously published primers(16). A CpG island of the SUFU gene was also identified, and its methylation status analysed by MSP(24). COL1A2 methylation status was assessed by bisulphite sequencing, and has previously been reported(25). Bisulphite treatment, methylated and unmethylated controls have been described previously(23). Primers and conditions for analysis of PTCH1 CpG island 1a are shown in Supplementary Table 2. Methylation status was designated as methylated or unmethylated, as previously described(26). For loci assessed by MSP, any sample showing a visible PCR product using primers specific for the methylated sequence was classed as showing evidence of methylation (i.e. methylated). For loci assessed by bisulphite sequencing, the relative peak intensities at each CpG residue were determined. Samples where the methylated peak represented >25% of the total peak height, in greater than 25% of the analysed CpG sites were classed as showing evidence of methylation (i.e. methylated).
Loss of hetererozygosity (LOH) analysis
LOH of chromosome regions 9q22.3 and the p-arm of chromosome 17 were analysed using polymorphic micro-satellite markers D9S1689, D9S1816, D9S287, D9S1809, D9S1786, D9S1851 and D17S2196, D17S936, D17S969, D17S974, D17S1866, D17S1308 respectively (www.ncbi.nlm.nih.gov/genome/sts), as previously described(27). LOH of chromosome 6 was analysed using previously described markers and methods(14).
A multi-gene mRNA expression signature to identify SHH and WNT pathway activated tumours
A 13-gene multiplex mRNA expression assay (GeXP; Beckman Coulter, Fullerton, CA, USA) (28) was developed and used to test tumours for membership of the SHH or WNT medulloblastoma subgroups in 39 cases. Detailed clinical and pathological data for individual cases are summarised in Table 1. Two previously reported, independent, medulloblastoma expression microarray data sets(12, 13), were used to design SHH and WNT subgroup signatures. Data were respectively downloaded from the St Jude Research website (http://www.stjuderesearch.org/site/data/medulloblastoma) and the Gene Expression Omnibus (GEO)(29). Data were rma normalised(30) using R and Bioconductor. Using t tests, we identified probes differential for the WNT (group A) or SHH (group B) subgroups, defined by Kool et al (12), which were significant in both datasets. Previous work had validated 3 SHH-associated genes (GLI1, PTCH2 and SFRP1) and 2 WNT-associated genes (DKK2, WIF1) by quantitative RT-PCR(13) and these genes were also considered for inclusion in the signatures. Putative signature genes were assessed for assay suitability, and assays were subsequently performed using 50ng total RNA per replicate, according to manufacturer’s instructions. To eliminate the possibility of detecting genomic DNA, PCR products were designed across exon boundaries; products were also designed where possible to overlap the Affymetrix probes from which they were derived. For genes with multiple transcripts, amplicons were designed that detected all known transcripts. Test gene expression was normalised to 28S rRNA, and results displayed are means based on independent assessment in triplicate. The presence of SHH or WNT expression signatures was used to identify sample subgroup membership (see next section). Signature gene selection is summarised in Supplementary Table 3, and PCR primer sequences are listed in Supplementary Table 4.
Table 1. Associations between molecular subgroups and medulloblastoma genomic, epigenomic and clinical disease features.
Cases are shown arranged by signature status (SHH, WNT, WNT/SHH independent; determined in Figure 11), then by ascending age. 2Mutation status (FS, frameshift mutation; MS, missense mutation). 3Hypermethylation status (black, methylated; white, unmethylated). 4Chromosomal loss (black, allelic loss; white, no loss). Age is shown in years and categorised into infants (≤3 years, black) and non-infants (>3 years, white). Pathology variant is indicated by a white square (classic), black square (DN) or grey square (LCA). Gender is indicated by black squares (F, female) and white squares (M, male). M stage 0/1 is indicated by a white square and M stage 2/3 by a black square. Raw ‘p’and ‘p’ values corrected for multiple hypothesis testing are shown for relationships between molecular/clinical correlates and subgroup membership (chi-squared tests, Bonferroni correction). Significant* (p<0.05) and marginally significant** (p=0.051 to 0.10) associations are marked (ns, not significant). Diagonally hatched boxes indicate unavailable data.
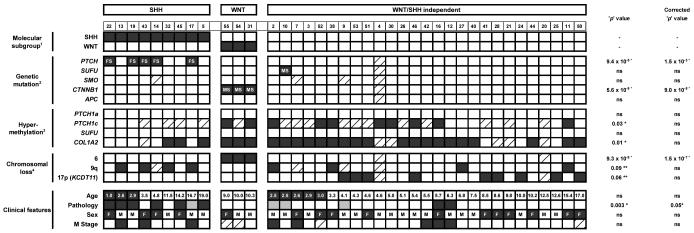
|
Integration and analysis of combined gene expression data sets
An additional medulloblastoma expression microarray data set, reported by Fattet et al(15), was normalised as described above. All three publicly available data sets have linked clinical (age at diagnosis, pathology sub-type and gender) and PTCH1 / CTNNB1 mutational data available (except PTCH1 mutation data not available for the Fattet et al(15) dataset). Expression data of signature genes from all three datasets, together with our own, were integrated. First, our GeXP expression data were log2 transformed as for the expression microarray data and, prior to joining, all data sets were scaled separately, on a per-gene basis, to give a mean of 0, with variance of 1. Unsupervised hierarchical clustering, supported by principal component analysis (PCA), was used to assign subgroup membership status (Figure 1), and these data were used in conjunction with stacked barplots (Figure 2) and silhouette plots (31) (Supplementary Figure 1) to assess performance of the signatures. There was some sample overlap between studies: 11 samples were assessed by expression array by Thompson et al(13) and GeXP; 3 samples were assessed by expression array by Kool et al(12) and GeXP, which enabled the comparison of subgroup assignment in individual samples using our signatures, when evaluated using different gene expression assays. Clinical and genomic correlates were also combined for the joined cohort. Duplicate analyses were removed from all correlative investigations.
Figure 1. Diagnostic WNT and SHH subgroup gene expression signatures recognize equivalent molecular subgroups across multiple medulloblastoma cohorts.

Data from four independent datasets are shown (A, primary investigation cohort; B, Kool et al (n=62; (12)); C, Thompson et al (n=46, (13)); D, Fattet et al (n=40, (15)). WNT and SHH subgroup expression signature positivity demonstrates close concordance with (i) underlying molecular defects in the respective pathways and (ii) discrete sample clusters identified on independent clustering of the most variable probes in each array dataset (Supplementary Figure 2). Each panel (A-D) shows hierarchical clustering of signature genes (WNT subgroup, red; SHH subgroup, blue; WNT/SHH-independent, grey). Mutational information for CTNNB1 and PTCH1 is shown (coloured boxes, mutation; grey checked boxes, missing data). Array clusters which show concordance with the detected SHH and WNT subgroup signatures (derived by clustering the most variable probes of each whole array dataset) are shown (purple, SHH subgroup; orange, WNT subgroup (Supplementary Figure 2)). Biplots show PCA for each signature geneset. Arrows show projections of expression axes for each gene (SHH signature genes, blue; WNT signature genes, red; WNT/SHH-independent cases, grey).
Figure 2. Identification of SHH and WNT subgroup medulloblastomas using diagnostic expression signatures.

Data are illustrated from the four independent datasets using stacked barplots (A, primary investigation cohort; B, Kool et al (n=62; (12)); C, Thompson et al (n=46, (13)); D, Fattet et al (n=40, (15)). WNT signature genes (red) and SHH signature genes (blue) in combination clearly define subgroup membership. Vertical dashed lines delineate sample groups positive for WNT and SHH signatures by hierarchical clustering and PCA analysis in Figure 1. Right-hand panel indicates stacked order of genes for each signature. Prior to generation of barplots, expression data from each cohort was scaled on a per gene basis to a mean of zero and a variance of one. Samples expressing all or most signature genes at above average levels will show bars of greater positive magnitude.
Statistical analysis
Fisher’s exact and chi-squared tests were used to identify relationships between expression signatures, molecular and clinical features of the disease, in the primary investigative (GeXP) cohort (n=39) and in the combined cohort (n=173). Bonferroni corrections for multiple hypothesis testing were applied where appropriate. Additional patient age data was kindly provided by Dr. Marcel Kool (Academic Medical Centre, Amsterdam, Netherlands).
Results
A diagnostic multi-gene mRNA expression signature assay for SHH and WNT medulloblastoma subgroups
We first developed and validated multi-gene mRNA expression signatures to facilitate routine identification of the SHH and WNT medulloblastoma molecular disease subgroups, and assessment of their molecular basis, associated biomarkers, and clinical relevance. The 8-gene SHH (BCHE, GLI1, ITIH2, MICAL1, PDLIM3, PTCH2, RAB33A and SFRP1) and 5-gene WNT (CCDC46, DKK2, PYGL, TNC and WIF1) subgroup signatures developed were initially assessed by GeXP assay in our primary sample cohort (n=39) and were unequivocal for all samples, independent of data analysis method used (stacked bar plots, PCA or unsupervised hierarchical clustering; Figures 1 & 2). Validation of these signatures in three independent medulloblastoma expression microarray datasets (Thompson et al(13), (n=46), Kool et al(12),(n=62) and Fattet et al(15), (n=40)) showed the signatures could be successfully interrogated by hierarchical cluster analysis and PCA, were diagnostic in all cases, independent of cohort or gene expression assay used (Figure 1), and showed close consistency with stacked bar plot data (Figure 2). Signature positivity was concordant with the disease subgroups apparent after independent clustering of the most variable probes within each entire array dataset, and correctly classified disease subgroup (i.e. as SHH, WNT or WNT/SHH-independent) in 99% (146/148) of cases overall (Figure 1). Datasets similarly showed complete concordance in sub-group assignment, using all analytical methods, of the 14 cases (4 SHH, 2 WNT and 8 WNT/SHH independent cases) where signatures were assessed in parallel by microarray and GeXP assays.
Integration of cohorts and gene expression datasets: molecular subgroup incidence
Based on our validated gene expression signatures and data analysis methods, the molecular subgroup data from all four cohorts could be combined for meta-analysis with the mutational and clinical data which were consistently reported for all studies. A total cohort of 173 cases were assessed in this analysis (Supplementary Table 5). Clinical demographics available for the combined cohort were consistent with previously reported group-wide estimates for medulloblastoma(1); male:female ratio (1.42:1), histology (115 Classic (68%), 39 (23%) DN, 16 (9%) LCA, 3 cases with data not available; pathological classifications were those reported in the original publications (12,13,15)) and age at diagnosis (median 6.45, range 0.3-35.3 years, 34 (20%) infant, 138 (80%) non-infant cases, 1 case with data not available). An overall incidence of 21 (12%) WNT and 42 (24%) SHH subgroup cases was observed. Sub-classification of the remainder of cases based on their transcriptomic patterns has not produced consistent findings in previous studies(12, 13). Remaining WNT/SHH-independent medulloblastomas were therefore classified as a single group, representing 110 (64%) cases.
Genetic and epigenetic mechanisms of SHH and WNT activation
Genetic mutations in PTCH1 and CTNNB1 were exclusively detected in SHH and WNT subgroup cases, respectively, across all four cohorts, where data was available (combined cohort, p=5.3 × 10−8 and p=0 (chi-squared test), respectively; Figure 1), further validating the fidelity of the gene expression signatures developed. CTNNB1 mutation was the primary mechanism and correlate of WNT pathway activation; in 20 cases where combined expression and mutation data were available, all except one WNT subgroup case (19/20; 95%) had concurrent CTNNB1 mutation. Consistent with this, no APC mutations were found in the 55 cases tested within our primary cohort.
PTCH1 mutation was a major mechanism of SHH pathway activation; 34% (11/32) of SHH subgroup cases investigated harboured a PTCH1 mutation (Figure 1). We therefore next undertook a systematic investigation of alternative genetic mechanisms of pathway activation in the remaining majority of SHH cases in our primary cohort. Mutational analysis encompassed all pathway genes in which mutations have previously been reported; the complete coding sequences of PTCH1 (including exon 1B, which has been shown to code for protein(32), but has not been analysed in previous studies(3, 20, 21)), SUFU and SMO (which has only been previously been assessed for mutation at specific residues(18)). In addition to PTCH1 truncating mutations (n=5), only one further potentially pathogenic missense SUFU mutation was identified, with no evidence of SMO mutation found (Supplementary Table 6). Additional non-pathogenic variants discovered (e.g. polymorphisms or intronic changes) are summarised in Supplementary Table 7. Allelic loss of chromosome 17p, targeting REN (KCTD11) at 17p13.2, has also been previously associated with SHH activation in medulloblastoma(19, 33), and was observed in 24% (9/37) of cases.
Epigenetic mechanisms of pathway activation were additionally investigated as an alternative to genetic determinants. Two predicted promoter-associated CpG islands spanning exons 1a and 1c of PTCH1, and a promoter-associated CpG island within SUFU, were identified and investigated for evidence of DNA hypermethylation, which may lead to epigenetic transcriptional silencing. No evidence of DNA methylation of the PTCH1 exon 1a-associated or the SUFU CpG island was seen in any tumour analysed (n=39; Table 1), indicating no major role in disease development. Mixed patterns of methylation of the PTCH1 exon 1c CpG island were observed in 12/27 of tumours successfully analysed (44%; Table 1), suggesting a potential role for this epigenetic mechanism.
Unlike PTCH1 mutations, other defects identified (PTCH1 exon 1c methylation, SUFU missense mutation and 17p allelic loss (REN (KCTD11))) were not associated with the SHH subgroup (Table 1), indicating that any role these mechanisms may play in medulloblastoma development is independent of the SHH pathway.
Genomic biomarkers of SHH and WNT pathway activation
We next investigated selected medulloblastoma chromosomal abnormalities (chromosome 6, 9q and 17p loss) and epigenomic defects (COL1A2 status) of biological and/or prognostic significance (1, 25), for their associations with the SHH and WNT disease subgroups, and each other, to assess any utility as biomarkers of pathway activation.
Chromosome 6 loss, CTNNB1 mutation and the absence of chromosome 9 and 17 abnormalities, were observed in all WNT cases in our primary cohort (Table 1), consistent with previous findings (12-15). Across the combined cohort, evidence of loss of an entire copy of chromosome loss 6 was associated with 88% (14/16) of WNT cases with available data (p<3×10−16, Fisher’s Exact), however this relationship was not exclusive; chromosome 6 loss was also detected in occasional non-WNT cases (2/145). Eight of 35 tumours tested (23%) showed evidence of genetic loss at the 9q22.3 region surrounding the PTCH1 locus in our primary cohort (Table 1). Four of eight were in the SHH subgroup and 2/4 tumours with PTCH1 mutations showed LOH of 9q22.3, however neither association reached significance (p=0.09 and 0.22 respectively, Fisher’s Exact test). A significant inverse association between 17p loss and membership of the SHH and WNT subgroups was observed; 17p losses were exclusively observed in WNT/SHH-independent cases (17p LOH in 0/12 SHH or WNT cases vs. 9/25 WNT/SHH-independent cases (p=0.02, Fisher’s Exact test)). In addition, COL1A2 hypermethylation was detected in 76% (25/33) of cases; an absence of COL1A2 methylation was significantly associated with the SHH subgroup (p=0.01, chi-squared test), but this relationship was not maintained when a correction for multiple hypothesis testing was applied (Table 1).
Medulloblastoma molecular subgroups display distinct clinical features
Analysis of the medulloblastoma molecular subgroups defined by our gene expression signatures, in all 173 cases of the combined cohort, revealed marked differences in their clinical disease features. WNT/SHH-independent tumours made up the majority of cases, and had their peak incidence in the 3 to 6 year age group, but were extremely rare in the first two years of life (Figure 3). In contrast, SHH subgroup tumours had their major peak of incidence in infancy (50% (21/42) of SHH cases were ≤3 years of age). The SHH subgroup represented the majority of the infant clinical group (62% (21/34) of cases ≤3 years of age), but were less common in non-infant children (>3 to 15 years; 15% (16/127), and had a further increased incidence in adult cases (≥16 years (45% (5/11); overall p=5.8 × 10−9, chi-squared test)). Almost all cases <2 years of age (92% (11/12) were SHH-positive. WNT subgroup cases were not observed in infants (minimum age observed, 5 years), and had a bi-modal age distribution with major and minor peaks at 10 and 20 years respectively.
Figure 3. Medulloblastoma molecular subgroups show distinct age of incidence distributions.

Data for the WNT (grey), SHH (black) and WNT/SHH-independent (hatched) subgroups are shown, based on a combined cohort of 173 medulloblastomas. (A) Density plots show subgroup-dependent ages of incidence. Case density represents the smoothed frequency of incidence within each of the three subgroups. Grey dotted line is plotted at 3 years of age. (B) Barplots show age distribution of dataset. (C) Barplot showing age distribution of cases aged ≤6 years at diagnosis. F, frequency.
Significant differences were also observed in the distribution of medulloblastoma histological subtypes between the molecular subgroups (p=3.1 × 10−11, chi-squared test; Figures 4,5). WNT subgroup cases exclusively displayed classic histology (p=0.0003, Fisher’s Exact test), and WNT/SHH-independent tumours were also predominantly of the classic subtype, but DN and LCA cases were also observed. Consistent with previous studies(3, 12, 13, 21), SHH cases were overall strongly associated with DN histology (p=1.1 × 10−9, Fisher’s exact test). However, this relationship was not absolute and LCA and classic cases were also observed in the SHH group. Most notably, examination of this large cohort revealed the relationship between SHH activation and DN pathology to be age dependent (Figures 4,5); DN cases made up the majority of infant (≤ 3 years old) SHH subgroup cases; all DN cases in this infant group displayed SHH activation. DN pathology may therefore serve as a surrogate marker of SHH activation in the infant group. In contrast, there were almost equal proportions of DN, LCA and classic cases in SHH-expressing non-infant cases, and the majority of non-infant DN tumours were not SHH activated (p=8.6 × 10−5, Fisher’s Exact test). No significant evidence for differences in metastatic stage (WNT 6% (1/16) M stage 2/3, SHH 16% (5/32) and WNT/SHH-independent 24% (20/82)) were observed between the different expression subgroups (p=0.20, chi-squared test).
Figure 4. Molecular subgroups show relationships to medulloblastoma histological variants.

Subgroup and histological information was available for 170/173 cases (see Supplementary Table 5). (A) WNT subgroup (n=20), (B) SHH subgroup (n=42), (C) WNT/SHH-independent cases (n=108; p=3.1 × 10−11, chi-squared test). White, classic (CLAS); grey, LCA; black DN histological variants.
Figure 5. Associations between SHH subgroup medulloblastomas and DN histology are age-dependent.

(A) Histological variants (white, classic (CLAS); grey, LCA; black DN) show significantly different distributions (p=0.05; chi-squared test) in SHH subgroup cases arising in infants (≤3 years at diagnosis (n=21); A1) and non-infants (>3 years (n=21); A2). (B). Infant (n=16; B1) and non-infant (n=23; B2) DN medulloblastomas show significantly different relationships to the SHH subgroup (p=8.6 × 10−5; Fisher’s exact test) (SHH subgroup, white; non-SHH, black).
Discussion
The identification of distinct medulloblastoma molecular subgroups(12, 13, 15) offers significant potential to improve our understanding of disease biology and clinical management. Here, we report the development and validation of minimal diagnostic gene expression signatures which can be routinely applied to identify the SHH, WNT and WNT/SHH-independent medulloblastoma disease subgroups. These gene expression signatures are robust and informative for subgroup identification in RNA extracted from snap-frozen tumour material, and using different gene expression assays. In particular, the GeXP multiplex expression assay reported offers a number of significant advantages over microarray methodologies for the routine assignment of subgroup affiliation; analysis can be undertaken straightforwardly, rapidly (in one working day, compared to 2-3 days for a microarray experiment) and cost-effectively (approximately one-tenth of microarray analysis costs) and, importantly, can be performed on small amounts of total RNA (150 ng, compared with 500ng to 5μg for a typical microarray expression analysis), a common limitation when only small amounts of biopsy material are available. This removal of the need for relatively time-consuming, complex and expensive array analysis platforms for subgroup assignment provides a strong basis for their clinical application; these methods are feasible for investigation in real-time across multiple treatment centres during clinical treatment and in future clinical trials.
We have shown that the disease subgroups recognised by these signatures are equivalent and consistently identified in four independent medulloblastoma cohorts, allowing their assembly into a large combined cohort. Coupled with an extensive analysis of our novel primary cohort, this has allowed significant insights into the underlying molecular mechanisms, associated biomarkers, and clinical characteristics of these molecular disease subgroups.
Our systematic investigation of specific medulloblastoma genetic and epigenetic defects in this study has informed their roles as determinants or correlates of the molecular subgroups identified. Consistent with previous studies(12-14), CTNNB1 mutations were identified as the primary pathway activating event present in almost all WNT subgroup tumours, with chromosome 6 losses also affecting the majority of these cases. PTCH1 mutation was the major mechanistic correlate of SHH activation, identified in ~34% of SHH cases. SHH-associated PTCH1 mutations were detected both in conjunction with chromosome 9q loss, and in the heterozygous state, indicating disruption of a single PTCH1 allele can be sufficient to cause SHH pathway disruption in medulloblastoma. An absence of COL1A2 hypermethylation was also significantly associated with SHH subgroup medulloblastomas, most strongly in infant cases. Notably, a number of the previously reported determinants of SHH activation that we examined (PTCH1 exon 1c methylation(16), SUFU missense mutation(17) and 17p (REN (KCTD11)(19) allelic loss) were not specifically associated with the SHH subgroup, indicating any role they may play in medulloblastoma is SHH-independent. It is similarly unclear whether the mixed methylation patterns observed for PTCH1 exon 1c in the present study have functional significance, as this was not assessed. Additionally, other SHH pathway defects examined (PTCH1 exon 1a hypermethylation), including events previously reported in medulloblastoma (SMO mutations(18) or SUFU truncating mutations(17)) were not observed at all, suggesting their roles are either less common than previously thought, or are restricted to limited tumour subsets less well represented in our mutation screening cohort. This is likely the case for SUFU mutations, which appear to be associated with germline inheritance and have their peak incidence in infants(17, 23, 34). Further mechanisms of pathway activation remain to be identified in the majority of SHH cases. Chromosome 17 defects were the only events significantly correlated with the most common WNT/SHH-independent subgroup, suggesting a role for chromosome 17 genes in these cases. This disease subgroup however remains the least well characterised at the molecular level. Sub-division of this group has been proposed on the basis of its transcriptomic and genomic patterns however, unlike the SHH and WNT groups, inconsistent results have been reported from different studies(12, 13), and the identification of specific genes and pathways associated with its pathogenesis will be critical to future advances in our understanding of its molecular basis and any underlying heterogeneity.
The significant associations observed between medulloblastoma molecular subgroups and specific gene, pathway and chromosomal defects (i) strongly support the existence of molecularly distinct SHH and WNT subgroups, (ii) inform the contributory mechanisms involved in their pathogenesis, and (iii) provide potential biomarkers for subgroup identification. When assessed for suitability as primary biomarkers, only CTNNB1 mutations, which were specifically observed in all but one WNT subgroup case, have sufficient sensitivity and specificity to have utility. Nuclear localisation of β-catenin has also been widely reported as a positive marker of WNT pathway activation(5, 8, 14), although its relationship to our WNT expression signature and CTNNB1 mutations could not be investigated in the present study due to tissue limitations. Likewise, COL1A2 status may have utility in the identification of SHH subgroup infant desmoplastic medulloblastomas (this study and (25)), particularly in cases where biopsy limitations do not allow assessment of the DN pathological variant. The remainder of gene and chromosomal defects investigated were not suitable as primary biomarkers for positive subgroup discrimination, as a result of either their (i) non-exclusivity, (ii) limitation to sub-sets of subgroup cases, or (iii) inverse correlation with pathway activation. In comparison, gene expression signatures positively identified all subgroup cases and provide an accurate diagnostic test for subgroup membership. The genomic markers examined may therefore provide useful secondary or confirmatory markers, when used in conjunction with these signatures. It is not presently clear whether the expression signatures reported translate into subgroup-specific protein expression. Protein expression markers, which are testable by immunohistochemistry, may have utility for routine subgroup assignment in the diagnostic setting, however careful investigation and validation of their sensitivity, robustness and reflection of expression array subgroup data, will be essential prior to their application.
The observation of 2/148 cases which were not consistently classified using the presently reported signatures and their respective array datasets (Figure 1) indicates potential difficulties in the robust classification of a small subset (<2%) of cases. Inspection of the microarray expression data for these 2 cases revealed markedly reduced expression of pathway signature genes relative to the other pathway positive cases in their respective cohorts, despite their subgroup positivity using our signatures (12, 13, 15) (Figure 2). Additionally, the stacked bar plot analyses revealed 2 further SHH-positive samples (T27, K452; Figure 2) which, although clustered consistently between signature and array on hierarchical cluster and PCA analysis, could be classified as indeterminate based on the application of quantitative criteria to the individual expression data in stacked bar plot analysis (i.e. cumulative stacked bar plot expression score >8 (for WNT expression signature positive cases) or >4 (SHH cases)). For such cases, additional markers of pathway activation (CTNNB1, PTCH1 mutation) could aid definitive subgroup assignment, and it is notable that 1 of these 2 samples also harboured a PTCH1 mutation, further supporting SHH subgroup membership. Silhouette analysis supported the robust assignment of subgroup status using our signatures (Supplementary Figure 1) but, consistent with the other analytical methods applied, did not support the subgroup assignment made for 3 of the 4 discrepant cases described above, further highlighting difficulties in their classification. Thus, in diagnostic applications, a ‘non-classified’ designation could be reserved for these rare cases which do not classify consistently across all analyses performed on the signature data, particularly where subgroup assignment may impact clinical or therapeutic decisions.
The combination of molecular and clinico-pathological data from four independent cohorts for meta-analysis, totalling 173 cases, has facilitated clear and significant insights to the clinical features of the medulloblastoma molecular subgroups, which have either not been apparent or not shown statistical significance in individual analyses of the smaller component cohorts reported to date(12, 13, 15). The SHH (24% of cases), WNT (12%) and WNT/SHH-independent (64%) groups show different age distributions and relationships to disease histopathology. SHH subgroup tumours peak in infancy and are intimately correlated with DN pathology in this group, to the extent that DN pathology may be considered as a surrogate marker for SHH activation in medulloblastomas arising in infants <3 years old at diagnosis, although classic and LCA cases also constitute a minority of SHH subgroup cases in this age group. This relationship breaks down in non-infants (≥3 years at diagnosis), where SHH tumours are less common, and show equivalent proportions of DN, classic and LCA disease; SHH-independent DN cases are also commonly observed in this age group. These data strongly indicate that (i) SHH subgroup and (ii) DN tumours, arising in the infant and non-infant age groups, have different biological and clinical characteristics, and that SHH-positive DN tumours of infancy represent a unique disease subgroup associated with a favourable clinical behaviour(35-37), and a characteristic molecular pathogenesis (COL1A2 unmethylated (25)) and mutational spectrum (SUFU(17, 34)). Conversely, WNT tumours display classic pathology and occur in non-infants. Notably, both the SHH and WNT subgroups show at least two different incidence peaks in their age distribution (both have second peaks in adults), suggesting additional clinical and molecular heterogeneity within these groups. The inclusion of cohort-wide central pathology review in future studies may aid the further refinement of the associations observed.
The lack of association between M stage and molecular subgroups (p=0.20) is in disagreement with the previous study by Kool et al(12), who reported metastatic tumours being less common in WNT and SHH pathway activated medulloblastomas. This could be due to the different measurement criteria for metastasis between the studies (the present study compared M0/1 versus M2/3, while Kool et al compared M0 versus M1/M2/M3). Alternatively, the increased numbers in this study (130 versus 58 with M stage data in the Kool et al study) may have enabled a more accurate characterisation of the relationship between signalling pathway activation and metastasis, and future large clinically controlled studies which include central review of metastatic status should be informative in this regard.
The identification of medulloblastoma molecular subgroups has significant prognostic and predictive potential to improve therapeutic delivery and disease outlook in the clinical setting, and could represent a first step in the molecular diagnostic triage of medulloblastomas, to guide treatment decisions. In addition to distinct clinical features, molecular subgroups also appear to have characteristic clinical behaviours; the favourable prognosis of WNT subtype medulloblastomas, is now established in multiple clinical cohorts(8, 9, 15, 38), and will form the basis of treatment reductions in forthcoming international molecularly-driven clinical trials(1). Combined data from this and other studies indicate SHH-positive DN tumours arising in infants represent a similarly favourable prognosis subgroup with a distinct molecular basis(25, 35-37). The non-availability of outcome data for the four cohorts assessed in our meta-analysis have limited any direct assessment of survival associations in these cohorts. Moreover, their retrospective, heterogeneously-treated nature would likely confound such analyses. Robust assessment of the prognostic impact of the medulloblastoma molecular subgroups will therefore now be essential, ideally within the context of adequately powered, centrally-reviewed and uniformly-treated clinical trials cohorts, to determine their utility to direct the selection of adjuvant therapy. The molecular signatures we report will have significant utility in this regard. Molecularly targeted SHH inhibitors are also currently under pre-clinical and clinical development, and have shown early evidence of activity in medulloblastoma(10, 11). The ability to accurately diagnose the SHH molecular subgroup will thus be important for the targeted delivery of these novel agents, and our findings have identified SHH-positive subgroups of medulloblastomas which would be predicted to benefit most from SHH inhibition strategies. However, the SHH pathway plays a key role in normal, including cerebellar, development, and its transient inhibition in young mice causes permanent defects in growth plate formation and bone structure(39). In view of such potential toxicities, our data recommend caution in their application, particularly in the infant age group where SHH subgroup tumours predominate.
Statement of translational relevance.
Understanding the molecular basis of medulloblastoma will be essential to improve clinical outcomes, through guidance of molecular risk-adapted adjuvant therapies, and delivery of molecularly targeted agents. Expression microarray studies indicate the existence of discrete medulloblastoma molecular subgroups associated with activation of specific developmental signalling pathways (i.e. SHH, WNT). However, any translational utility will require definition of subgroup clinical and molecular correlates in large cohorts, alongside biomarkers and assays for routine subgroup assignment prior to adjuvant therapy selection. We report development of diagnostic gene expression signatures, which can be applied rapidly and cost-effectively in small biopsies, using array-independent methods, to assign sub-group status. Using these signatures in 173 medulloblastomas, we demonstrate disease subgroups are robust and reproducible, and have distinct clinical, molecular and outcome characteristics of therapeutic importance. These findings provide strong rationale and methodologies to support subgroup assessment as a basis for future medulloblastoma clinical trials and biological studies investigations.
Supplementary Material
Acknowledgements
Medulloblastomas investigated in this study include samples provided by the UK Children’s Cancer and Leukaemia Group (CCLG) as part of CCLG-approved biological study BS-2007-04. This study was conducted with ethics committee approval from Newcastle / North Tyneside REC (study reference 07/Q0905/71).
Grant information:
This work was supported by grants from The Katie Trust (to S.C. Clifford), The Samantha Dickson Brain Tumour Trust (to S.C. Clifford) and Cancer Research UK (to S.C. Clifford and D.W. Ellison).
References
- 1.Pizer BL, Clifford SC. The potential impact of tumour biology on improved clinical practice for medulloblastoma: progress towards biologically driven clinical trials. British journal of neurosurgery. 2009;23:364–75. doi: 10.1080/02688690903121807. [DOI] [PubMed] [Google Scholar]
- 2.Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Molecular and cellular biology. 2000;20:9055–67. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raffel C, Jenkins RB, Frederick L, et al. Sporadic medulloblastomas contain PTCH mutations. Cancer research. 1997;57:842–5. [PubMed] [Google Scholar]
- 4.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science (New York, NY. 1997;277:1109–13. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 5.Eberhart CG, Tihan T, Burger PC. Nuclear localization and mutation of beta-catenin in medulloblastomas. Journal of neuropathology and experimental neurology. 2000;59:333–7. doi: 10.1093/jnen/59.4.333. [DOI] [PubMed] [Google Scholar]
- 6.Huang H, Mahler-Araujo BM, Sankila A, et al. APC mutations in sporadic medulloblastomas. The American journal of pathology. 2000;156:433–7. doi: 10.1016/S0002-9440(10)64747-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dahmen RP, Koch A, Denkhaus D, et al. Deletions of AXIN1, a component of the WNT/wingless pathway, in sporadic medulloblastomas. Cancer research. 2001;61:7039–43. [PubMed] [Google Scholar]
- 8.Ellison DW, Onilude OE, Lindsey JC, et al. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005;23:7951–7. doi: 10.1200/JCO.2005.01.5479. [DOI] [PubMed] [Google Scholar]
- 9.Gajjar A, Chintagumpala M, Ashley D, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. The lancet oncology. 2006;7:813–20. doi: 10.1016/S1470-2045(06)70867-1. [DOI] [PubMed] [Google Scholar]
- 10.Romer JT, Kimura H, Magdaleno S, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer cell. 2004;6:229–40. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 11.Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. The New England journal of medicine. 2009;361:1173–8. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kool M, Koster J, Bunt J, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE. 2008;3:e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–31. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 14.Clifford SC, Lusher ME, Lindsey JC, et al. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell cycle (Georgetown, Tex. 2006;5:2666–70. doi: 10.4161/cc.5.22.3446. [DOI] [PubMed] [Google Scholar]
- 15.Fattet S, Haberler C, Legoix P, et al. Beta-catenin status in paediatric medulloblastomas: correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. The Journal of pathology. 2009;218:86–94. doi: 10.1002/path.2514. [DOI] [PubMed] [Google Scholar]
- 16.Diede SJ, Guenthoer J, Geng LN, et al. DNA methylation of developmental genes in pediatric medulloblastomas identified by denaturation analysis of methylation differences. Proceedings of the National Academy of Sciences of the United States of America. 2009;107:234–9. doi: 10.1073/pnas.0907606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor MD, Liu L, Raffel C, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31:306–10. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- 18.Reifenberger J, Wolter M, Weber RG, et al. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer research. 1998;58:1798–803. [PubMed] [Google Scholar]
- 19.Di Marcotullio L, Ferretti E, De Smaele E, et al. REN(KCTD11) is a suppressor of Hedgehog signaling and is deleted in human medulloblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10833–8. doi: 10.1073/pnas.0400690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pietsch T, Waha A, Koch A, et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer research. 1997;57:2085–8. [PubMed] [Google Scholar]
- 21.Wolter M, Reifenberger J, Sommer C, Ruzicka T, Reifenberger G. Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer research. 1997;57:2581–5. [PubMed] [Google Scholar]
- 22.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta neuropathologica. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott DK, Straughton D, Cole M, Bailey S, Ellison DW, Clifford SC. Identification and analysis of tumor suppressor loci at chromosome 10q23.3-10q25.3 in medulloblastoma. Cell cycle (Georgetown, Tex. 2006;5:2381–9. doi: 10.4161/cc.5.20.3360. [DOI] [PubMed] [Google Scholar]
- 24.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderton JA, Lindsey JC, Lusher ME, et al. Global analysis of the medulloblastoma epigenome identifies disease-subgroup-specific inactivation of COL1A2. Neuro-oncology. 2008;10:981–94. doi: 10.1215/15228517-2008-048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindsey JC, Lusher ME, Anderton JA, et al. Identification of tumour-specific epigenetic events in medulloblastoma development by hypermethylation profiling. Carcinogenesis. 2004;25:661–8. doi: 10.1093/carcin/bgh055. [DOI] [PubMed] [Google Scholar]
- 27.Langdon JA, Lamont JM, Scott DK, et al. Combined genome-wide allelotyping and copy number analysis identify frequent genetic losses without copy number reduction in medulloblastoma. Genes Chromosomes Cancer. 2006;45:47–60. doi: 10.1002/gcc.20262. [DOI] [PubMed] [Google Scholar]
- 28.Rai AJ, Kamath RM, Gerald W, Fleisher M. Analytical validation of the GeXP analyzer and design of a workflow for cancer-biomarker discovery using multiplexed gene-expression profiling. Analytical and bioanalytical chemistry. 2009;393:1505–11. doi: 10.1007/s00216-008-2436-7. [DOI] [PubMed] [Google Scholar]
- 29.Barrett T, Troup DB, Wilhite SE, et al. NCBI GEO: mining tens of millions of expression profiles--database and tools update. Nucleic acids research. 2007;35:D760–5. doi: 10.1093/nar/gkl887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics (Oxford, England) 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 31.Rousseeuw PJ. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J Comput Appl Math. 1987;20:53–65. [Google Scholar]
- 32.Kogerman P, Krause D, Rahnama F, et al. Alternative first exons of PTCH1 are differentially regulated in vivo and may confer different functions to the PTCH1 protein. Oncogene. 2002;21:6007–16. doi: 10.1038/sj.onc.1205865. [DOI] [PubMed] [Google Scholar]
- 33.De Smaele E, Di Marcotullio L, Ferretti E, Screpanti I, Alesse E, Gulino A. Chromosome 17p deletion in human medulloblastoma: a missing checkpoint in the Hedgehog pathway. Cell cycle (Georgetown, Tex. 2004;3:1263–6. doi: 10.4161/cc.3.10.1200. [DOI] [PubMed] [Google Scholar]
- 34.Brugieres L, Pierron G, Chompret A, et al. Incomplete penetrance of the predisposition to medulloblastoma associated with germ-line SUFU mutations. Journal of medical genetics. 47:142–4. doi: 10.1136/jmg.2009.067751. [DOI] [PubMed] [Google Scholar]
- 35.Rutkowski S, Bode U, Deinlein F, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. The New England journal of medicine. 2005;352:978–86. doi: 10.1056/NEJMoa042176. [DOI] [PubMed] [Google Scholar]
- 36.Garre ML, Cama A, Bagnasco F, et al. Medulloblastoma variants: age-dependent occurrence and relation to Gorlin syndrome--a new clinical perspective. Clin Cancer Res. 2009;15:2463–71. doi: 10.1158/1078-0432.CCR-08-2023. [DOI] [PubMed] [Google Scholar]
- 37.McManamy CS, Pears J, Weston CL, et al. Nodule formation and desmoplasia in medulloblastomas-defining the nodular/desmoplastic variant and its biological behavior. Brain pathology (Zurich, Switzerland) 2007;17:151–64. doi: 10.1111/j.1750-3639.2007.00058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korshunov A, Remke M, Werft W, et al. Adult and Pediatric Medulloblastomas Are Genetically Distinct and Require Different Algorithms for Molecular Risk Stratification. J Clin Oncol. doi: 10.1200/JCO.2009.25.7121. [DOI] [PubMed] [Google Scholar]
- 39.Kimura H, Ng JM, Curran T. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer cell. 2008;13:249–60. doi: 10.1016/j.ccr.2008.01.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.