

Abstract
The diverse biological activities of γ-hydroxyalkenal phospholipids and their involvement in disease are the subject of intense study. Phospholipid aldehydes, such as the 4-hydroxy-7-oxohept-5-enoic acid ester of 2-lyso-phosphatidylcholine (HOHA-PC), the 5-hydroxy-8-oxo-6-octenoic acid ester of 2-lyso-PC (HOOA-PC), and the 9-hydroxy-12-oxododec-10-enoic acid ester of 2-lyso-PC (HODA-PC), are generated by oxidative cleavage of polyunsaturated fatty acyl phospholipids. To facilitate investigations of their chemistry and biology, we now report efficient total syntheses of HOOA, HODA, and HOHA phospholipids. Because the target γ-hydroxyalkenals readily decompose through oxidation of the aldehyde group to a carboxylic acid or through cyclization to furans, these syntheses generate the sensitive functional array of the target phospholipids under mild conditions from acetal derivatives that are suitable for long-term storage.
Keywords: γ-Hydroxy-α, β-unsaturated aldehyde, oxidized phospholipid, HOHA-PC, HOOA-PC, HODA-PC, organic synthesis, cardiovascular disease, atherosclerosis
1. Introduction
Free radical-induced oxidative cleavage of phospholipids containing docosahexaenoate (C22), arachidonate (C20), or linoleate (C18) generates truncated, biologically active aldehydes including γ-hydroxyalkenal phospholipids, i.e., the 4-hydroxy-7-oxohept-5-enoic acid ester of 2-lysophosphatidylcholine (HOHA-PC), the 5-hydroxy-8-oxo-6-octenoic acid ester of 2-lyso-PC (HOOA-PC), and the 9-hydroxy-12-oxododec-10-enoic acid ester of 2-lyso-PC (HODA-PC) respectively (Scheme 1). These γ-hydroxyalkenals react with protein lysyl ε-amino residues to generate biologically active carboxyalkylpyrrole derivatives (Scheme 1).1,2 HOHA-PC, HOOA-PC and HODA-PC and the derived protein adducts 2-(ω-carboxyethyl)pyrroles (CEPs), 2-(ω-carboxypropyl)pyrroles (CPPs) and 2-(ω-carboxyheptyl)pyrroles (CHPs), are present in human atherosclerotic lesions.3–5 The γ–hydroxyalkenal phospholipids contribute to the generation of atherosclerotic plaques. Thus, they promote the conversion of monocyte macrophages into foam cells by serving as ligands for the scavenger receptor CD36 that mediates endocytosis of oxidized low-density lipoprotein particles by macrophage cells.6,7 Binding of these phospholipids, and their more oxidized derivatives, to platelet CD36 receptors induces aggregation leading to thrombosis.8 Their binding to the macrophage scavenger receptor class B, type I (SR-BI) prevents binding of its physiological ligand, high-density lipoprotein, to SR-BI and consequently interferes with SR-BI-mediated selective uptake of cholesteryl esters in hepatocytes.9 Thus, oxidative stress resulting in the accumulation of specific oxidized phospholipids in plasma may have an inhibitory effect on reverse cholesterol transport. HOOA-PC induces monocyte binding to endothelial cells10–12 and this may promote infiltration of monocyte macrophages into the subendothelial space where they become foam cells through unregulated endocytosis of oxidized low-density lipoprotein. Also, HOOA-PC inhibits lipopolysaccharide-induced expression of E-selectin a major endothelial cell adhesion molecule involved in neutrophil binding.10 Specifically, HOOA-PC induces expression of chemokines that promote interaction with monocytes.13 Thus, HOOA-PC is important as a putative proinflammatory molecule regulating leukocyte endothelial interactions.11,14 Furthermore, covalent modification of proteins by HOOA-PC and HODA-PC results, inter alia, in impaired proteolytic degradation of internalized macromolecules by mouse peritoneal macrophages and inhibition of cathepsin B, a lysosomal protease. HOHA-PC also inhibits posttranslational processing of the nascent 25 kDa membrane fusion protein Rab5a to produce the active 23 kDa “mature” Rab5a in phagosomal membranes.15 Most recently, proteins containing CEPs, that are generated in vivo from HOHA-PC, were shown to initiate destruction of the retina in age-related macular degeneration.16 We previously reported total syntheses of HOHA-, HOOA- and HODA-PC.1,3 However, those syntheses were lengthy and involved reactions that are difficult to perform. Herein, we report superior syntheses of HOOA, HODA, and HOHA derivatives that will facilitate investigation of their biological involvements.
Scheme 1.

Oxidation of polyunsaturated fatty acids generates γ-hydroxy-α,β-unsaturated aldehydes, that react with proteins and form 2-(ω-carboxylalkyl)pyrroles.
2. Results and Discussion
2.1. Synthetic Design
γ-Hydroxyalkenals readily decompose through oxidation of the aldehyde group to a carboxylic acid or cyclization to furans. Therefore, a synthetic strategy was designed that generates the sensitive functional array of the target phospholipids 1 under mild conditions from stable acetal precursors 2 that are suitable for long-term storage, and that might be readily converted to the target γ-hydroxyalkenal phospholipids under mild conditions (Scheme 2). Masking of the aldehyde carbonyl as an acetal also allows selective reduction of the ketone carbonyl in the ketoalkenal derivatives 3. The precursors 3 might be assembled through cross-coupling of the known vinyl tin 4 and acid chlorides 5. Alternatively, keto alkenal precursors 6 are readily available via oxidation of furans 7.
Scheme 2.
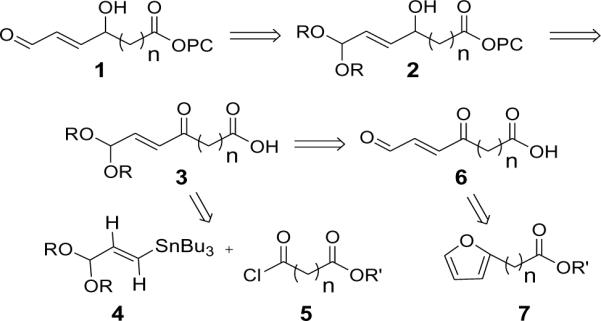
Retrosyntheses of γ-hydroxyalkenal phospholipids.
2.2. HOHA Ethyl Ester
To test the feasibility of generating the requisite γ-hydroxyalkenal functional array in 2 byselective reduction of a ketonefrom an ester of 3 (n = 2, R = Me), we synthesized (E)-ethyl-4-hydroxy-7-oxohept-5-enoate (11) (Scheme 3). The furyl ester 7a was oxidatively ring opened with N-bromosuccinimide (NBS).17 The resulting keto aldehyde 8 was selectively protected using trimethyl orthoformate and Montmorillonite K1018 to give 9. Reduction of the ketone carbonyl in 9 was readily accomplished in excellent yield to provide the masked γ-hydroxyalkenal 10. The target γ-hydroxyalkenal 11 could be generated by hydrolysis of of the dimethyl acetal in 10 promoted by the weak acid, pyridinium p-toluenesufonic acid (PPTs). However, concomitant transesterification delivered the lactone 12 in excellent yield when the hydrolysis was performed in the presence of stronger acid catalysts, such as Amberlyst-15 or TFA.
Scheme 3.
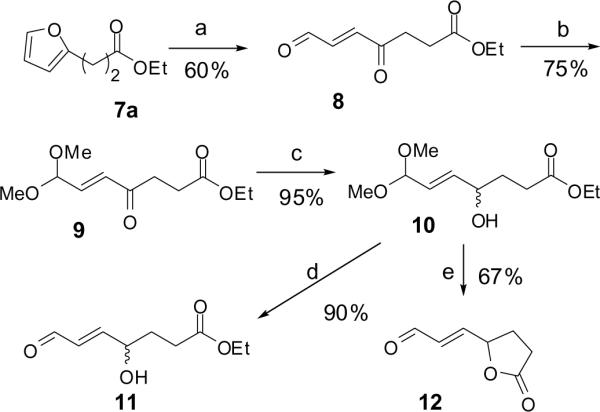
Synthesis of HOHA ethyl ester. Reagents and conditions: (a) NBS, THF/acetone/ H2O(5:4:1), pyridine; (b) K 10 / CH(OCH3)3; (c) NaBH4, MeOH; (d) PPTS, acetone/H2O (2:1); (e) Amberlyst-15, acetone/H2O (2:1), 67% or TFA/H2O (95:5), rt, 61%
2.3. HOOA Methyl Ester
In an alternative approach (Scheme 4), the requisite carbon skeleton was assembled by regio- and stereoselective tributylstannylcupration of 3,3-diethoxyprop-1-yne (13) with the Lipshutz reagent19, Bu3SnCu(Bu)CNLi2, followed by PdCl2(CH3CN)2 catalyzed acylation of the resuting vinyltin 420 with glutaric acid monomethyl ester chloride (5b).21 Reduction of the resulting ketone 14b with sodium borohydride gave the stable masked γ-hydroxyalkenal 15b in excellent yield. The target γ-hydroxyalkenal 16 could be generated by hydrolysis of 15b promoted by a weak acid. However, as for the 10 → 11 conversion above, concomitant transesterification delivered the lactone 17 when the hydrolysis was performed in the presence of stronger acid catalysts.
Scheme 4.
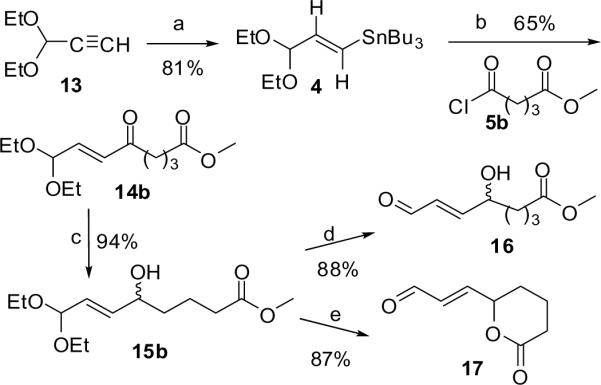
Synthesis of HOOA methyl ester. Reagents and conditions: (a) Bu3SnCu(Bu)CNLi2, THF, −78 °C, then H2O; (b) PdCl2(MeCN)2 (1 mol%), DMF, 0 °C; (c) NaBH4, EtOH, 0 °C; (d) PPTS, acetone/H2O (2:1), rt; (e) Amberlyst 15, TFA /H2O (95:5), rt.
2.4. HOHA-PC
Enzymatic hydrolysis of ethyl ester 9 with porcine pancreatic lipase (PPL) in phosphate buffer saline (PBS) buffer afforded key intermediate 2 (Scheme 5), that is precursor for many oxidized phospholipids.22 When lithium hydroxide was applied to hydrolyze methyl ester group, the diethyl acetal moiety was also deprotected. Therefore, selective enzymatic hydrolysis of the ester at pH 7 was considered. Synthesis of PC-ester 18 was efficiently accomplished using the coupling reagents 2,6-dichlorobenzoyl chloride and 1-methylimidazole,23,24 resulting in 50% shorter reaction time and improved yield of the ester coupling product, as compared to using general coupling reagents such as dicyclohexylcarboimide and 4-N,N-dimethylaminopyridine, which adhere to silica gel resulting in laborious separation via chromatography. Selective reduction of ketone 18a using NaBH4 in methanol and subsequent mild deprotection of the dimethylacetal 2a gave HOHA-PC 1a in excellent yield.
Scheme 5. Synthesis of HOHA-PC.

Reagents and conditions: (a) porcine pancreatc lipase, phosphate buffer; (b) L-α-lysophosphatidylcholine (HO-PC), 2,6-dichlorobenzoyl chloride, 1-methylimidazole, CH2Cl2; (c) NaBH4, MeOH, (d) PPTS, THF/acetone/H2O (5:4:1).
2.5. HOOA and HODA-PCs
HOOA-PC (1b) and HODA-PC (1c) were assembled by PdCl2(CH3CN)2-catalyzed acylation of vinylstanane 420 with glutaric acid monomethyl ester chloride (5b) or azelaeic acid monomethyl ester chloride (5c)25–27 to generate ketones 14b or 14c, respectively (Scheme 6). Acylation of L-α-lysophosphatidylcholine (HO-PC) with the carboxylic acids produced by hydrolysis of the methyl esters 3 followed by selective reduction of the ketone carbonyl delivered stable precursors 2 of the γ-hydroxyalkenal phospholipids 1.
Scheme 6. Syntheses of HOOA and HODA-PCs.

Reagents and conditions: (a) oxalyl chloride, benzene; (b) PdCl2(MeCN)2 (1 mol%), DMF, 0 °C; (c) porcine pancreating lipase, PBS; (d) L-α-lysophosphatidylcholine (HO-PC), 2,6-dichlorobenzoyl chloride, 1-methylimidazole, CH2Cl2; (e) NaBH4, MeOH; (f) PPTS, THF/acetone/H2O (5:4:1)
3. Conclusion
New total syntheses readily deliver the γ-hydroxy-α,β-unsaturated aldehydic esters HOHA-PC (1a), HOOA-PC (1b), and HODA-PC (1c) of 2-lysophosphatidylcholine from the corresponding acetals 2. These relatively stable precursors are protected against oxidative degradation of the aldehyde and cyclization to furans. However, our model studies with 11 and 16 revealed a potential pitfall, acid-catalyzed lactonization to 12 or 17, respectively (Scheme 7). Since L-α-lysophosphatidylcholine (HO-PC) – that would be released by such lactonization of HOHA-PC (1a) or HOOA-PC (1b) – is biologically active, it is important to avoid strongly acidic conditions during the generation of these oxidatively truncated phospholipids from the stable acetal precursors.
Scheme 7.
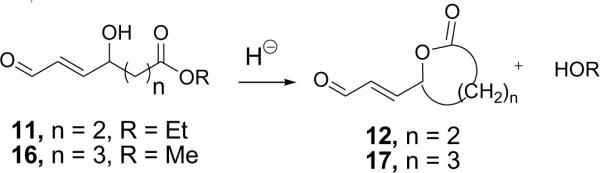
Lactone formation is promoted by strong acid.
Experimental Section
4.1. Ethyl (E)-4, 7-dioxohept-5-enoate (8)
N-Bromosuccinimide (NBS, 1.59 g, 8.9 mmol) was dissolved in tetrahydrofuran-acetone-H2O (5:4:1, 12 mL) and slowly added to a solution of ethyl-(3-(2-furyl)propanoate (7a, 1 g, 5.9 mmol) and pyridine (962 μL, 11.9 mmol) in tetrahydrofuran-acetone-H2O (5:4:1, 8 mL) at −20 °C. The solution was stirred for 1 h at −20 °C and 3 h at room temperature. The solvents were then evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (30% ethyl acetate/hexanes, TLC: Rf = 0.3) to give 8 (653 mg, 60%). 1H NMR (400 MHz, CDCl3) δ 9.76 (d, J = 7.1 Hz, 1H), 6.89 (d, J = 16.2 Hz, 1H), 6.79 (dd, J = 16.9, 7.0 Hz, 1H), 4.11 (q, J = 7.2 Hz, 2H), 2.99 (t, J = 6.2 Hz, 2H), 2.65 (t, J = 6.1 Hz, 2H), 1.22 (t, J = 6.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) 198.2, 193.4, 172.3, 144.5, 137.7, 60.9, 35.8, 27.9, 14.2. HRMS (FAB): m/z calcd for C9H13O4 (MH+), 185.0814; found, 185.0818.
4.2. Ethyl (E)-7, 7-dimethoxy-4-oxohept-5-enoate (9)
Aldehyde 8 (299 mg, 1.6 mmol) was stirred with suspension of the Montmorillonite K-10 (500 mg) and trimethyl orthoformate (500 μL) in dry dichloromethane (3 mL) at room temperature for 1 h. The mixture was then filtered through a pad of Celite 521, followed by washing the residue with dichloromethane (10 mL). The solvents were evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (10% ethyl acetate/hexanes) to give 9 (280 mg, 75%). TLC (30% ethyl acetate/hexanes, Rf = 0.32): 1H NMR (400 MHz, CDCl3) δ 6.62 (dd, J = 19.5, 3.9 Hz, 1H), 6.37 (dd, J = 16.2, 1.1 Hz, 1H), 4.93 (m, 1H), 4.11 (q, J = 7.2 Hz, 2H), 3.32 (s, 6H), 2.89 (t, J = 6.6 Hz, 2H), 2.60 (t, J = 6.6 Hz, 2H), 1.23 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) 198.1, 172.8, 140.8, 131.9, 101.1, 60.8, 53.1, 35.3, 28.1, 14.4. HRMS (FAB): m/z calcd for C11H19O5 (MH+, 231.1232; found, 231.1235.
4.3. Ethyl (E)-4-hydroxy-7, 7-dimethoxyhept-5-enoate (10)
Sodium borohydride (9.4 mg, 0.25 mmol) was added in small portions over 5 min at 0 °C to a stirred solution of ester 9 (47.7 mg, 0.2 mmol) in ethanol (1.5 mL). The reaction mixture was stirred for 4 h and then quenched by addition of methanol to destroy excess sodium borohydride. The mixture was neutralized to pH 7 by addition of saturated sodium bicarbonate solution, followed by addition of brine. The aqueous layer was extracted with ethyl acetate. Solvents were evaporated from the combined organic extracts under reduced pressure, and the residue purified by flash chromatography on a silica gel column (20% ethyl acetate/hexanes) to give 10 (45.7 mg, 95%). TLC (45% ethyl acetate/hexanes, Rf = 0.21): 1H NMR (400 MHz, CDCl3) δ 5.88 (ddd, J = 15.8, 5.7, 1.0 Hz, 1H), 5.69 (ddd, J = 15.8, 4.7, 1.3 Hz, 1H), 4.79 (d, J = 4.8 Hz, 1H), 4.22 (m, 1H), 4.12 (q, J = 7.2 Hz, 2H), 3.31 (s, 6H), 2.45 (t, J = 6.6 Hz, 2H), 2.06 (s, -OH), 2.00–1.74 (m, 2H), 1.23 (t, J = 7.2 Hz, 3H). HRMS (FAB): m/z calcd for C11H20NaO5 (MNa+), 255.1209; found, 255.1206.
4.4. Ethyl (E)-4-hydroxy-7-oxohept-5-enoate (11)
Acetal 10 (5 mg, 0.02 mmol) was dissolved in acetone/water = 2:1 (0.5 mL) and pyridinium p-toluenesulfonate (PPTS, 5.9 mg, 0.022 mmol) was added and the mixture was stirred at room temperature for 2 h. Solvents were then evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (20% ethyl acetate/hexanes) to give 11 (3.6 mg, 90%). TLC (45% ethyl acetate/hexanes, R = f = 0.20): 1H NMR (400 MHz, CDCl3) δ 9.60 (d, J = 7.8 Hz, 1H), 6.80 (dd, J = 15.7, 4.4 Hz, 1H), 6.35 (ddd, J = 15.7, 7.8, 1.7 Hz, 1H), 4.54 (m,1H), 4.15 (q, J = 7.1 Hz, 3H), 2.59 (d, J = 4.8 Hz, 1H), 2.50 (ddd, J =1 7.1, 10.4, 7.6 Hz, 3H), 2.14–1.98 (m, 1H), 1.89 (dt, J = 14.4, 6.6 Hz, 2H), 1.27 (t, J = 7.1 Hz, 6H). HRMS (FAB): m/z calcd for C9H13O3 (MH+ -H2O), 169.0865; found, 169.0867.
4.5. (E)-3-(5-Oxotetrahydrofuran-2-yl)acrylaldehyde (12)
Method (A)
Amberlyst-15 (15 mg) was added in small portions to a stirred solution of acetal 10 (15 mg, 0.06 mmol) in acetone/water = 3:1 (1.5 mL) and the mixture was stirred at room temperature for 2 h. Solvents were then evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (40% ethyl acetate/hexanes) to give lactone 12 (6 mg, 67%). TLC (50% ethyl acetate/hexanes, Rf = 0.15).
Method (B)
Acetal 10 was dissolved in TFA/H2O = 95:5 (1 mL) and stirred for 1.5 h. Solvents were then evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (40% ethyl acetate/ hexanes) to give lactone 12 (5.5 mg, 61%). 1H NMR (400 MHz, CDCl3) δ 9.60 (d, J = 7.6 Hz, 1H), 6.80 (dd, J = 15.8, 4.7 Hz, 1H), 6.32 (ddd, J= 15.8, 7.6, 1.6 Hz, 1H), 5.31–5.10 (m, 1H), 2.69–2.43 (3H), 2.20–1.97 (m, 1H). HRMS (FAB): m/z calcd for C7H9O3 (MH+), 141.0551; found, 141.0473. The 1H NMR spectral data for 12 is in agreement with that reported previously.28,29
4.6. (E)-7, 7-Dimethoxy-4-oxohept-5-enoic acid (3a)
To increase solubility at room temperature ester 9 (205 mg, 0.89 mmol) was dissolved in PBS (pH 7.4, 50 mM, 7 mL) and methanol (1 mL). To this rapidly stirred solution, was then added porcine pancreatic lipase (type II, crude, 50 mg), sodium chloride (5 mg), and calcium chloride (10 mg). The pH of the reaction mixture was maintained at 7.0–7.2 by addition of sodium hydroxide solution (0.1 N). Upon completion, the solution was evaporated under reduced pressure, the residue extracted with chloroform/methanol (2:1, v/v), filtered, and dried with anhydrous magnesium sulfate. Solvents were then evaporated from the combined organic extracts under reduced pressure and the residue purified by flash chromatography on a silica gel column (chloroform/methanol = 20:1) to give 3a (90 mg, 50%). TLC (chloroform/methanol/H2O = 80:19:1, Rf = 0.44): 1H NMR (400 MHz, CDCl3) δ 6.63 (dd, J = 16.4, 4 Hz, 1H), 6.38 (dd, J = 16.4, 1.2 Hz, 1H), 4.95 (dd, J = 4.0, 1.6 Hz, 1H), 3.33 (s, 6H), 2.90 (t, J = 6.4 Hz, 2H), 2.61 (t, J = 6.4 Hz, 2H). 13C NMR (100 MHz, CDCl3) 198.2, 178.4, 140.1, 131.8, 101.1, 53.2, 35.3, 28.1. HRMS (FAB): m/z calcd for C9H14NaO5 (MNa+), 225.0740; found, 225.0746.
4.7. (E)-(7, 7-Dimethoxy-4-oxohept-5-enoyl)-1-palmitoyl-sn-glycero-3-phosphatidylcholine (18a)
1-Methylimidazole (18.7 μL, 0.23 mmol) and 2, 6-dichlorobenzoyl chloride (37 μL, 0.26 mmol) were added to a solution of acid 3a (31.7 mg, 0.16 mmol) and L-α-lysophosphatidylcholine (38.8 mg, 78 μmol) in dry methylene chloride (4 mL). The resulting mixture was stirred for 20 h at room temperature and monitored by TLC for disappearance of starting material. Solvents were then evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (chloroform/methanol/H2O = 16:9:1, TLC: Rf = 0.23) to give 18a (41.5 mg, 78%). 1H NMR (400 MHz, CD3OD:CDCl3 =1:2) δ 6.61 (dd, J = 16.2, 3.9 Hz, 1H), 6.36 (d, J = 16.2 Hz, 1H), 5.19 (m, 1H), 4.93 (m, 1H), 4.37 (m, 1H), 4.34 (m, 1H), 4.14 (dd, J = 11.9,6.4 Hz, 2H), 3.98 (m, 2H), 3.83 (m, 2H), 3.37 (bs, 9H), 3.34 (s, 6H), 2.89 (m, 2H), 2.62 (m, 2H), 2.27 (t, J = 7.6 Hz, 2H), 1.56 (m, 2H), 1.24 (24H), 0.87 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CD3OD:CDCl3 =1:2) 198.1, 173.8, 172.3, 141.0, 131.8, 101.1, 76.9, 71.7, 71.6, 66.7, 63.7, 62.9, 59.5, 54.7, 53.3, 35.1, 34.3, 32.2, 29.9, 29.8, 29.7, 29.6, 29.5, 29.4, 28.1, 25.1, 22.3, 14.4. HRMS (FAB): m/z calcd for C33H63NO11 (M+), 680.4139; found, 680.4131.
4.8. (E)-2-(4-Hydroxy-7, 7-dimethoxyhept-5-enoyl)-1-palmitoyl-sn-glycero-3-phosphatidylcholine (2a)
Sodium borohydride (1.7 mg, 45.8 μmol) was slowly added at 4 °C to a stirred solution of ester 18a (26 mg, 38 μmol) in dry methylene chloride (0.5 mL) and methanol (1 mL). Once complete, the reaction mixture was neutralized with acetic acid, solvents were evaporated under reduced pressure, and the residue purified by flash chromatography on a silica gel column (chloroform/methanol/H2O = 11:9:1, TLC: Rf = 0.27) to give 2a (22.1 mg, 85%). 1H NMR (400 MHz, CD3OD:CDCl3 =1:2) δ 5.99 (dd, J = 16, 6 Hz, 1H), 5.76 (dd, J = 16.4, 4.8 Hz, 1H), 5.34 (m, 1H), 4.89 (d, 1H), 4.51 (m, 1H), 4.42 (m, 2H), 4.27 (dd, J = 12, 6.8 Hz, 2H), 4.12 (m, 2H), 3.82 (m, 2H), 3.48(s, 1H), 3.44 (bs, 9H), 3.37 (s, 6H), 2.56 (m, 2H), 2.43 (m, 2H), 1.93 (m, 2H), 1.70(m, 2H), 1.41 (24H), 0.98 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CD3OD:CDCl3 =1:2) 173.8, 173.0, 137.8, 128.6, 101.0, 76.9, 71.8, 71.6, 70.2, 66.4, 63.6, 62.9, 59.4, 54.7, 53.4, 35.1, 34.3, 32.1, 29.9, 29.8, 29.7, 29.6, 29.5, 29.4, 28.3, 25.2, 22.3, 14.4. HRMS (FAB): m/z calcd for C33H63NNaO11P ((M-H)Na+), 704.4115; found, 704.4119.
4.9. (E)-2-(4-Hydroxy-7-oxohept-5-enoyl)-1-palmitoyl-sn-glycero-3-phosphatidylcholine (HOHA-PC, 1a)
PPTS (1.3 mg, 5.1 μmol) was added to a stirred solution of ester 2a (3.2 mg, 4.6 μmol) in tetrahydrofuran-acetone-H2O = 5:4:1 (1 mL). The resulting mixture was stirred for 2 h at room temperature and monitored by TLC for disappearance of starting material. Upon completion, the solvents were evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (chloroform/methanol/H2O = 11:9:1, TLC: Rf = 0.2) to give 1a (2.9 mg, 97%). 1H NMR (400 MHz, CD3OD:CDCl3 =1:2) δ 9.54 (d, J = 8 Hz, 1H), 6.92 (dd, J = 15.6, 4.4 Hz, 1H), 6.29 (dd, J = 16, 8 Hz, 1H), 5.21 (m, 1H), 4.35 (m, 1H), 4.28 (m, 2H), 4.13 (m, 2H), 3.98 (m, 2H), 3.65 (m, 2H), 3.30 (s, 9H), 2.43–2.62 (m, 2H), 2.27–2.34 (m, 2H), 1.80–1.97 (m, 2H), 1.57 (m, 2H), 1.23 (24H), 0.85 (t, J = 6.8 Hz, 3H). HRMS (FAB): m/z calcd for C31H59NNaO10P (M+H+), 636.3876; found, 636.3850
4.10. 1-Tributylstannyl-3, 3-diethoxy-prop-1-ene (4)
According to the published procedure,21 3, 3-diethoxy-1-propyne (0.97 mL, 6.8 mmol) was added dropwise via syringe to a THF solution of Bu3SnCu(Bu)CNLi2, and the reaction mixture was stirred for 2 h under −78 °C before quenching with water. The dark solution was extracted with ether (3 × 20 mL), followed by drying of the combined extracts over anhydrous sodium sulfate. The solvents were evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (ethyl actate/hexanes = 3:97, TLC: Rf = 0.28) to give 4 (2.56 g, 81%). 1H NMR (400 MHz, CDCl3) δ 6.40 – 6.26 (m, 1H), 6.03 – 5.88 (m, 1H), 4.89 – 4.72 (m, 1H), 3.63 (dq, J = 9.5, 7.1 Hz, 3H), 3.49 (dq, J = 9.5, 7.1 Hz, 3H), 1.57 – 1.40 (9H), 1.36 – 1.25 (9H), 1.24 – 1.14 (6H), 0.93 – 0.80 (9H). The 1H NMR spectral data for stannane 4 is in agreement with that reported previously.30
4.11. Methyl (E)-8,8-diethoxy-5-oxooct-6-enoate (14b)
The procedure was modified from that reported.21 Glutaric acid monomethyl ester chloride (371 μL, 2.68 mmol) was added dropwise via syringe to a solution of stannane 4 (1.12 g, 2.68 mmol) and PdCl2(MeCN)2 (6.9 mg, 26.8 μmol) in DMF (3 mL). The reaction mixture was then cooled in an ice bath and stirred for 1 h, and then for 4 h at room temperature. Saturated aqueous sodium fluoride and acetone were added to the brown-red reaction mixture to remove tributyltin chloride. The mixture was extracted with ethyl acetate (3 × 20 mL), followed by drying of the combined extracts over anhydrous sodium sulfate. Solvent was evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (ethyl acetate:hexanes = 7:93) to give 14b (450 mg, 65%). TLC: (30% ethyl acetate/hexanes; Rf = 0.38). 1H NMR (400 MHz, CDCl3) δ 6.62 (dd, J = 16, 4.4 Hz, 1H), 6.32 (dd, J = 16.4, 1.2 Hz, 1H), 5.02 (m, 1H), 3.64 (s, 3H), 3.59–3.64 (m, 2H), 3.48–3.54(m, 2H), 2.64 (t, J = 6.6 Hz, 2H), 2.34 (t, J = 7.2 Hz, 2H), 1.92 (tt, J = 7.2, 7.2 Hz, 2H), 1.20 (t, J = 7.2 Hz, 6H). 13C NMR (100 MHz, CDCl3) 199.6, 173.8, 141.5, 131.6, 99.7, 61.7, 51.8, 39.4, 33.2, 19.1, 15.4. HRMS (FAB): m/z calcd for C13H23O5 (MH+), 259.1545; found, 259.1504.
4.12. Methyl (E)-8,8-diethoxy-5-hydroxyoct-6-enoate (15b)
Sodium borohydride (1.5 mg, 0.03 mmol) was added in small portions over 2 min at 0 °C to a stirred solution of ester 14b (10 mg, 0.03 mmol) in ethanol (1 mL). The reaction mixture was stirred for 4 h and then quenched by addition of methanol to destroy excess sodium borohydride. The mixture was neutralized to pH 7 by addition of saturated sodium bicarbonate solution, followed by addition of brine. The aqueous layer was extracted with ethyl acetate, solvents were evaporated from the combined organic extracts under reduced pressure, and the residue purified by flash chromatography on a silica gel column (20% ethyl acetate/hexanes) to give 15b (9.4 mg, 94%). TLC (45% ethyl acetate/hexanes, Rf = 0.24): 1H NMR (400 MHz, CDCl3) δ 5.86 (dd, J = 15.7, 6.0 Hz, 1H), 5.70 (ddd, J = 15.7, 5.0, 1.2 Hz, 1H), 4.89 (d, J = 5.0 Hz, 1H), 4.12 (m, 1H), 3.68 – 3.58 (5H), 3.55 – 3.41 (m, 2H), 2.34 (m, 2H), 1.77 – 1.62 (3H), 1.62 – 1.50 (2H), 1.29 – 1.13 (6H). HRMS (FAB): m/z calcd for C13H24NaO5 (MNa+), 283.1522; found, 283.1523
4.13. Methyl (E)-5-hydroxy-8-oxooct-6-enoate (16)
Acetal 15b (9.4 mg, 0.03 mmol) was dissolved in acetone/water = 2:1 (1 mL), PPTS (10 mg, 0.033 mmol) was added, and the mixture stirred at room temperature for 2 h. After completion, solvents were evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (20% ethyl acetate/hexanes) to give 16 (5.9 mg, 88%). TLC (45% ethyl acetate/hexanes, Rf= 0.20): 1H NMR (400 MHz, CDCl3) δ 9.58 (d, J = 7.8 Hz, 1H), 6.81 (dd, J = 15.7, 4.5 Hz, 1H), 6.32 (ddd, J = 15.7, 7.8, 1.6 Hz, 1H), 4.45 (bs, -OH), 4.13 (q, J = 7.1 Hz, 1H), 3.67 (s, 3H), 2.47 – 2.26 (2H), 1.86 – 1.51 (2H), 1.35 – 1.08 (2H). HRMS (FAB): m/z calcd for C9H13O3 (MH+-H2O), 167.0709; found, 167.0722.
4.14. (E)-3-(6-Oxotetrahydro-2H-pyran-2-yl)acrylaldehyde (17)
Amberlyst-15 (10 mg) was added in small portions to a stirred solution of acetal 15b (16 mg, 0.06 mmol) in TFA/water =95:5 (1.5 mL) and the mixture was stirred at room temperature for 1.5 h. Solvents were then evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (45% ethyl acetate/hexanes) to give lactone 17 (8.2 mg, 87%). TLC (50% ethyl acetate/hexanes, Rf = 0.1): 1H NMR (400 MHz, CDCl3) δ 9.61 (d, J = 7.5 Hz, 1H), 6.76 (dd, J = 15.8, 4.3 Hz, 1H), 6.37 (ddd, J = 15.8, 7.6, 1.7 Hz, 1H), 5.19–5.04 (m, 1H), 2.76–2.62(m, 1H), 2.55 (m, 1H), 2.18–2.05 (m, 1H), 2.06–1.87 (m, 2H), 1.83–1.64 (m, 1H). HRMS (TOF-MS/ESI): m/z calcd for C8H10NaO3 (MNa+), 177.0528; found, 177.0529. 1H NMR spectral data for 17 is in agreement with that reported previously.28
4.15. (E)-8,8-Diethoxy-5-oxooct-6-enoic acid (3b)
The acid 3b was prepared using a solution of ester 14b (118.5 mg, 0.45 mmol) in PBS (pH 7.4, 50 mM, 8 mL) and methanol (1 mL) to increase solubility at room temperature. Porcine pancreatic lipase (PPL, type II, crude, 100 mg), then sodium chloride (20 mg) and calcium chloride (40 mg) were added. The pH of the reaction mixture was maintained at 7.0–7.2 by addition of sodium hydroxide solution (0.1 N). After completion, solvents were evaporated under reduced pressure, the residue extracted with chloroform:methanol (2:1, v/v), filtered, and dried using anhydrous magnesium sulfate. Solvents were evaporated from the combined organic extracts under reduced pressure and the residue purified by flash chromatography on a silica gel column (chloroform/methanol = 20:1) to give 3b (65 mg, 58%). TLC (chloroform/methanol/H2O = 80:19:1, Rf = 0.51): 1H NMR (400 MHz, CDCl3) δ 6.65 (dd, J = 16.2, 4 Hz, 1H), 6.34 (dd, J = 16, 1.2 Hz, 1H), 5.05 (dd, J = 4.2, 1.6 Hz, 1H), 3.63–3.69 (m, 2H), 3.50–3.56 (m, 2H), 2.69 (t, J = 7.2 Hz, 2H), 2.43 (t, J = 6.8 Hz, 2H), 1.95 (tt, J = 7.2, 7.2 Hz, 2H), 1.24 (t, J = 7.2 Hz, 6H). 13C NMR (100 MHz, CDCl3) 199.7, 179.2, 141.6, 131.6, 99.7, 61.8, 39.3, 33.2, 18.8, 15.4. HRMS (FAB): m/z calcd for C12H21O5 (MH+), 245.1389; found, 245.1382.
4.16. (E)-2-(8,8-Diethoxy-5-oxooct-6-enoyl)-1-palmitoyl-sn-glycero-3-phosphatidylcholine (18b)
1-Methylimidazole (11.5 μL, 0.14 mmol) and 2,6-dichlorobenzoyl chloride (22.7 μL, 0.16 mmol) were added to a solution of acid 3b (31.7 mg, 0.16 mmol) and L-2-lysophophatidylcholine (23.5 mg, 96 μmol) in dry methylene chloride (4 mL). The resulting mixture was stirred for 17 h at room temperature and monitored by TLC for disappearance of the starting material. Solvents were then evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (chloroform/methanol/H2O = 16:9:1, TLC: Rf = 0.34) to give 18b (25.6 mg, 74%). 1H NMR (400 MHz, CD3OD:CDCl3 =1:2) δ 6.64 (dd, J = 16.2, 4.0 Hz, 1H), 6.33 (dd, J = 16.0, 1.2, 1H), 5.21 (m, 1H), 5.04 (m, 1H), 4.39 (m, 1H), 4.25 (m, 2H), 4.13 (dd, J = 12.0, 5.2 Hz, 2H), 4.00 (m, 2H), 3.61–3.69 (4H), 3.49–3.57 (m, 2H), 3.30 (bs, 9H), 2.70 (t, J = 6.8 Hz, 2H), 2.37 (t, J = 6.8 Hz, 2H), 2.29 (t, J = 7.2 Hz, 2H), 1.89(m, 2H), 1.57 (m, 2H), 1.22 (30H), 0.85 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CD3OD:CDCl3 =1:2) 200.5, 174.1, 172.9, 141.8, 131.3, 99.8, 70.8, 66.5, 63.9, 62.6, 61.9, 59.3, 54.0, 39.2, 34.1, 33.2, 32.0, 29.8, 29.7, 29.6, 29.5, 29.4, 29.3, 29.2, 24.9, 22.7, 18.9, 14.9, 13.8. HRMS (FAB): m/z calcd for C33H69NO11P (MH+), 722.4608; found, 722.4589.
4.17. (E)-2-(5-Hydroxy-8,8-diethoxyoct-6-enoyl)-1-palmitoyl-sn-glycero-3-phosphatidylcholine (2b)
Sodium borohydride (1.9 mg, 50 μmol) was slowly added at 4 °C to a stirred solution of ester 18b (33.4 mg, 46 μmol) in dry methylene chloride (1 mL) and methanol (2 mL). Once complete, the reaction mixture was neutralized with acetic acid, solvents were evaporated under reduced pressure, and the residue purified by flash chromatography on a silica gel column (chloroform/methanol/H2O = 11:9:1, TLC: Rf = 0.22) to give 2b (32.8 mg, 98%). 1H NMR (400 MHz, CD3OD:CDCl3 =1:2) δ 5.81 (dd, J = 15.6, 6 Hz, 1H), 5.62 (dd, J = 15.6, 6 Hz, 1H), 5.21 (m, 1H), 4.86 (d, 1H), 4.37 (m, 1H), 4.22 (m, 2H), 4.13 (dd, J = 12.4, 7.2 Hz, 2H), 4.07 (m, 1H), 3.64 (m, 2H), 3.47–3.59 (4H), 3.16 (bs, 9H), 2.27–2.36 (4H), 1.68 (m, 2H), 1.52–1.57 (4H), 1.16–1.26 (30H), 0.85 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CD3OD:CDCl3 =1:2) 174.1, 173.4, 137.1, 127.3, 101.6, 70.8, 70.6, 66.5, 63.8, 62.6, 61.5, 59.2, 54.0, 36.3, 36.2, 34.0, 33.9, 32.0, 29.7, 29.5, 29.4, 29.3, 29.2, 24.9, 22.7, 20.9, 14.9, 13.9. HRMS (FAB): m/z calcd for C36H70NNaO11P (MNa+), 746.4584; found, 746.4584
4.18. (E)-2-(5-Hydroxy-8-oxooct-6-enoyl)-1-palmitoyl-sn-glycero-3-phosphatidylcholine (HOOA-PC, 1b)
PPTS (4.6 mg, 18.4 μmol) was added to a stirred solution of ester 2b (11.1 mg, 15.3 μmol) in tetrahydrofuran/acetone/H2O = 5:4:1 (2 mL), the resulting mixture was stirred for 4 h at room temperature, and monitored by TLC for disappearance of starting material. Upon completion, solvents were evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (chloroform/methanol/H2O = 11:9:1, TLC: Rf = 0.18) to give 1b (8.4 mg, 85%). 1H NMR (400 MHz, CD3OD:CDCl3 =1:2) δ 9.53 (d, J = 8 Hz, 1H), δ 6.93 (dd, J = 15.6, 4.4 Hz, 1H). 6.29 (ddd, J = 15.6, 8, 1.6 Hz, 1H), 5.21 (m, 1H), 4.36 (m, 2H), 4.11–4.26 (4H), 3.99 (m, 2H), 3.58 (m, 2H), 3.30 (s, 9H), 2.39 (m, 2H), 2.31 (m, 2H), 1.57–1.77 (6H), 1.57(m, 2H), 1.26 (24H), 0.85 (t, J = 6.8 Hz, 3H). HRMS (FAB): m/z calcd for C32H61NO10P (MH+), 650.4033; found, 650.4030.
4.19. Methyl 9-(chlorocarbonyl)octanoate (5c)
Monomethyl azelate (500 mg, 2.4 mmol) was added dropwise via syringe to a solution of oxalyl chloride (300 mg, 3.5 mmol, 1.5 eq) in benzene (3 mL). The reaction mixture was stirred for 30 min at room temperature and then refluxed for 2 h, after which time no acid starting material was detected by IR. Upon completion, the solvent was evaporated under reduced pressure and the residue purified by vacuum distillation to give 5c (491 mg, 90%). IR (film, cm−1) 2943, 2861, 1811, 1733, 1462, 1434, 1251, 1205. 1H NMR (400 MHz, CDCl3) δ 3.66 (s, 3H), δ 2.87 (m, 2H), 2.30 (m, 2H), 1.65 (m, 2H), 1.60 (m, 2H), 1.35 (6H).
4.20. Methyl (E)-12,12-diethoxy-9-oxododec-10-enoate(14c)
The procedure was modified from that reported.21 1-Tributylstannyl-3,3-diethoxy-prop-1-ene (4, 380 mg, 0.9 mmol) was added dropwise via syringe to a solution of acyl chloride 5c (200 mg, 0.9 mmol) and PdCl2(MeCN)2 (2.3 mg, 9 μmol) in DMF (1.5 mL) and the reaction mixture stirred first for 1 h in an ice bath and then 3 h at room temperature. Saturated aqueous sodium fluoride and acetone were added to the brown-red reaction mixture to remove tributyltin chloride. The mixture was extracted with ethyl acetate (3 × 20 mL), followed by drying of the combined extracts over anhydrous sodium sulfate. Solvents were evaporated from the organic extracts under reduced pressure and the residue purified by flash chromatography on a silica gel column (ethyl acetate/hexanes = 7:93) to give 14c (171 mg, 60%). TLC (30% ethyl acetate/hexanes; Rf= 0.51). 1H NMR (400 MHz, CDCl3) δ 6.62 (dd, J = 16.1, 4.3 Hz, 1H), 6.33 (dd, J = 16.1, 1.3 Hz, 1H), 5.04 (dd, J = 4.3, 1.3 Hz, 1H), 3.76 – 3.59 (m, 5H), 3.59 – 3.43 (m, 2H), 2.64 (t, J = 6.6 Hz, 2H), 2.34 (t, J = 7.2 Hz, 2H), 1.92 (p, J = 7.2 Hz, 2H), 1.20 (t, J = 7.2 Hz, 6H). 13C NMR (100 MHz, CDCl3) 200.8, 174.5, 141.1, 131.8, 99.8, 61.7, 51.7, 40.6, 34.3, 29.2, 25.09, 24.06, 15.54, 15.43. HRMS (FAB): m/z calcd for C17H31O5 (MH+), 315.2171; found, 315.2175
4.21. (E)-12,12-Diethoxy-9-oxododec-10-enoic acid (3c)
The acid was prepared using a solution of ester 14c (88.7 mg, 0.28 mmol) in PBS (pH 7.4, 50 mM, 8 mL) and methanol (1 mL) to increase solubility at room temperature. Porcine pancreatic lipase (type II, crude, 100 mg), sodium chloride (20 mg), and calcium chloride (40 mg) were added. The pH of the reaction mixture was maintained at 7.0–7.2 by addition of sodium hydroxide solution (0.1 N). Upon completion, solvents were evaporated under reduced pressure, the residue extracted with chloroform/methanol (2:1, v/v), filtered, and dried with anhydrous magnesium sulfate. Solvents were evaporated from the combined organic extracts under reduced pressure and the residue purified by flash chromatography on a silica gel column (chloroform/methanol = 15:1) to give 3c (56 mg, 67%). TLC (chloroform/methanol/H2O = 80:19:1, Rf = 0.70): 1H NMR (400 MHz, CDCl3) δ 6.61 (dd, J = 16, 4.4 Hz, 1H), 6.33 (dd, J = 16, 1.2 Hz, 1H), 5.04 (dd, J = 4.4, 1.2 Hz, 1H), 3.65–3.66 (m, 2H), 3.50–3.56 (m, 2H), 2.56 (t, J = 7.6 Hz, 2H), 2.33 (t, J = 7.2 Hz, 2H), 1.60 (m, 2H), 1.30 (6H), 1.22 (6H). 13C NMR (100 MHz, CDCl3) 200.9, 179.8, 141.2, 131.8, 99.8, 61.7, 40.5, 34.2, 29.2, 29.1, 29.0, 24.8, 24.1, 15.4. HRMS (FAB): m/z calcd for C16H28O5 (M+), 300.1937; found, 300.1930.
4.22. (E)-2-(12,12-Diethoxy-9-oxododec-10-enoyl)-1-palmitoyl-sn-glycero-3-phosphatidylcholine (18c)
1- Methylimidazole (10.6 μL, 0.13 mmol) and 2,6-dichlorobenzoyl chloride (20.9 μL, 0.14 mmol) were added to a solution of acid 3c (20 mg, 0.06 mmol) and L-α-lysophophatidylcholine (22 mg, 0.04 mmol) in dry methylene chloride (4 mL). The resulting mixture was stirred for 24 h at room temperature and monitored by TLC for disappearance of starting material. The mixture was evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (chloroform/methanol/H2O = 16:9:1, TLC: Rf = 0.34) to give 18c (24.5 mg, 71%). 1H NMR (400 MHz, CD3OD:CDCl3 =1:2) δ 6.61 (dd, J = 16.1, 4.4 Hz, 1H), 6.32 (dd, J = 16.1, 1.2 Hz, 1H), 5.20 (m, 1H), 5.04 (m, 1H), 4.39 (m, 1H), 4.23 (dt, J = 11.8, 6.0 Hz, 2H), 4.13 (dd, J = 11.9, 6.8 Hz, 1H), 3.95 (m, 2H), 3.72 – 3.46 (6H), 3.26 (bs, 9H), 2.59 (t, J = 7.4 Hz, 2H), 2.30 (4H), 1.58 (6H), 1.33 – 1.09 (36H), 0.85 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CD3OD:CDCl3 =1:2) 200.9, 174.1, 173.6, 141.5, 141.1, 131.9, 99.8, 68.3, 67.0, 62.6, 62.1, 61.9, 57.5, 54.0, 40.5, 35.6, 34.2, 34.1, 32.0, 29.7, 29.6, 29.4, 29.3, 29.1, 29.0, 28.9, 24.9, 24.8, 23.9, 22.7, 17.7, 14.9, 13.9. HRMS (FAB): m/z calcd for C40H77NO11P(M+), 778.5229; found, 778.5124.
4.23. (E)-2-(9-Hydroxy-12,12-diethoxydodec-10-enoyl)-1-palmitoyl-sn-glycero-3-phosphatidylcholine (2c)
Sodium borohydride (1.4 mg, 37 μmol) was slowly added at 4 °C to a stirred solution of ester 18c (24.5 mg, 31.4 μmol) in dry methylene chloride (1 mL) and methanol (2 mL). Upon completion, the reaction mixture was neutralized with glacial acetic acid. Solvents were evaporated under reduced pressure, and residue purified by flash chromatography on a silica gel column (chloroform/methanol/H2O = 11:9:1, TLC: Rf = 0.30) to give 2c (20.5 mg, 87%). 1H NMR (400 MHz, CD3OD:CDCl3 =1:2) δ 5.80 (dd, J = 15.6, 6.4 Hz, 1H), 5.60 (dd, J = 15.6, 6.4 Hz, 1H), 5.21 (m, 1H), 4.86 (m, 1H), 4.41 (m, 1H), 4.26 (m, 2H), 4.13 (dd, J = 12, 6.8 Hz, 1H), 4.04 (m, 1H), 3.96 (m, 2H), 3.71–3.41 (6H), 3.18 (bs, 9H), 2.27–2.33 (4H), 1.58 (8H), 1.16–1.29 (36H), 0.85 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CD3OD:CDCl3 =1:2) 174.1, 173.6, 137.6 127.0, 101.7, 71.4, 70.5, 66.5, 63.8, 62.7, 61.4, 59.2, 54.0, 40.3, 37.1, 34,2, 34.1, 32.0, 29.7, 29.6, 29.4, 29.3, 29.1, 29.0, 28.9, 24.9, 24.8, 22.7, 15.0, 14.9, 13.9. HRMS (FAB): m/z calcd for C40H78NNaO11P(MNa+), 802.5210; found, 802.5217.
4.24. (E)-(9-Hydroxy-12-oxodec-10-enoyl)-1-palmitoyl-sn-glycero-3-phosphatidylcholine (HODA-PC, 1c)
PPTS (5.6 mg, 22.2 μmol) was added to a stirred solution of ester 2c (14.5 mg, 18.6 μmol) in tetrahydrofuran/acetone/H2O = 5:4:1 (2 mL). The resulting mixture was stirred for 5 h at room temperature and monitored by TLC for disappearance of starting material. Upon completion, solvents were evaporated under reduced pressure and the residue purified by flash chromatography on a silica gel column (chloroform/methanol/H2O = 11:9:1, TLC: Rf = 0.4) to give 1c (11.8 mg, 90%). 1H NMR (400 MHz, CD3OD + CDCl3) δ 9.52 (d, J = 8 Hz, 1H), 6.91 (dd, J = 15.6, 4.8 Hz, 1H), 6.26 (ddd, J = 15.6, 8, 1.6 Hz, 1H), 5.20 (m, 1H), 4.38 (m, 1H), 4.31 (m, 1H), 4.22 (m, 2H), 4.14 (m, 1H), 3.97 (m, 2H), 3.58 (m, 2H), 3.30 (s, 9H), 2.27–2.33 (4H), 1.57(7H), 1.23–1.31 (31H), 0.85 (t, J = 6.8 Hz, 3H). HRMS (FAB): m/z calcd for C36H67NO9P(MH+ - H2O), 688.4548; found, 688.4583.
Supplementary Material
Acknowledgment
We are grateful for support of this work by National Institutes of Health Grants RO1-GM021249, RO1-EY016813, and RO1-HL053315.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data Supplementary data associated with this article can be found, in the online version, at?
References
- (1).Gu X, Sun M, Gugiu B, Hazen S, Crabb JW, Salomon RG. J Org Chem. 2003;68:3749–61. doi: 10.1021/jo026721t. [DOI] [PubMed] [Google Scholar]
- (2).Sun M, Deng Y, Batyreva E, Sha W, Salomon RG. J Org Chem. 2002;67:3575–84. doi: 10.1021/jo0105383. [DOI] [PubMed] [Google Scholar]
- (3).Deng Y, Salomon RG. J Org Chem. 1998;63:7789–94. [Google Scholar]
- (4).Kaur K, Salomon RG, O'Neil J, Hoff HF. Chem Res Toxicol. 1997;10:1387–96. doi: 10.1021/tx970112c. [DOI] [PubMed] [Google Scholar]
- (5).Sayre LM, Sha W, Xu G, Kaur K, Nadkarni D, Subbanagounder G, Salomon RG. Chem Res Toxicol. 1996;9:1194–201. doi: 10.1021/tx960094j. [DOI] [PubMed] [Google Scholar]
- (6).Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, Silverstein RL, Hoff HF, Salomon RG, Hazen SL. J Biol Chem. 2002;277:38517–23. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- (7).Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Gugiu B, Fox PL, Hoff HF, Salomon RG, Hazen SL. J Biol Chem. 2002;277:38503–16. doi: 10.1074/jbc.M203318200. [DOI] [PubMed] [Google Scholar]
- (8).Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, Chen J, Zhang R, Silverstein RL, Hazen SL. Nat Med. 2007;13:1086–95. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ashraf MZ, Kar NS, Chen X, Choi J, Salomon RG, Febbraio M, Podrez EA. J Biol Chem. 2008;283:10408–14. doi: 10.1074/jbc.M710474200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Subbanagounder G, Deng Y, Borromeo C, Dooley AN, Berliner JA, Salomon RG. Vascul Pharmacol. 2002;38:201–9. doi: 10.1016/s1537-1891(02)00170-2. [DOI] [PubMed] [Google Scholar]
- (11).Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witztum JL, Palinski W, Schwenke D, Salomon RG, Sha W, Subbanagounder G, Fogelman AM, Berliner JA. J Biol Chem. 1997;272:13597–607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- (12).Leitinger N, Tyner TR, Oslund L, Rizza C, Subbanagounder G, Lee H, Shih PT, Mackman N, Tigyi G, Territo MC, Berliner JA, Vora DK. Proc Natl Acad Sci U S A. 1999;96:12010–5. doi: 10.1073/pnas.96.21.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lee H, Shi W, Tontonoz P, Wang S, Subbanagounder G, Hedrick CC, Hama S, Borromeo C, Evans RM, Berliner JA, Nagy L. Circ Res. 2000;87:516–21. doi: 10.1161/01.res.87.6.516. [DOI] [PubMed] [Google Scholar]
- (14).Subbanagounder G, Watson AD, Berliner JA. Free Radic Biol Med. 2000;28:1751–61. doi: 10.1016/s0891-5849(00)00233-1. [DOI] [PubMed] [Google Scholar]
- (15).Hoff HF, O'Neil J, Wu Z, Hoppe G, Salomon RL. Arterioscler Thromb Vasc Biol. 2003;23:275–82. doi: 10.1161/01.atv.0000051407.42536.73. [DOI] [PubMed] [Google Scholar]
- (16).Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, Ufret RL, Salomon RG, Perez VL. Nat Med. 2008;14:194–8. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kobayashi Y, Kishihara K, Watatani K. Tetrahedron Lett. 1996;37:4385–4388. [Google Scholar]
- (18).Taylor EC, Chiang C-S. Synthesis. 1977:467. [Google Scholar]
- (19).Beaudet I, Parrain J, Quintard J. Teterahedron Lett. 1991;32:6333–6336. [Google Scholar]
- (20).Lipshutz BH, Ellsworth EL, Dimock SH, Reuter DC. Tetrahedron Lett. 1989;30:2065–2068. [Google Scholar]
- (21).Parrain J, Beaudet I, Duchene A, Watrelot S, Quintard J. Teterahedron Lett. 1993;34:5445–48. [Google Scholar]
- (22).Rodriguez A, Nomen M, Spur B, Godfroid J. Eur. J. Org. Chem. 1999:2655–62. [Google Scholar]
- (23).Acharya HP, Kobayashi Y. Synlett. 2005;13:2015–18. [Google Scholar]
- (24).Gaffney PR, Reese CB. J. Chem. Soc., Perkin Trans. 2001;1:192–205. [Google Scholar]
- (25).Abell AD, Morris KB, Litten JC. J. Org. Chem. 1990;55:5217–21. [Google Scholar]
- (26).Micovic IV, Ivanovic MD, Piatak DM. J. Serb. Chem. Soc. 1988;53:419–26. [Google Scholar]
- (27).Uno T, Ku J, Prudent JR, Huang A, Schultz PG. J. Am. Chem. Soc. 1996;118:3811–17. [Google Scholar]
- (28).Yeh MP, Chuang L, Hsieh Y, Tsai M. Chem. Commun. 1999:805–06. [Google Scholar]
- (29).Gu X, Zhang W, Salomon RG. J. Am. Chem. Soc. 2007;129:6088–89. doi: 10.1021/ja0689785. [DOI] [PubMed] [Google Scholar]
- (30).Parrain J, Duchene A, Quintard J. J. Chem. Soc., Perkin Trans. 1990;1:187–89. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.