

Background
Efficient antigen (Ag) delivery is a major challenge for dendritic cell (DC)-based immunotherapy of solid organ malignancies.1 Endogenous Ag delivery has important limitations. Electrofusion of DC and tumor cells is both laborious and time consuming.2 Transduction with DNA or mRNA via viral vectors is complicated by interference with the Ag presentation machinery of DC and generation of anti-viral antibodies.3 Exogenous Ag delivery has a separate set of problems. External pulsing of DC by peptides results in a brief stimulation of T cells because of rapid turnover of class I-peptide complexes on the cell surface. Besides, this approach can only be used in patients bearing specific major histocompatibility (MHC) haplotypes.4, 5 Whole tumor lysates are potentially complex enough to overcome the ability of tumors to down-regulate targeted antigens. However, the use of soluble tumor lysates is confounded by inherent instability of soluble macromolecules, poor internalization of soluble materials by DC and inefficient cross-presentation to cytotoxic T cells (CTL).6
Particulate delivery systems made from biocompatible and degradable polymers such as poly(lactic-co-glycolic acid) (PLGA) may overcome many of the problems encountered during transfer of Ag to DC.7 Encapsulation within particles has the potential to prevent Ag degradation by proteolytic enzymes8, increase efficiency of Ag loading9,10 and prolong release, all of which might lead to enhanced presentation of MHC-peptide complexes.11 Encapsulation is also associated with improved shelf life of Ag and reduced need for adjuvants.12 Because particles provide targeted delivery 13, many authors have suggested that they could be used in DC-based cancer vaccines.14–16 While peptides or single antigens encapsulated within PLGA preparations have been shown to induce specific CTL responses17,18, only a few studies have examined encapsulation of complex cellular material.19,20 There is no published literature describing delivery of encapsulated complex panoply of tumor associated antigens (TAA) derived from patients afflicted by cancers and its effect on stimulation of CTL. The present study was performed to generate this crucial evidence prior to using nanoparticles (NP) in clinical trials of DC-based immunotherapy.
We hypothesized that anti-tumor T cell responses could be enhanced by encapsulating TAA within NP prior to delivery to DC. In initial experiments, we used cell lysates made from rodent FAT7 and human FaDu head and neck squamous cell carcinoma (HNSCC) lines. We optimized formulation conditions to ensure protein integrity and sustained delivery of representative tumor antigens for 7–10 days, which is the probable life span of an Ag-loaded DC. The results were adopted when encapsulating fresh tumor tissue from patients with advanced HNSCC. Ag containing-NP were delivered to patient-derived DC and tested for their ability to stimulate autologous cluster of differentiation (CD) 8+ T cells. Based on the production of immunostimulatory interferon (IFN)-γ and immunoinhibitory interleukin (IL)-10 cytokines, TAA-loaded NP were found to be more efficient in delivering antigens when compared to conventional tumor lysates. This is the first report describing CD8+ T cell responses evoked by degradable NP-loaded with TAA derived from autologous human tumor tissue. It provides critical parameters that would facilitate clinical translation of this technology.
Methods
Cell Lines
Cells from FaDu and FAT7 cell lines [American Type Culture Collection (ATCC), Manassas, VA, USA] were grown to more than 90% confluence in monolayers in ATCC recommended media in a humidified atmosphere containing 5% CO2. FaDu was grown in 1% sodium pyruvate (Sigma-Aldrich, St. Louis, MO, USA) + Eagle’s Minimal Essential Medium + 10% fetal bovine serum + 1% non-essential amino acids + 1.5 mg/ml sodium bicarbonate + 1% penicillin/streptomycin (all reagents from Invitrogen, Carlsbad, CA, USA). FAT7 cells were grown using Ham’s F12K medium + 10% fetal bovine serum (non-heat inactivated) + 1% penicillin/streptomycin (all reagents from Invitrogen, Carlsbad, CA, USA) + 0.1% insulin-HEPES + 0.5% hydrocortisone (both from Sigma-Aldrich, St. Louis, MO, USA) and 0.5% Transferrin (Calbiochem, San Diego, CA, USA).
Lysate preparation
Cell pellets were re-suspended in ice cold phosphate buffered saline (PBS) (Sigma-Aldrich, St. Louis, MO, USA) at concentrations ranging from 2–4 × 108/ml and subjected to four cycles of freeze-thaw cycles (alternating liquid nitrogen and 37 °C water bath treatment) followed by sonication for 15 seconds (s) (Ultrasonic Processor, Tekmar, Cincinnati, OH, USA) on ice at 38% amplitude to break the cell membranes. Cell death and lysis were confirmed by trypan blue exclusion and haemocytometry. Lysates were spun at 12,000 rpm for 20 minutes (min) at 4 °C to remove cellular debris. Supernatants were collected and stored at −20 °C. Protein content of lysate preparations was measured using the Bicinchonic acid (BCA) protein assay kit (Thermo Scientific, Rockford, IL, USA) as per manufacturer’s protocol. Absorbance at 562 nm was measured using a spectrophotometer and protein concentrations determined by comparing absorbance readings to a standard calibration curve of bovine serum albumin (BSA) in the concentration range between 5μg/ml and 250μg/ml.
Lyophilized lysate
Portions of concentrated lysates were converted to an Ag powder by flash freezing to −70 °C for 1 hour, followed by overnight lyophilization (Labconco Freezone 2.5 Plus, Kansas City, MO, USA) at 0.120 mBar at −87 °C . The lyophilized lysate was immediately used for encapsulation.
Nanoparticle fabrication
PLGA copolymers (50% lactide: 50% glycolide) with inherent viscosity (i.v.) of 0.59dL/g (80 kDa) or 0.39 dL/g (45 kDa) (DURECT Corp., Pelham, AL, USA) were dissolved in methylene chloride (Fisher Scientific, Pittsburgh, PA, USA) prior to being used for encapsulation. Polyvinyl alcohol (0.5% PVA) (Sigma-Aldrich, St. Louis, MO, USA) was used as a stabilizer. All reagents were of analytical grade. Protein loading was altered by either using differing concentrations of soluble lysate or by utilizing concentrated Ag powder to increase protein concentration. PLGA NP were fabricated from aqueous protein solutions using the solvent evaporation method from a water/oil/water (W2/O/W1) emulsion, as described previously.6 Alternatively, a lyophilized pellet was crushed and then transferred directly to the polymer-containing organic solution while continuously being vortexed to form an oil-in-water (O/W1) emulsion. Both forms of NP were then evaporated by identical methodologies. Control NP (blanks) were synthesized by using sterile PBS instead of aqueous protein solutions.
Nanoparticle characterization
Assessment of morphology
Scanning electron microscopy (SEM) was used to characterize NP in terms of size and morphology. SEM was also to evaluate the lyophilized lysate itself. A thin film of test sample was smeared onto a metal stub with double-sided adhesive carbon tape (Nisshin EM. Co. Ltd, Tokyo, Japan) and coated with gold (Cressington Sputter Coater 108 Auto, Redding, CA, USA). Imaging was completed using a XL-30 Environmental Scanning Electron Microscope-FEG (FEI, Hillsboro, OR, USA) with an acceleration voltage of 10 kV in a high vacuum mode to achieve magnifications between 9000x and 22,000x. Diameters from representative images of test samples were analyzed using Image J (image analysis software developed by National Institutes of Health, USA).
Assessment of protein encapsulation
Extent of protein encapsulation was calculated based on the amount of protein extracted from a known mass of NP. Five mg of NP were degraded in 1 mL of 100mM NaOH + 0.05% SDS (Ricca Chemicals, Arlington, TX, USA) by incubation at 37 °C with continuous agitation in an orbital shaker. Samples were centrifuged at 16,100 × g for 5 min at room temperature (RT). Supernatants were analyzed for total protein as described above. Encapsulation efficiency was calculated using the formula: (mass protein extracted/mass protein used in the encapsulation procedure) × 100%.
Measurement of protein release rate
The rate of release of proteins from NP was measured during incubation under controlled conditions. Five mg of NP were suspended in 1 ml of PBS and incubated at 37 °C with continuous agitation in an orbital shaker. At designated time points, the suspension was centrifuged at 16,100 × g for 5 min at RT. Supernatants were collected and stored at −20°C for future studies. The particles were re-suspended in 1 ml of fresh PBS in the original tube for further incubation. Supernatant samples were analyzed for total protein using the micro BCA protein assay as per manufacturer’s protocol. Absorbance at 562nm was measured using a micro-plate reader (Molecular Probes Inc, USA); protein concentrations were determined by comparing absorbance readings to standards prepared from BSA in the concentration range between 0.5μg/ml and 200 μg/ml. Cumulative release was studied for 7 days. Release efficiency was calculated using the formula: (cumulative protein released/total protein content of NP) × 100%.
Determining protein composition and integrity
To assess whether a representative selection of tumor-associated proteins was encapsulated and released, protein composition of pre- and post-encapsulation samples were compared on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels by silver staining and western blotting. A portion of the original tumor lysate served as the pre-encapsulation sample. Post-encapsulation samples were obtained as described above. Supernatants containing released proteins were concentrated on YM-10 Ultracel YM membrane Microcon filtration units (Millipore, Billerica, MA, USA) by centrifugation at 12,000 × g for 30 min at 4 °C. An additional spin at 12,000 × g for 5 min at 4 °C was completed before storing samples at −20 °C for future assays. Protein samples were then run on SDS-PAGE gels (Protean 11 Pre-cast gel 8–16% Tris HCl 15 well, Bio-Rad, Hercules, CA, USA) and stained with SilverXpress Silver staining kit (Invitrogen, Carlsbad, CA, USA) to visualize protein bands. Differences in staining of lanes were analyzed using Image J. Western Blot analysis was also carried out to estimate the structural integrity of proteins released from NP. Samples were transferred to nitrocellulose membranes (0.2 μm, Invitrogen, Carlsbad, CA, USA) and probed for the HNSCC-associated protein p53. This was done using a combination of polyclonal goat anti-p53 antibody (AF1355, R&D Systems, Minneapolis, MN, USA) and horse radish peroxidase-conjugated donkey anti-goat immunoglobulin (Ig) (HAF 109, R&D Systems, Minneapolis, MN, USA) before detection by the enhanced chemiluminescence method (GE Healthcare, Amersham, Buckinghamshire, U.K.).
Human samples
Ethical permission from the Yale School of Medicine Human Investigation Committee was obtained to conduct in vitro experiments on tumor and blood samples. Procedures followed were in accordance with institutional guidelines. Five patients with stage III/IV HNSCC disease were recruited from the clinics of the Section of Otolaryngology, Yale School of Medicine, USA. An informed consent was obtained to procure fresh tumor tissue which was used to make tumor lysates by methodologies similar to those described above for HNSCC lines. Peripheral blood drawn on the same day was used for generation of DC from blood monocyte precursors as well as to isolate CD8+ T cells.
Tumor lysate for encapsulation
Fresh tumor tissue was washed three times in modified FaDu medium (Eagle’s Minimal Essential Medium + 10% fetal bovine serum + 1% sodium pyruvate + 1% non-essential amino acids + 1.5 mg/ml sodium bicarbonate + 10% penicillin/streptomycin), dissected under sterile conditions prior to admixing with tumor digest solution (collagenase, dispase and hyaluronidase) for 2 hours (hrs) in an incubator at 37 °C. After removal of the undigested tumor material by filtration through a cell strainer (BD Falcon™ 70 μm filter; BD, Franklin Lakes, NJ, USA), the cell solution was washed twice in modified FaDu medium, counted, and re-suspended in sterile ice cold PBS for production of NP as described above. Cell solutions were brought up in minimum volumes of PBS (depending on original tumor volume and number of cells isolated) to ensure that tumor lysate concentrations were as high as possible. Protein content was determined using the BCA assay as described above. When feasible, lyophilized versions of tumor lysates were encapsulated in parallel to the concentrated aqueous solutions. In most cases, 100 μl of the soluble lysate was lyophilized (using the technique described above) to fabricate NP using 50mg of 80K PLGA with 0.5% PVA. Characterization by SEM, studies of encapsulation and release profile, composition and structural integrity of proteins post-encapsulation were performed as described above.
DC generation and isolation of CD8+ T cells
Whole blood collected in heparin-containing 50 ml conical tubes (BD, Franklin Lakes, NJ, USA ) was diluted 1:1 in sterile PBS, layered over Isolymph gradient solution (CTL Scientific Supply Corp, Deer Park, NY, USA) and centrifuged at 1500 × g for 30 min at RT. The cells at the interface were collected and washed twice with sterile Hanks’ BSS (Invitrogen, Carlsbad, CA, USA). Cells were counted and their viability checked by Trypan blue exclusion prior to suspension in complete RPMI (RPMI 1640 plus 10% FBS, 2% Pen/Strep and 25 mM HEPES) at 2 × 106 cells/ml. Cells were plated in 5 ml aliquots into 6-well polystyrene flat-bottom cell culture plates. Monocytes were allowed to adhere for 1hr at 37° C in a 5% CO2 atmosphere; floating cells were isolated, washed with complete RPMI, counted, and re-suspended in 90% complete RPMI/10% DMSO prior to storage in cryovials at −80 °C for subsequent use. Adherent monocytes were incubated with complete RPMI plus 0.25 ng/ml of IL-4 and 0.8 ng/ml of granulocyte-macrophage colony-stimulating factor (GM-CSF) (Peprotech, Rocky Hill, NJ, USA) with medium changes on days 1, 3 and 5. On day 6, the cells were harvested by scraping, washed and counted. A portion of the DC was used immediately in Ag presentation assays while the remainder was cryopreserved as described above. CD8+ T cells were isolated from non-adherent cells by negative selection utilizing a magnetic activated cell sorting system (MACS) (Miltenyi Biotech, Auburn, CA, USA) as per manufacturer’s protocol. Cells were suspended in T cell medium (IMDM + 5% human AB serum + 1% L-glutamine + 0.2% Gentamicin + 100 U/ml of IL-2) at 0.5 × 106 cells per ml, in preparation for co-culture with DC.
DC/T cell co-culture
The effectiveness of NP-mediated TAA delivery to DC was established by assessing CD8+ T cell stimulation after DC/T cell co-culture. DC were plated in 6-well plates after which they were loaded with equimolar quantities of Ag in the form of either soluble lysate or NP. NP were initially suspended in DC growth medium before loading 0.5 mg of NP per 1 × 106 DC. This ratio was previously determined to minimize the presence of unincorporated NP following co-culture. Four to six hours later, lipopolysaccharide (LPS; Sigma-Aldrich, St. Louis, MO, USA) was added at 100 ng/ml to trigger maturation of cells. After 24 hrs, DC were harvested, washed twice, and re-plated in 12-well polystyrene flat-bottom cell culture plates. Two to three hours after plating, freshly isolated CD8+ T cells were added to the wells containing DC in a ratio between 1:3 and 2:3. The medium was replaced on days 3 and 5. On day 6, autologous frozen DC were thawed, counted and re-plated as above for a second round of CD8+ T cell stimulation. They were loaded with soluble TAA, TAA-containing NP or control NP and matured as described above. To determine whether NP-mediated antigen delivery was more efficient and effective than delivery in the soluble form, DC were loaded with soluble lysate containing antigen five-fold in excess of that within NP. DC and pre-stimulated CD8+ T cells were co-incubated in IL-2-free medium at a ratio of 2:1 while unstimulated CD8+ T cells acted as negative controls; experiments were done in triplicate. After 24 hrs, the supernatants were harvested and stored at −20° C for future assays.
Cytometric bead array
Cytokine profiling of DC/T cell stimulation was performed on supernatants using a human T-helper (Th1/Th2) cytometric bead array (CBA) cytokine kit (BD Biosciences, San Diego, CA, USA) as per manufacturer’s instructions. This used the sensitivity of amplified fluorescence detection by flow cytometry to measure levels of IL-2, IL-4, IL-6, IL-10, Tumor necrosis factor (TNF)-α and IFN- γ
SEM of NP loading onto DC
NP-loaded DC were deposited on a cover slip housed within wells in a 12-well polystyrene flat-bottom cell culture plate and allowed to adhere over 1–2 hrs. For each of the subsequent steps, the cover slip was transferred to a fresh well. Cells were gently washed twice with PBS, fixed in a solution containing 1% Glutaraldehyde in 0.1 M Cacodylate buffer, pH 7.0 (Polysciences, Inc, Warrington, PA, USA) for five minutes, washed for five minutes in distilled water and then dehydrated in graded ethanol. Cells were dried rapidly by immersion in Hexamethyldisilazane (Polysciences, Inc, Warrington, PA, USA) for 15 min after which they were air dried and stored at RT for future analysis. SEM was done as described above.
Statistics
Statistical analysis was done using Statistical Package for the Social Sciences (SPSS) version 17.0 (SPSS, Surrey UK). Spearman’s rho (rank correlation) was used to assess relation between encapsulation and lysate concentration, and that between release and protein content of NP. Differences in size of NP made from either soluble or lyophilized versions of lysate were compared using the 2-tailed Student’s t-test. Mann-Whitney U and Kruskal-Wallis tests were used to compare cytokine production by CD8+ T cells under different conditions. A level of p<0.05 was deemed to be statistically significant.
Results
Encapsulation profile
Soluble lysate
Graded concentrations of lysates from cell lines (such as FaDu) were encapsulated within two commonly used molecular weight (MW) fractions of PLGA in order to determine the effects of protein concentration on encapsulation of tumor lysate proteins (Table 1). Higher protein concentration within the lysate led to higher amounts of protein encapsulation; Pearson’s coefficient of correlation was 0.92 for 45K PLGA and 0.95 for 80K PLGA. We found no definitive trend in encapsulation efficiency. Increasing encapsulation with increasing protein loading was also observed when encapsulating tumor proteins from freshly isolated tumor explants (Table 2). Lysate concentration of greater than 25 mg/ml resulted in significantly greater encapsulation (p<0.001; Spearman’s rho) than lower concentrations.
Table 1.
Effect of concentration of cell line-lysate on encapsulation profile. Differing numbers of cells were deliberately dissolved within a finite volume of PBS. When keeping PLGA MW constant, extent of encapsulation increased with increasing concentration of lysate.
| Protein concentration of lysate (mg/ml) | PLGA MW (K) | Protein encapsulated (μg/mg) | Efficiency of encapsulation (%) |
|---|---|---|---|
| 60 | 45 | 40 | 66.7 |
| 50 | 45 | 26 | 52 |
| 25 | 45 | 21.2 | 84.8 |
| 12.5 | 45 | 6 | 48 |
| 6.25 | 45 | 5.4 | 88.2 |
| 60 | 80 | 46 | 76.7 |
| 50 | 80 | 36 | 72 |
| 25 | 80 | 26 | ≈100 |
| 12.5 | 80 | 6.6 | 52.8 |
| 6.25 | 80 | 5.4 | 85 |
Table 2.
Effect of concentration of fresh tumor-derived lysate on encapsulation profile. Size of tissue samples available determined the number of cells dissolved within a finite volume of PBS. Encapsulation increased with increasing lysate concentration.
| Protein concentration of lysate (mg/ml) | PLGA MW (K) | Protein encapsulated (μg/mg) | Efficiency of encapsulation (%) |
|---|---|---|---|
| 14.2 | 80 | 8 | 56.3 |
| 32.5 | 80 | 14 | 43.1 |
Lyophilized lysate
To further increase protein loading in NP, larger volumes of the aqueous lysate were lyophilized prior to encapsulation so that a single emulsion method could be used for NP fabrication. While there was an increase in total protein encapsulation, there was a decrease in efficiency of encapsulation (Table 3). A similar trend was also observed with the more dilute lysate obtained from a fresh human HNSCC sample of limited size (Table 4).
Table 3.
Effect of lyophilization of cell line-lysate on encapsulation. Despite using similar concentrations, lyophilization of lysate enabled increased encapsulation when compared to conventional soluble form. This was due to increased antigen per unit of polymer.
| Protein concentration of lysate (μg/μl) | Volume of lysate (μl) | Type of lysate* | Protein content starting lysate (μg) | Weight of NP used for encapsulation (mg) | Protein content of NP (μg/mg) | Encapsulation efficiency (%) |
|---|---|---|---|---|---|---|
| 24 | 50 | S | 1200 | 50 | 34 | ≈100 |
| 24 | 150 | L | 3600 | 50 | 54 | 75 |
| 24 | 450 | L | 10800 | 50 | 140 | 64.8 |
S= Soluble; L=Lyophilized
Table 4.
Effect of lyophilization of fresh tumor-derived lysate on encapsulation. Limited availability of fresh tumor material resulted in a relatively dilute lysate. However lyophilization enabled increase in the extent of encapsulation.
| Protein concentration of lysate (μg/μl) | Volume of lysate (μl) | Type of lysate* | Protein content of starting lysate (μg) | Weight of NP used for encapsulation (mg) | Protein content of NP (μg/mg) | Encapsulation efficiency (%) | |
|---|---|---|---|---|---|---|---|
| 32.5 | 50 | S | 1625 | 50 | 14 | 43.1% | |
| 23 | 100 | L | 2300 | 50 | 19.6 | 42.6% |
S= Soluble; L=Lyophilized
Kinetics of protein release
To estimate the total amount of Ag delivered from nanoparticles as a function of time, cumulative release during incubation in buffered saline was measured over a seven day period. This period was selected because it is the approximate functional lifespan of an Ag-loaded DC. Protein release was biphasic: a “burst release” was observed over the first 24 hours while a more sustained release was observed over the subsequent days. Higher initial protein content in the NP led to greater cumulative release (Figure 1). A similar trend was observed for NP made from either lyophilized lysate or from aqueous solutions. As expected from their greater total protein content, lyophilization of lysate led to a significant increase in cumulative protein release (p = 0.049; Mann-Whitney U) (Figure 2). This increase was also associated with a faster rate of release (slope of release vs. time curve) during the sustained release phase. Similar results were obtained with lysates made from a cell line (Figure 2, A) and from human tumor tissue (Figure 2, B). Release was significantly higher from NP with a protein content greater than 46 μg/mg (p= 0.03; Spearman’s rho). This degree of loading was only achievable when lyophilized lysate was used for fabrication of NP.
Figure 1.
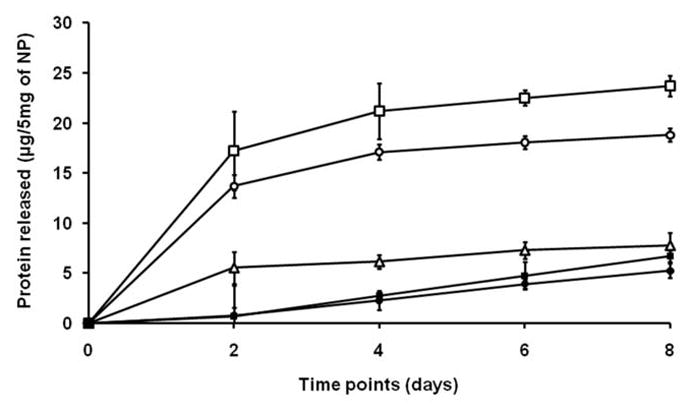
Effect of protein content of nanoparticles on cumulative release for PLGA 80 MW. The initial burst of protein release and total amount of protein release over 7 days both increased with the quantity of encapsulated protein (μg) per unit of polymer (mg). Open square: 46 μg/mg; open circle: 36 μg/mg; open triangle: 26 μg/mg; filled square: 6.6 μg/mg and filled square: 5.4 μg/mg.
Figure 2.

Comparison of cumulative protein release from NP fabricated from soluble or lyophilized lysates from (a) HNSCC cell lines and (b) freshly isolated human tumors. Lyophilization of lysate enabled greater release. The protein content (μg) per unit of polymer (mg) for (a) was: 140 μg/mg (open square) (lyophilized lysate-NP), 54 μg/mg (open circle) (lyophilized lysate-NP) and 34 μg/mg (open triangle) (soluble lysate –NP); for (b) was: 19.6 μg/mg (filled square) (lyophilized lysate-NP) and 14 μg/mg (filled circle) (soluble lysate –NP).
Documentation of tumor antigen encapsulation and release
To confirm that the typical panoply of tumor-associated proteins liberated from whole cell lysates was encapsulated and released, pre- and post-encapsulation samples were subjected to electrophoretic separation on SDS-PAGE gels followed by silver staining. Prominent bands across a spectrum of protein molecular weights indicated uniform tumor protein encapsulation and release. A higher starting concentration of lyophilized lysates led to more intense staining of protein bands in the 30K to 66.4K range (Figure 3, A).
Figure 3.

(a) Silver stained SDS/PAGE gel demonstrating a representative selection of tumor associated proteins prior to and after encapsulation. A relative paucity of bands between 46K and 66.4K was noted in NP-released samples with an apparent stacking at 97.4K. Staining was more intense in the lane containing release sample from the lyophilized version of NP. Both post-encapsulation samples were obtained from equal amounts (5mg/lane) of 80K PLGA/0.5% PVA NP. Lane 1: Lyophilized lysate; lane 2: Soluble lysate; lane 3: Pre-encapsulation lysate (3.25μg); lane 4: MW ladder. b) Western blot of membrane probed with anti-p53 antibody. Lane 1: post-encapsulation release sample (80K PLGA); lane 2: lysate (pre-encapsulation); lane 3: 46kDa labeled band from MW ladder.
Immunodetection of p53
To further demonstrate that tumor lysate proteins survived encapsulation and release, western blotting was performed to detect the mutant p53 protein previously described to be associated with squamous malignancies.21 Pre- and post-encapsulation material were probed using a monoclonal antibody to p53; continued presence of p53 implied lack of significant degradation during NP fabrication (Figure 3, B).
NP morphology
SEM was used to compare morphological appearances of the NP derived from either soluble or lyophilized lysates. All NP preparations were spherical with a wide size distribution (Figure 4). NP formed with lyophilized lysate (Figure 4, A) were larger (392 ± 304 nm) than those made using soluble lysate (Figure 4, B) (310 ± 280 nm) although the difference was not statistically significant. NP from lyophilized lysate also contained a higher percentage of irregularly shaped particles, most likely the result of encapsulation of whole non-spherical protein crystals of significant size (441 ± 142 nm) (Figure 4, C). Uptake of NP by dendritic processes of DC was observed by SEM (Figure 4, D).
Figure 4.

Scanning electron micrographs of: (a) 80K/0.5% soluble lysate NP, (b) 80K/0.5% lyophilized lysate NP, (c) lyophilized lysate protein powder pre-encapsulation, and d) Day 6 human DC exposed to soluble lysate NP in the process of phagocytic engulfment.
Biological activity of encapsulated human tumor-derived antigens
To determine whether NP with encapsulated tumor lysates could deliver Ag to patient-derived DC, a series of experiments was performed using tumor material and peripheral blood CD8+ T cells. Freshly isolated tumor material and patient blood were obtained from HNSCC patients (after appropriate consent, see Methods). Protein lysates were obtained from the tumor samples and encapsulated. These encapsulated lysates were compared to raw lysate preparations. DC were obtained from peripheral blood using GM-CSF/IL-4; CD8+ T-cell populations were isolated by antibody-based magnetic separation. DC in culture were exposed to either 1) Ag in soluble tumor lysate (in a 5-fold molar excess) or 2) the same Ag encapsulated into NP. Exposed DC were used to stimulate autologous CD8+ T cells. After allowing for overnight interaction, supernatants from co-incubated DC and CD8+ T cells were collected and analyzed for cytokine production. In three out of the five patients (numbers 1, 2 and 3), NP-mediated Ag delivery caused increased IFN-γ and decreased IL-10 production; in the other two patients (numbers 4 and 5) either increased IFN-γ or decreased IL-10 production was observed (Figure 5). Data from four out of five patients with advanced HNSCC were pooled. There was either a significantly increased IFN-γ production (p=0.0071; Kruskal-Wallis test) or significantly decreased IL-10 production (p=0.0004; Kruskal-Wallis test) by CD8+ T cells following co-culture with DC exposed to TAA-NP as compared to that with DC exposed to unencapsulated lysate. IFN-γ production by CD8+ T cells was significantly greater (p=0.028; Mann-Whitney U test) and IL-10 production significantly lesser (p=0.049; Mann-Whitney U test) after stimulation with Ag containing-NP as compared to blank NP, thereby excluding non-specific stimulation by PLGA nanoparticles alone. However there were no significant differences in levels of IL-2, IL-4, IL-6 and TNF-α production following CD8+ stimulation by either of the antigen versions.
Figure 5.

Cytokine analysis after DC/CD8+ T cell co-incubation: each panel represents the results from an individual patient (# 1, 2, 3, 4 and 5). In 3 out of 5 patients both increased IFN-γ production and decreased IL-10 production were observed. In the other 2 patients either of these but not both were seen. Black bar: T cells + DC + blank NP; gray bar: T cells + DC + lysate; open bar: T cells + DC + lysate-NP.
Discussion
PLGA-based micro- or nanoparticles have been successfully used for the controlled release of macromolecular drugs such as proteins, peptides, genes, single Ags, and human growth hormone.22–24 In the only prior study done to evaluate tumor lysate encapsulation within PLGA spheres, we reported findings using tumors from mice.6 There are no prior reports of encapsulation and delivery of an Ag mixture derived from solid organ malignancies in humans. The superior CTL responses associated with delivery in PLGA25 could potentially overcome limitations of current DC-based adoptive immunotherapies.26
The present study provides evidence supporting the use of PLGA for efficient Ag delivery of TAA to DC. In this work, we have optimized the encapsulation protocol, defined parameters for Ag delivery and confirmed biological potency. We believe that this pre-clinical evaluation would satisfy regulatory requirements surrounding the use of novel biological products in humans27,28 and facilitate future testing of PLGA particles in clinical trials.
We optimized encapsulation techniques for lysates using single cell suspensions of continuous murine and human HNSCC cell lines. The aims of these initial studies were to: A) maximize liberation of TAA into the lysate used for encapsulation, B) enhance extent of encapsulation and C) increase release of antigen payload within 7 days, the approximate life span of an immature DC. The cell lines provided unrestricted supplies of tumor Ag for these studies. In later experiments, we tested our refined protocols for encapsulation of fresh surgical specimens from HNSCC patients. The experimental design of these studies with human tissues mimicked vaccination protocols for Phase I/II clinical trials of DC-based immunotherapy.
Total protein entrapment increased with increasing concentration (loading) of lysate (Tables 1 and 2). This pattern of encapsulation was observed in another study wherein efficiencies between 25.0 and 80.0% were recorded for low loading and between 65.8 and 94.5% for high loading.29 Likewise, loading efficiency was seen to increase with rising Fluorescein isothiocyanate-albumin in the internal water phase during double-emulsion fabrication.30 Earlier reports have attributed this to lower percentage of protein lost due to adsorption to the preparation vessel or aerosolization during sonication.29 We defined a threshold concentration leading to optimal encapsulation of a complex mixture of tumor Ags (Table 1). Concentration of the lysate depended on the number of cells per unit volume of PBS; increasing number of cells increased the concentration. In an effort to maximize encapsulation, we dissolved cells from patient samples in as low a volume of PBS as possible. The maximum achievable concentration was 60mg/mL (Table 1) because cells had to be pelleted within a minimum volume of PBS prior to lysis. This minimum volume was the major limitation for protein loading into NP. Nevertheless, improving the extent of encapsulation enabled using just 0.5 mg of NP per million DC during in vitro experiments. This ratio could be used as the dose for primary injection in a pilot protocol of NP-based DC vaccines.
Release was found to increase with Ag (protein) content of the NP at a constant polymer molecular weight (Figure 1). This trend was also observed in previous studies reporting rapid release due to the large concentration gradient between the Ag-rich NP and the outer water phase.31,32 The biphasic release pattern (short burst followed by a period of sustained release) (Figure 1) is potentially useful for delivery of Ag to DC because it provides a continuous supply of Ag complexes on the DC surface for CTL stimulation.33 Unlike most prior studies in which single defined Ags were used, our NPs contained tumor lysate-derived Ags of varied molecular weights. We observed that with increasing lysate concentration, there was a more than equal rise in the initial burst (Figures 1 and 2, A). Further studies are needed to find out whether this could be due to increased encapsulation and release of higher molecular weight proteins when using more concentrated lysates within NP.
Lyophilized tumor lysates were also used for encapsulation. Lyophilization improved encapsulation (Table 3) because a greater amount of protein became available for loading. This strategy overcame the loading constraint observed when employing soluble lysate. This technique has been employed previously although only for entrapment of model single Ags such as BSA.34 Lyophilized lysate-NP demonstrated superior encapsulation (Table 4) and release (Figure 2, B) when compared to conventional soluble version. To increase quantities of Ag released within DC for surface presentation, we used this technique to produce NP when encapsulating patient tumor tissues for DC/T cell co-culture experiments when possible.
Increased immunostimulatory IFN-γ or decreased immunoinhibitory IL-10 was seen in cells from four out of five patients (Figure 5) after PLGA-mediated Ag delivery. Since the polymers contained 5-fold less Ag than their lysate counterparts, polymer-based delivery was more efficient than conventional lystae. Unlike previous work in which tumor cell lines expressing the immuno-dominant peptides have been used for encapsulation and T cell lines tailored for certain specific epitopes used for analysis, we employed whole tumor material from freshly excised surgical specimens and freshly isolated T cells. Because of this approach, we observed variations in cytokine profiles among samples from different patients. NP delivery resulted in greater immunostimulatory IFN-γ and lesser immunoinhibitory IL-10 production in only 3 out of the 5 patients (Figure 5; patients 1–3). In the other two, either of these but not both were observed (Figure 5; patients 4 and 5). Such variations in human T cell responses have been reported earlier. In a clinical trial of gp 100 209-2M, MART1 M26 and tyrosinase 370D peptide-based DC immunotherapy for patients with malignant melanoma, mixed results were obtained despite administering similar vaccination dosages and schedules. In 4 patients CD8+ T-cells showed increased secretion of IFN-γ and TNF-α while in 6 others there was an increase in production of either but not both cytokines resulting in variations in disease-free survival.35 Taken together however, our data indicate that NP encapsulation of whole tumor lysates results in favorable and efficient T cell stimulation when compared to conventional soluble lysate. This could enable overcoming immune resistance often seen in patients with solid organ cancers.36 In turn it could enable better control of disease, reduce chances of recurrence of malignancies and eventually help improve overall survival. Future clinical trials are needed to confirm whether this is true.
In summary, we aimed to optimize parameters surrounding the efficacy of Ag encapsulation within NP made from a US Food and Drug Administration-approved polymer, to be used as part of a DC-based adoptive immunotherapy for HNSCC. The extent of encapsulation of tumor Ag increased with increasing lysate concentration. Lyophilization of cell containing-lysates enabled superior encapsulation and release of a spectrum of tumor Ags. This was the method of choice when generating lysates for NP fabrication when sufficient protein is available. PLGA-mediated Ag delivery was found to be more efficient than conventional soluble lysate in evoking favorable cytolytic CD8+ T cell driven anti-tumor responses in vitro. This strategy holds promise for use in Phase I/II clinical trials of patients with advanced or recurrent solid organ malignancies.
Acknowledgments
This work was partially funded by an intra-mural grant available to the Department of Dermatology, Yale University School of Medicine, a Cancer Research-UK grant and the Virginia Alden Wright Fund, Yale New Haven Hospital. No support came from any commercial association. There is no potential, perceived or real conflict of interest.
List of abbreviations
- Ag
Antigen
- ATCC
American Type Culture Collection
- BCA
Bicinchonic acid
- BSA
Bovine serum albumin
- CBA
Cytometric bead array
- CD
Cluster of differentiation
- CTL
Cytotoxic T cells
- DC
Dendritic cell
- GM-CSF
Granulocyte macrophage-colony stimulating factor
- HNSCC
Head and neck squamous cell carcinoma
- IFN-γ
Interferon-γ
- IL
Interleukin
- LPS
Lipopolysaccharide
- MACS
Magnetic activated cell sorting
- MHC
Major Histocompatibility
- MW
Molecular weight
- NP
Nanoparticles
- PBS
Phosphate buffered saline
- PLGA
Poy (lactic-co-glycolic acid)
- PVA
Polyvinyl alcohol
- RT
Room temperature
- SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SEM
Scanning electron microscopy
- SPSS
Statistical Package for the Social Sciences
- TAA
Tumor associated antigen
- Th
T-helper
- TNF-α
Tumor necrosis factor-α
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Figdor CG, de Vries JM, Lesterhuis WJ, Melief CJM. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;10:475–80. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Ma B, Zhou Y, Zhang M, Qiu X, Sui Y, et al. Dendritic cell fused with allogeneic breast cancer cell line induces tumour antigen-specific CTL responses against autologous breast cancer cells. Breast Cancer Res Treat. 2007;105:277–86. doi: 10.1007/s10549-006-9457-8. [DOI] [PubMed] [Google Scholar]
- 3.Jenne L, Schuler G, Steinkasserer A. Viral vectors for dendritic cell based immunotherapy. Trends Immunol. 2001;22:102–07. doi: 10.1016/s1471-4906(00)01813-5. [DOI] [PubMed] [Google Scholar]
- 4.Jager E, Ringhoffer M, Karbach J, Arand M, Oesch F, Knuth A. Inverse relationship of melanocyte differentiation antigen expression in melanoma tissues and CD8+ cytotoxic- T-cell responses: evidence for immunoselection of antigen-loss variants in vivo. Int J Cancer. 1996;66:470–76. doi: 10.1002/(SICI)1097-0215(19960516)66:4<470::AID-IJC10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 5.Riker A, Cormier J, Panelli M, Kammula U, Wang E, Abati A, et al. Immune selection after antigen-specific immunotherapy of melanoma. Surgery. 1999;126:112–20. [PubMed] [Google Scholar]
- 6.Solbrig CM, Saucier-Sawyer JK, Cody V, Saltzman WM, Hanlon DJ. Polymer nanoparticles for immunotherapy from encapsulated tumor associated antigens and whole tumor cells. Mol Pharm. 2007;4:47–57. doi: 10.1021/mp060107e. [DOI] [PubMed] [Google Scholar]
- 7.Scholl I, Boltz-Nitulescu G, Jensen-Jarolim E. Review of novel particulate antigen delivery systems with special focus on treatment of type I allergy. J Control Rel. 2005;104:1–27. doi: 10.1016/j.jconrel.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 8.Kovacsovics-Bankowski M, Rock KL. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC Class I molecules. Sci. 1995;267:243–46. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- 9.Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;28:2723–730. [PubMed] [Google Scholar]
- 10.Waeckerle-Men Y, Groettrup M. PLGA microspheres for improved antigen delivery to dendritic cells as cellular vaccines. Adv Drug Deliv Rev. 2005;57:475–82. doi: 10.1016/j.addr.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 11.Gupta RK, Rost BE, Relyvelt E, Siber GR. Adjuvant properties of aluminium and calcium compounds. In: Powell MF, Newman MJ, editors. Vaccine design: the subunit and adjuvant approach. New York: Plenum Press; 1995. pp. 229–48. [DOI] [PubMed] [Google Scholar]
- 12.Nakoaka R, Tabata Y, Ikada Y. Adjuvant effect of biodegradable poly (D, L-lactic acid) granules capable for antigen release following intraperitoneal injection. Vaccine. 1996;14:1671–676. doi: 10.1016/s0264-410x(96)00098-9. [DOI] [PubMed] [Google Scholar]
- 13.Peppas LB, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2004;56:1649–659. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 14.Diwan M, Elamanchili P, Lane H, Gainer A, Samuel J. Biodegradable Nanoparticle Mediated Antigen Delivery to Human Cord Blood Derived Dendritic Cells for Induction of Primary T Cell Responses. J Drug Targ. 2003;11:495–507. doi: 10.1080/10611860410001670026. [DOI] [PubMed] [Google Scholar]
- 15.Kwon YJ, Standley SM, Goh SL, Frechet JMJ. Enhanced antigen presentation and immunostimulation of dendritic cells using acid-degradable cationic nanoparticles. J Control Rel. 2005;105:199–212. doi: 10.1016/j.jconrel.2005.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levine MM, Sztein MB. Vaccine development strategies for improving immunization: the role of modern immunology. Nat Immunol. 2004;5:460–64. doi: 10.1038/ni0504-460. [DOI] [PubMed] [Google Scholar]
- 17.Partidos CD, Vohra P, Jones D, Farrar G, Steward MW. CTL responses induced by a single immunization with peptide encapsulated in biodegradable microparticles. J Immunol Methods. 1997;206:143–51. doi: 10.1016/s0022-1759(97)00102-6. [DOI] [PubMed] [Google Scholar]
- 18.Elamanchili P, Lutsiak CME, Hamdy S, Diwan M, Samuel J. “Pathogen-mimicking” nanoparticles for vaccine delivery to dendritic cells. J Immunother. 2007;30:378–95. doi: 10.1097/CJI.0b013e31802cf3e3. [DOI] [PubMed] [Google Scholar]
- 19.Murillo M, Grillo MJ, Rene J, Marin CM, Barberan M, Blasco JM, et al. A Brucella ovis antigenic complex bearing (poly-e-caprolactone) microparticles confer protection against experimental brucellosis in mice. Vaccine. 2001;19:4099–106. doi: 10.1016/s0264-410x(01)00177-3. [DOI] [PubMed] [Google Scholar]
- 20.Ren JM, Zou QM, Wang FK, He Q, Chen W, Zen WK. PELA microspheres loaded H Pylori lysates and their mucosal immune response. World J Gastroenterol. 2002;8:1098–102. doi: 10.3748/wjg.v8.i6.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balz VSK, Gotte K, Bockmuhl U, Petersen I, Bier H. Is the p53 inactivation frequency in squamous cell carcinomas of the head and neck underestimated? Analysis of p53 exons 2–11 and human papilloma virus 16/18 E6 transcripts in 123 unselected tumor specimens. Cancer Res. 2003;63:1188–191. [PubMed] [Google Scholar]
- 22.Carrasquillo KG, Stanley A, Aponte-Carro JC, De Jesus P, Costantino HR, Bosques CJ, et al. Non-aqueous encapsulation of excipient-stabilized spray-freeze dried BSA into poly(lactide-coglycolide) microspheres results in release of native protein. J Control Rel. 2001;76:199–208. doi: 10.1016/s0168-3659(01)00430-8. [DOI] [PubMed] [Google Scholar]
- 23.Ravi Kumar MNV, Bakowsky U, Lehr CM. Preparation and characterization of cationic PLGA nanospheres as DNA carriers. Biomat. 2004;25:1771–777. doi: 10.1016/j.biomaterials.2003.08.069. [DOI] [PubMed] [Google Scholar]
- 24.Bala I, Hariharan S, Ravi Kumar MNV. PLGA nanoparticles in drug delivery: the state of the art. Crit Rev Ther Drug Carr Syst. 2004;21:387–422. doi: 10.1615/critrevtherdrugcarriersyst.v21.i5.20. [DOI] [PubMed] [Google Scholar]
- 25.Johansen P, Gomez JMM, Gander B. Development of synthetic biodegradable microparticulate vaccines: a roller coaster story. Exp Rev Vaccines. 2007;6:471–74. doi: 10.1586/14760584.6.4.471. [DOI] [PubMed] [Google Scholar]
- 26.Nestle FO, Farkas A, Conrad C. Dendritic-cell-based therapeutic vaccination against cancer. Curr Opinion Immunol. 2005;17:163–69. doi: 10.1016/j.coi.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 27.Johansen P, Gander B, Merkle HP, Sesardic D. Ambiguities in the preclinical quality assessment of microparticulate vaccines. Trends Biotechnol. 2000;18:203–11. doi: 10.1016/s0167-7799(00)01437-2. [DOI] [PubMed] [Google Scholar]
- 28.Sesardic D, Dobbelaer R. European Union regulatory developments for new vaccine adjuvants and delivery systems. Vaccine. 2004;22:2452–456. doi: 10.1016/j.vaccine.2003.11.071. [DOI] [PubMed] [Google Scholar]
- 29.Sandor M, Enscore D, Weston P, Mathiowitz E. Effect of protein molecular weight on release from micron-sized PLGA microspheres. J Control Rel. 2001;76:297–311. doi: 10.1016/s0168-3659(01)00446-1. [DOI] [PubMed] [Google Scholar]
- 30.Sun SW, Jeong YI, Jung SW, Kim SH. Characterization of FITC-albumin encapsulated poly (D, Lactide-co-glycolide) microspheres and its release characteristics. J Microencapsul. 2003;20:479–88. doi: 10.1080/0265204031000093014. [DOI] [PubMed] [Google Scholar]
- 31.O'Hagan DT, Jeffery H, Davis SS. The preparation of poly (lactide-co-glycolide) microparticles. III. Microparticle/polymer degradation rates and the in vitro release of a model protein. Int J Pharm. 1994;103:37–45. [Google Scholar]
- 32.Hans ML, Lowman AM. Biodegradable nanoparticles for drug delivery and targeting. Curr Opin Solid State and Mater Sci. 2002;6:319–27. [Google Scholar]
- 33.Audran R, Peter K, Dannull J, Men Y, Scandella E, Groettrup M, et al. Encapsulation of peptides in biodegradable microspheres prolongs their MHC class-I presentation by dendritic cells and macrophages in vitro. Vaccine. 2003;21:1250–255. doi: 10.1016/s0264-410x(02)00521-2. [DOI] [PubMed] [Google Scholar]
- 34.Carrrasquillo KG, Carro JCA, Alejandro A, Toro DD, Griebenow K. Reduction of structural perturbations in bovine serum albumin by non-aqueous microencapsulation. J Pharm Pharmacol. 2001;53:115–20. doi: 10.1211/0022357011775091. [DOI] [PubMed] [Google Scholar]
- 35.Chen DS, Soen Y, Stuge TB, Lee PP, Weber JS, Brown PO, et al. Marked Differences in Human Melanoma Antigen-Specific T Cell Responsiveness after Vaccination Using a Functional Microarray. P LoS Med. 2005;2:e265. doi: 10.1371/journal.pmed.0020265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhardwaj N. Harnessing the immune system to treat cancer. J Clin Invest. 2007;117:1130–136. doi: 10.1172/JCI32136. [DOI] [PMC free article] [PubMed] [Google Scholar]