

Summary
The U1 small nuclear ribonucleoprotein particle (snRNP) is a target of autoreactive B cells and T cells in several rheumatic diseases including systemic lupus erythematosus (SLE) and mixed connective tissue disease (MCTD). We propose that inherent structural properties of this autoantigen complex, including common RNA-binding motifs, B and T-cell epitopes, and a unique stimulatory RNA molecule, underlie its susceptibility as a target of the autoimmune response. Immune mechanisms that may contribute to overall U1-snRNP immunogenicity include epitope spreading through B and T-cell interactions, apoptosis-induced modifications, and Toll-like receptor (TLR) activation through stimulation by U1-snRNA. We conclude that understanding the interactions between U1-snRNP and the immune system will provide insights into why certain patients develop anti-U1-snRNP autoimmunity, and more importantly how to effectively target therapies against this autoimmune response.
Keywords: U1-snRNP, U1-70K, lupus, mixed connective tissue disease, autoimmunity, epitope spreading
Introduction
Chronic inflammatory rheumatic diseases are often characterized by serum autoantibodies directed against ubiquitous nuclear components. RNA-associated molecules are a class of autoantigens that includes small nuclear ribonucleoprotein particles (snRNPs), RNA polymerases, SS-A/Ro, SS-B/La, tRNA synthetases, signal recognition particle (SRP), and others (1). Autoantibodies that recognize snRNPs have been described in a cadre of rheumatic diseases including systemic lupus erythematosus (SLE), mixed connective tissue disease (MCTD), inflammatory myositis, and systemic sclerosis (SS). In fact, snRNPs were originally identified as factors that bound to autoantibodies in SLE and MCTD patient sera (2–4). There has been a major effort to elucidate the role of anti-snRNP autoimmunity in these rheumatic diseases. While we have learned a great deal about RNA-associated autoantigens over the last few decades, it is still not clear whether snRNP complexes initiate or drive autoimmunity or if reactivity to snRNPs is a product of misguided immune processes. Importantly, these questions are not unique to autoantibodies that recognize RNA-containing antigens. Rather the role of antigenic properties in driving autoimmunity is relevant across many self-antigens that are involved in autoimmune disease. For example, in early stages of rheumatoid arthritis (RA), autoantibodies are detected against proteins that are citrullinated, which is a posttranslational modification (5). Increases in citrullinated proteins are observed in inflamed joints in the collagen-induced arthritis (CIA) model for RA and in brain tissue in the experimental autoimmune encephalomyelitis (EAE) model for MS, suggesting that citrullination is involved in these autoimmune responses (6). Understanding how specific examples of autoantigen properties and modifications evoke autoantibody responses is important for informing our understanding of autoantibody development in general.
Here we focus on the autoimmune response against components of the U1-snRNP complex, with an emphasis on the protein U1-70K (also called U1-68K, U1-70, and U1-70 kD). Recently Pomeranz-Krummel et al. (7) determined the X-ray crystal structure of U1-snRNP to 5 Å resolution. This work motivates a fresh look into how structural properties of U1-snRNP contribute to the particles' autoantigenicity. We propose that inherent properties of U1-snRNP in part explain how it becomes a target of autoreactive immune cells: (i) motifs common to more than one protein within the U1-snRNP particle are B and T-cell targets; epitope spreading within the particle and to other RNA-associated antigens with similar motifs expands the autoimmune response; (ii) specific modifications during apoptosis alter structural features of the antigen, creating new epitopes to which the immune system has not been tolerized; and (iii) the RNA component of the U1-snRNP stimulates cells through Toll-like receptors (TLRs), leading to secretion of type I interferons (IFN-I) and autoantibody production.
In this review we explore the structure and modifications of U1-snRNP and the interaction between this splicing complex and the immune system during apoptosis and in cases of disease. Through this analysis, we can begin to understand the mechanisms that underlie autoimmunity to RNA-containing antigens and speculate about how to specifically and effectively design antigen-specific therapies to avoid anti-U1-snRNP pathogenic responses.
Structure and assembly of the U1-snRNP complex
The basic molecular biology and structure of the snRNPs informs our understanding of how the U1-snRNP becomes an antigenic target. This section provides background regarding snRNP structure and function. U1-snRNP is one of five snRNPs that comprise the mammalian spliceosome. The spliceosome is a large macromolecular machine responsible for processing pre-messenger RNA (pre-mRNA), whereby intronic sequences are removed, and protein coding sequences are ligated together to form mature RNA that is ready for translation into proteins (8). The five snRNPs of the spliceosome are termed U1, U2, U4, U5, and U6. Each snRNP consists of a unique small nuclear RNA molecule, specific associated proteins, and seven common core proteins called Smith (Sm) proteins (Sm-B/Sm-B′, Sm-D1, Sm-D2, Sm-D3, Sm-E, Sm-F, and Sm-G), named for the patient whose serum contained antibodies specific for the Sm complex (9). Autoantibodies against Sm and what is referred to in clinical testing as ‘RNP’, which refers to U1-specific proteins and the U1-snRNA, are directed against distinct molecular entities. How the distinction between Sm and RNP was discovered is discussed later in more detail. Briefly, autoantibodies against Sm precipitate all the snRNP RNA molecules, as the Sm proteins are common to all five snRNPs, whereas anti-RNP autoantibodies precipitate only the U1 specific RNA but not the other unique RNA molecules (10).
U1-snRNP is composed of U1-snRNA (also called the U1-RNA), the seven common core Sm proteins, and three U1-specific proteins (U1-70K, U1-A, and U1-C) (9). The crystal structure of the U1-snRNP complex (7), together with previous structural and biochemical data, reveals how the molecules of this complex are assembled.
The U1-RNA molecule is 165 nucleotides in length and forms four stem loops that resemble an asymmetrical X-shape (7, 11). The seven Sm proteins are bound to the Sm binding site on U1-RNA, which is located between stem loops 3 and 4, forming the particle core (7). The X-ray crystal structures of heteromeric Sm proteins (D1-D2) and (B-D3) led to an early model where the Sm proteins form a ring around the central RNA molecule (12). This model was supported by a single particle electron microscopy (EM) structure of the U1-snRNP complex at 10 Å resolution that revealed a ‘doughnut’ shape composed of Sm proteins in a circular arrangement (13). The recent crystal structure of U1-snRNP also supports the ring model, with interactions between the RNA backbone and Sm proteins stabilizing the core (7). The implications of the immunogenicity of this large protein and nucleic acid complex will be discussed in further detail below.
U1-70K is a 437 amino acid polypeptide chain that contains an N-terminal domain of approximately 90 residues, an RNA binding domain roughly between residues 100–180, and a C-terminal domain containing serine/arginine (SR) repeats (14, 15). U1-A is 282 amino acids in length and is comprised of two RNA-recognition motifs and an intervening proline-rich domain (16, 17). Both U1-70K and U1-A contain a conserved 80 amino acid RNA binding motif (RNP-80), and have the capacity to directly bind U1-snRNA as individual molecules (18–20). U1-70K binds to stem loop 1 of U1-RNA, and the N-terminal RNA binding domain of U1-A binds to stem loop 2 (7, 21). The U1-C protein is a 159 amino acid polypeptide chain with an N-terminal zinc finger-like sequence and a C-terminal region rich in methionine and proline residues. Unlike U1-70K and U1-A, U1-C does not contain an RNA recognition motif and cannot bind to U1-RNA alone. Rather, the N-terminal portion of U1-70K is necessary and sufficient for U1-C binding to the core (15).
Early EM studies have suggested that U1-70K is positioned closer to the 5′ end of the U1-RNA molecule within the U1-snRNP complex (22). These observations are consistent with the most recent U1-snRNP structure, where U1-70K wraps around the core domain, with the N-terminal domain located near the 5′ end of U1-RNA. The N-terminal portion of U1-70K together with the C-terminal domain of Sm-D3 creates a binding groove for the U1-C helix (7). The physical associations between U1-70K, U1-A, U1-C, and U1-RNA will be important later in this review for understanding how autoantibody and T-cell recognition spreads between these molecules.
In 1980, Lerner and Steitz (23) hypothesized that U1-RNA recognized pre-mRNA and was involved in splicing, based on sequence similarity between U1-RNA and sequences in splice junctions. They observed that snRNPs were highly conserved from human to insects and were most abundant in metabolically active cells (23). Interestingly, in the crystal structure of the U1-snRNP complex, the 5′ end of U1-RNA from one complex forms base pairs with the 5′ end of U1-RNA in an adjacent complex (7). The authors speculated that this might represent a snapshot of how U1-RNA interacts with the 5′ splice site on pre-mRNA. In addition, one of the helices of U1-C binds across the minor groove of the RNA-RNA interaction between the 5′ ends of adjacent U1-RNA molecules, suggesting a role for U1-C in stabilizing base pairing (7).
How the snRNP complexes assemble to form the spliceosome and catalyze the splicing of pre-mRNA is beyond the scope of this review, and has been extensively reviewed elsewhere (8, 24–26). Briefly, U1-snRNP initiates assembly of the spliceosome onto pre-mRNA through binding to the 5′ pre-mRNA splice site, followed by the ATP-dependent binding of U2-snRNP at the branch site where cleavage occurs (8, 26). The tri-snRNP complex consisting of U4, U5, and U6 is then recruited, followed by removal of U1 and U4 snRNPs (25, 27). The complex rearranges to become catalytically active, and a two-step mechanism excises the intronic sequence as a lariat product and ligates the exon ends to generate a mature mRNA (8). EM studies have been important in understanding the structures of spliceosome intermediates, which are difficult to crystallize due to sample heterogeneity, limited amounts of material, and instability over time (28). Recent reviews have described what has been learned from EM structures of the spliceosome (28, 29).
Another class of splicing factors, the serine/arginine rich (SR) proteins, can interact with the U1-snRNP complex and are found in co-immunoprecipitates from patient sera (30). SR proteins are a highly-conserved family of splicing factors (31) that are involved in spliceosome assembly onto pre-mRNA, and are thought to enhance U1-snRNP binding to the 5′ splice site (8). The N-termini of SR proteins contain one or more RNA recognition motifs characteristic of many RNA binding proteins. The C-terminus contains at least one arginine/serine domain similar to those found in U1-70K (8). SR proteins can interact with U1-70K and U2AF35 of the U2-snRNP complex, possibly acting as a bridge between components bound to the 5′ and 3′ splice sites on pre-mRNA (32). SR proteins have been shown to have distinct abilities to promote U1-snRNP interactions with alternative 5′ splice sites, and are involved in the regulation of alterative splicing (33, 34). SR proteins have themselves been identified as autoantigens in SLE. Their recognition is in part determined by the presence or absence of phosphate groups on serine residues in the SR domains (35).
Detection of U1-snRNP autoantibodies
In 1971, Mattioli et al. (36) described sera from SLE patients that contained antibodies that recognized an RNase- and trypsin- sensitive ‘ribonucleoprotein’ isolated from calf thymus extract in immunodiffusion assays. In 1972, Sharp and colleagues (37) at Stanford prepared extractable nuclear antigens (ENA) from calf thymus and tested for ENA-reactivity in serum from patients with a variety of rheumatic conditions. They developed a hemagglutination assay, which was thought to be more sensitive than previous assays (37). The hemmagglutination assay involved treating sheep red blood cells with tannic acid and passively adsorbing ENA or denatured DNA from purified calf thymus. Addition of serum with ENA reactivity resulted in agglutination of the red blood cells. In this seminal paper, reactivity to ENA was detected in 38 out of 71 (53.5%) SLE patients, and in all MCTD patients 25/25 (100%) (37). The authors also noted that RNase abolished ENA reactivity in MCTD serum, while trypsin only partially reduced reactivity (37). The subset of SLE sera that exhibited ENA reactivity did not recognize an RNase- or DNase-sensitive component, but the reactivity was modestly sensitive to trypsin (37). Sharp and colleagues (37) observed a speckled anti-nuclear antibody pattern when testing serum samples from all MCTD patients that was also RNase-sensitive. These experiments identified an RNase-sensitive antigen recognized by serum from patients with a newly defined clinical entity, which they termed MCTD. The RNase-sensitive component of ENA was later defined as ribonuleoprotein (RNP or nRNP), and the RNase-resistant component was defined as the Sm antigen (38).
These early studies were based on immunodiffusion, immunofluorescence, and hemagglutination assays (Fig. 1-A,B,C). One obvious limitation of these approaches is that they analyze reactivity to a large complex rather than specific polypeptides. Autoantibodies directed against many other antigens can produce a speckled nuclear staining pattern (39). Although hemagglutination and immunodiffusion have generally been surpassed by more sensitive and specific assays, immunofluorescence patterns are used both in research and clinically to aid in the initial differential diagnosis of various rheumatic conditions (9).
Fig. 1. Immunoassays for detecting autoantibodies against U1-snRNP.
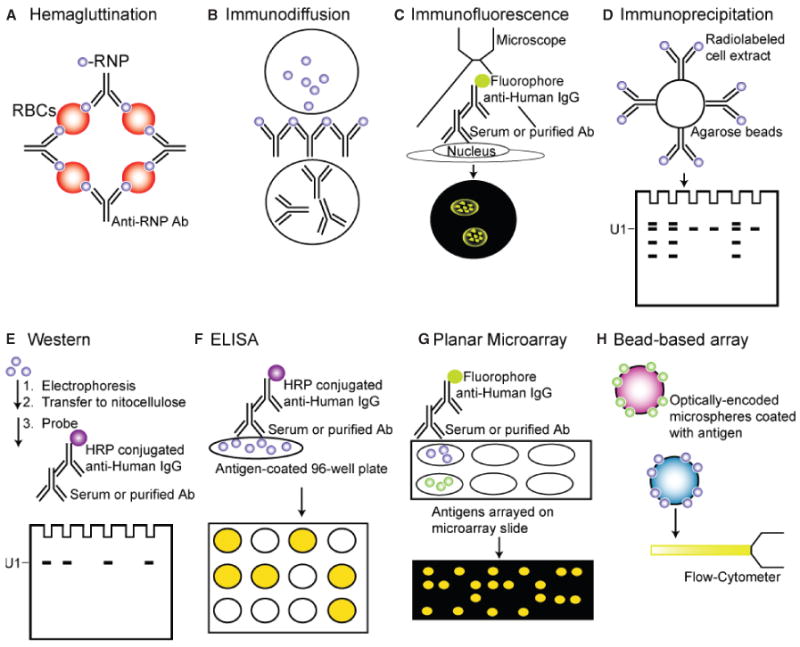
(A) Hemagluttination, (B) The Immunodiffusion assay, (C) Immunofluorescence, (D) Immunoprecipitation, (E) Western blot, (F) ELISA, (G) Planar microarrays, (H) Bead-based arrays.
ENA reactivity was further characterized by immunoprecipitation of nuclear extracts from 32P and 35S-labeled cells to reveal the distinct reactivities attributable to anti-Sm and anti-RNP autoantibodies (10) (Fig. 1D). Based on its reactivity to the U1-snRNP complex, the anti-RNP serum became known as anti-U1-RNP, anti-nuclear RNP (anti-nRNP), or small nuclear ribonucleoprotein (anti-U1-snRNP) (40, 41). Several years later, Pettersson and colleagues (41) used immunoprecipitation and western blotting to demonstrate that three proteins, U1-70K, U1-A, and U1-C, contained the antigenic determinants of the U1-RNP. The anti-Sm patient sera and a mouse monoclonal anti-Sm antibody reacted with proteins Sm-B/B′, Sm-D, and to a lesser extent Sm-E in the Smith complex (41). These fundamental studies laid the foundation for future studies aimed at defining specific reactivity to individual snRNP antigens.
Early studies using enzyme-linked immunosorbent assays (ELISA) to detect antibodies to affinity-purified RNP, Sm, Ro (SSA), and La (SSB) predictably demonstrated increased sensitivity compared to previous methods (42, 43). Subsequent studies demonstrated that recombinantly expressed antigens could also be used for ELISAs (44). Commercial sources of antigens currently market both purified and recombinant protein preparations as well as synthetic peptides. Regardless of the technique used, assays for anti-RNP have demonstrated sensitivity ranging from 71% to 100% for the diagnosis of MCTD, depending on the criteria used for diagnosis (45). Many clinical laboratories currently use either ELISA or western blot (Fig. 1E,F), and this will likely be the trend for some time (9).
Anti-U1-snRNP autoantibodies in disease
The prevalence of anti-RNP autoantibodies varies depending on the disease. Anti-RNP autoantibodies are detected in 30–40% of SLE patients and in nearly all MCTD patients, as anti-RNP reactivity is a criteria for the diagnosis of MCTD (9, 40, 46). Antibodies specifically directed against U1-70K are found in 75–90% of MCTD patients and represent the most commonly detected U1-snRNP component; anti-U1-70K antibodies are found in only 20–50% of SLE patients whose serum is positive for anti-RNP (9, 47–49). Antibodies against the RNA component of U1-snRNP are found in 38% of anti-RNP positive patient sera (9, 50). In one study, anti-U1-A antibodies were detected in greater than 90% of anti-U1-snRNP sera from patients with rheumatic disease (50). Overall, clinical testing for anti-RNP has been determined to be a good predictor of MCTD but not of SLE (45).
MCTD overlaps clinically with SLE, SS, inflammatory myopathy, and rheumatoid arthritis (RA). A detailed description of the pathogenesis of MCTD can be found in recent reviews (39, 40). Briefly, clinical features of MCTD include Raynaud's phenomenon, arthritis, acrosclerosis, hand edema, mild myositis, and pulmonary involvement, with the primary cause of disease-related death resulting from pulmonary hypertension (39, 40, 51). In the first description of MCTD, patients lacked serious renal or central nervous system (CNS) involvement but demonstrated inflammatory muscle disease, symptoms associated with SS, and responsiveness to corticosteroids (37).
In patients with SS, the frequency of anti-RNP ranges from 2 to 14%, most often associated with overlapping connective tissue disease syndromes and a favorable response to corticosteroids (52). SS patients with anti-RNP antibodies usually exhibit Raynaud's phenomenon (53).
Anti-RNP autoantibodies are also found in patients with inflammatory myositis, a condition where skeletal muscle is damaged due to inflammation (54). In a study comparing SLE patients with biopsy-proven myositis to SLE patients without myositis, a significant difference in the percentage of anti-RNP antibodies (80% and 21%, respectively) was observed (55). Of the SLE/myositis patients, clinical features included Raynaud's phenomenon at the initial presentation (100%), interstitial lung disease (ILD) (100%), and erosive joint disease (60%).
It has been reported that myositis occurs in MCTD with varying frequencies ranging between 20 and 70% (56). When patients with myositis and anti-RNP antibodies were compared to patients without anti-RNP, the anti-RNP positive group had a more favorable outcome, with symptoms of myositis subsiding with corticosteroid treatment (56). While this particular cohort of patients with anti-RNP and myositis met diagnostic criteria for MCTD (56), in another small series of five patients that had anti-RNP and inflammatory myositis, no patient met the MCTD diagnostic criteria (54). None of the five patients exhibited Raynaud's phenomenon, three had destructive arthritis involving distal joints, and three had pulmonary manifestations (54). As this is a very small series of patients, it is difficult to draw a conclusion about the relationship of anti-RNP, myositis, and MCTD. Nevertheless, these studies highlight the conflicting evidence for associations between autoantibodies and disease diagnoses, signs, and symptoms, and the need to better understand the clinical significance of anti-RNP autoantibodies.
One approach to explore the potential relationship between anti-U1-snRNP antibodies and disease activity has been to evaluate patient's samples over extended periods of time. In a longitudinal study of 47 MCTD patients followed over a period of 3–29 years (mean duration of followup was 15 years), all patients had autoantibodies against RNP and U1-70K. Of the patient's sera, 81%, 48%, 79%, and 14% recognized U1-A, U1-C, Sm- B/B′, and Sm-D, respectively during periods of active disease. In periods of remission, there was a reduction in autoantibodies against U1-70, U1-A, Sm-B/B′, and U1-C (51). Antibodies against U1-70K, which were once detectable by ELISA, were no longer measurable during periods of long-term remission or improvement (51). This study supports a correlation between the presence of anti-U1 autoantibodies and disease activity.
In a longitudinal study evaluating SLE serum samples for autoantibody reactivity prior to clinical presentation of disease up through presentation, anti-RNP autoantibodies were first detected close to the time of clinical disease onset (57). Anti-snRNP autoantibodies were detected on average 1.2 years before diagnosis (compared to 3.4 years for autoantibodies against Ro or La), in a phase the authors term ‘pathogenic autoimmunity’ (57). How the presence of these autoantibodies contributes to pathogenesis in connective tissue diseases is still poorly understood. Are anti-RNP antibodies themselves pathogenic? What clinical symptoms develop because of the anti-RNP response, and what disease processes are potentially causing anti-RNP autoimmunity? While we do not directly address these questions here, as this is an extensive topic and longstanding discussion in the field, some studies suggest that clinical features can influence the autoantigenicity of U1-snRNP, which is relevant to our discussion.
A correlation between anti-RNP reactivity and the presence of Raynaud's phenomenon has been reported (58, 59). Sera from patients with Raynaud's phenomenon recognize oxidative fragments of U1-70K (60). This observation led to the hypothesis that Raynaud's phenomenon results in ischemia-reperfusion with production of reactive oxygen species that modify U1-70K (60). In another study, a link between anti-RNP autoantibodies and lung injury was investigated in a murine model. Mice normally resistant to lung damage were injected with anti-RNP-containing serum, and subjected to mesenteric ischemia/reperfusion-induced injury. Mice exposed to anti-RNP sera exhibited a dose-dependent increase in lung damage compared to controls (61). These data begin to describe how clinical events might induce alterations in autoantigen immunogenecity.
Autoantibody and T-cell epitopes
B-cell epitopes
A number of studies have mapped precisely which epitopes within the U1-snRNP particle, specifically within U1-70K, are recognized by autoantibodies (Table 1). The crystal structure of the U1-snRNP complex reveals the structural elements of U1-70K that interact with U1-RNA (7). Not surprisingly, a large portion of U1-70K is accessible to the immune system and maps to linear domains where many autoantibody reactivities are found (Fig. 2A,C,D).
Table 1. U1-70K autoantibody epitopes.
| Sequence | Species | Disease, % Reactivity, (N) | Reference |
|---|---|---|---|
| 52–77 | Human | MCTD, 53%, (17); SLE, 63%, (16); Scleroderma, 0% (6) | (64) |
| 63–84 | Human | RNP + SLE, 100%, (3) | (160) |
| 94–194 | Human/mouse | RNP+, 90%, (113)/MRL/lpr, 85% (7) | (161) |
| 100–156 | Human | MCTD, 94% (17); SLE, 81%, (16); Scleroderma, 83%, (6) | (64) |
| 119–126 | Human | RNP+, (25) | (162) |
| 124–372 | Human | RNP+ (SLE/MCTD), 100%, (17) | (63) |
| 131–151 | Human/mouse | SLE, 66%, (3)/MRL/lpr | (67) |
| 143–195 | Human | RNP+ CTD, 93%, (16) | (66) |
| 195–259 | Human/mouse | RNP+, 0.1%, (113)/MRL/lpr, 0.12%, (8) | (161) |
| 196–259 | Human | MCTD, 24%, (17); SLE, 94%, (16); Scleroderma, 50%, (6) | (64) |
| 217–234 | Human | RNP+ SLE, 66%, (3) | (160) |
| 234–253 | Human | RNP+, 10%, (180) | (65) |
| 276–297 | Human | RNP+ (SLE/MCTD), 90%, (17) | (63) |
| 276–371 | Human | RNP+, 100%, (180) | (65) |
| 282–296 | Human | RNP+ SLE, 90%, (10) | (163) |
| 386–437 | Human | MCTD, 35%, (17); SLE, 74%, (16); Scleroderma, 33%, (6) | (64) |
MCTD, mixed connective tissue disease; SLE, systemic lupus erythematosus; RNP, ribonuleoprotein.
Fig. 2. Modifications and autoimmune epitopes on U1-70K.
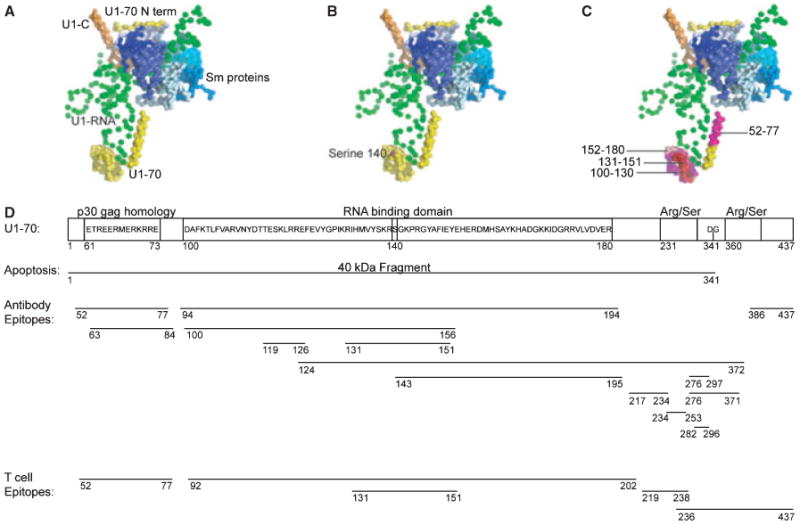
(A) The U1-snRNP crystal structure (PDB ID: 3CW1) (7) was visualized using Pymol. In this depiction of the structure, U1-RNA is depicted in green, U1-70K in yellow, U1-C in orange, and the Sm proteins are displayed in blues. U1-A is not included in this depiction, as it was not part of the crystal complex in this structure. (B) The phosphorylation of serine residue 140 is a modification that occurs during apoptosis, and is shown in magenta. (C) The domain (100–180) of U1-70K is highlighted and is the RNA-binding domain of the protein. Notably the B-cell and T-cell epitope 131–151 is shown in red. The residues around this epitope are highlighted: 100–130 in magenta, and 152–180 in light pink. These regions contain additional B-cell and T-cell epitopes. (D) The domains of U1-70K include an N-terminal domain, RNA-binding domain, and serine/arginine repeats. During apoptosis, a 40 kDa fragment is cleaved at residue 341. B-cell and T-cell epitopes on U1-70K are shown.
The first identification of a B-cell epitope within the U1-70K protein was by Query and Keene in 1987 (62). Autoantibodies from SLE patients recognized a core consensus sequence ETPEEREERRR in the N-terminal domain of U1-70K (Fig. 2D). The sequence was also found in the p30gag mammalian retrovirus, introducing the idea that molecular mimicry may be a mechanism underlying autoreactivity to U1-70K (62). Another group reported that serum autoantibodies derived from SLE patients recognized an epitope located between amino acids 124–372 from U1-70K, with further studies narrowing the region to residues 276–297 (63). In this study, patients displayed a polyclonal response to multiple epitopes of U1-70K (63). In another study, epitopes from U1-70K were identified between residues 52–77, 100–156, and 196–259 in patients with SLE, MCTD, and SS (64). Heterogeneity was observed between individual patients when comparing reactivity to each of these linear epitopes (64). Additional epitopes have been defined between residues 99–167 (65), and 135–194 (66); these epitopes are located in the portion of U1-70K that interacts with U1-RNA (Fig. 2D). An overlapping peptide screen in the MRL/lpr mouse revealed that epitope 131–151 of U1-70K is immunogenic for both B cells and T cells (67). This epitope is within other reported epitopes and is also centrally located within the RNA binding domain of U1-70K (Fig. 2C,D).
Although many autoantibody epitopes within U1-70K have been reported, it is not surprising that epitopes within the RNA-binding domain consistently appear across studies (68). Similarly, autoantibody epitopes identified within the U1-A protein are contained within the RNA binding domains, suggesting that intramolecular epitope spreading may occur between U1-70K and U1-A (68, 69). The RNP consensus sequence found in U1-70K is also found in U1-A between residues 52–59. In a study that mapped U1-A epitopes using overlapping peptides, a peptide within the RNA binding domain, 35–58, was recognized by 94% of MCTD patient sera and by 19% of SLE sera (69). Other shared epitopes between U1-70K and U1-A have been reported and include a region of U1-A localized between residues 103–108, and in U1-70K between residues 68–72. In one study, 10 of 13 SLE patient sera were bound both of these epitopes (70). It is possible that patient sera contain autoantibodies that are cross-reactive to epitopes on U1-70 and U1-A, or that autoantibodies to RNP epitopes result from epitope spreading and T-cell help.
Autoantibody epitopes have been described for the protein U1-C. Major regions identified include epitopes at the N-terminus of the protein from amino acids 5–48 and at the C-terminus between residues 97–133 (71). In a study that isolated monoclonal autoantibody fragments against U1-C from either semi-synthetic or patient-derived antibody libraries, both U1-C specific and crossreactive antibodies were identified (72). Four of the U1-C specific antibodies recognized the N-terminal region between residues 30–63. Two crossreactive and one specific antibody bound to the C-terminal portion of U1-C from 98 to 126, where the crossreactive antibodies also recognized the proline-rich region of U1-A from residues 187–204 (72).
T-cell epitopes
T-cell epitopes have been defined within the U1-snRNP complex using overlapping peptide screens (73), recombinant portions of individual RNP polypeptides, and elution of peptides from MHC molecules (74) (Table 2, Fig. 2D). One approach has been to measure T-cell proliferation by thymidine incorporation following in vitro stimulation with peptides or portions of candidate antigens. The first U1-snRNP-reactive T cells were described in peripheral blood mononuclear cells from patients with SLE and MCTD. T cells proliferated in response to the C-terminal portion of U1-70K containing amino acid residues 336–437 (75). In MRL/lpr mice, the epitope between amino acids 131–151 from U1-70K was sufficient to induce CD4+ T-cell proliferation in vitro and B-cell IgG production in an APC-dependent manner (67). Further, administration of the peptide 131–151 in vivo induced epitope spreading within U1-70K to new epitopes, including residues 57–77 and 219–238. These epitopes had the capacity to stimulate T-cell proliferation in vitro (76). Likewise, T-cell reactivity to peptide 131–151 was tested in (NZB × NZW)F1 mice by administering the peptide in vivo. (NZB × NZW)F1 mice do not typically produce autoantibodies against the U1-snRNP. CD4+ T cells isolated from treated mice proliferated in response to in vitro stimulation with the peptide 131–151 (77). In addition, autoantibodies directed against U1-70K were detected at later stages of disease in these mice to at least 25 weeks of age (77).
Table 2. U1-70K T-cell epitopes.
| Sequence | Species | Disease, assay | Reference |
|---|---|---|---|
| 57–77 | Mouse | MRL/lpr, proliferation following immunization with U1-70K 131–151 | (76) |
| 92–202 | Human | CTD, T-cell clones | (82) |
| 131–151 | Mouse/human | MRL/lpr, NZB × NZW, proliferation/SLE patients, proliferation | (67, 77, 146) |
| 131–151 (P140)* | Mouse/human | MRL/lpr, proliferation/SLE patients, proliferation | (144, 146) |
| 219–238 | Mouse | MRL/lpr, proliferation following immunization with U1-70K 131–151 | (76) |
| 336–437 | Human | SLE/MCTD, proliferation | (75) |
131–151 (P140) is a phosphorylation modification to serine residue 140 on U1-70K.
SLE, systemic lupus erythematosus; MCTD, mixed connective tissue disease.
Another strategy to define T-cell epitopes has been to generate T-cell clones from human patients. This approach has proven useful for analyzing TCR α and β chain usage. In general, conserved TCR gene usage and common motifs provide evidence of antigen-driven T-cell autoimmunity. For example, in Type 1 Diabetes (T1D), over half of the T-cell clones expanded from pancreatic draining lymph nodes from patients were found to express the same Vβ chain (78). In comparison, very few T-cell clones from controls utilized the same V genes, suggesting that clonal expansion of T cells had occurred in the T1D patients (78). In patients with connective tissue diseases (CTD), U1-70K specific T-cell clones have been isolated and displayed limited Vβ gene usage (79). In other studies, T-cell clones recognizing U1-70K were isolated and found to utilize a range of TCRVα and Vβ genes; however, common motifs were found in the CDR3 of the Vα chain, suggesting selection of T cells by a common epitope (80). Across another panel of U1-70K specific T-cell clones, a significant double-glycine motif emerged in the J region of the α chain in 23 out of 32 clones (81). The authors hypothesized that this particular sequence may be important for flexibility in binding the antigen epitope (81).
T-cell clones have also been useful for defining T-cell epitopes, cytokine production, and the frequency of autoreactive T cells. T-cell clones generated from CTD patients recognized peptides within the RNA binding domain of U1-70K located between residues 92–202 (82). Many of these T-cell clones showed restriction to HLA-DRB1*0401 based on blocking experiments with monoclonal antibodies against the HLA framework (82). T-cell clones against U1-70K that were isolated from CTD patients produced interferon γ (IFNγ), interleukin-2 (IL-2), and variable amounts of IL-4 and IL-10 (83).
Autoreactive T cells that recognize peptides from U1-A have been detected in MRL/lpr mice. T cells proliferated in response to bone marrow-derived dendritic cells pulsed with a peptide from residues 31–50 of U1-A. Interestingly, the main responding T cells were negative for surface expression of CD4 and CD8 (84); these double negative T cells are characteristic of the lymphoproliferation observed in MRL/lpr mice (85). In another study, bone marrow-derived dendritic cells pulsed with U1-A were administered in vivo to non-autoimmune mice, resulting in activated autoreactive T cells and anti-U1-A autoantibodies (86). In humans, T cells isolated from MCTD patients that recognized U1-A were found at an estimated frequency of 1 in 4065 to 1 in 23 256 PBMCs (87). In this study, all 12 MCTD patients, and 5 out of 6 SLE patients, all of whom were positive for anti-RNP, responded to recombinant U1-A in T cell proliferation assays (87).
Based on these studies, T-cell reactivity is consistently observed against the RNA-binding domain of U1-70K and U1-A (67, 77, 82, 84, 88). This RNA-binding region in both molecules is also a critical target for autoantibody reactivity, suggesting it may promote epitope spreading within the T-cell and B-cell compartments (89).
Mechanisms of U1-snRNP immunogenicity: epitope spreading
It is well known that T cells provide help to antigen-specific B cells through a cognate interaction, whereby B cells process whole antigen and present fragments of the antigen to T cells in an MHC-restricted manner (90). This cognate interaction plays an important role in how autoantibodies are generated and how T-cell tolerance is broken to certain antigens. Such a mechanism was demonstrated in vivo for U1-snRNP. B cells were obtained from mice immunized with an snRNP cross-reactive antigen and transferred into naïve mice boosted with native snRNP. These B cells induced autoreactive T cell activation in the naive hosts (91). In MRL/lpr mice deficient for CD40L, there is no detectable anti-RNP response (92), providing further evidence that anti-RNP autoimmunity is T-cell dependent. It was recently shown that CD4+ T cells are sufficient to induce anti-RNP autoimmunity and tissue damage in a disease model of MCTD, in part depending upon the APCs co-transferred with the T cells (93).
The importance of T-cell help is also supported in the human SLE literature. T-cell help through secreted factors was required for autoantibody production against heterogenous nuclear RNP (hnRNP) in in vitro co-cultures of autologous B and T cells (94). In another study, autoreactive T-cell clones derived from SLE patients responded to autologous B cells in an HLA class II-restricted manner and provided help to produce both antigen-specific antibodies and to support polyclonal B-cell activation through the production of IL-6 (95).
Evidence exists supporting the concept that B-cell or T-cell reactivity to a single epitope of an autoantigen can result, through multiple mechanisms, in recognition of many epitopes (68). The spread of reactivity can occur within a single antigen to multiple epitopes on the same protein (intramolecular spreading). Reactivity initially against one polypeptide can also extend to other polypeptides or proteins within a greater macromolecular complex (intermolecular epitope spreading) (68). Epitope spreading, both within a protein and within a larger complex, are observed in the anti-U1-snRNP response (68, 96). Why would the U1-snRNP particle be particularly susceptible to mechanisms of epitope spreading? We have seen that both B-cell and T-cell epitopes exist within the U1-snRNP particle. In addition, the molecules that comprise the U1-snRNP are physically attached, and common motifs, like RNA-binding sequences, are found in multiple proteins within the particle. These intrinsic features of the U1-snRNP help to explain why spreading of autoantibody reactivity to multiple epitopes within the particle is observed.
How the spread of reactivity occurs is the topic of a number of reviews on epitope spreading in autoimmunity (68, 96–98). Some mechanisms thought to be involved in the spread of reactivity include molecular mimicry, modified forms of antigens, cross-reactive B-cell epitopes, immune complexes, and ‘intrastructural’ T-cell help (98). In this section, we review the intrastructural T-cell help hypothesis, which requires that molecules within a complex be physically attached, and we discuss how sequence homology between proteins within U1-snRNP could lead to the spread of autoreactivity.
In the intrastructural T-cell help hypothesis, B cells process and present peptides to T cells, where the peptides are derived from an antigen complex with components that are physically linked. B cells reactive to one portion of the complex receive help from T cells that recognize another component. Evidence for intrastructural spreading was demonstrated in mice immunized with human U1-A and intact snRNP particles. Immunized mice developed antibodies against U1-A, U1-70K, and Sm-B/B′, suggesting that initial reactivities against U1-A spread to other proteins in the snRNP particle. When snRNP particles were disrupted by digestion with ribonuclease and administered with human U1-A, antibody reactivities were only observed against U1-A, and not against U1-70 or Sm-B/B′ (99). In another study, immunization with the various proteins from the U1-snRNP complex yielded distinct patterns of autoantibody reactivity. Immunization with U1-A resulted in autoantibodies against U1-A, U1-70, Sm-B, but not to Sm-D. Immunization with Sm-D generated autoantibodies specific for U1-A and Sm-B (100). Taken together, these data indicate that the spead of reactivity between proteins physically linked in the U1-snRNP is not a random process.
The order in which autoantibodies against U1-snRNP appear has been studied in mice and humans and was different between species, In MRL/lpr mice the first anti-U1-snRNP autoantibodies were against U1-A, followed by U1-70, Sm-B/B′, and Sm-D (101). Anti-RNP antibodies were of the IgG2a isotype, indirectly implicating T-helper cells in their generation (101). In contrast, in a cohort of over 3000 SLE patients, the first IgG autoantibodies to appear were directed against U1-70K and Sm-B/B′, followed by U1-A, U1-C, and Sm-D1 (102). The difference in order of anti-U1-snRNP reactivities in mice and humans suggests there may be species-specific mechanisms involved that affect autoantigen immunogenicity and epitope spreading.
Another way in which orderly epitope spreading could occur within the U1-snRNP complex is due to sequence homology. The RNA binding motif (RNP1) is thought to be central for autoreactivity against U1-snRNP (68). RNP1 motifs are found in U1-70K, U1-A, and in other RNA-containing antigens, including the U2-snRNP specific protein B″, and the 60 kDa SS-B (La) antigen (69). In fact, U1-A contains 77% and 86% sequence homology with U2-snRNP B″ between U1-A residues 4–98 and B″ 7–101, and between U1-A 206–283 and B″147–225, respectively (69). Reactivity initially directed against the RNA-binding domain of U1-70K for example, could spread to additional epitopes on U1-70K, and to other proteins that have the RNP1 motif within the U1-sRNP particle, or to other polypeptides that contain and RNP1 domain (68). Antigen-presenting cells (APCs) that present epitopes containing the RNP1 motif to T cells would in turn activate B cells to produce antibodies against the RNP1 domain (Fig. 3A,B). As described above in the intrastructural T-cell help hypothesis, B cells that recognize one component of the particle may receive help from T cells that are specific for another epitope (98) (Fig. 3C). It is also possible that B cells internalize the antigen complex through the B-cell receptor (BCR) and process and present new epitopes to T cells in an MHC-restricted manner (68) (Fig. 3D). Monneaux and Muller (68) proposed that T cells specific for even one epitope within the RNP1 motif could initiate epitope spreading and would be considered ‘driver T-cell clones’. Such a T cell would express a high affinity T-cell receptor (TCR) and produce pro-inflammatory products upon activation (68).
Fig. 3. Epitope spreading within the U1-snRNP complex.
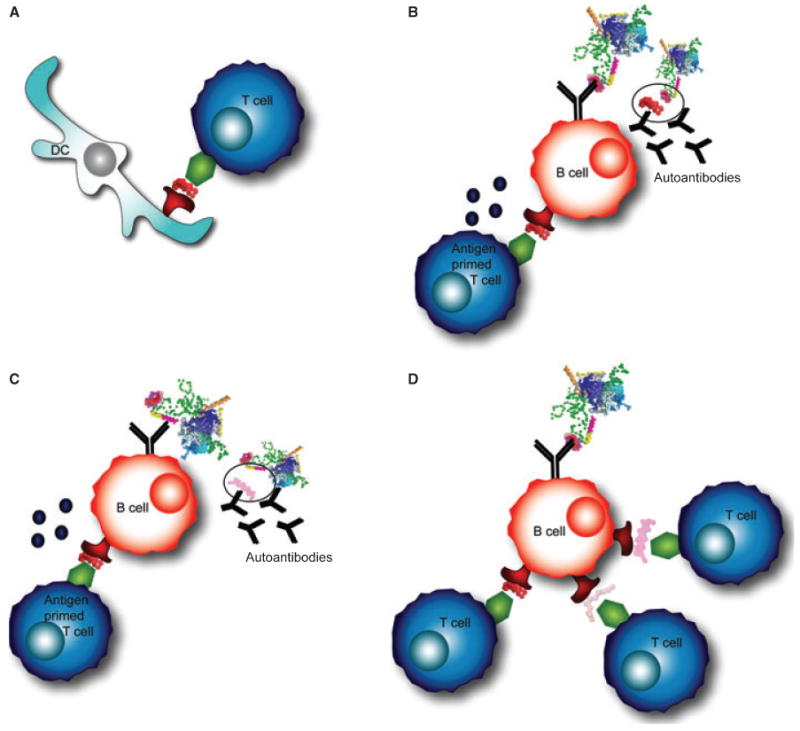
(A) Professional antigen-presenting cells, such as dendritic cells (DCs), prime antigen-specific T cells. DCs process the antigen complex and present peptides in MHC class II molecules on their surface. (B) The B-cell receptor (BCR) on a B cell recognizes the same complex, processes the antigen, and presents peptides to T cells. An antigen-primed T cell provides help to a cognate B cell, allowing the B cell to secrete antibodies specific for the same epitope as the BCR. (C) A B cell recognizing one portion of the antigen complex may present a peptide to T cells from another portion of the complex and receive T-cell help. This interaction results in autoantibody production to an epitope on the complex that is different from the T cell epitope. (D) A B cell that recognizes one portion of the complex could internalize, process, and present peptides to T cells that recognize epitopes from other portions of the U1-snRNP particle.
Apoptosis-induced modifications
It is thought that proteins modified during apoptosis can be presented to the immune system in ways that bypass tolerance to self-proteins (103, 104). Our laboratory recently reviewed the possible sources of autoantigens and the mechanisms leading to their recognition by the immune system in autoimmune diseases (105). Briefly, the generation of new forms of autoantigens can occur through cleavage by caspases or granzymes, interferon-inducible expression of untolerized forms of self-antigens, alternative mRNA splicing, and phosphorylation or oxidative events, among others (105).
The U1-snRNP complex may become immunogenic through a number of mechanisms, several of which may be intrinsic to the structure of the particle itself. Alterations to U1-snRNP during apoptosis are one such mechanism. As described below, modifications to U1-snRNP are in some cases unique to this particle (Table 3).
Table 3. Modifications to U1-70K during apoptosis.
| Modification | Description | Reference |
|---|---|---|
| Location | UV-induced localization to plasma membrane | (106, 107) |
| Phosphorylation | Serine 140 on U1-70K | (107) |
| Phosphorylation | U1 associated SR proteins | (108) |
| Cleavage | Caspase-3 mediated cleavage resulting in 40 kDa fragment | (109, 111) |
| New complex formation | UV-induced RNA/U1-70K complexes | (112) |
A seminal experiment by Casciola-Rosen and colleagues (106) demonstrated that in the setting of UV-induced apoptosis, autoantigens cluster at the surface of the cell membrane in distinct apoptotic blebs containing snRNPs. This work revealed that the relocalization from the nucleus to the cell membrane during apoptosis could make autoantigens more available for recognition by B cells, T cells, and APCs that engulf and present apoptotic material (103). Cells stained with antibodies directed against a phosphorylated form of U1-70K showed localization of the modified version of U1-70K in large clusters of apoptotic bodies at the membranes (107), demonstrating that modified forms of U1-70K relocalize during apoptosis.
While relocalization is not unique to U1-70K, protein-specific modifications of U1-70K occur during apoptosis. The structure of the U1-snRNP reveals that U1-70K wraps around the core of the particle and contains a portion that protrudes outward where it binds to U1-RNA (7). Phosphorylation of U1-70K occurs at serine residue 140 and is an early apoptotic event (107). Serine 140 is located within the RNA binding domain of U1-70K and is thought to be central to the autoimmune response in SLE and MCTD (107) (Fig. 2B,D). Other phosphorylation events associated with the U1-snRNP have been reported. Our group tested a panel of sera from patients with SLE for the ability to precipitate phosphoproteins from apoptotic Jurkat T-cell lysates. Sera known to react with U1-snRNP precipitated a phosphoprotein complex (pp54, pp42, pp34, and pp32) from cells undergoing apoptosis (30). These proteins subsequently were identified as serine/arginine-rich splicing factors (SR proteins) that are phosphorylated during apoptosis, associate with the U1-snRNP, and are commonly recognized by patient sera (108).
In addition to phosphorylation events, U1-70K is specifically cleaved in apoptotic cells by caspase-3 between residues Asp341 and Gly342 (109, 110). This results in the generation of a 40 kDa fragment of U1-70K that remains associated with the U1-snRNP complex (109, 111) (Fig. 2D). Proteolysis of U1-70K is concordant with the appearance of cells that have apoptotic morphology (109). Another specific alteration of U1-70K was demonstrated when keratinocytes were subjected to ultraviolet irradiation, resulting in the formation of covalently-associated complexes of U1-RNA and U1-70K that were recognized by autoantibodies from patient sera (112). In SLE patients, photosensitivity is a diagnostic criterion, and UV exposure can initiate and exacerbate skin lesions (113). The dose of UV radiation used to induce the covalent U1-70K/U1-RNA species was physiologic, suggesting that these new complexes may occur in vivo in response to normal UV exposure. In addition, the U1-RNA molecule undergoes a specific modification during apoptosis that removes the first 5–6 nucleotides (111). No obvious apoptosis-specific modifications to U1-A or U1-C have been described (111).
Autoantibodies in patient sera have been shown to recognize apoptotically modified forms of U1-70K. Recombinant single chain autoantibody fragments were generated from patient-derived phage display antibody libraries and preferentially bound to the 40 kDa apoptotic cleavage product of U1-70K (114). In another study, sera were tested against intact versus apoptotic forms of U1-70K in the presence of competing full-length protein. Fifteen of 29 patient sera recognized the apoptotic form, indicating that the apoptotic form of U1-70K is a distinct target of the autoimmune response (115). In a study of 53 MCTD patients, serum from 54% of patients had autoantibodies that preferentially targeted the apoptotic form of U1-70K over the intact form, in particular during early stages of the disease (116). It is unclear yet whether recognition of apoptotic forms of U1-70K have implications for diagnosis, prognosis, or response to therapy.
Given that cellular mechanisms during apoptosis can generate immunogenic autoantigens, it is not surprising that abnormalities in apoptosis have been observed in SLE (103, 117). The percentage of apoptotic cells from 34 SLE patients compared to RA patients and controls was found to be higher directly ex vivo, and in vitro lymphocytes underwent apoptosis at a faster rate (118). Another study demonstrated a significant increase in the percentage of early apoptotic cells in peripheral blood of SLE patients compared to normal healthy controls (13.68% and 5.62%, respectively) (119). Autoantibody reactivity to modified forms of U1-70K has been shown to correlate with clinical disease manifestations. Serum autoantibodies from patients with lupus skin disease recognized apoptotically modified U1-70K to a greater extent than sera from patients without skin disease; sera from patients with Raynaud's phenomenon contained autoantibodies that recognized U1-70K modified by oxidative stress (60).
In summary, apoptotic mechanisms can modify autoantigens in ways that make them recognizable to autoreactive adaptive immune cells.
U1-RNA as a TLR ligand
The inflammatory immune response in SLE and other CTDs is in part mediated by TLRs. The role of TLRs in autoimmune disease has been reviewed in great detail elsewhere (120, 121). Importantly, TLR9 and TLR7 interact directly with DNA and RNA molecules, respectively (122, 123). TLRs also interact in specific ways with the nucleotide content of DNA- and RNA-containing autoantigens. In lupus-prone mice, the production of anti-dsDNA and anti-chromatin autoantibodies requires TLR9 and engagement through the BCR (124, 125). This ‘two receptor paradigm’ was also demonstrated for RNA-containing antigens which stimulate autoreactive B cells to produce autoantibodies through BCR and TLR7 engagement, a response that was enhanced by the cytokine IFN-α (125). The fact that U1-snRNP contains its own unique RNA molecule suggests this mechanism may be involved in the autoimmune response against the U1-snRNP, and is closely connected with the production of type-I interferons (IFN-I).
Levels of IFNs are increased in the serum of SLE patients and show a good correlation with disease activity (126). Gene expression profiling of PBMCs derived from SLE patients reveals an interferon inducible gene signature that correlates with SLEDAI scores (127). IFN-induced chemokines were also recently shown to correlate with disease activity, organ damage, and autoantibody patterns in SLE (128). Interestingly, the expression of IFN-α induced genes significantly correlated with the titer of antibodies against RNA binding proteins in SLE patients (129–131).
The role of TLR7 and interferons in the development of anti-U1-snRNP autoimmunity has been studied in a number of mouse models of lupus. TLR7 is required for the generation of antibodies against RNA-containing antigens in MRL/lpr mice (132). In the pristane model of lupus, mice that are injected with the mineral oil pristane develop anti-U1-snRNP autoantibodies and inflammatory kidney disease (133). Our laboratory has shown that STAT1 and IRF9, critical mediators of downstream IFN-I signaling, are required for the production of IgG autoantibodies against components of the U1-snRNP in pristane-treated mice (134). In particular, the IFN-I signaling pathway was critical for isotype switching to the pathogenic IgG2a isotype (134). The defects in isotype switching were not attributable to global defects in B-cell activation, signaling, or isotype switching, as ovalbumin (OVA) in complete Freund's adjuvant (CFA) induced isotype-switched, anti-OVA antibodies (134). B cells from IRF9−/− and STAT1−/− mice had specific defects in activation through TLRs, suggesting defects in TLR7 may account for the lack of isotype-switched autoantibodies (134). In addition, IFN-I receptor deficient mice (IFNAR1−/−) do not produce anti-RNP autoantibodies (135), and IFNAR2−/− mice treated with pristane fail to develop autoantibodies against RNA-associated antigen complexes (136). B cells deficient in IFNAR2 fail to upregulate TLR7 in response to IFN-I (136). These data suggest a role for IFN-I in driving the autoimmune response to RNA-associated autoantigens through TLR-dependent mechanisms.
The involvement of TLRs in anti-U1-snRNP autoimmunity is likely explained by the ability of the antigen itself to be stimulatory. U1-snRNA acts as an endogenous adjuvant in mice, and is capable of inducing dendritic cell (DC) maturation as well as B and T-cell activation, resulting in antigen-specific autoimmune responses. Pretreatment of U1-RNA with RNase or co-administration with a TLR-7 antagonist inhibits this effect, suggesting that TLR7 is directly involved in the U1-RNA adjuvant effect (137). In addition, it has been shown that U1-RNA induces IFN-α production in monocytes isolated from PBMCs, possibly through immune complex formation (138) (Fig. 4A). U1-RNA induced activation of a human cell line transfected with TLR3, a different RNA sensor. The activity was abolished with RNase treatment of the antigen (139). Immunization of C57BL/6 mice transgenic for HLA-DR4 with the U1-70K peptide 63–205 and U1-RNA, led to the development of autoantibodies specific for U1-70K, and the development of pulmonary inflammation (140). The dependence on TLRs was investigated in TLR3−/− mice, where lung disease did not develop. However, the mice unexpectedly developed anti-dsDNA autoantibodies and nephritis. Further studies suggested that U1-RNA was activating proinflammatory cascades through TLR7 (140). These data suggest that the tissue-specific inflammatory milieu somehow also contributes to end-organ damage, perhaps explaining why SLE and MCTD patients develop nephritis, interstitial lung disease, or myositis.
Fig. 4. T-cell-independent and T-cell-dependent mechanisms of autoantibody production.
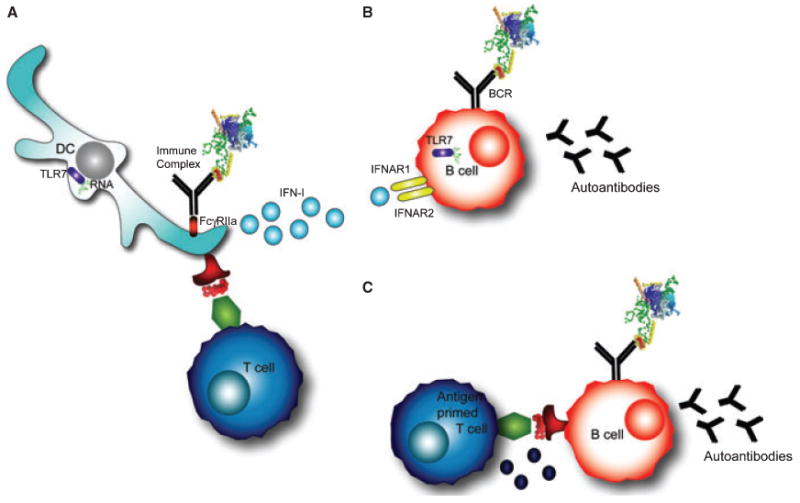
(A) Dendritic cells internalize immune complexes through FcγRIIα. RNA contained in the immune complex stimulates TLR7, resulting in type-I interferon production (IFN-I). In addition, DCs present peptides from the processed antigen complex on their surface to T cells. (B) T-cell-independent B-cell activation occurs in B cells that are stimulated through the B-cell receptor (BCR) and TLR7 by RNA-containing antigens. In addition, interferons signal through IFNAR receptors on the B-cell surface. These signals can induce autoantibody production against the RNA-containing protein complex in some models. (C) T-cell-dependent B-cell activation occurs through the typical T-B cognate interaction that provides T-cell help and leads to autoantibody production.
In many of these studies, B cells had the intrinsic capacity to produce autoantibodies through stimulation of the BCR and TLRs in a T-cell-independent manner (Fig. 4A,B). In the AM14 transgenic B-cell system, for example, B cells recognize RF, and T-cell-independent autoantibody responses occur in extrafollicular sites through a MyD88-dependent mechanism (141). While T cells may not be required in some cases to generate autoantibodies, these examples do not exclude a role for T cells in the anti-U1-snRNP response. The interaction between B and T cells may still be important for the diverse response that results from epitope spreading (Fig. 4C). Another role for T cells may be to enable autoreactive B cells to overcome anergy (142). This mechanistic hypothesis was explored in a recent review (143). Most likely, multiple mechanisms, both T-cell dependent and independent, contribute to the immune events that result in U1-snRNP autoreactivity.
Future directions
Antigen-specific therapies
A major goal in the field is to apply what has been learned about B-cell and T-cell specificities to the generation of antigen-specific therapies that will directly target inappropriate immune responses. To this end, Muller et al. (144) identified a phosphorylation modification of residue serine 140 of the U1-70K peptide 131–151 (P140) that tolerized MRL/lpr mice. Mice administered the P140 peptide intravenously had decreased proteinuria and anti-dsDNA antibody production, and prolonged survival (144). Tolerization may extend to affect intermolecular epitope spreading (89). T cells from mice that were administered P140 no longer proliferated ex vivo to recall antigens, including a peptide from Sm-D1, but retained the ability to respond to an influenza epitope (89). The mechanism of P140-induced tolerance is not defined. A recent study identified a receptor on the surface of spleen and lymph node cells called the heat-shock cognate HSC70 protein that binds weakly to P140 with a kDa of 7.32 μM (145). In addition, administration of P140 induced apoptosis of CD4+ T cells in MRL/lpr mice, which was dependent on the presence of γδ T cells (145). It is not clear if P140 acts via γδ T cells or how exactly the HSC70 protein is involved. Experiments measuring gene expression changes in P140 treated mice revealed downregulation of genes involved in inflammation including, il5, il15, csf2, tnfsf10, cdk4, icam1, foxp2, and itch, and upregulation of the Nfκb1 and stat6 genes (145). Additional experiments to elucidate the mechanism of P140 tolerance will be important.
The P140 peptide was also shown to induce tolerance in human patients. CD4+ T cells from SLE patients proliferated in response to the wildtype peptide 131–151 but not to P140 (146). Recently the P140 peptide entered human clinical trials. P140 was administered subcutaneously at 2-week intervals as part of a phase I/II clinical trial designed to demonstrate safety and to identify dosing regimens that could modulate autoreactive B and T cells. It was observed that IgG anti-dsDNA decreased by at least 20% in 7 of 10 patients in the lower dose group (3 × 200 μg), and in 1 of 10 patients in the higher dose group (3 × 1000 μg) (147). SLE disease activity index (SLEDAI) scores were lower in the low dose group, and it was concluded that the treatment was safe, tolerable, and potentially efficacious (147). If this therapeutic approach proves successful, it will be a meaningful step towards antigen-specific therapy in SLE and MCTD.
The Sm proteins have also been candidates for tolerizing therapies. In a series of in vivo tolerizing studies, the peptide 83–119 from the Sm-D protein was administered in (NZB × NZW)F1 mice. Mice treated with the peptide demonstrated a delay in autoantibody production and prolonged survival (148). Cytokine expression in T-cell cultures was altered following peptide therapy, showing a decrease in IFNγ and IL-4, and an increase in TGFβ. When tolerized T cells were adoptively transferred, naive T cells proliferated less in response to anti-CD3 stimulation. In addition, a high proportion of IL-10-expressing T cells was observed, which suppressed autoantibody production by B cells in co-culture experiments (148). These studies demonstrate the potential for antigen-specific therapies that target T-cell responses, and the need to understand the immune mechanisms that tolerizing therapies employ.
DNA vaccination with lupus autoantigens, and in particular with the Sm antigens, has shown potential in the induction of antigen-specific tolerance. In these experiments, mice treated with pristane were administered a DNA plasmid encoding Sm proteins, together with either IL-10 or IFNγ encoding plasmids. Mice treated with the DNA vaccine and IL-10 produced lower levels of anti-U1-RNP and anti-Sm autoantibodies and had significantly less proteinuria (B.T. Marquez et al., personal communication). DNA vaccines as potential therapies in autoimmune disease have been reviewed elsewhere (149) and remain an area of active research in the field.
Tetramers to detect antigen-specific T cells
Although we have learned a great deal about antigen-specific T-cell responses, a peptide-MHC tetramer reagent to detect antigen-specific T cells in SLE or MCTD has not been described. Generating recombinant MHC class II molecules loaded with relevant autopeptides has proven difficult due to the challenges in making stable complexes in high concentration. However, such a reagent will allow single cell studies and sorting of antigen-specific T cells by FACS, which will be important for answering questions of frequency, phenotype, and function. A tetramer reagent will be particularly useful for tracking T-cell responses to antigen-specific therapies, including tolerizing peptide therapies and DNA vaccines that are thought to impact the T-cell compartment. Once a single peptide-MHC tetramer is validated, an obvious extension will be to generate a panel of peptide-MHC tetramers, each loaded with a different peptide epitope. Newell et al. (150) described a method for the simultaneous detection of multiple T-cell specificities using combinations of tetramers. This work was demonstrated in humans, and while MHC class I molecules were used, it may be possible to extend this approach to class II tetramers.
Autoantigen arrays to monitor autoantibody reactivities
Given the clinical and serologic overlap of many rheumatic diseases, a comprehensive patient evaluation involves screening for reactivity against multiple antigens. The need to test for autoantibodies to multiple autoantigens served as a natural motivation for the development of autoantigen arrays (151, 152). Autoantigen array methodology has been extensively reviewed elsewhere (153, 154). Briefly, planar autoantigen arrays involve printing, immobilizing, or synthesizing in situ hundreds to thousands of potential autoantigens on a slide, where an antigen is identified by its spatial address on the array. The arrays can then be probed with any sample containing Ig, including serum, cerebrospinal fluid, synovial fluid, or purified monoclonal antibodies. After washing, slides are probed with a fluorophore-labeled secondary antibody to detect bound Ig, thus identifying an autoantibody ‘signature’ for that sample (Fig. 1G). Autoantigen arrays including U1-A, U1-C, U1-70K, Sm, dsDNA, and many other autoantigens have been previously described (152). Although there are still technical aspects that need optimization (133, 155), planar autoantigen arrays are an invaluable research tool that may evolve into a clinical tool. Overall, autoantigen arrays use less serum (on the order of 1–2 μl) than serial single-plex assays, identify multiple reactivities simultaneously, permit isotyping, and allow for identification of novel reactivities (151–154, 156). In the future, arrays of modified synthetic peptides may facilitate better understanding of the post-translational modifications targeted by autoantibodies. Our laboratory has recently detected autoantibodies specific for post-translationally modified autoantigens in SLE sera using peptide arrays (C. L. Liu and P. J. Utz, unpublished results).
While the first iterations of autoantigen microarray technology involved autoantigens arrayed onto a coated microscope slide, bead-based arrays have subsequently emerged (157, 158). In contrast to the planar array format, which identifies antigens by a coordinate on the slide, bead-based arrays use optically encoded microspheres to identify individual antigens that are coupled to the bead. Current bead array technologies can measure hundreds of analytes simultaneously, although in practice they are more typically approximately 20-plex assays. Bead-based platforms have their own set of challenges (157–159), but the main theoretical advantages of this technology include improved coating of the antigen and the ability to perform all steps in fluid-phase without antigen dessication. There is currently an FDA-approved, multiplex bead-based, flow-cytometry assay that measures antibodies to RNP, Sm, and dsDNA, among other autoantigens (Fig. 1H). While this is a move toward multiplex assays for diagnosis of rheumatic disorders, there is still much progress to be made.
Assays to detect anti-U1-snRNP autoantibodies are used routinely in rheumatology clinics to assist in the diagnosis of SLE and are central to the diagnosis of MCTD. Therefore the accuracy and precision of these assays are important considerations for clinicians and scientists. Technical issues will still need to be addressed before moving multiplex assays to detect autoantibody reactivities firmly into the clinical arena, including development of appropriate reference sera for more accurate and reproducible measures.
Conclusions
Autoimmunity against the U1-snRNP particle is prevalent in patients with rheumatic diseases, yet it is still not clear why it becomes an immune target in some patients and not others. Based on understanding the structure of this antigen and the ways in which it becomes immunogenic, we propose that the U1-snRNP plays a central role in autoimmune pathogenesis in a subset of SLE patients and in most if not all patients with MCTD. Both the U1-RNA component and the specific proteins, particularly U1-70K, engage immune cells and their receptors in a complex network of interactions that ultimately lead to widespread autoimmunity, inflammation, and tissue injury.
Based on the complexity and overlapping features of these diseases, we find that large-scale efforts to study many factors within the peripheral blood of individual patients in a high-throughput fashion are incredibly important. However, developing multiplex assays to characterize autoantibodies in the clinic or to detect autoreactive T-cell specificities will not be trivial. It may also be a useful exercise to focus on reactivities and responses against one particular antigen to gain a better understanding of the mechanisms underlying antigen specific autoreactivities. Due to the inherent structural properties of the U1-snRNP that make it immunogenic and the correlations cited between U1-snRNP and certain clinical settings, this RNP complex is an excellent candidate for extensive evaluation. In addition, the population of MCTD patients, although rare, guarantees a patient group with prominent anti-U1-snRNP reactivity. If we begin to understand the nuances in interactions between an immunogenic autoantigen and an individual's immune system across many patients within a cohort, we will gain insight into designing antigen-specific therapies that could effectively target autoimmune responses, and guide the immune system back toward a tolerized state.
Acknowledgments
The authors would like to thank lab members A. Chu, I. Balboni, and D. Thibault, for their insights and critical review of the manuscript, and R. Cheung, J. Price, and C.L. Liu for helpful discussions. N.H.K. is funded by a fellowship from the National Science Foundation (NSF). M.G.K. is funded through the NIH's Medical Scientist Training Program (MSTP) at Stanford University. P.J.U. is the recipient of a Donald E. and Delia B. Baxter Foundation Career Development Award and was supported by the the Floren Family Trust, the Northern California Chapter of the Arthritis Foundation, US National Institutes of Health Grants DK61934, AI50854, AI50865, and AR49328 and National Heart, Lung, and Blood Institute Proteomics Contract N01-HV-28183. NHK and MGK would like to thank their mentor for submitting this article for them on their wedding day, August 8, 2009.
References
- 1.von Muhlen CA, Tan EM. Autoantibodies in the diagnosis of systemic rheumatic diseases. Semin Arthritis Rheum. 1995;24:323–358. doi: 10.1016/s0049-0172(95)80004-2. [DOI] [PubMed] [Google Scholar]
- 2.Holman H, Deicher HR. The reaction of the lupus erythematosus (L.E.) cell factor with deoxyribonucleoprotein of the cell nucleus. J Clin Invest. 1959;38:2059–2072. doi: 10.1172/JCI103984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan EM, Kunkel HG. Characteristics of a soluble nuclear antigen precipitating with sera of patients with systemic lupus erythematosus. J Immunol. 1966;96:464–471. [PubMed] [Google Scholar]
- 4.Sharp GC, et al. Association of autoantibodies to different nuclear antigens with clinical patterns of rheumatic disease and responsiveness to therapy. J Clin Invest. 1971;50:350–359. doi: 10.1172/JCI106502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Venrooij WJ, Zendman AJ, Pruijn GJ. Autoantibodies to citrullinated antigens in (early) rheumatoid arthritis. Autoimmun Rev. 2006;6:37–41. doi: 10.1016/j.autrev.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 6.Kidd BA, et al. Epitope spreading to citrullinated antigens in mouse models of autoimmune arthritis and demyelination. Arthritis Res Ther. 2008;10:R119. doi: 10.1186/ar2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pomeranz Krummel DA, Oubridge C, Leung AK, Li J, Nagai K. Crystal structure of human spliceosomal U1 snRNP at 5.5 A resolution. Nature. 2009;458:475–480. doi: 10.1038/nature07851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kramer A. The structure and function of proteins involved in mammalian pre-mRNA splicing. Annu Rev Biochem. 1996;65:367–409. doi: 10.1146/annurev.bi.65.070196.002055. [DOI] [PubMed] [Google Scholar]
- 9.Lutz CS, James J. Antibodies to Spliceosomal Components Dubois' Lupus Erythematosus. Lippincott: Williams & Wilkins; 2007. [Google Scholar]
- 10.Lerner MR, Steitz JA. Antibodies to small nuclear RNAs complexed with proteins are produced by patients with systemic lupus erythematosus. Proc Natl Acad Sci USA. 1979;76:5495–5499. doi: 10.1073/pnas.76.11.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krol A, Westhof E, Bach M, Luhrmann R, Ebel JP, Carbon P. Solution structure of human U1 snRNA. Derivation of a possible three-dimensional model. Nucleic Acids Res. 1990;18:3803–3811. doi: 10.1093/nar/18.13.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kambach C, et al. Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs. Cell. 1999;96:375–387. doi: 10.1016/s0092-8674(00)80550-4. [DOI] [PubMed] [Google Scholar]
- 13.Stark H, Dube P, Luhrmann R, Kastner B. Arrangement of RNA and proteins in the spliceosomal U1 small nuclear ribonucleoprotein particle. Nature. 2001;409:539–542. doi: 10.1038/35054102. [DOI] [PubMed] [Google Scholar]
- 14.Theissen H, et al. Cloning of the human cDNA for the U1 RNA-associated 70K protein. EMBO J. 1986;5:3209–3217. doi: 10.1002/j.1460-2075.1986.tb04631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelissen RL, Will CL, van Venrooij WJ, Luhrmann R. The association of the U1-specific 70K and C proteins with U1 snRNPs is mediated in part by common U snRNP proteins. EMBO J. 1994;13:4113–4125. doi: 10.1002/j.1460-2075.1994.tb06729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lutz CS, McClain MT, Harley JB, James JA. Anti-U1A monoclonal antibodies recognize unique epitope targets of U1A which are involved in the binding of U1 RNA. J Mol Recognit. 2002;15:163–170. doi: 10.1002/jmr.569. [DOI] [PubMed] [Google Scholar]
- 17.Faig OZ, Lutz CS. Novel specificity of anti-U1A autoimmune patient sera. Scand J Immunol. 2003;57:79–84. doi: 10.1046/j.1365-3083.2003.01186.x. [DOI] [PubMed] [Google Scholar]
- 18.Query CC, Bentley RC, Keene JD. A common RNA recognition motif identified within a defined U1 RNA binding domain of the 70K U1 snRNP protein. Cell. 1989;57:89–101. doi: 10.1016/0092-8674(89)90175-x. [DOI] [PubMed] [Google Scholar]
- 19.Query CC, Bentley RC, Keene JD. A specific 31-nucleotide domain of U1 RNA directly interacts with the 70K small nuclear ribonucleoprotein component. Mol Cell Biol. 1989;9:4872–4881. doi: 10.1128/mcb.9.11.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scherly D, Boelens W, van Venrooij WJ, Dathan NA, Hamm J, Mattaj IW. Identification of the RNA binding segment of human U1 A protein and definition of its binding site on U1 snRNA. EMBO J. 1989;8:4163–4170. doi: 10.1002/j.1460-2075.1989.tb08601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. Crystal structure at 1.92 A resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature. 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- 22.Kastner B, Kornstadt U, Bach M, Luhrmann R. Structure of the small nuclear RNP particle U1: identification of the two structural protuberances with RNP-antigens A and 70K. J Cell Biol. 1992;116:839–849. doi: 10.1083/jcb.116.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lerner MR, Boyle JA, Mount SM, Wolin SL, Steitz JA. Are snRNPs involved in splicing? Nature. 1980;283:220–224. doi: 10.1038/283220a0. [DOI] [PubMed] [Google Scholar]
- 24.Staley JP, Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998;92:315–326. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- 25.Murray HL, Jarrell KA. Flipping the switch to an active spliceosome. Cell. 1999;96:599–602. doi: 10.1016/s0092-8674(00)80568-1. [DOI] [PubMed] [Google Scholar]
- 26.Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 27.Bessonov S, Anokhina M, Will CL, Urlaub H, Luhrmann R. Isolation of an active step I spliceosome and composition of its RNP core. Nature. 2008;452:846–850. doi: 10.1038/nature06842. [DOI] [PubMed] [Google Scholar]
- 28.Stark H, Luhrmann R. Cryo-electron microscopy of spliceosomal components. Annu Rev Biophys Biomol Struct. 2006;35:435–457. doi: 10.1146/annurev.biophys.35.040405.101953. [DOI] [PubMed] [Google Scholar]
- 29.Luhrmann R, Stark H. Structural mapping of spliceosomes by electron microscopy. Curr Opin Struct Biol. 2009;19:96–102. doi: 10.1016/j.sbi.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 30.Utz PJ, Hottelet M, van Venrooij WJ, Anderson P. Association of phosphorylated serine/arginine (SR) splicing factors with the U1-small ribonucleoprotein (snRNP) autoantigen complex accompanies apoptotic cell death. J Exp Med. 1998;187:547–560. doi: 10.1084/jem.187.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zahler AM, Lane WS, Stolk JA, Roth MB. SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev. 1992;6:837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]
- 32.Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 33.Zahler AM, Neugebauer KM, Lane WS, Roth MB. Distinct functions of SR proteins in alternative pre-mRNA splicing. Science. 1993;260:219–222. doi: 10.1126/science.8385799. [DOI] [PubMed] [Google Scholar]
- 34.Zahler AM, Roth MB. Distinct functions of SR proteins in recruitment of U1 small nuclear ribonucleoprotein to alternative 5′ splice sites. Proc Natl Acad Sci USA. 1995;92:2642–2646. doi: 10.1073/pnas.92.7.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neugebauer KM, Merrill JT, Wener MH, Lahita RG, Roth MB. SR proteins are autoantigens in patients with systemic lupus erythematosus. Importance of phospho-epitopes. Arthritis Rheum. 2000;43:1768–1778. doi: 10.1002/1529-0131(200008)43:8<1768::AID-ANR13>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 36.Mattioli M, Reichlin M. Characterization of a soluble nuclear ribonucleoprotein antigen reactive with SLE sera. J Immunol. 1971;107:1281–1290. [PubMed] [Google Scholar]
- 37.Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed connective tissue disease-an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA) Am J Med. 1972;52:148–159. doi: 10.1016/0002-9343(72)90064-2. [DOI] [PubMed] [Google Scholar]
- 38.Sharp GC, et al. Association of antibodies to ribonucleoprotein and Sm antigens with mixed connective-tissue disease, systematic lupus erythematosus and other rheumatic diseases. N Engl J Med. 1976;295:1149–1154. doi: 10.1056/NEJM197611182952101. [DOI] [PubMed] [Google Scholar]
- 39.Venables PJ. Mixed connective tissue disease. Lupus. 2006;15:132–137. doi: 10.1191/0961203306lu2283rr. [DOI] [PubMed] [Google Scholar]
- 40.Hoffman RW, Maldonado ME. Immune pathogenesis of Mixed Connective Tissue Disease: a short analytical review. Clin Immunol. 2008;128:8–17. doi: 10.1016/j.clim.2008.03.461. [DOI] [PubMed] [Google Scholar]
- 41.Pettersson I, Hinterberger M, Mimori T, Gottlieb E, Steitz JA. The structure of mammalian small nuclear ribonucleoproteins. Identification of multiple protein components reactive with anti-(U1)ribonucleoprotein and anti-Sm autoantibodies. J Biol Chem. 1984;259:5907–5914. [PubMed] [Google Scholar]
- 42.Delpech A, Gilbert D, Daliphard S, Le Loet X, Godin M, Tron F. Antibodies to Sm, RNP and SSB detected by solid-phase ELISAs using recombinant antigens: a comparison study with counter immunoelectrophoresis and immunoblotting. J Clin Lab Anal. 1993;7:197–202. doi: 10.1002/jcla.1860070402. [DOI] [PubMed] [Google Scholar]
- 43.Maddison PJ, Skinner RP, Vlachoyiannopoulos P, Brennand DM, Hough D. Antibodies to nRNP, Sm, Ro(SSA) and La(SSB) detected by ELISA: their specificity and inter-relations in connective tissue disease sera. Clin Exp Immunol. 1985;62:337–345. [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia Lerma JG, Mendoza AZ, Ramos MJ, Sequi J. Evaluation of recombinant Ro/SSA, La/SSB, Sm, and U1 RNP autoantigens in clinical diagnosis. J Clin Lab Anal. 1995;9:52–58. doi: 10.1002/jcla.1860090110. [DOI] [PubMed] [Google Scholar]
- 45.Benito-Garcia E, Schur PH, Lahita R. Guidelines for immunologic laboratory testing in the rheumatic diseases: anti-Sm and anti-RNP antibody tests. Arthritis Rheum. 2004;51:1030–1044. doi: 10.1002/art.20836. [DOI] [PubMed] [Google Scholar]
- 46.Takada K, et al. Clinical characteristics of patients with both anti-U1RNP and anti-centromere antibodies. Scand J Rheumatol. 2008;37:360–364. doi: 10.1080/03009740802116190. [DOI] [PubMed] [Google Scholar]
- 47.St Clair EW, et al. Expression of autoantibodies to recombinant (U1) RNP-associated 70K antigen in systemic lupus erythematosus. Clin Immunol Immunopathol. 1990;54:266–280. doi: 10.1016/0090-1229(90)90088-8. [DOI] [PubMed] [Google Scholar]
- 48.Faria AC, Barcellos KS, Andrade LE. Longitudinal fluctuation of antibodies to extractable nuclear antigens in systemic lupus erythematosus. J Rheumatol. 2005;32:1267–1272. [PubMed] [Google Scholar]
- 49.Habets WJ, et al. Antibodies against distinct nuclear matrix proteins are characteristic for mixed connective tissue disease. Clin Exp Immunol. 1983;54:265–276. [PMC free article] [PubMed] [Google Scholar]
- 50.van Venrooij WJ, Hoet R, Castrop J, Hageman B, Mattaj IW, van de Putte LB. Anti-(U1) small nuclear RNA antibodies in anti-small nuclear ribonucleoprotein sera from patients with connective tissue diseases. J Clin Invest. 1990;86:2154–2160. doi: 10.1172/JCI114954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burdt MA, Hoffman RW, Deutscher SL, Wang GS, Johnson JC, Sharp GC. Long-term outcome in mixed connective tissue disease: longitudinal clinical and serologic findings. Arthritis Rheum. 1999;42:899–909. doi: 10.1002/1529-0131(199905)42:5<899::AID-ANR8>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 52.Ho KT, Reveille JD. The clinical relevance of autoantibodies in scleroderma. Arthritis Res Ther. 2003;5:80–93. doi: 10.1186/ar628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chung L, Utz PJ. Antibodies in scleroderma: direct pathogenicity and phenotypic associations. Curr Rheumatol Rep. 2004;6:156–163. doi: 10.1007/s11926-004-0061-9. [DOI] [PubMed] [Google Scholar]
- 54.Coppo P, Clauvel JP, Bengoufa D, Oksenhendler E, Lacroix C, Lassoued K. Inflammatory myositis associated with anti-U1-small nuclear ribonucleoprotein antibodies: a subset of myositis associated with a favourable outcome. Rheumatology (Oxford) 2002;41:1040–1046. doi: 10.1093/rheumatology/41.9.1040. [DOI] [PubMed] [Google Scholar]
- 55.Dayal NA, Isenberg DA. SLE/myositis overlap: are the manifestations of SLE different in overlap disease? Lupus. 2002;11:293–298. doi: 10.1191/0961203302lu186oa. [DOI] [PubMed] [Google Scholar]
- 56.Lundberg I, Nennesmo I, Hedfors E. A clinical, serological, and histopathological study of myositis patients with and without anti-RNP antibodies. Semin Arthritis Rheum. 1992;22:127–138. doi: 10.1016/0049-0172(92)90006-y. [DOI] [PubMed] [Google Scholar]
- 57.Arbuckle MR, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 58.Reichlin M, Van Venrooij WJ. Autoantibodies to the URNP particles: relationship to clinical diagnosis and nephritis. Clin Exp Immunol. 1991;83:286–290. doi: 10.1111/j.1365-2249.1991.tb05629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thompson D, Juby A, Davis P. The clinical significance of autoantibody profiles in patients with systemic lupus erythematosus. Lupus. 1993;2:15–19. doi: 10.1177/096120339300200104. [DOI] [PubMed] [Google Scholar]
- 60.Greidinger EL, Casciola-Rosen L, Morris SM, Hoffman RW, Rosen A. Autoantibody recognition of distinctly modified forms of the U1-70-kd antigen is associated with different clinical disease manifestations. Arthritis Rheum. 2000;43:881–888. doi: 10.1002/1529-0131(200004)43:4<881::AID-ANR20>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 61.Keith MP, et al. Anti-ribonucleoprotein antibodies mediate enhanced lung injury following mesenteric ischemia/reperfusion in Rag-1(−/−) mice. Autoimmunity. 2007;40:208–216. doi: 10.1080/08916930701262986. [DOI] [PubMed] [Google Scholar]
- 62.Query CC, Keene JD. A human autoimmune protein associated with U1 RNA contains a region of homology that is cross-reactive with retroviral p30gag antigen. Cell. 1987;51:211–220. doi: 10.1016/0092-8674(87)90148-6. [DOI] [PubMed] [Google Scholar]
- 63.Guldner HH, Netter HJ, Szostecki C, Lakomek HJ, Will H. Epitope mapping with a recombinant human 68-kDa (U1) ribonucleoprotein antigen reveals heterogeneous autoantibody profiles in human autoimmune sera. J Immunol. 1988;141:469–475. [PubMed] [Google Scholar]
- 64.Cram DS, Fisicaro N, Coppel RL, Whittingham S, Harrison LC. Mapping of multiple B cell epitopes on the 70-kilodalton autoantigen of the U1 ribonucleoprotein complex. J Immunol. 1990;145:630–635. [PubMed] [Google Scholar]
- 65.Netter HJ, Guldner HH, Szostecki C, Will H. Major autoantigenic sites of the (U1) small nuclear ribonucleoprotein-specific 68-kDa protein. Scand J Immunol. 1990;32:163–176. doi: 10.1111/j.1365-3083.1990.tb02906.x. [DOI] [PubMed] [Google Scholar]
- 66.Nyman U, Lundberg I, Hedfors E, Pettersson I. Recombinant 70-kD protein used for determination of autoantigenic epitopes recognized by anti-RNP sera. Clin Exp Immunol. 1990;81:52–58. doi: 10.1111/j.1365-2249.1990.tb05290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Monneaux F, Briand JP, Muller S. B and T cell immune response to small nuclear ribonucleoprotein particles in lupus mice: autoreactive CD4(+) T cells recognize a T cell epitope located within the RNP80 motif of the 70K protein. Eur J Immunol. 2000;30:2191–2200. doi: 10.1002/1521-4141(2000)30:8<2191::AID-IMMU2191>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 68.Monneaux F, Muller S. Epitope spreading in systemic lupus erythematosus: identification of triggering peptide sequences. Arthritis Rheum. 2002;46:1430–1438. doi: 10.1002/art.10263. [DOI] [PubMed] [Google Scholar]
- 69.Barakat S, Briand JP, Abuaf N, van Regenmortel MH, Muller S. Mapping of epitopes on U1 snRNP polypeptide A with synthetic peptides and autoimmune sera. Clin Exp Immunol. 1991;86:71–78. doi: 10.1111/j.1365-2249.1991.tb05776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.James JA, Harley JB. Human lupus anti-spliceosome A protein autoantibodies bind contiguous surface structures and segregate into two sequential epitope binding patterns. J Immunol. 1996;156:4018–4026. [PubMed] [Google Scholar]
- 71.Halimi H, Dumortier H, Briand JP, Muller S. Comparison of two different methods using overlapping synthetic peptides for localizing linear B cell epitopes in the U1 snRNP-C autoantigen. J Immunol Methods. 1996;199:77–85. doi: 10.1016/s0022-1759(96)00171-8. [DOI] [PubMed] [Google Scholar]
- 72.Hoet RM, Kastner B, Luhrmann R, van Venrooij WJ. Purification and characterization of human autoantibodies directed to specific regions on U1RNA; recognition of native U1RNP complexes. Nucleic Acids Res. 1993;21:5130–5136. doi: 10.1093/nar/21.22.5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Monneaux F, Muller S. Key sequences involved in the spreading of the systemic autoimmune response to spliceosomal proteins. Scand J Immunol. 2001;2:45–54. doi: 10.1046/j.1365-3083.2001.00942.x. [DOI] [PubMed] [Google Scholar]
- 74.Freed JH, Marrs A, VanderWall J, Cohen PL, Eisenberg RA. MHC class II-bound self peptides from autoimmune MRL/lpr mice reveal potential T cell epitopes for autoantibody production in murine systemic lupus erythematosus. J Immunol. 2000;164:4697–4705. doi: 10.4049/jimmunol.164.9.4697. [DOI] [PubMed] [Google Scholar]
- 75.O'Brien RM, Cram DS, Coppel RL, Harrison LC. T-cell epitopes on the 70-kDa protein of the (U1)RNP complex in autoimmune rheumatologic disorders. J Autoimmun. 1990;3:747–757. doi: 10.1016/s0896-8411(05)80041-1. [DOI] [PubMed] [Google Scholar]
- 76.Monneaux F, Parietti V, Briand JP, Muller S. Intramolecular T cell spreading in unprimed MRL/lpr mice: importance of the U1-70k protein sequence 131-151. Arthritis Rheum. 2004;50:3232–3238. doi: 10.1002/art.20510. [DOI] [PubMed] [Google Scholar]
- 77.Monneaux F, Dumortier H, Steiner G, Briand JP, Muller S. Murine models of systemic lupus erythematosus: B and T cell responses to spliceosomal ribonucleoproteins in MRL/Fas(lpr) and (NZB × NZW)F(1) lupus mice. Int Immunol. 2001;13:1155–1163. doi: 10.1093/intimm/13.9.1155. [DOI] [PubMed] [Google Scholar]
- 78.Kent SC, et al. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature. 2005;435:224–228. doi: 10.1038/nature03625. [DOI] [PubMed] [Google Scholar]
- 79.Hoffman RW, et al. Human T-cell clones reactive against U-small nuclear ribonucleoprotein autoantigens from connective tissue disease patients and healthy individuals. J Immunol. 1993;151:6460–6469. [PubMed] [Google Scholar]
- 80.Talken BL, Lee DR, Caldwell CW, Quinn TP, Schafermeyer KR, Hoffman RW. Analysis of T cell receptors specific for U1-70kD small nuclear ribonucleoprotein autoantigen: the alpha chain complementarity determining region three is highly conserved among connective tissue disease patients. Hum Immunol. 1999;60:200–208. doi: 10.1016/s0198-8859(98)00117-7. [DOI] [PubMed] [Google Scholar]
- 81.Talken BL, Bailey CW, Reardon SL, Caldwell CW, Hoffman RW. Structural analysis of TCRalpha and beta chains from human T-Cell clones specific for small nuclear ribonucleoprotein polypeptides Sm-D, Sm-B and U1-70 kDa: TCR complementarity determining region 3 usage appears highly conserved. Scand J Immunol. 2001;2:204–210. doi: 10.1046/j.1365-3083.2001.00930.x. [DOI] [PubMed] [Google Scholar]
- 82.Greidinger EL, et al. T cell immunity in connective tissue disease patients targets the RNA binding domain of the U1-70kDa small nuclear ribonucleoprotein. J Immunol. 2002;169:3429–3437. doi: 10.4049/jimmunol.169.6.3429. [DOI] [PubMed] [Google Scholar]
- 83.Holyst MM, Hill DL, Hoch SO, Hoffman RW. Analysis of human T cell and B cell responses against U small nuclear ribonucleoprotein 70-kd, B, and D polypeptides among patients with systemic lupus erythematosus and mixed connective tissue disease. Arthritis Rheum. 1997;40:1493–1503. doi: 10.1002/art.1780400818. [DOI] [PubMed] [Google Scholar]
- 84.Yang MH, Suen JL, Li SL, Chiang BL. Identification of T-cell epitopes on U1A protein in MRL/lpr mice: double-negative T cells are the major responsive cells. Immunology. 2005;115:279–286. doi: 10.1111/j.1365-2567.2005.02139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol. 1991;9:243–269. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- 86.Suen JL, Chuang YH, Chiang BL. In vivo tolerance breakdown with dendritic cells pulsed with U1A protein in non-autoimmune mice: the induction of a high level of autoantibodies but not renal pathological changes. Immunology. 2002;106:326–335. doi: 10.1046/j.1365-2567.2002.01438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Okubo M, et al. Detection and epitope analysis of autoantigen-reactive T cells to the U1-small nuclear ribonucleoprotein A protein in autoimmune disease patients. J Immunol. 1993;151:1108–1115. [PubMed] [Google Scholar]
- 88.Greidinger EL, Zang YJ, Jaimes K, Martinez L, Nassiri M, Hoffman RW. CD4+ T cells target epitopes residing within the RNA-binding domain of the U1-70-kDa small nuclear ribonucleoprotein autoantigen and have restricted TCR diversity in an HLA-DR4-transgenic murine model of mixed connective tissue disease. J Immunol. 2008;180:8444–8454. doi: 10.4049/jimmunol.180.12.8444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Monneaux F, Parietti V, Briand JP, Muller S. Importance of spliceosomal RNP1 motif for intermolecular T-B cell spreading and tolerance restoration in lupus. Arthritis Res Ther. 2007;9:R111. doi: 10.1186/ar2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature. 1985;314:537–539. doi: 10.1038/314537a0. [DOI] [PubMed] [Google Scholar]
- 91.Mamula MJ, Fatenejad S, Craft J. B cells process and present lupus autoantigens that initiate autoimmune T cell responses. J Immunol. 1994;152:1453–1461. [PubMed] [Google Scholar]
- 92.Peng SL, et al. alpha beta T cell regulation and CD40 ligand dependence in murine systemic autoimmunity. J Immunol. 1997;158:2464–2470. [PubMed] [Google Scholar]
- 93.Greidinger EL, et al. Tissue targeting of anti-RNP autoimmunity: effects of T cells and myeloid dendritic cells in a murine model. Arthritis Rheum. 2009;60:534–542. doi: 10.1002/art.24256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Greidinger EL, Gazitt T, Jaimes KF, Hoffman RW. Human T-cell clones specific for heterogeneous nuclear ribonucleoprotein A2 autoantigen from connective tissue disease patients assist in autoantibody production. Arthritis Rheum. 2004;50:2216–2222. doi: 10.1002/art.20287. [DOI] [PubMed] [Google Scholar]
- 95.Takeno M, et al. Autoreactive T-cell clones from patients with systemic lupus erythematosus support polyclonal autoantibody production. J Immunol. 1997;158:3529–3538. [PubMed] [Google Scholar]
- 96.Mamula MJ. Epitope spreading: the role of self peptides and autoantigen processing by B lymphocytes. Immunol Rev. 1998;164:231–239. doi: 10.1111/j.1600-065x.1998.tb01223.x. [DOI] [PubMed] [Google Scholar]
- 97.James JA, Harley JB. B-cell epitope spreading in autoimmunity. Immunol Rev. 1998;164:185–200. doi: 10.1111/j.1600-065x.1998.tb01220.x. [DOI] [PubMed] [Google Scholar]
- 98.Deshmukh US, Bagavant H, Lewis J, Gaskin F, Fu SM. Epitope spreading within lupus-associated ribonucleoprotein antigens. Clin Immunol. 2005;117:112–120. doi: 10.1016/j.clim.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 99.Fatenejad S, Mamula MJ, Craft J. Role of intermolecular/intrastructural B- and T-cell determinants in the diversification of autoantibodies to ribonucleoprotein particles. Proc Natl Acad Sci USA. 1993;90:12010–12014. doi: 10.1073/pnas.90.24.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Deshmukh US, Kannapell CC, Fu SM. Immune responses to small nuclear ribonucleoproteins: antigen-dependent distinct B cell epitope spreading patterns in mice immunized with recombinant polypeptides of small nuclear ribonucleoproteins. J Immunol. 2002;168:5326–5332. doi: 10.4049/jimmunol.168.10.5326. [DOI] [PubMed] [Google Scholar]
- 101.Fatenejad S, Brooks W, Schwartz A, Craft J. Pattern of anti-small nuclear ribonucleoprotein antibodies in MRL/Mp-lpr/lpr mice suggests that the intact U1 snRNP particle is their autoimmunogenic target. J Immunol. 1994;152:5523–5531. [PubMed] [Google Scholar]
- 102.Greidinger EL, Hoffman RW. The appearance of U1 RNP antibody specificities in sequential autoimmune human antisera follows a characteristic order that implicates the U1-70 kd and B′/B proteins as predominant U1 RNP immunogens. Arthritis Rheum. 2001;44:368–375. doi: 10.1002/1529-0131(200102)44:2<368::AID-ANR55>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 103.Utz PJ, Anderson P. Posttranslational protein modifications, apoptosis, and the bypass of tolerance to autoantigens. Arthritis Rheum. 1998;41:1152–1160. doi: 10.1002/1529-0131(199807)41:7<1152::AID-ART3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 104.Utz PJ, Gensler TJ, Anderson P. Death, autoantigen modifications, and tolerance. Arthritis Res. 2000;2:101–114. doi: 10.1186/ar75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Graham KL, Utz PJ. Sources of autoantigens in systemic lupus erythematosus. Curr Opin Rheumatol. 2005;17:513–517. doi: 10.1097/01.bor.0000171215.87993.6b. [DOI] [PubMed] [Google Scholar]
- 106.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dieker J, et al. Apoptosis-linked changes in the phosphorylation status and subcellular localization of the spliceosomal autoantigen U1-70K. Cell Death Differ. 2008;15:793–804. doi: 10.1038/sj.cdd.4402312. [DOI] [PubMed] [Google Scholar]
- 108.Utz PJ, Hottelet M, Schur PH, Anderson P. Proteins phosphorylated during stress-induced apoptosis are common targets for autoantibody production in patients with systemic lupus erythematosus. J Exp Med. 1997;185:843–854. doi: 10.1084/jem.185.5.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Casciola-Rosen LA, Miller DK, Anhalt GJ, Rosen A. Specific cleavage of the 70-kDa protein component of the U1 small nuclear ribonucleoprotein is a characteristic biochemical feature of apoptotic cell death. J Biol Chem. 1994;269:30757–30760. [PubMed] [Google Scholar]
- 110.Casciola-Rosen L, et al. Apopain/CPP32 cleaves proteins that are essential for cellular repair: a fundamental principle of apoptotic death. J Exp Med. 1996;183:1957–1964. doi: 10.1084/jem.183.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Degen WG, Aarssen Y, Pruijn GJ, Utz PJ, van Venrooij WJ. The fate of U1 snRNP during anti-Fas induced apoptosis: specific cleavage of the U1 snRNA molecule. Cell Death Differ. 2000;7:70–79. doi: 10.1038/sj.cdd.4400617. [DOI] [PubMed] [Google Scholar]
- 112.Andrade F, Casciola-Rosen LA, Rosen A. Generation of novel covalent RNA-protein complexes in cells by ultraviolet B irradiation: implications for autoimmunity. Arthritis Rheum. 2005;52:1160–1170. doi: 10.1002/art.20992. [DOI] [PubMed] [Google Scholar]
- 113.Kuhn A, Beissert S. Photosensitivity in lupus erythematosus. Autoimmunity. 2005;38:519–529. doi: 10.1080/08916930500285626. [DOI] [PubMed] [Google Scholar]
- 114.Degen WG, Pieffers M, Welin-Henriksson E, van den Hoogen FH, van Venrooij WJ, Raats JM. Characterization of recombinant human autoantibody fragments directed toward the autoantigenic U1-70K protein. Eur J Immunol. 2000;30:3029–3038. doi: 10.1002/1521-4141(200010)30:10<3029::AID-IMMU3029>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 115.Greidinger EL, Foecking MF, Ranatunga S, Hoffman RW. Apoptotic U1-70 kd is antigenically distinct from the intact form of the U1-70-kd molecule. Arthritis Rheum. 2002;46:1264–1269. doi: 10.1002/art.10211. [DOI] [PubMed] [Google Scholar]
- 116.Hof D, et al. Autoantibodies specific for apoptotic U1-70K are superior serological markers for mixed connective tissue disease. Arthritis Res Ther. 2005;7:R302–R309. doi: 10.1186/ar1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kaplan MJ. Apoptosis in systemic lupus erythematosus. Clin Immunol. 2004;112:210–218. doi: 10.1016/j.clim.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 118.Emlen W, Niebur J, Kadera R. Accelerated in vitro apoptosis of lymphocytes from patients with systemic lupus erythematosus. J Immunol. 1994;152:3685–3692. [PubMed] [Google Scholar]
- 119.Perniok A, Wedekind F, Herrmann M, Specker C, Schneider M. High levels of circulating early apoptic peripheral blood mononuclear cells in systemic lupus erythematosus. Lupus. 1998;7:113–118. doi: 10.1191/096120398678919804. [DOI] [PubMed] [Google Scholar]
- 120.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Marshak-Rothstein A, Rifkin IR. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol. 2007;25:419–441. doi: 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]
- 122.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 123.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 124.Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lau CM, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979;301:5–8. doi: 10.1056/NEJM197907053010102. [DOI] [PubMed] [Google Scholar]
- 127.Bennett L, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fu Q, et al. Association of elevated transcript levels of interferon-inducible chemokines with disease activity and organ damage in systemic lupus erythematosus patients. Arthritis Res Ther. 2008;10:R112. doi: 10.1186/ar2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005;52:1491–1503. doi: 10.1002/art.21031. [DOI] [PubMed] [Google Scholar]
- 130.Zhuang H, et al. Association of anti-nucleoprotein autoantibodies with upregulation of Type I interferon-inducible gene transcripts and dendritic cell maturation in systemic lupus erythematosus. Clin Immunol. 2005;117:238–250. doi: 10.1016/j.clim.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 131.Hua J, Kirou K, Lee C, Crow MK. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 2006;54:1906–1916. doi: 10.1002/art.21890. [DOI] [PubMed] [Google Scholar]
- 132.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 133.Kattah MG, Alemi GR, Thibault DL, Balboni I, Utz PJ. A new two-color Fab labeling method for autoantigen protein microarrays. Nat Methods. 2006;3:745–751. doi: 10.1038/nmeth910. [DOI] [PubMed] [Google Scholar]
- 134.Thibault DL, et al. IRF9 and STAT1 are required for IgG autoantibody production and B cell expression of TLR7 in mice. J Clin Invest. 2008;118:1417–1426. doi: 10.1172/JCI30065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nacionales DC, et al. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis Rheum. 2007;56:3770–3783. doi: 10.1002/art.23023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Thibault DL, Graham KL, Lee LY, Balboni I, Hertzog PJ, Utz PJ. Type I IFN receptor controls B cell expression of nucleic acid sensing toll-like receptors and autoantibody production in a murine model of lupus. Arthritis Res Ther. 2009;11:R112. doi: 10.1186/ar2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kelly-Scumpia KM, et al. In vivo adjuvant activity of the RNA component of the Sm/RNP lupus autoantigen. Arthritis Rheum. 2007;56:3379–3386. doi: 10.1002/art.22946. [DOI] [PubMed] [Google Scholar]
- 138.Lovgren T, Eloranta ML, Kastner B, Wahren-Herlenius M, Alm GV, Ronnblom L. Induction of interferon-alpha by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and Sjogren's syndrome autoantigen-associated RNA. Arthritis Rheum. 2006;54:1917–1927. doi: 10.1002/art.21893. [DOI] [PubMed] [Google Scholar]
- 139.Hoffman RW, et al. U1 RNA induces innate immunity signaling. Arthritis Rheum. 2004;50:2891–2896. doi: 10.1002/art.20428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Greidinger EL, et al. A murine model of mixed connective tissue disease induced with U1 small nuclear RNP autoantigen. Arthritis Rheum. 2006;54:661–669. doi: 10.1002/art.21566. [DOI] [PubMed] [Google Scholar]
- 141.Herlands RA, Christensen SR, Sweet RA, Hershberg U, Shlomchik MJ. T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity. 2008;29:249–260. doi: 10.1016/j.immuni.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Seo SJ, et al. The impact of T helper and T regulatory cells on the regulation of anti-double-stranded DNA B cells. Immunity. 2002;16:535–546. doi: 10.1016/s1074-7613(02)00298-4. [DOI] [PubMed] [Google Scholar]
- 143.Shlomchik MJ. Sites and stages of autoreactive B cell activation and regulation. Immunity. 2008;28:18–28. doi: 10.1016/j.immuni.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 144.Monneaux F, Lozano JM, Patarroyo ME, Briand JP, Muller S. T cell recognition and therapeutic effect of a phosphorylated synthetic peptide of the 70K snRNP protein administered in MR/lpr mice. Eur J Immunol. 2003;33:287–296. doi: 10.1002/immu.200310002. [DOI] [PubMed] [Google Scholar]
- 145.Page N, et al. The spliceosomal phosphopeptide P140 controls the lupus disease by interacting with the HSC70 protein and via a mechanism mediated by gammadelta T cells. PLoS ONE. 2009;4:e5273. doi: 10.1371/journal.pone.0005273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Monneaux F, et al. Selective modulation of CD4+ T cells from lupus patients by a promiscuous, protective peptide analog. J Immunol. 2005;175:5839–5847. doi: 10.4049/jimmunol.175.9.5839. [DOI] [PubMed] [Google Scholar]
- 147.Muller S, et al. Spliceosomal peptide P140 for immunotherapy of systemic lupus erythematosus: results of an early phase II clinical trial. Arthritis Rheum. 2008;58:3873–3883. doi: 10.1002/art.24027. [DOI] [PubMed] [Google Scholar]
- 148.Riemekasten G, et al. Intravenous injection of a D1 protein of the Smith proteins postpones murine lupus and induces type 1 regulatory T cells. J Immunol. 2004;173:5835–5842. doi: 10.4049/jimmunol.173.9.5835. [DOI] [PubMed] [Google Scholar]
- 149.Ferrera F, La Cava A, Rizzi M, Hahn BH, Indiveri F, Filaci G. Gene vaccination for the induction of immune tolerance. Ann N Y Acad Sci. 2007;1110:99–111. doi: 10.1196/annals.1423.012. [DOI] [PubMed] [Google Scholar]
- 150.Newell EW, Klein LO, Yu W, Davis MM. Simultaneous detection of many T-cell specificities using combinatorial tetramer staining. Nat Methods. 2009;6:497–499. doi: 10.1038/nmeth.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Joos T, et al. A microarray enzyme-linked immunosorbent assay for autoimmune diagnostics. Electrophoresis. 2000;21:2641–2650. doi: 10.1002/1522-2683(20000701)21:13<2641::AID-ELPS2641>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 152.Robinson W, et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nature Med. 2001;8:295–301. doi: 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- 153.Balboni I, Chan SM, Kattah M, Tenenbaum JD, Butte AJ, Utz PJ. Multiplexed protein array platforms for analysis of autoimmune diseases. Annu Rev Immunol. 2006;24:391–418. doi: 10.1146/annurev.immunol.24.021605.090709. [DOI] [PubMed] [Google Scholar]
- 154.Chan SM, Utz PJ. The challenge of analyzing the proteome in humans with autoimmune diseases. Ann N Y Acad Sci. 2005;1062:61–68. doi: 10.1196/annals.1358.009. [DOI] [PubMed] [Google Scholar]
- 155.Balboni I, Limb C, Tenenbaum JD, Utz PJ. Evaluation of microarray surfaces and arraying parameters for autoantibody profiling. Proteomics. 2008;8:3443–3449. doi: 10.1002/pmic.200800146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Graham KL, Vaysberg M, Kuo A, Utz PJ. Autoantigen arrays for multiplex analysis of antibody isotypes. Proteomics. 2006;6:5720–5724. doi: 10.1002/pmic.200600345. [DOI] [PubMed] [Google Scholar]
- 157.Rouquette AM, Desgruelles C, Laroche P. Evaluation of the new multiplexed immunoassay, FIDIS, for simultaneous quantitative determination of antinuclear antibodies and comparison with conventional methods. Am J Clin Pathol. 2003;120:676–681. doi: 10.1309/GJHK-0D24-YDDX-W0NF. [DOI] [PubMed] [Google Scholar]
- 158.Martins TB, Burlingame R, von Muhlen CA, Jaskowski TD, Litwin CM, Hill HR. Evaluation of multiplexed fluorescent microsphere immunoassay for detection of autoantibodies to nuclear antigens. Clin Diagn Lab Immunol. 2004;11:1054–1059. doi: 10.1128/CDLI.11.6.1054-1059.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ray CA, et al. Development, validation, and implementation of a multiplex immunoassay for the simultaneous determination of five cytokines in human serum. J Pharm Biomed Anal. 2005;36:1037–1044. doi: 10.1016/j.jpba.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 160.Poole BD, et al. Early targets of nuclear RNP humoral autoimmunity in human systemic lupus erythematosus. Arthritis Rheum. 2009;60:848–859. doi: 10.1002/art.24306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Takeda Y, et al. Antigenic domains on the U1 small nuclear ribonucleoprotein-associated 70K polypeptide: a comparison of regions selectively recognized by human and mouse autoantibodies and by monoclonal antibodies. Clin Immunol Immunopathol. 1991;61:55–68. doi: 10.1016/s0090-1229(06)80007-3. [DOI] [PubMed] [Google Scholar]
- 162.Welin Henriksson E, Wahren-Herlenius M, Lundberg I, Mellquist E, Pettersson I. Key residues revealed in a major conformational epitope of the U1-70K protein. Proc Natl Acad Sci USA. 1999;96:14487–14492. doi: 10.1073/pnas.96.25.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.James JA, Scofield RH, Harley JB. Basic amino acids predominate in the sequential autoantigenic determinants of the small nuclear 70K ribonucleoprotein. Scand J Immunol. 1994;39:557–566. doi: 10.1111/j.1365-3083.1994.tb03413.x. [DOI] [PubMed] [Google Scholar]