

Abstract
The design and general synthesis of enantiopure isoxazolidinone monomers as precursors for the preparation of enantiopure N-terminal hydroxylamine—β3-oligopeptides, which may be used as reaction partners with α-ketoacids in the decarboxylative amide ligation reaction, is described.
Keywords: Enantiopure isoxazolidinone monomers, β3-oligopeptides, Amide ligation
1. Introduction
β3-Oligopeptides have emerged as one of the most powerful and versatile classes of foldamers1 for the design of molecules with defined higher order structures and properties. Pioneering studies by Seebach2 and Gellman3 established the β3-oligopeptide motif as a promising venue in which to design molecules that possess predictable secondary and tertiary structure4 that arise from a discrete sequence of β3-amino acid residues. These principles have been widely exploited in the design, synthesis, and study of β3-oligopeptides, or structures incorporating β3-amino acids, with a wide range of biological properties including antimicrobial activity,5 the disruption of protein–protein interactions,6 and inhibition of γ-secretase.7 Recently, Schepartz has demonstrated that a designed β3-oligopeptide possesses not only secondary and tertiary structure but quaternary structure as well, and forms helical bundles.8 Gell-man and Hilvert have prepared a β3-oligopeptide that functions as an artificial enzyme.9 Taken together, these studies establish synthetic β3-oligopeptides as the most promising platform for the design of artificial macromolecules that can offer the properties and activities of natural α-oligopeptides with advantages including stable secondary structure with short peptide sequences, improved metabolic stability, and ease of incorporating unnatural functional groups for engineering novel reactivity.
One of the greatest obstacles to the continued development of this exciting field is the synthetic access to higher order β3-oligopeptides. For example, the Zwit-1F protein designed by Schepartz10 consists of 28 β3-amino acid residues and must be synthesized using harsh and often inefficient peptide coupling and Fmoc-deprotection conditions as the peptide increases in length.1b Fragment couplings of medium size (5–10 β-amino acid residues) are an attractive alternative to the direct synthesis of longer peptides, but poor solubility of the protected peptide fragments and concomitant inefficiency in the key amide-forming reaction detracts from this approach. The logical alternative, native chemical ligation11 of unprotected β-oligopeptide fragments, has been explored by Seebach12 and has utility in certain cases. It is not applicable, however, to the synthesis of β-oligopeptides that lack a thiol-containing side chain. Furthermore, the synthesis of the necessary enantiopure β3-cysteine amino acid is lengthy and complicated.13
As part of our efforts to improve synthetic access to β3-oligopeptides, we have previously reported an alternative and orthogonal route to their preparation by the chemoselective amide-forming ligation of α-ketoacids and cyclic hydroxylamines (Fig. 1).14 This reaction does not require any coupling reagents or side chain protection during peptide bond formation, operates under aqueous conditions, and produces CO2 as the only reaction byproduct. It proceeds in the inverse, N→C synthetic direction.15 Furthermore, the necessary monomers are synthetically accessible by a convenient diastereoselective nitrone cycloaddition. In our published work,15 we demonstrated the solution phase synthesis of short β3-oligopeptides using this method, and our ongoing work has extended this to the solid-supported preparation of longer, fully unprotected peptide fragments. We have also greatly improved and optimized the synthesis of the requisite isoxazolidine monomers.
Figure 1.

Iterative, aqueous synthesis of β3-oligopeptides via α-ketoacid–isoxazolidine couplings.
2. Results and discussion
In the course of our studies we recognized that the unique N→C synthetic direction of this amide-ligation approach to β3-oligopeptide synthesis offers an exciting opportunity to interface with the traditional Fmoc-based peptide coupling approach to β3-amino acid synthesis. The products of our ketoacid–isoxazolidine-based synthesis are side-chain unprotected C-terminal β3-peptide α-ketoacids I that would be ideally suited to react with β3-oligopeptide containing an N-terminal hydroxylamine II in a fragment ligation strategy by α-ketoacid–hydroxylamine ligation (Scheme 1). Importantly, we have demonstrated, in the context of α-peptide synthesis,16 that this ligation reaction meets all the criteria for a truly chemoselective peptide-forming reaction and can operate at low reactant concentrations (10 mM) and with equimolar reactant stochiometries, which are usually encountered during attempted fragment couplings.
Scheme 1.

Decarboxylative amide formation reaction between a terminal α-ketoacid and a hydroxylamine.
In order to design a practical β3-peptide ligation strategy, we required a suitable method for the synthesis, protection, incorporation, and deprotection of enantiomerically pure β3-peptide hydroxylamines onto the N-terminus of a synthetic β3-oligopeptide. As part of ongoing work on the synthesis of enantiopure N-hydroxy-α-amino acids we have employed several approaches that involve the hydrolysis of nitrone intermediates.17 In considering an extension of this strategy to the synthesis of the corresponding β3-oligopeptides, we were concerned about the potential for nitrone elimination via retro-Michael reaction with the β3-amino acids and deterred by the need for a multi-step transformation of the precious, enantiomerically pure β3-amino acid derivatives. We there-fore sought a de novo synthesis of enantiomerically pure N-hydroxy-β3-amino acid monomers as well as new methods to incorporate them onto the N-terminus of a β3-oligopeptide chain.
This article documents our successful strategy for the asymmetric synthesis of enantiopure isoxazolidinone monomers that correspond to common β-amino acids, a method for their direct coupling with amine and peptide nucleophiles, and their deprotection to give the desired β-N-hydroxyamino peptides (Scheme 2). These procedures allow access to the requisite fragments for our ongoing work on developing a general strategy for the synthesis of β3-oligopeptides by the combination of Fmoc-based peptide synthesis, iterative α-ketoacid–isoxazolidine peptide-formation, and α-ketoacid–hydroxylamine ligations for fragment condensations.
Scheme 2.

Retrosynthesis of the terminal N-hydroxylamine β3-oligopeptide.
At the outset of our studies, we recognized that three distinct but interconnected successes would be necessary for achieving our goal of a general, convenient method for the synthesis of N-terminal β3-peptide hydroxylamines: (1) a suitable protecting group strategy for masking the sensitive hydroxylamine, (2) a method for the introduction of a suitable monomer onto the peptide chain via amide-bond formation and (3) a reliable method for the preparation of such monomers with >99% ee.
With these goals in mind, we were initially attracted to a 2007 report by Cordova on the enantioselective addition of carbamate-protected hydroxylamines to α,β-unsaturated aldehydes (Scheme 3), catalyzed by (S)-O-TMS-diphenylprolinol 10.18 The resulting cyclic acetal was oxidized to the isoxazolidinone carbamate, which appeared to be ideal precursors to β3-peptide hydroxylamines.
Scheme 3.

Organocatalytic formation of enantioenriched isoxazolidinones from α,β-unsaturated aldehydes (TPAP=tetrapropylammonium perruthenate, NMO N-methyl-morpholine N-oxide).
We were pleased to find that Cbz-protected isoxazolidinone 11 could be readily coupled to an amine by stirring the two reactants in CH2Cl2 or DMF (Scheme 4). Despite this promising result, two obstacles remained. First, the benzyl carbamate was an unsuitable protecting group for our eventual goal as it required hydrogenolysis for its deprotection, which we feared could cleave the N–O bond. Second, despite numerous attempts to improve the enantiose-lectivity or enrich the enantiopurity of the isoxazolidines, we were unable to prepare these monomers in >95% ee. This was particularly true of the aliphatic substituted variants that were essential to our eventual β3-peptide ligation strategy.
Scheme 4.

Coupling of N-Boc isoxazolidinone with an N-terminal amine.
Our studies did establish the use of carbamate protected isoxazolidinones for the protection and introduction of the β3-N-hydroxyamino acid-residues into peptides, and our goals subsequently shifted to a reliable preparation of such monomers in enantiomerically pure form (Fig. 2).
Figure 2.

Boc protected isoxazolidinone monomers.
In our parallel work on the synthesis and use of isoxazolines for β-peptide synthesis, we had outstanding results with several variations of Vasella’s d-mannose derived hydroxylamine19 as a chiral auxiliary for diastereoselective nitrone cycloadditions.20 Most importantly, most of the cycloadducts were crystalline solids that could be enriched to diastereo- and enantiopurity by re-crystallization. The d-mannose derived auxiliary, however, afforded the absolute stereochemistry opposite of that found in the ‘pseudonatural’ β3-amino acids (Fig. 3). The corresponding l-mannose is prohibitively expensive for use as a chiral auxiliary. Kibayashi21 has reported the use of d-gulose as a convenient surrogate for l-mannose and we had already adopted this auxiliary in our ongoing studies. The use of gulose derived auxiliaries brings the additional benefits of higher diastereoselectivities in the nitrone cycloadditions, increased crystallinity of the cycloadducts, and the ready availability of both d- and l-gulose-derived lactones as starting materials. As we were already using and producing this auxiliary on scale, seeking its application to the preparation of these enantiopure monomers seemed ideal.
Figure 3.

Absolute configurations of β3-amino acids prepared from d-mannose and d-gulose-derived chiral auxiliaries.
Several cycloaddition strategies were explored. The most attractive, from asynthetic point of view, was the use of commercially available substituted acrylonitriles 19 or 20.22,23 Indeed, the combination of d-gulose derived hydroxylamine 1, an aldehyde, and either of these acrylates afforded cycloadducts 21 or 22 in respectable yield. In both cases, treatment of these products with NEt3 and H2O afforded isoxazolidinones 23 or 24 in good yield. Following recrystallization, these products were obtained as single diastereomers in enantiomerically pure form (Scheme 5).
Scheme 5.

Preparation of isoxazolidinone monomers starting from a cyanoacrylate.
The only problem with this approach was the limited availability of both α-acetoxyacrylonitrile (19) and α-chloroacrylonitrile (20). These restricted substances are not available to research laboratories on scale and commercial supplies of even small quantities were unreliable. The hazardous nature of their synthesis, while conducted at times in our lab, effectively prohibited their preparation in the quantities we desired. Therefore, despite these encouraging results we were forced to consider alternatives.
An alternative was inexpensive, widely available vinyl acetate (26).24 Its low cost allowed us to use an excess of this olefin (10 equiv) as the reaction solvent (Scheme 6). Following nitrone cycloaddition, acetate hydrolysis and lactol oxidation afforded identical isoxazolidinones as were prepared from the substituted acrylates.
Scheme 6.

Preparation of isoxazolidinone monomers from vinyl acetate.
Unfortunately, this cycloaddition afforded a much poorer ratio of diastereomers and it was difficult to separate and isolate the desired cycloadduct in pure form. In some cases, this made it difficult or impossible to prepare the isoxazolidinone in enantiomerically pure form. Furthermore, although the vinyl acetate itself was an inexpensive reagent, this approach required additional acetate hydrolysis and lactol oxidation steps that detracted from its overall appeal.
We therefore explored an alternative cycloaddition strategy based on our prior experience with α-ketoacids and their derivatives. We believed that monomers of the type 34 or 35, which were structurally very similar to those we had prepared for our iterative β3-peptide synthesis,15 could be converted by oxidative decarboxylation to the desired isoxazolidinones (Scheme 7).
Scheme 7.

Decarboxylative oxidation to the isoxazolidinone monomers.
Following the preparation of cycloadducts derived from either acrylate 32 or 33, we explored conditions for hydrolysis and oxidative decarboxylation under basic conditions (Table 1). With 34, derived from acrylate 32, only trace amounts of the desired product were obtained upon treatment with basic peroxide. Increasing the reaction time or changing the base did not improve the results. In contrast, cycloadducts 35 derived from acrylate 33 could be oxidatively decarboxylated in good yield upon stirring overnight with potassium carbonate and hydrogen peroxide (Scheme 8). This general procedure was applied to the monomers corresponding to β3-leucine, valine, phenylalanine, and alanine in good to excellent yields.
Table 1.
Screening of conditions for the decarboxylative oxidation
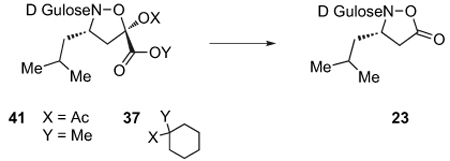 | |||||
|---|---|---|---|---|---|
| Entry | Acetal | Solvent | T/°C | Time/h | Yielda |
| 1 | 41 | NaOH/H2O2 | rt | 1 h | Trace |
| 2 | 41 | K2CO3/H2O2 equiv 5:5 |
rt→50°C | 16 h | No reac |
| 3 | 41 | NaOH/H2O2 | rt | 16 h | decomp. |
| 4 | 37 | NaOH/H2O2 | rt | 16 h | decomp. |
| 5 | 37 | K2CO3/H2O2b equiv 5:5 |
rt | 16 h | 82% |
| 6 | 37 | NaOH/H2O2 | rt | 3 h | 53% |
Isolated yield.
After 1 h an additional 5 equiv K2CO3/5 equiv H2O2 are added and then stirred overnight.
Scheme 8.

Decarboxylative oxidation to the isoxazolidinone monomers.
The key advantage of this approach overall of the others considered is the ease of securing the cycloadducts in enantiomerically pure form.25 Cycloadditions of nitrones generated in situ from an aldehyde and the d-gulose-derived chiral auxiliary are generally cleaner, higher yielding, and more diastereoselective with cyclic acrylate 33. In almost all cases, the cycloadducts are obtained as single enantiomers following a facile recrystallization.
These cycloadditions with many different aldehydes and the utility of the resulting enantiomerically pure products for β3-amino acid synthesis are the subject of a separate publication.25 For the purpose of our studies on the incorporation of β3-N-hydroxyamino acids into peptides, we selected the aliphatic substituted monomers (leucine, valine, phenylalanine, and alanine) for further investigations.
Removal of the carbohydrate auxiliary from the isoxazolidinone proved to be initially more difficult than expected. These lactones are hydrolytically unstable, and the standard conditions for auxiliary removal, perchloric acid in MeOH,15 both cleaved the auxiliary and opened the lactone to give the corresponding methyl esters (Table 2, entries 1–3). Decreased reaction times and lower reaction temperatures allowed us to isolate some of the desired product (entries 7–8).
Table 2.
Cleavage of the sugar auxiliary
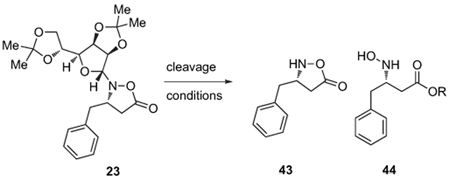 | |||||
|---|---|---|---|---|---|
| Entry | equiv HClO4 | Solvent | T/°C | Time/h | Conversion (43/44)a |
| 1 | 2 | MeOH | 60°C | 3 h | 99% (0/100) |
| 2 | 0.5 | MeOH | rt | 3 h | 99% (0/100) |
| 3 | 2 | MeOH | rt | 3 h | 99% (0/100) |
| 4 | 2 | iPrOH | 60°C | 3 h | 99% (0/100) |
| 5 | 2 | iPrOH | 60°C | 30 min | 23 |
| 6 | 2 | EtOH | 60°C | 30 min | Trace |
| 7 | 2 | MeOH | rt | 1 h | 56% (60/40) |
| 8 | 2 | MeOH | rt | 35–50 min | 53% (70/30) |
| 9 | 2 | THF/H2O 4:1 | rt | 3 h | Trace |
| 10 | 2 | Dioxane/H2O 4:1 | rt | 3 h | Trace |
| 11 | 2 | DME/H2O 4:1 | rt | 3 h | Trace |
| 12 | 3 | MeCN/H2O 3:1 | rt | 3 h | 99%b [100/0] |
| 13 | 3 | MeCN | rt | 3 h | 99%c [100/0] |
Conversion determined by 1H NMR spectroscopy. Ratio of products 43/44 is given in brackets.
The isolated yield after column chromatography was 62%.
The isolated yield after column chromatography was 84%.
The use of other alcohols, however, required higher temperatures and did not favor the auxiliary removal (entries 4–6). Eventually, we found that conducting the auxiliary cleavage in CH3CN at room temperature afforded clean removal without lactone opening. This convenient procedure has proven to be generally useful for removal of the d-gulose-derived auxiliary from a number of cyclic hydroxylamine derivatives. It was applied to our targeted monomers to give the desired products in good yield (Fig. 4).
Figure 4.

Free isoxazolidinones prepared by auxiliary removal.
Although our NMR evidence clearly showed only a single diastereomer, we carefully checked the enantiopurity of the isoxazolidinones prepared by this cycloaddition route. Direct assay of the enantiopurity of the sensitive unprotected isoxazolidines by HPLC or SFC analysis on chiral columns was complicated by decomposition. We therefore elected to convert them to their corresponding N-phenylethylamide esters by ligation with phenylpyruvic acid followed by methyl ester formation. Analysis by SFC on chiral columns and comparison to a racemic sample established the enantiopurity (Scheme 9). The absolute configuration was confirmed by single crystal X-ray analysis of the cycloadduct ent-37.25
Scheme 9.

Amide ligation of the isoxazolidinone monomers with α-ketoacids.
The final step of the monomer synthesis was the introduction of the Boc-group. This group served both as a protecting group for the hydroxylamine and also activated the isoxazolidinone toward amide-formation with an amine or peptide. In all cases examined, this was accomplished with Boc anhydride and DMAP in moderate to good yield (Scheme 10). These products were purified by column chromatography and were stable for weeks when stored at 4 °C.
Scheme 10.

Boc protection of the isoxazolidinone monomers.
As a model study for our intended application of these monomers to the preparation of N-terminal β3-peptide hydroxylamines and their ligation with α-ketoacids, we sought to introduce the β-N-hydroxyamino acid onto a β3-dipeptide (Scheme 11). This study was aimed to both identify conditions for the coupling of the monomer and to establish that the Boc-deprotection could be effected without damage to the hydroxylamine.
Scheme 11.

Preparation of an N-hydroxylamine terminal dipeptide and ligation with an α-ketoacid.
With simple amines, mixing the Boc-protected monomer in CH2Cl2 afforded the desired amide. With the more sterically demanding β3-peptides, however, this protocol was not effective. Fortunately, a brief course of optimization identified the combination of DMF and 0.5 equiv of DMAP at 50°C as suitable for direct introduction of the N-hydroxylamino acid. Removal of the Boc-group with TFA proceeded smoothly and ligation with phenyl-pyruvic acid afforded the expected triamide 56.
3. Conclusion
In summary, we have designed and implemented a scalable route to enantiopure protected isoxazolidinone monomers, which serve as precursors to the N-terminal hydroxylamine–β3-oligopetides that can be used in amide ligation reaction with α-ketoacids–β3-oligopetides. Our four-step route to the monomers takes advantage of a general 1,3-cycloaddition procedure to access enantiopure cycloadducts that may be readily oxidized to the crystalline isoxazolidinones. This robust methodology for the preparation of N-isoxazolidinone amino acid derivatives makes possible our ongoing efforts to access β3-oligopeptides by chemoselective ligation of two independently prepared β3-oligopeptide units.
4. Experimental
4.1. General methods
All reactions utilizing air- or moisture-sensitive reagents were performed in oven-dried glassware under an atmosphere of dry N2. CH2Cl2 was distilled from CaH2. THF and Et2O were distilled from Na/benzophenone. Thin-layer chromatography (TLC) was performed on EMD precoated plates (silica gel 60 F254, Art 5715, 0.25 mm) and were visualized by fluorescence quenching under UV light and by staining with phosphomolybdic acid or potassium permanganate. Preparative thin-layer chromatography (PTLC) was performed using plates prepared from silica gel EMD 60 PF254 (Art 7749). Column chromatography was performed on EMD Silica Gel 60 (230–400 mesh) using a forced flow of 0.5–1.0 bar. 1H NMR (500 MHz) and 13C NMR (125 MHz) were measured on a Bruker Avance AVII-500 spectrometer. Chemical shifts are expressed in parts per million (ppm) and coupling constants are reported as Hertz (Hz). Splitting patterns are indicated as follows: br, broad; s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Infrared (IR) spectra were recorded on a JASCO FT/IR-4100 spectrophotometer and are reported as wavenumber (cm−1). Enantiomeric purity was determined by preparation of both enantiomers of the peptide followed by analysis on chiral SFC.
4.1.1. SFC (supercritical fluid chromatography) conditions
Column: Daicel Chiralpak OJ-H (4.6×250 mm). Eluents: gradient 5–80% iPrOH (0.1% TFA v/v) in CO2, rate 3%/min or 5%/min, Flow rate 2.0 ml/min; isocratic iPrOH (0.1% TFA v/v) in CO2, Flow rate 2.0 ml/min. Detection: 220 nm.
4.1.2. 2,3,5,6-O-Diisopropylidene-d-gulose hydroxylamine (1)
The d-gulose hydroxylamine 1 was prepared following a previously published literature procedure and scaled up to 1 mol.21,26–29 Spectroscopic data was consistent with literature results.
4.1.3. Synthesis of 3-methylene-1,4-dioxaspiro[4.5]decan-2-one (33)
The acrylate was prepared following a previously published literature procedure and scaled up to 1 mol. Spectroscopic data was consistent with literature results.
4.1.4. Synthesis of methyl 2-acetoxyacrylate (32)
The acrylate was prepared following a previously published literature procedure30 and scaled up to 1 mol. Spectroscopic data was consistent with literature results.
4.2. General procedure A. Nitrone cycloadditions with the gulose derived chiral auxiliary
A mixture of d-gulose oxime 1 (14.5 mmol, 1.00 equiv), aldehyde (43.5 mmol, 3.00 equiv), and vinyl acetate 26 (0.14 mol, 10.00 equiv) was heated at 110 °C in a sealed tube for 16 h. With stirring, in some cases a minimal amount of toluene (1.0–2.0 mL) was added to form a solution. After cooling to room temperature, the resulting crude product was purified by flash chromatography to obtain a mixture of three diastereomers in a ratio of 9:6:2, determined by 1H NMR. In some cases the crude solid (or viscous oil) was recrystallized from heptane (50 mL/g) to give a single diastereomer of the pure cycloadduct.
4.3. General procedure B. Nitrone cycloadditions with the gulose derived chiral auxiliary
A 0.3 M solution of d-gulose oxime 1 (7.26 mmol, 1.00 equiv), aldehyde (14.5 mmol, 2.00 equiv), and spiroacrylate 33 (8.71 mmol, 1.20 equiv) in toluene was heated to reflux with a Dean–Stark trap fitted with a reflux condenser overnight. The reaction was monitored by TLC for the disappearance of the UV active nitrone spot. After cooling to room temperature the mixture was concentrated under reduced pressure. The crude product was purified by flash chromatography and all the observed diastereomers were separated (three of the four possible diastereomers, except in the case of the alanine derivative, in which all of the diastereomers were observed). The resulting solid (or sometimes viscous oil) was recrystallized from heptane (50 mL/g) to give the cycloadduct as a single diastereomer.
4.4. General procedure C. Nitrone cycloadditions with the gulose derived chiral auxiliary
A 0.3 M solution of d-gulose oxime 1 (3.63 mmol, 1.00 equiv), aldehyde (4.00 mmol, 1.10 equiv), and acetoxy 2-methoxy acrylate 32 (7.26 mmol, 2.00 equiv) in toluene was heated to reflux with a Dean–Stark trap fitted with a reflux condenser. The reaction was monitored by TLC for the disappearance of the UV active nitrone spot. After cooling to room temperature the solvent was removed under reduced pressure. The crude product was purified by flash chromatography and two of the four possible diastereomers, were isolated as a 6:1 mixture. The resulting solid (or sometimes viscous oil) was recrystallized from heptane (50 mL/g) to give the cycloadduct as a single diastereomer.
4.5. General procedure D. Nitrone cycloadditions with the gulose derived chiral auxiliary
A 1 M solution of d-gulose oxime 1 (0.73 mmol, 1.00 equiv), aldehyde (1.46 mmol, 2.00 equiv), and 2-chloroacrylonitrile 19 (2.18 mmol, 3.00 equiv) was dissolved in toluene and heated at 100 °C in a sealed tube overnight. After cooling to room temperature the resulting crude residue was purified by flash chromatography to obtain a 4:1 mixture of two of the four possible diastereomers. The resulting solid (or sometimes viscous oil) was recrystallized from heptanes (50 mL/g) to give the cycloadduct as a pure single diastereomer.
4.6. General procedure E. Nitrone cycloadditions with the gulose derived chiral auxiliary
A 1 M solution of d-gulose oxime 1 (0.73 mmol, 1.00 equiv), aldehyde (1.46 mmol, 3.00 equiv), and α-acetoxyacrylonitrile 20 (2.18 mmol, 3.00 equiv) was dissolved in toluene and heated at 115 °C in a sealed tube overnight. After cooling to room temperature the resulting crude residue was purified by flash chromatography to give a 1:1 mixture of two of the four possible diastereomers.
4.7. General procedure F. Auxiliary cleavage with HClO4
To a 0.1 M solution of the gulose-isoxazolidinone (0.32 mmol, 1.00 equiv) in MeCN was slowly added HClO4 (0.97 mmol, 70% w/w in H2O, 3.00 equiv), and the solution was stirred at room temperature for 3 h. The resulting mixture was quenched by the slow addition of saturated NaHCO3 solution and extracted with EtOAc (3×). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography to afford the free isoxazolidinone.
4.8. General procedure G1. Oxidation of the spirocyclohexanone acetal to the isoxazolidinone
A solution of d-gulose-isoxazolidine cyclohexanone acetal 35 (1.55 mmol, 1.00 equiv) in THF/H2O (6:1 v/v, 0.1 M) was cooled to 0 °C. Then K2CO3 (7.77 mmol, 5.00 equiv) and H2O2 (30% w/w, 5.00 equiv) were slowly added. After stirring the reaction for 1 h at room temperature, another 5.00 equiv (7.77 mmol) of both K2CO3 and H2O2 were added. The reaction was monitored by TLC, and in some cases it was stirred overnight. The resulting mixture was quenched by the addition of saturated Na2S2O7 (2×) and extracted with EtOAc (3×). The combined organic layers were washed with NaHCO3 (3×) and NH4Cl (3×), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography to afford the isoxazolidinone as a white solid.
4.9. General procedure G2. Oxidation of the spirocyclohexanone acetal to the isoxazolidinone
A 0.1 M solution of d-gulose-isoxazolidine cyclohexanone acetal 35 (1.55 mmol, 1.00 equiv) in THF was cooled down to 0 °C and 2 M NaOH (7.77 mmol, 5.00 equiv) and H2O2 (30% w/w, 5.00 equiv) were slowly added. After the reaction was stirred for 1 h at room temperature, another 5.00 equiv of both NaOH and H2O2 were added. The reaction was monitored by TLC, but was never stirred longer than 3 h because it started to decompose. After completion the resulting mixture was quenched by the addition of saturated Na2S2O7 (2×) and extracted with EtOAc (3×). The combined organic layers were washed with NaHCO3 (3×) and NH4Cl (3×), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography to afford the isoxazolidinone as a white solid.
4.10. General procedure H. Deprotection of the acetate to the isoxazolidine acetal
To a solution of d-gulose-isoxazolidine acetate (3.22 mmol, 1.00 equiv) in MeOH/H2O (10:1 v/v, 0.1 M) was added K2CO3 (1.00 equiv), and the solution was stirred for 1–2 h at room temperature. The resulting reaction mixture was quenched by the addition of saturated NH4Cl (3×) and extracted with a 9:1 mixture of CHCl3/iPrOH (3×), the combined organic layers were washed with brine (3×), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography to afford the isoxazolidine acetal.
4.11. General procedure I. Oxidation of the acetate to the isoxazolidinone
NMO (2.12 mmol, 1.20 equiv) and TPAP (0.17 mmol, 10 mol %) was added to a 0.1 M solution of d-gulose-isoxazolidine acetal (1.76 mmol, 1.00 equiv) and preactivated molecular sieves (4 Å) in dry CH2Cl2. After stirring for 1 h at room temperature, hexanes (×1/2 V of CH2Cl2) was then added to the reaction and stirred for another 15 min. The resulting mixture was filtered through silica gel, washed with EtOAc and the combined organic solvents were concentrated under reduced pressure. The resulting residue was purified by column chromatography to afford the isoxazolidinone.
4.12. General procedure J. Deprotection of the cyanoacetate to the isoxazolidinone
To a solution of d-gulose-isoxazolidine cyanoacetate (1.00 equiv) in THF/H2O (4:1 v/v, 0.5 M) was added Et3N (2.00 equiv), and the mixture was stirred overnight at room temperature. The resulting reaction mixture was quenched by the addition of saturated NH4Cl (3×) and extracted with EtOAc (3×). The combined organic layers were washed with brine (3×), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography to afford the isoxazolidinone (62–75% yield).
4.13. General procedure K. Boc protection of the free isoxazolidinone
A 0.1 M solution of the free isoxazolidinone (0.34 mmol, 1.00 equiv) in dry MeCN was cooled to 0 °C. Then Boc2O (0.51 mmol, 1.50 equiv) and DMAP (0.25–0.62 mmol, 0.50–1.20 equiv; it is important not to add an excess to avoid decomposition) were added and stirred at room temperature. The reaction was stirred for 1–3 h. After completion by TLC, the mixture was concentrated under reduced pressure and the resulting residue was purified by column chromatography to afford the N-Boc protected isoxazolidinone.
4.14. General procedure L. Cbz-protected isoxazolidinone from the α,β unsaturated aldehyde11
To a 0.1 M solution of cat 10 (0.11 mmol, 20 mol %) in CH2Cl2 were added α,β-unsaturated cynnamaldehyde 9 (0.57 mmol, 1.00 equiv) and CbzNHOH (0.68 mmol, 1.00 equiv) at 0 °C. After stirring the reaction for 3 h, it was concentrated down under reduced pressure and purified by column chromatography to afford the isoxazolidinone in 94% yield as a yellow oil. The compound was oxidized to the corresponding isoxazolidinone according general procedure I.
4.15. General procedure M. Isoxazolidinone opening to the amide bond and Cbz protected hydroxylamine
To a 0.1 M solution of Cbz-protected isoxazolidinone 11 (40.0 mg, 0.135 mmol) in dry CH2Cl2 was added 1,2-phenylethyl amine 12 (0.019 mL, 0.148 mmol). After 2 h stirring at room temperature, the reaction mixture was concentrated under reduced pressure. The resulting residue was purified by column chromatography to afford coupled N-Cbz hydroxylamine 13 as a white solid.
4.16. General procedure N. Isoxazolidinone opening to the amide bond and Cbz protected hydroxylamine
To a 0.01 M solution of the amino-β-Phe-β-Ala-OEt 54 (0.034 mmol, 1.00 equiv) in DMF, Boc-Leu isoxazolidinone 3 (0.068 mmol, 2.00 equiv) was added and stirred at 60 °C. After 16 h, the reaction mixture was concentrated under reduced pressure and the resulting residue was purified by column chromatography to afford the N-Boc-terminal-hydroxylamine tripeptide 55 as a yellow oil.
4.17. General procedure O. Deprotection and coupling with phenylpyruvic acid to the tripeptide
HO-Boc-N-β-Leu-β-Phe-β-Ala-OEt 55 (0.066 mmol, 1.00 equiv) was stirred in a 0.01 M solution of 1:1 CH2Cl2/TFA for 1 h at room temperature. The reaction mixture was then concentrated under reduced pressure. The TFA salt of the hydroxylamine 4 (0.066 mmol, 1.00 equiv) was then dissolved in a 0.1 M solution of 1:1 tBuOH/H2O, and phenylpyruvic acid 48 (0.066 mmol, 1.00 equiv) was added. The reaction was stirred overnight at 50 °C and the resulting residue was concentrated down and purified by column chromatography to afford the N-benzyl tripeptide 56.
4.18. General procedure P. Coupling of deprotected isoxazolidines with phenylpyruvic acid
To a 0.15 M solution of isoxazolidinone (0.068 mmol, 1.00 equiv) in tBuOH/H2O (1:1) was added phenylpyruvic acid 48 (0.074 mmol, 1.10 equiv) and heated at 45 °C for 16 h. The reaction mixture was directly concentrated under reduced pressure. The resulting residue was dissolved in MeOH 0.1 M. After cooling to 0 °C, TMSCHN2 2.0 M in Et2O (0.14 mmol, 2.00 equiv) was slowly added and stirred for another 10 min. The reaction mixture was then concentrated and purified by column chromatography to afford the amide product. These reactions were not optimized and were performed only to obtain material suitable for determining the enantiopurity by SFC analysis.
4.18.1. d-Gulose-(S)-3-isobutylisoxazolidin-5-yl acetate (29)
Prepared according to general procedure A. Purification by column chromatography using hexanes/EtOAc (6:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 74% yield. Rf=0.25 (hexanes/EtOAc 4:1); recrystallization from heptane provided a single diastereomer. mp149–150 °C; 1H NMR (500 MHz, CDCl3) δ 0.91 (dd, J=6.3, 13.6 Hz, 6H), 1.11 (m, 1H), 1.26 (s, 3H), 1.37 (s, 3H), 1.41 (s, 3H), 1.43 (s, 3H), 1.58 (m, 1H), 1.68 (m, 1H), 2.07 (s, 3H), 2.17 (m, 1H) 2.50 (dd, J=7.5, 13.2 Hz, 1H), 3.72 (m, 2H), 4.07 (m, 1H), 4.37 (q, J=4.2 Hz, 1H), 4.65 (m, 1H), 4.80 (m, 1H), 4.91 (d, J=5.6 Hz, 1H), 6.39 (d, J=5.2 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 170.4, 112.8, 109.8, 99.0, 98.3, 84.2, 84.1, 80.6, 75.6, 66.2, 59.4, 44.3, 40.9, 26.8, 26.2, 25.6, 25.4, 25.1, 23.2, 22.1, 21.4; IR (thin film) ν 3420, 2955, 1745, 1653, 1372, 1236, 1111, 1088, 1066, 938, 850; HRMS (ESI): m/z: calcd for C21H35NO8: 429.2363; found: 452.2254 [M+Na]+.
4.18.2. d-Gulose-(S)-3-isobutylisoxazolidin-5-ol (7)
Prepared according to general procedure H. Purification by column chromatography using hexanes/EtOAc (2:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 82% yield (5:1 dr). Rf=0.23 (hexanes/EtOAc 2:1); mp135–136 °C; 1H NMR (500 MHz, CDCl3) δ 0.88 (qd, J=10.1, 6.4 Hz, 6H), 1.07 (m, 1H), 1.26 (s, 3H), 1.35 (s, 3H), 1.39 (s, 3H), 1.42 (s, 3H), 1.52 (m, 1H), 1.62 (m, 1H), 2.02 (m, 1H), 2.39 (m, 1H), 3.70 (m, 2H), 3.90 (m, 1H), 4.05 (m, 1H), 4.17 (m, 1H), 4.37 (m, 1H), 4.66 (m, 1H), 4.92 (m, 1H), 5.60 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 112.9, 109.8, 99.6, 99.4, 84.4, 84.0, 80.7, 75.6, 66.2, 59.6, 44.6, 42.3, 26.8, 26.2, 25.7, 25.3, 25.1, 23.1, 22.4; IR (thin film) ν 3436, 2986, 2955, 2872, 1456, 1371, 1211, 1163, 1086, 849; HRMS (ESI): m/z: calcd for C19H33NO7: 387.2257; found: 388.2216 [M+Na]+.
4.18.3. d-Gulose-(S)-3-isobutylisoxazolidin-5-one (23)
Prepared according to general procedures I, G1, G2, J. Purification by column chromatography using hexanes/EtOAc (6:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 82% yield. It was recrystallized if necessary from heptane to afford a single diastereomer. Rf=0.25 (hexanes/EtOAc 4:1); mp142–143 °C; + 21.1 (c 1.06, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 0.94 (dd, J=12.3, 6.3 Hz, 6H), 1.29 (s, 3H), 1.37 (s, 3H), 1.41 (s, 3H), 1.45 (s, 3H), 1.71 (m, 2H), 2.39 (dd, J=17.6, 4.9 Hz, 1H), 2.87 (dd, J=17.6, 7.9 Hz, 1H), 3.72 (dd, J=8.6, 6.7 Hz, 1H), 3.94 (m, 1H), 4.02 (dd, J=8.5, 4.0 Hz, 1H), 4.20 (dd, J=8.6, 6.8 Hz, 1H), 4.36 (q, J=8.4 Hz, 1H), 4.72 (m, 2H), 4.90 (d, J=6.1 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 176.2, 113.2, 110.0, 98.0, 85.4, 84.2, 80.3, 75.7, 66.1, 58.9, 42.8, 33.8, 26.8, 26.1, 25.4, 25.2, 24.8, 22.9, 22.3; IR (thin film) ν 2987, 2956, 1792, 1381, 1211, 1165, 1077, 1044, 892, 846; HRMS (ESI): m/z: calcd for C19H31NO7: 385.2101; found: 408.2004 [M+Na]+.
4.18.4. (S)-3-Isobutylisoxazolidin-5-one (46)
Prepared according to general procedure F. Purification by column chromatography using hexanes/EtOAc (3:1 v/v) as the eluent afforded the isoxazolidinone 46 as a yellow oil in 73% yield. Rf=0.33 (hexanes/EtOAc 2:1);. −13.9 (c 0.775, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.92 (m, 6H), 1.40 (m, 1H), 1.53 (m, 1H), 1.69 (m, 1H), 2.39 (dd, J=17.1, 8.9 Hz, 1H), 2.77 (m, 1H), 3.90 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 178.4, 58.0, 41.1, 36.5, 25.4, 22.5, 22.4; IR (thin film) ν 3428, 2961, 2091, 1773, 1647, 1200; HRMS (ESI): m/z: calcd for C7H13NO2: 143.0946; found: 143.0925 [M+H]+.
4.18.5. (S)-tert-Butoxycarbonyl-3-isobutylisoxazolidin-5-one (3)
Prepared according to general procedure K. Purification by column chromatography using hexanes/EtOAc (6:1 v/v) as the eluent afforded the carbamate as a yellow oil in 63% yield. Rf=0.35 (hexanes/EtOAc 4:1); +93.4 (c 1.00, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.95 (dd, J=16.1, 6.4 Hz, 6H), 1.28 (m, 1H), 1.50 (s, 9H), 1.74 (m, 1H), 2.38 (dd, J=17.6, 2.1 Hz, 1H), 2.96 (dd, J=17.6, 8.7 Hz, 1H), 4.60 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 173.7, 156.3, 84.0, 58.9, 43.2, 34.6, 28.1, 24.9, 22.7, 21.9; IR (thin film) ν 2959, 1807, 1745, 1469, 1370, 1302, 1134; HRMS (ESI): m/z: calcd for C12H21NO4: 243.1471; found: 244.1538 [M+H]+.
4.18.6. (S)-Methyl-5-methyl-3-(2-phenylacetamido)-hexanoate (58)
Prepared according to general procedure P. Purification by column chromatography using hexanes/EtOAc (2:1 v/v) as the eluent afforded the carbamate as a yellow oil in 46% yield. Rf=0.23 (hexanes/EtOAc 2:1); −42.7 (c 0.94, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.85 (d, J=6.6 Hz, 3H), 0.87 (d, J=6.5 Hz, 3H) 1.22 (m, 1H), 1.36 (m, 1H), 1.48 (m, 1H), 2.46 (m, 2H), 3.55 (s, 2H), 3.60 (s, 3H), 4.29 (m, 1H), 5.80 (d, J=8.46 Hz, 1H), 7.30 (m, 3H), 7.35 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 172.2, 170.4, 135.0, 129.5, 129.1, 127.4, 51.7, 44.4, 44.1, 43.1, 38.8, 25.2, 22.9, 22.3; IR (thin film) ν 3434, 2956, 2362, 2099, 1646, 1559, 1437, 1263; HRMS (ESI): m/z: calcd for C16H23NO3: 277.1678; found: 278.1747 [M+H]+.
4.18.7. d-Gulose-(S)-5-chloro-3-isobutylisoxazolidine-5-carbonitrile (21)
Prepared according to general procedure D. Purification by column chromatography using hexanes/EtOAc (8:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 68% yield (6:1 dr). Rf=0.40 (hexanes/EtOAc 4:1); recrystallization from heptane provided a single diastereomer. Mp 129 °C; −115.2 (c 0.95, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 0.94 (dd, J=6.6, 1.6 Hz, 6H), 1.32 (m, 4H), 1.38 (s, 3H), 1.42 (s, 3H), 1.47 (s, 3H), 1.63 (m, 1H), 1.75 (m, 1H), 2.94 (dd, J=13.8, 6.8 Hz, 1H), 3.23 (dd, J=13.8, 6.5 Hz, 1H), 3.73 (dd, J=8.5, 6.8 Hz, 1H), 3.98 (m, 1H), 4.04 (dd, J=8.5, 3.9 Hz, 1H), 4.21 (dd, J=8.5, 6.9 Hz, 1H), 4.37 (q, J=8.4 Hz, 1H), 4.70 (dd, J=6.0, 4.0 Hz, 1H), 4.92 (d, J=6.1 Hz, 1H), 5.03 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 114.7, 113.3, 110.0, 98.5, 38.8, 88.8, 84.7, 84.1, 80.2, 75.5, 66.1, 61.3, 53.5, 42.3, 26.8, 26.1, 25.9, 25.3, 24.9, 22.7, 22.6; IR (thin film) ν 3425, 2985, 2959, 2936, 2872, 1785, 1643, 1455, 1373, 1211, 1092, 1034, 876, 848; HRMS (ESI): m/z: calcd for C21H35NO8: 430.1871; found: 431.1938 [M+H]+.
4.18.8. d-Gulose-(S)-methyl-5-acetoxy-3-isobutylisoxazolidine-5-carboxylate (41)
Prepared according to general procedure C. Purification by column chromatography using hexanes/EtOAc (5:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 60% yield (4:1 dr). Rf=0.28 (hexanes/EtOAc 4:1); recrystallization from heptane provided a single diastereomer. Mp 131–132 °C; 1H NMR (500 MHz, CDCl3) δ 0.91 (d, J=6.6 Hz, 3H), 0.93 (d, J=6.6 Hz, 3H), 1.17 (m, 1H), 1.28 (m, 3H), 1.36 (s, 3H), 1.40 (s, 3H), 1.44 (s, 3H), 1.67 (m, 1H), 1.78 (m, 1H), 2.10 (s, 3H), 2.34 (dd, J=14.0, 2.5 Hz, 1H), 2.86 (dd, J=14.0, 7.8 Hz, 1H), 3.70 (m, 1H), 3.79 (s, 3H), 4.03 (dd, J=8.5, 4.0 Hz, 1H), 4.20 (m, 1H), 4.35 (m, 1H), 4.63 (s, 1H), 4.66 (dd, J=5.9, 4.2 Hz, 1H), 5.05 (d, J=6.1 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 169.5, 168.1, 112.8, 109.8, 104.8, 97.5, 84.6, 84.0, 80.2, 75.7, 66.1, 59.4, 53.5, 43.5, 42.6, 26.7, 26.2, 25.7, 25.3, 25.0, 23.3, 22.0, 21.0; IR (thin film) ν 3402, 2954, 2361, 1757, 1648, 1456, 1371, 1208, 1088; HRMS (ESI): m/z: calcd for C21H37NO10: 487.2417; found: 510.2299 [M+Na]+.
4.18.9. d-Gulose-(S)-5-cyano-3-benzylisoxazolidin-5-yl acetate (22)
Prepared according to general procedure E. Purification by column chromatography using hexanes/EtOAc (5:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 54% yield (~1:1 dr). Rf=0.42 (hexanes/EtOAc 4:1); 1H NMR (500 MHz, CDCl3) δ 1.26 (s, 3H), 1.29 (s, 3H), 1.35 (s, 3H), 1.36 (s, 3H), 1.38 (m, 6H), 1.41 (s, 3H), 1.44 (s, 3H), 2.14 (s, 3H), 2.19 (s, 3H), 1.32 (m, 4H), 2.67 (dd, J=14.3, 3.7 Hz, 1H), 2.74 (m, 2H), 2.85 (d, J=5.6 Hz, 2H), 2.96 (dd, J=14.3, 7.8 Hz, 1H), 3.08 (m, 2H), 3.58 (q, J=7.1 Hz, 1H), 3.63 (dd, J=8.4, 4.1 Hz, 1H), 3.74 (m, 1H), 3.99 (m, 1H), 4.04 (m, 1H), 4.13 (m, 2H), 4.28 (q, J=7.6 Hz, 2H), 4.60 (m, 2H), 4.67 (s, 1H), 4.70 (s, 1H), 4.84 (d, J=6.0 Hz, 1H), 4.93 (d, J=6.0 Hz, 1H), 7.26–7.32 (m, 10H); 13C NMR (125 MHz, CDCl3) δ 168.2, 167.9, 137.7, 129.4, 128.8, 127.0, 126.9, 113.2, 113.1, 109.9, 109.8, 98.3, 97.5, 96.7, 95.3, 84.5, 84.3, 83.9, 80.3, 80.1, 75.6, 75.5, 66.0, 63.2, 62.6, 47.0, 45.8, 39.9, 39.3, 26.9, 26.1, 26.0, 25.5, 25.4, 24.9, 24.8, 21.1, 21.0; IR (thin film) ν 3422, 2987, 2936, 1769, 1654, 1455, 1372, 1211, 1162, 1087, 847, 701; HRMS (ESI): m/z: calcd for C25H32N2O8: 488.2159; found: 511.2042 [M+Na]+.
4.18.10. d-Gulose-(S)-3-benzylisoxazolidin-5-yl acetate (27)
Prepared according to general procedure A. Purification by column chromatography using hexanes/EtOAc (6:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 68% yield (3:1.2:1 dr). Rf=0.39 (hexanes/EtOAc 2:1); major diastereomer 1H NMR(500 MHz, CDCl3) δ 1.27 (s, 3H), 1.36 (s, 3H), 1.40 (s, 3H), 1.43 (s, 3H), 2.50 (s, 3H), 2.32 (m, 1H), 2.46 (m, 1H), 2.81 (dd, J=13.4, 8.7 Hz, 1H), 3.12 (dd, J=13.5, 6.8 Hz, 1H), 3.61 (m, 1H), 3.84 (m, 2H), 4.14 (m, 2H), 4.30 (m, 1H), 4.63 (m, 1H), 4.93 (d, J=6.2 Hz, 1H), 6.43 (d, J=5.9 Hz, 1H), 7.17–7.22 (m, 3H), 7.23–7.33 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 170.1, 138.8, 129.5, 129.1, 128.6, 126.6, 112.8, 109.7, 99.2, 98.0, 96.7, 87.1, 84.2, 84.1, 80.4, 75.7, 66.1, 61.6, 40.0, 37.9, 27.0, 26.0, 25.4, 24.8, 21.5; IR (thin film) ν 2986, 2934, 1746, 1454, 1372, 1212, 1163, 1088, 1039, 979, 934, 848, 701; HRMS (ESI): m/z: calcd for C24H33NO8: 463.2206; found: 486.2104 [M+Na]+.
4.18.11. d-Gulose-(S)-3-benzylisoxazolidin-5-ol (59)
Prepared according to general procedure H. Purification by column chromatography using hexanes/EtOAc (2:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 54% yield (3:1.2:1 dr). Rf=0.38 (hexanes/EtOAc 1:1); major diastereomer 1H NMR(500 MHz, CDCl3) δ 1.28 (s, 3H), 1.37 (s, 3H), 1.39 (s, 3H), 1.45 (s, 3H), 2.18 (m, 1H), 2.21 (m, 1H), 2.60 (dd, J=13.3, 8.3 Hz, 1H), 3.03 (m, 1H), 3.81 (m, 1H), 3.84 (m, 2H), 4.21 (m, 2H), 4.65 (m, 1H), 4.93 (m, 1H), 5.06 (m, 1H), 5.64 (m, 1H), 7.17–7.22 (m, 3H), 7.23–7.33 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 138.9, 129.6, 128.6, 126.7, 112.9, 109.8, 99.9, 99.2, 84.4, 83.8, 80.6, 75.7, 66.1, 62.7, 41.3, 40.0, 27.0, 26.4, 25.4, 25.0, 24.9, 21.5; IR (thin film) ν 3432, 2986, 2936, 1642, 1454, 1372, 1264, 1211, 1163, 1088, 1035, 979, 894, 848, 736, 702; HRMS (ESI): m/z: calcd for C22H31NO7: 421.2101; found: 422.2162 [M+H]+.
4.18.12. d-Gulose-(S)-3-benzylisoxazolidin-5-one (24)
Prepared according to general procedure I, G1, G2, J. Purification by column chromatography using hexanes/EtOAc (6:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 84% yield. It was recrystallized if necessary from heptane to give a single diastereomer. Rf=0.20 (hexanes/EtOAc 4:1); mp 124–125 °C; +11.9 (c 0.75, CHCl3); 1H NMR (500 MHz, CDCl3) δ 1.27 (s, 3H), 1.36 (s, 3H), 1.40 (s, 3H), 1.44 (s, 3H), 2.52 (dd, J=17.8, 5.3 Hz, 1H), 2.77 (m, 2H), 3.07 (dd, J=13.7, 7.3 Hz, 1H), 3.57 (dd, J=8.3, 7.0 Hz, 1H), 3.63 (dd, J=8.4, 4.1 Hz, 1H), 4.11 (m, 2H), 4.28 (q, J=8.3 Hz, 1H), 4.63 (dd, J=5.93, 4.28 Hz, 1H), 4.72 (s, 1H), 4.87 (d, J=6.0 Hz, 1H), 7.17–7.27 (m, 3H), 7.28–7.33 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 175.5, 136.9, 129.4, 128.7, 126.9, 113.1, 109.8, 97.8, 84.8, 84.1, 80.0, 75.6, 65.9, 61.4, 39.8, 33.1, 26.9, 26.0, 25.3, 24.6; IR (thin film) ν 34,535, 2988, 1793, 1643, 1372, 1264, 1210, 1163, 1087, 1039, 891, 847, 702; HRMS (ESI): m/z: calcd for C22H29NO7: 419.1944; found: 442.1857 [M+Na]+.
4.18.13. (S)-3-Benzylisoxazolidin-5-one (43)
Prepared according to general procedure F. Purification by column chromatography using hexanes/EtOAc (3:1 v/v) as the eluent afforded the isoxazolidinone 43 as a yellow oil in 84% yield. Rf=0.30 (hexanes/EtOAc 2:1);. −24.1 (c 0.96, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 2.53 (dd, J=17.1, 7.7 Hz, 1H), 2.75 (dd, J=17.1, 6.8 Hz, 1H), 2.87 (m, 1H), 3.00 (dd, J=13.9, 6.9 Hz, 1H), 4.08 (m, 1H), 7.19 (m, 2H), 7.28 (m, 1H), 7.33 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 177.1, 129.2, 129.0, 127.3, 60.4, 35.5; IR (thin film) ν 3235, 3062, 3028, 2922, 1780, 1603, 1497, 1454, 1414, 1174, 1031, 997, 898, 746, 700; HRMS (ESI): m/z: calcd for C10H11NO2: 177.0790; found: 178.0876 [M+H]+.
4.18.14. (S)-tert-Butoxycarbonyl-3-benzylisoxazolidin-5-one (51)
Prepared according to general procedure K. Purification by column chromatography using hexanes/EtOAc (6:1 v/v) as the eluent afforded the carbamate as a yellow oil in 58% yield. Rf=0.29 (hexanes/EtOAc 4:1); +44.4 (c 1.04, CHCl3); 1H NMR (500 MHz, CDCl3) δ 1.49 (s, 9H), 2.56 (dd, J=17.8, 3.0 Hz, 1H), 2.81–2.91 (m, 2H), 3.20(dd, J=13.8, 5.9 Hz, 1H), 4.75 (m, 1H), 7.21 (m, 2H), 7.26 (m, 1H), 7.31 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 172.6, 155.6, 135.9, 129.5, 128.9, 127.3, 84.2, 61.1, 40.0, 33.6, 28.2; IR (thin film) ν 2980, 2930, 1806, 1741, 1455, 1370, 1320, 1254, 1142, 1082, 846, 701; HRMS (ESI): m/z: calcd for C15H19NO4: 277.1314; found: 300.1204 [M+Na]+.
4.18.15. (S)-Methyl-4-phenyl-3-(2-phenylacetamido)-butanoate (60)
Prepared according to general procedure P. Purification by column chromatography using hexanes/EtOAc (2:1 v/v) as the eluent afforded the carbamate as a yellow oil in 52% yield. Rf=0.15 (hexanes/EtOAc 2:1); mp 79 °C; –28.7 (c 0.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ 2.45 (m, 2H), 2.75 (m, 1H), 2.82 (m, 1H), 3.50 (s, 1H), 3.61 (s, 1H), 4.45 (m, 1H), 5.90 (d, J=8.8 Hz, 1H), 7.03 (m, 2H), 7.16 (m, 2H), 7.23 (m, 3H), 7.23 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 172.1, 170.4, 137.4, 134.8, 129.5, 129.4, 129.1, 128.7, 127.4, 126.8, 51.8, 47.5, 44.1, 39.8, 37.2; IR (thin film) ν 3285, 3063, 3029, 2924, 2337, 1736, 1647, 1547, 1496, 1437, 1206, 1152, 729, 700; HRMS (ESI): m/z: calcd for C16H23NO3: 311.1521; found: 334.1421 [M+Na]+.
4.18.16. d-Gulose-(S)-3-isopropylisoxazolidin-5-yl acetate (28)
Prepared according to general procedure A. Purification by column chromatography using hexanes/EtOAc (6:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 61% yield. Rf=0.20 (hexanes/EtOAc 4:1); recrystallization from heptane provided a single diastereomer. Mp 154–155 °C; 1H NMR (500 MHz, CDCl3) δ 0.90 (dd, J=6.7, 11.0 Hz, 6H), 1.13 (s, 3H), 1.26 (s, 3H), 1.37 (s, 3H), 1.42 (s, 3H), 1.67 (m, 1H), 2.06 (s, 3H), 2.36 (m, 2H), 3.42 (m, 1H), 3.74 (m, 1H), 4.09 (dd, J=3.5, 8.5 Hz, 1H), 4.19 (m, 1H), 4.36 (m, 1H), 4.65 (m, 1H), 4.80 (s, 1H), 4.91 (d, J=6.0 Hz, 1H), 6.36 (d, 1H); 13C NMR (125 MHz, CDCl3) δ 170.4, 112.7, 109.8, 99.5, 98.7, 84.0, 83.9, 80.6, 75.7, 67.2, 66.2, 37.4, 31.9, 26.7, 26.2, 25.6, 25.4, 25.1, 21.4, 19.7, 18.8; IR (thin film) ν 3439, 2983, 2872, 1752, 1654, 1374, 1237, 1211, 1164, 1237, 1211, 1066, 851; HRMS (ESI): m/z: calcd for C20H33NO8: 415.2206; found: 438.2104 [M+Na]+.
4.18.17. d-Gulose-(S)-3-isopropylisoxazolidin-5-ol (61)
Prepared according to general procedure H. Purification by column chromatography using hexanes/EtOAc (2:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 72% yield (5:1 dr). Rf=0.25 (hexanes/EtOAc 2:1); mp 145–146 °C; 1H NMR (500 MHz, CDCl3) δ 0.89 (d, J=6.7 Hz, 3H), 0.91 (d, J=6.7 Hz, 3H), 1.30 (s, 3H), 1.38 (s, 3H), 1.41 (s, 3H), 1.46 (s, 3H), 1.69 (m, 1H), 2.27 (m, 2H), 3.16 (d, J=3.1 Hz, 1H), 3.42 (m, 1H), 3.75 (dd, J=8.6, 6.5 Hz, 1H), 4.11 (dd, J=8.6, 3.8 Hz, 1H), 4.21 (dd, J=8.5, 7.0 Hz, 1H), 4.38 (q, J=8.5 Hz, 1H), 5.02 (m, 2H), 5.61 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 112.9, 109.8, 99.8, 84.2, 83.7, 80.7, 75.6, 67.2, 66.1, 39.1, 31.8, 26.7, 26.2, 25.4, 25.2, 19.9, 18.6; IR (thin film) ν 3434, 2987, 1642, 1372, 1210, 1082, 848; HRMS (ESI): m/z: calcd for C18H31NO7: 374.2101; found: 374.2166 [M+H]+.
4.18.18. d-Gulose-(S)-3-isopropylisoxazolidin-5-one (30)
Prepared according to general procedures I, G1, G2, J. Purification by column chromatography using hexanes/EtOAc (6:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 90% yield. It was recrystallized if necessary from heptane to give a single diastereomer. Rf=0.28 (hexanes/EtOAc4:1); mp 118 °C; +35.0 (c 1.14, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.94 (m, 6H), 1.28 (s, 3H), 1.37 (s, 3H), 1.40 (s, 3H), 1.44 (s, 3H), 1.83 (m, 1H), 2.49 (dd, J=18.0, 3.7 Hz, 1H), 2.82 (dd, J=18.0, 8.9 Hz, 1H), 3.63 (m, 1H), 3.74 (dd, J=8.6, 6.5 Hz, 1H), 4.05 (dd, J=8.5, 3.9 Hz, 1H), 4.20 (m, 1H), 4.35 (q, J=8.3 Hz, 1H), 4.65 (s, 1H), 4.72 (m, 1H), 4.90 (d, J=6.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 176.4, 113.2, 110.0, 98.1, 85.2, 84.0, 80.3, 75.7, 66.1, 65.2, 31.4, 30.2, 26.8, 26.1, 25.4, 24.8, 18.8, 18.7; IR (thin film) ν 3434, 2987, 1791, 1645, 1372, 1210, 1162, 1078, 847; HRMS (ESI): m/z: calcd for C18H29NO7: 371.1944; found: 394.1851 [M+Na]+.
4.18.19. (S)-3-Isopropylisoxazolidin-5-one (45)
Prepared according to general procedure F. Purification by column chromatography using hexanes/EtOAc (3:1 v/v) as the eluent afforded the isoxazolidinone 45 as a yellow oil in 67% yield. Rf=0.13 (hexanes/EtOAc 4:1);. +9.1 (c 1.28, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.97 (dd, J=10.9, 6.7 Hz, 6H), 1.77 (m, 1H), 2.49 (dd, J=17.2, 8.8 Hz, 1H), 2.73 (dd, J=17.2, 6.9 Hz, 1H), 3.55 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 177.6, 65.2, 34.3, 31.6, 19.3, 19.0; IR (thin film) ν 3223, 2964, 2935, 2877, 1782, 1782, 1469, 1416, 1299, 1190, 1145, 1079, 1014, 904, 850; HRMS (ESI): m/z: calcd for C6H11NO2: 129.0790; found: 130.0862 [M+H]+.
4.18.20. (S)-tert-Butoxycarbonyl-3-isopropylisoxazolidin-5-one (53)
Prepared according to general procedure K. Purification by column chromatography using hexanes/EtOAc (8:1 v/v) as the eluent afforded the carbamate as a yellow oil in 60% yield. Rf=0.38 (hexanes/EtOAc 4:1); +183.9 (c 0.44, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.98 (m, 6H), 1.50 (s, 9H), 1.91 (m, 1H), 2.54 (dd, J=18.0, 2.1 Hz, 1H), 2.92 (dd, J=18.0, 9.6 Hz, 1H), 4.35 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 173.7, 156.5, 83.9, 65.1, 32.1, 31.5, 28.2, 18.1, 17.9; IR (thin film) ν 2975, 2935, 2878, 1807, 1744, 1717, 1471, 1371, 1338, 1254, 1144, 1053, 958, 877, 849, 772; HRMS (ESI): m/z: calcd for C12H21NO4: 229.1314; found: 252.1213 [M+Na]+.
4.18.21. (S)-Methyl-4-methyl-3-(2-phenylacetamido)-pentanoate (62)
Prepared according to general procedure P. Purification by column chromatography using hexanes/EtOAc (2:1 v/v) as the eluent afforded the carbamate as a yellow oil in 52% yield. Rf=0.29 (hexanes/EtOAc 1:1); −57.5 (c 0.55, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.78 (d, J=6.7 Hz, 3H), 0.83 (d, J=6.7 Hz, 3H), 1.72 (m, 1H), 2.44 (m, 1H), 3.56 (s, 2H), 3.59 (s, 3H), 4.03 (m, 1H), 5.84 (d, J=8.4 Hz, 1H), 7.29 (m, 3H), 7.34 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 172.2, 170.5, 135.1, 129.5, 129.1, 127.4, 51.8, 51.6, 44.1, 36.5, 31.4, 19.4, 18.7; IR (thin film) ν 3293, 3064, 3030, 2962, 2875, 1739, 1645, 1549, 1437, 1371, 1280, 1195, 728, 696; HRMS (ESI): m/z: calcd for C15H21NO3: 263.1521; found: 264.1591 [M+H]+.
4.18.22. d-Gulose-(S)-3-methylisoxazolidin-5-one (42)
Prepared according to general procedures G1, G2. Purification by column chromatography using hexanes/EtOAc (6:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 52% yield (8:1 dr). Rf=0.37 (hexanes/EtOAc 4:1); mp 113–114 °C; +40.6 (c 1.09, CHCl3); 1H NMR (500 MHz, CDCl3) δ 1.26 (s, 3H), 1.35 (m, 6H), 1.39 (s, 3H), 1.43 (s, 3H), 2.51 (dd, J=17.2, 9.1 Hz, 1H), 2.77 (dd, J=17.3, 7.5 Hz, 1H), 3.70 (dd, J=8.6, 6.5 Hz, 1H), 3.83 (m, 1H), 4.02 (dd, J=8.4, 4.0 Hz, 1H), 4.16 (dd, J=8.6, 6.8 Hz, 1H), 4.31 (q, J=8.3 Hz, 1H), 4.70 (m, 1H), 4.71 (s, 1H), 4.88 (d, J=6.1 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 174.7, 113.1, 109.8, 98.7, 85.2, 84.0, 80.2, 77.4, 77.2, 76.9, 75.8, 66.0, 58.9, 36.3, 26.8, 26.0, 25.4, 24.7, 18.8; IR (thin film) ν 3434, 2987, 2938, 1793, 1641, 1382, 1262, 1211, 1163, 1087, 894, 847, 620; HRMS (ESI): m/z: calcd for C16H25NO7: 343.1631; found: 366.1515 [M+Na]+.
4.18.23. (S)-3-Methylisoxazolidin-5-one (47)
Prepared according to general procedure F. Purification by column chromatography using hexanes/EtOAc (1:1 v/v) as the eluent afforded the isoxazolidinone 47 as a yellow oil in 42% yield. Rf=0.09 (hexanes/EtOAc 2:1); −6.2 (c 0.26, CHCl3); 1H NMR (500 MHz, CDCl3) δ 1.32 (d, J=6.4 Hz, 3H), 2.40 (dd, J=17.1, 8.7 Hz, 1H), 2.81 (dd, J=16.8, 5.8 Hz, 1H), 3.98 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 177.7, 55.4, 37.7, 29.8; IR (thin film) ν 3537, 3227, 2977, 2934, 1779, 1654, 1459, 1415, 1385, 1326, 1200, 1146, 1081, 1033, 942, 900, 829; HRMS (ESI): m/z: calcd for C4H7NO2: 101.0477; found: 102.0560 [M+H]+.
4.18.24. d-Gulose-(S)-3-isobutylisoxazolidin-5-carboxycyclohexan-1,1-acetal (37)
Prepared according to general procedure B. Purification by column chromatography using hexanes/EtOAc (7:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 81% yield. After recrystallization from heptane, it was obtained a single diastereomer. Rf=0.43 (hexanes/EtOAc 4:1); mp 193–194 °C; −25.9 (c 1.05, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.90 (d, J=6.6 Hz, 3H), 0.90 (d, J=6.6 Hz, 3H), 1.11 (m, 1H), 1.25 (s, 3H), 1.34 (s, 3H), 1.37 (m, 4H), 1.41 (s, 3H), 1.44 (m, 1H), 1.58–1.89 (m, 10H), 2.04 (d, J=13.8 Hz, 1H), 2.91 (dd, J=13.7, 7.7 Hz, 1H), 3.91 (m, 1H), 3.97 (dd, J=8.4, 3.8 Hz, 1H), 4.17 (m, 1H), 4.33 (q, J=7.8 Hz, 1H), 4.63 (m, 1H), 4.66 (s, 1H), 4.84 (d, J=6.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 169.5, 112.9, 111.7, 109.8, 105.9, 96.7, 84.5, 84.3, 80.3, 75.6, 66.0, 58.5, 42.6, 41.1, 37.6, 36.4, 26.6, 26.1, 25.3, 25.2, 25.0, 24.3, 23.5, 23.0, 22.9, 21.7; IR (thin film) ν 3430, 2988, 2943, 2870, 1802, 1454, 1376, 1259, 1237, 1166, 1087, 1034, 992, 923, 875, 848, 733; HRMS (ESI): m/z: calcd for C26H41NO9: 445.2625; found: 534.2679 [M+Na]+.
4.18.25. d-Gulose-(S)-3-benzylisoxazolidin-5-carboxycyclohexan-1,1-acetal (40)
Prepared according to general procedure B. Purification by column chromatography using hexanes/EtOAc (6:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 65% yield. After recrystallization from heptane, it was obtained a single diastereomer. Rf=0.32 (hexanes/EtOAc 4:1); mp 164–165 °C; −36.6 (c 1.05, CHCl3); 1H NMR (500 MHz, CDCl3) δ 1.27 (s, 3H), 1.36 (s, 3H), 1.42 (s, 3H), 1.43 (s, 3H), 1.48 (m, 2H), 1.65–1.84 (m, 6H), 1.91 (m, 2H), 2.20 (d, J=13.9 Hz, 1H), 2.81 (dd, J=13.9, 8.6 Hz, 2H), 3.06 (dd, J=13.6, 7.9 Hz, 1H), 3.53 (m, 1H), 3.57 (dd, J=8.4, 4.0 Hz, 1H), 4.12 (m, 2H), 4.27 (q, J=8.2 Hz, 1H), 4.55 (dd, J=5.8, 4.3 Hz, 1H), 4.69 (s, 1H), 4.84 (d, J=6.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 169.4, 138.6, 129.6, 128.6, 126.5, 112.9, 111.9, 109.7, 105.9, 96.4, 84.5, 84.0, 80.2, 75.7, 66.1, 61.5, 39.9, 33.4, 37.7, 36.5, 27.0, 26.1, 25.4, 24.9, 24.4, 23.2, 23.0; IR (thin film) ν 3432, 2986, 2938, 2866, 1803, 1638, 1453, 1372, 1233, 1179, 1158, 1116, 1088, 1036, 936, 882, 849, 735, 701; HRMS (ESI): m/z: calcd for C29H39NO9: 545.2625; found: 564.2517 [M+Na]+.
4.18.26. d-Gulose-(S)-3-methylisoxazolidin-5-carboxycyclohexan-1,1-acetal (39)
Prepared according to general procedure B. Purification by column chromatography using hexanes/EtOAc (7:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 73% yield (6:3:1:1 dr). After recrystallization from heptane, it was obtained a single diastereomer. Rf=0.30 (hexanes/EtOAc 4:1); +14.2 (c 0.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ 1.28 (s, 3H), 1.31 (d, J=6.6 Hz, 3H), 1.37 (s, 3H), 1.41 (m, 4H), 1.44 (m, 4H), 1.58–1.91 (m, 8H), 2.13 (dd, J=13.7, 3.6 Hz, 1H), 2.88 (dd, J=13.7, 3.6 Hz, 1H), 3.69 (dd, J=8.5, 6.6 Hz, 1H), 3.87(m, 1H), 4.04(dd, J=8.5, 3.8 Hz, 1H), 4.18 (dd, J=8.5, 6.7 Hz, 1H), 4.34(q, J=6.7 Hz, 1H), 4.66 (m, 2H), 4.86 (d, J=9.8 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 169.2, 113.0, 111.8, 109.8, 105.1, 97.5, 84.4, 84.2, 80.4, 75.8, 66.2, 57.0, 42.9, 37.6, 36.5, 26.9, 26.2, 25.5, 25.0, 24.4, 23.0, 22.9, 19.3; IR (thin film) ν 3433, 2986, 2942, 2096, 1804, 1642, 1456, 1372, 1186, 1087, 936, 848; HRMS (ESI): m/z: calcd for C23H35NO9: 469.2312; found: 490.2376 [M+H]+.
4.18.27. d-Gulose-(S)-3-isopropylisoxazolidin-5-carboxy-cyclohexan-1,1-acetal (38)
Prepared according to general procedure B. Purification by column chromatography using hexanes/EtOAc (7:1 v/v) as the eluent afforded the diastereomeric mixture as a colorless solid in 65% yield. Rf=0.16 (hexanes/EtOAc 7:1). After recrystallization from heptane, it was obtained a single diastereomer. Mp 173–174 °C; −21.1 (c 1.07, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.94 (d, J=6.7 Hz, 3H), 1.00 (d, J=6.5 Hz, 3H), 1.28 (s, 3H), 1.37 (s, 3H), 1.41 (s, 3H), 1.44 (m, 4H), 1.62–1.71 (m, 4H), 1.61–1.72 (m, 5H), 2.33 (d, J=14.0Hz, 1H), 2.79 (dd, J=14.0, 7.9 Hz, 1H), 3.43 (m, 1H), 3.72 (dd, J=8.6, 6.6 Hz, 1H), 4.05 (dd, J=8.5, 3.9 Hz, 1H), 4.20 (m, 1H), 4.35 (q, J=8.4 Hz, 1H), 4.66 (m, 2H), 4.87 (d, J=6.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 169.7, 113.0, 111.7, 109.9, 106.1, 96.8, 84.5, 84.3, 80.3, 75.7, 67.1, 66.2, 37.7, 36.5, 29.8, 26.8, 26.2, 25.4, 25.0, 24.4, 23.1, 23.0, 20.9, 19.8; IR (thin film) ν 3419, 2939, 2869, 2361, 1803, 1452, 1372, 1229, 1196, 1114, 1089, 982, 936, 849, 734; HRMS (ESI): m/z: calcd for C25H39NO9: 497.2625; found: 520.2518 [M+Na]+.
4.18.28. (S)-Benzyloxycarbonyl-phenyl-3-isoxazolidin-5-one (11)
Prepared according to general procedure L Purification by column chromatography using hexanes/EtOAc (5:1 v/v) as the eluent afforded the carbamate as a white solid in 89% yield. Rf=0.22 (hexanes/EtOAc 4:1); mp 83–84 °C; 1H NMR (500 MHz, CDCl3) δ 2.85 (dd, J=17.8, 3.7 Hz, 1H), 3.34 (dd, J=17.8, 9.5 Hz, 1H), 5.24 (m, 2H), 5.64 (dd, J=9.4, 3.7 Hz, 1H), 7.29–7.43 (m, 10H); 13C NMR (125 MHz, CDCl3) δ 171.6, 156.3, 138.4, 134.8, 129.2, 128.7, 128.4, 125.8, 69.0, 63.0, 37.2; IR (thin film) ν 2952, 1807, 1723, 1497, 1455, 1411, 1322, 1163, 1131, 750, 697; HRMS (ESI): m/z: calcd for C17H15NO4: 297.1001; found: 320.0897 [M+Na]+.
4.18.29. Benzylhydroxy(3-oxo-3-(phenethylamino)-1-phenylpropyl) carbamate (13)
Prepared according to general procedure M. Purification by column chromatography using hexanes/EtOAc (2:1 v/v) as the eluent afforded the carbamate as a colorless solid in 82% yield. Rf=0.31 (hexanes/EtOAc 1:1); 1H NMR(500 MHz, CDCl3) δ 2.56 (dd, J=14.1, 4.8 Hz, 1H), 2.70 (m, 2H), 3.02 (dd, J=13.9, 11.2 Hz, 1H), 3.40 (m, 2H), 5.12 (m, 2H), 5.51 (dd, J=11.1, 4.7 Hz, 1H), 6.14 (m, 1H), 7.10 (m, 2H), 7.22 (m, 1H), 7.28 (m, 10H), 7.26 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 171.0, 157.1, 139.3, 138.7, 136.0, 128.8, 128.7, 128.6, 128.2, 128.0, 127.0, 126.6, 68.0, 60.0, 41.0, 39.8, 35.4; IR (thin film) ν 3305, 2927, 2106, 1697, 1642, 1554, 1497, 1454, 1303, 1105, 748, 698; HRMS (ESI): m/z: calcd for C25H26N2O4: 418.1893; found: 417.1816 [M–H].
4.18.30. Ethoxy-β-Ala-β-Phe-N-Boc-β-Leu-hydroxylamine (55)
Prepared according to general procedure N. Purification by column chromatography using EtOAc as the eluent afforded a colorless oil. Rf=0.26 (EtOAc); −12.7 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.88 (d, J=6.5 Hz, 3H), 0.91 (d, J=6.5 Hz, 3H), 1.10 (m, 1H), 1.26 (m, 3H), 1.44 (s, 9H), 1.59 (m, 1H), 1.67 (m, 1H), 2.16 (dd, J=14.5, 6.2 Hz, 1H), 2.22 (dd, J=13.6, 3.6 Hz, 1H), 2.34 (dd, J=14.6, 3.9 Hz, 1H), 2.47–2.58 (m, 3H), 2.74 (dd, J=13.6, 8.9 Hz, 1H), 2.91 (m, 1H), 3.51 (m, 2H), 4.15 (q, J=7.1 Hz, 2H), 4.45 (m, 1H), 6.43 (m, 1H), 6.79 (d, J=8.8 Hz, 1H), 7.16 (m, 2H), 7.19–7.30 (m, 3H), 7.82 (br s, 1H); 13C NMR (125 MHz, CDCl3) δ 173.0, 172.0, 171.0, 157.1, 137.9, 129.3, 128.7, 126.8, 81.6, 61.2, 48.1, 41.4, 39.9, 39.8, 38.5, 35.1, 34.0, 29.8, 28.4, 24.7, 23.3, 22.0, 14.3; IR (thin film) ν 3414, 3297, 2956, 2925, 2870, 2366, 1654, 1540, 1456, 1368, 1253, 1166, 1095, 1030, 846, 701; HRMS (ESI): m/z: calcd for C27H43N3O7: 521.3103; found: 544.2994 [M+Na]+.
4.18.31. Ethoxy-β-Ala-β-Phe-β-Leu-benzylamide (56)
Prepared according to general procedure O. Purification by column chromatography using CHCl3/MeOH 20:1 as the eluent afforded a colorless oil in 32% yield over three steps. Rf=0.17 (CHCl3/MeOH 20:1); –21.1 (c 0.58, CHCl3); 1H NMR (500 MHz, CDCl3) δ 0.83 (m, 6H), 0.89 (m, 1H), 1.17–1.29 (m, 3H), 1.36 (m, 1H), 1.44 (m, 1H), 2.19 (m, 1H), 2.22 (m, 1H), 2.34 (m, 2H), 2.53 (m, 2H), 2,70 (dd, J=13.6, 8.6 Hz, 1H), 2.92 (dd, J=13.5, 6.3 Hz, 1H), 3.50 (m, 3H), 4.16 (q, J=7.1 Hz, 2H), 4.20 (m, 1H), 4.36 (m, 1H), 6.28 (m, 1H), 6.32 (d, J=8.8 Hz, 1H), 7.16 (d, J=7.5 Hz, 2H), 7.20–7.34 (m, 8H); 13C NMR (125 MHz, CDCl3) δ 172.6, 171.2, 170.8, 170.7, 138.1, 135.2, 129.4, 129.3, 128.9, 128.7, 127.2, 126.8, 61.0, 48.2, 45.2, 44.0, 43.2,40.9, 40.0, 38.6, 35.0, 34.1, 25.2, 22.9, 22.3, 14.3; IR (thin film) ν 3283, 2926, 1735, 1644, 1546, 1454, 1367, 1185, 698 HRMS (ESI): m/z: calcd for C30H41N3O5: 523.3046; found: 546.2944 [M+Na]+.
Supplementary Material
Acknowledgements
This work was supported by the Arnold and Mabel Beckman Foundation, the David and Lucille Packard Foundation, and Research Corporation (Cottrell Scholar Award to J.W.B). We appreciate protocols from Nancy Carrillo and Justin Russak as well as the helpful synthetic advice and discussions from Dr. Justin Struble and Dr. Hiroshi Ishida. We are grateful to BioBlocks, Inc. for a generous gift 1.
Footnotes
Dedicated to Professor Brian Stoltz in honor of his 2009 Tetrahedron Young Investigator Award
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.tet.2010.04.016. This data include MOL files and InChiKeys of the most important compounds described in this article.
References and notes
- 1.(a) Seebach D, Beck AK, Bierbaum DJ. Chem. Biodiv. 2004;1:1111–1239. doi: 10.1002/cbdv.200490087. [DOI] [PubMed] [Google Scholar]; (b) Cheng PR, Gellman SH, DeGrado WF. Chem. Rev. 2001;101:3219–3232. doi: 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]; (c) Goodman CM, Choi S, Shandler S, Degrado WF. Nat. Chem. Biol. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seebach D, Overhand M, Kühnle FNM, Martinoni B. Helv. Chim. Acta. 1996;79:913–941. [Google Scholar]
- 3.Appella DH, Christianson LA, Karle IL, Powell DR, Gellman SH. J. Am. Chem. Soc. 1996;118:13071–13072. [Google Scholar]
- 4.(a) Cheng RP, DeGrado WF. J. Am. Chem. Soc. 2001;123:5162–5163. doi: 10.1021/ja010438e. [DOI] [PubMed] [Google Scholar]; (b) Arvidsson PI, Rueping M, Seebach D. Chem. Commun. 2001:649–650. [Google Scholar]
- 5.Porter EA, Wang X, Lee H-S, Weisblum B, Gellman SH. Nature. 2000;404:565. doi: 10.1038/35007145. [DOI] [PubMed] [Google Scholar]
- 6.Sadowsky JD, Schmitt MA, Lee H-S, Umezawa N, Wang S, Tomita Y, Gellman SH. J. Am. Chem. Soc. 2005;127:11966–11968. doi: 10.1021/ja053678t. [DOI] [PubMed] [Google Scholar]
- 7.Imamura Y, Watanabe N, Umezawa N, Iwatsubo T, Kato N, Tomita T, Higuchi T. J. Am. Chem. Soc. 2009;131:7353–7359. doi: 10.1021/ja9001458. [DOI] [PubMed] [Google Scholar]
- 8.(a) Qiu XJ, Petersson EJ, Matthews EE, Schepartz A. J. Am. Chem. Soc. 2006;128:11338–11339. doi: 10.1021/ja063164+. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Daniels DS, Petersson EJ, Qiu XJ, Schepartz A. J. Am. Chem. Soc. 2007;129:1532–1533. doi: 10.1021/ja068678n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Petersson EJ, Craig CJ, Daniels DS, Qiu XJ, Schepartz A. J. Am. Chem. Soc. 2007;129:5344–5345. doi: 10.1021/ja070567g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mueller MM, Windsor MA, Pomerantz WC, Gellman SH, Hilvert D. Angew. Chem., Int. Ed. 2009;48:922–925. doi: 10.1002/anie.200804996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petersson EJ, Schepartz A. J. Am. Chem. Soc. 2008;130:821–823. doi: 10.1021/ja077245x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For recent reviews of peptide ligations, see: Dawson PE, Kent SBH. Annu. Rev. Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. Macmillan D. Angew. Chem., Int. Ed. 2006;45:7668–7672. doi: 10.1002/anie.200602945. Hackenberger CPR, Schwarzer D. Angew. Chem., Int. Ed. 2008;47:10030–10074. doi: 10.1002/anie.200801313.
- 12.Kimmerlin T, Seebach D, Hilvert D. Helv. Chim. Acta. 2002;85:1812–1826. [Google Scholar]
- 13.Seebach D, Kimmerlin T, Sebesta R, Campo MA, Beck AK. Tetrahedron. 2004;60:7455–7506. [Google Scholar]
- 14.Bode JW, Fox RM, Baucom KD. Angew. Chem., Int. Ed. 2006;45:1248–1252. doi: 10.1002/anie.200503991. [DOI] [PubMed] [Google Scholar]
- 15.Carrillo N, Davalos EA, Russak JA, Bode JW. J. Am. Chem. Soc. 2006;128:1452–1453. doi: 10.1021/ja057706j. [DOI] [PubMed] [Google Scholar]
- 16.(a) Ju L, Lippert AR, Bode JW. J. Am. Chem. Soc. 2008;130:4253–4255. doi: 10.1021/ja800053t. [DOI] [PubMed] [Google Scholar]; (b) Ju L, Bode JW. Org. Biomol. Chem. 2009;7:2259–2264. doi: 10.1039/b901198f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Tokuyama H, Kuboyama T, Amano A, Yamashita T, Fukuyama T. Synthesis. 2000:1299–1304. [Google Scholar]; (b) Tokuyama H, Kuboyama T, Fukuyama T. Org. Synth. 2003;80:207–208. [Google Scholar]
- 18.Ibrahem I, Rios R, Vesely J, Zhao G-L, Córdova A. Chem. Commun. 2007:849–851. doi: 10.1039/b613410f. [DOI] [PubMed] [Google Scholar]
- 19.(a) Vasella A. Helv. Chim. Acta. 1977;60:426–446. [Google Scholar]; (b) Mzengeza S, Whitney RA. J. Org. Chem. 1988;53:4074–4081. [Google Scholar]
- 20.For elegant work on the use of diastereoselective nitrile oxide cycloadditions for the synthesis of β-amino acids, see: Minter AR, Fuller AA, Mapp AK. J. Am Chem. Soc. 2003;125:6846–6847. doi: 10.1021/ja0298747. Fuller AA, Chen B, Minter AR, Mapp AK. J. Am. Chem. Soc. 2005;127:5376–5383. doi: 10.1021/ja0431713.
- 21.(a) Iida H, Kasahara K, Kibayashi C. J. Am. Chem. Soc. 1986;108:4647–4648. [Google Scholar]; (b) Kasahara K, Iida H, Kibayashi C. J. Org. Chem. 1989;54:2225–2233. [Google Scholar]
- 22.Ranganathan S, Ranganathan D, Mehrotra AK. Synthesis. 1977:289–296. [Google Scholar]
- 23.Keirs D, Moffat D, Overton K, Tomanek R. J. Chem. Soc., Perkins Trans. 1. 1991:1041–1051. [Google Scholar]
- 24.Chiacchio U, Corsaro A, Gumina G, Rescifina A, Iannazzo D, Piperno A, Romeo G, Romeo R. J. Org. Chem. 1999;64:9321–9327. [Google Scholar]
- 25.Yu S, Ishida H, Juarez-Garcia ME, Bode JW. submission in progress, see Supplementary data. [Google Scholar]
- 26.Purchased from Aldrich or Acros Organics.
- 27.Shing TKM, Gillhouley JG. Tetrahedron. 1994;50:8685–8698. [Google Scholar]
- 28.Buchanan JG, Lumbard KW, Sturgeon RJ, Thompson DK, Wightman RH. J. Chem. Soc., Perkin Trans. 1. 1990;3:699–706. [Google Scholar]
- 29.Mita N, Tamura O, Ishibashi H, Sakamoto M. Org. Lett. 2002;4:1111–1114. doi: 10.1021/ol0200044. [DOI] [PubMed] [Google Scholar]
- 30.Wolinsky J, Novak R, Vasileff R. J. Org. Chem. 1964;29:3596–3598. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.