

Abstract
Heme proteins are extremely diverse, widespread, and versatile biocatalysts, sensors, and molecular transporters. The chlorite dismutase family of hemoproteins received its name due to the ability of the first-isolated members to detoxify anthropogenic ClO2−, a function believed to have evolved only in the last few decades. Family members have since been found in fifteen bacterial and archaeal genera, suggesting ancient roots. A structure- and sequence-based examination of the family is presented, in which key sequence and structural motifs are identified and possible functions for family proteins are proposed. Newly identified structural homologies moreover demonstrate clear connections to two other large, ancient, and functionally mysterious protein families. We propose calling them collectively the CDE superfamily of heme proteins.
Keywords: heme, protein, bacteria, peroxidase, oxygen
1 Introduction
Unicellular organisms exhibit a remarkable ability to change. The natural environment offers diverse circumstances in which organisms, in the face of natural and man-made selection pressures, have little recourse other than to adapt or perish. The oxochlorates provide a compelling example. These toxic, highly oxidizing complex anions are used extensively in a variety of industries in the millions of tons. This extensive use, coupled to the high solubility and kinetic inertness of the salts, has made them serious fresh water pollutants of particular concern to the United States Environmental Protection Agency (1–4). They are primarily anthropogenic and relatively recent additions to the natural environment (≤ 50 years). Microbes have evolved not only to contend with polluting oxochlorates, but to exploit their oxidative power. Several diverse species of Proteobacteria are now known to use perchlorate (ClO4−) and chlorate (ClO3−) as terminal electron acceptors under anaerobic conditions (5, 6). The bacteria reduce perchlorate to chlorate and chlorate to chlorite via a molybdopterin-dependent perchlorate reductase, a homolog of bacterial respiratory nitrate reductases. Given the level of homology, the identical organization of protein subunits, and the similar array of cofactors, it appears likely that perchlorate reductase evolved from some member of the DMSO reductase superfamily via duplication of a gene set followed by divergence.
In contrast to perchlorate reductase, chlorite dismutase (Cld) has no obvious sequence relationships with well-characterized proteins. Its role in oxochlorate respirers is not to reduce but to detoxify chlorite, a by-product of perchlorate or chlorate respiration. As such, it also has no functional correlate in the analogous nitrate respiratory pathways, which reduce NO3− completely to NH3 or N2. There is additional evidence for independent transcriptional regulation and lateral transfer of the cld gene into chlorate and perchlorate reductase operons (6). Though the presence of a cld homolog has been used as a biomarker for perchlorate respiration (7, 8), it is now clear that sequences annotated as clds are extremely widespread in ~500 highly taxonomically diverse organisms, the overwhelming majority of which do not respire perchlorate or chlorate, and moreover have no expected capacity for chlorite detoxification (9). These genes have been placed into their own superfamily, and we refer herein to the gene products collectively (regardless of origin) as Cld family proteins (CFPs). Based on analyses of hundreds of complete bacterial genomes, the gene products specifically from non-perchlorate-respiring bacteria (NPRB) have been assigned to their own cluster of orthologs, COG3253S, where the “S” indicates that their biological function is unknown. Given the expected lateral transfer of the cld gene into chlorate and perchlorate reducing gene clusters (6), and the absence of any biological need for chlorite detoxification in the majority of cld-bearing bacteria and archaea, it would appear likely that efficient chlorite detoxification evolved from an enzyme family with a decidedly different role or roles. The following analyses of primary sequence, structure, and phylogeny are aimed at identifying key sequence motifs and structural elements of the cld gene family, relating these to known structures and protein families, and positing possible roles for gene products in the diverse species that harbor them. These analyses lead from the seemingly isolated issue of chlorite detoxification toward three distinct protein families, which share a common fold but variable active sites. These proteins could have fundamental roles in bacterial Fe biochemistry.
2. Chlorite dismutase from Dechloromonas aromatica
The chlorite-decomposing Cld from D. aromatica (DA-Cld) has been characterized structurally and mechanistically, and it serves as a point for comparisons among family members. Despite having been co-opted for its function relatively recently, the enzyme is remarkably fast and efficient at substantive chlorite detoxification. It has a kcat of 2.0 × 105 s−1 and kcat/KM near the diffusion limit (3.2 × 107 M−1 s−1, pH 5.2, 4 °C) (10). It is highly specific; even in the presence of a large excess of one or two electron reducing agents, the enzyme faithfully returns one molecule of O2 and one Cl− for each ClO2− consumed. While intermediates in the reaction with chlorite could not be detected by stopped flow methods at 4 °C, the ferryl (Fe(IV)=O) porphyrin cation radical (Compound I) commonly seen in many heme proteins forms rapidly in reactions with peracetic acid. A mechanism in which Compound I and a hypochlorite (ClO−) leaving group form and then recombine has consequently been proposed (Scheme 1) (11). Mechanisms involving homolytic bond cleavage and the direct generation of a ferryl species alone (Compound II), or a completely concerted mechanism, are also possible. The former has support from our more recent work with site-directed mutants and computations (12).
Scheme 1.

Proposed mechanisms for chlorite decomposition and O2 evolution catalyzed by DA-Cld. A Cld-chlorite Michaelis complex forms. (Top) Heterolytic bond cleavage yields the ferryl-porphyrin cation radical intermediate (Compound I) and hypochlorite as the leaving group. The oxygen atom of hypochlorite acts as a nucleophile toward the electron-deficient Compound I, forming a peroxychlorite anion that rapidly breaks down into products. (Bottom) The same Michaelis complex reacts via homolytic cleavage of the (O)Cl-O− bond, yielding the ferryl-porphyrin complex (Compound II) and the hypochloryl radical. Recombination of the radicals yields the peroxychlorite anion and then the reaction products.
The full gene from D. aromatica contains a sequence encoding a 26-amino acid leader peptide targeting the protein for export. In order to obtain the protein in soluble/folded form, this sequence was removed and only the mature enzyme coding region was expressed (13). Interestingly, the signal peptide appears to be of the SecB type, indicating that the protein is transported across the inner membrane in its unfolded form and presumably acquires heme in the periplasm itself. The functional enzyme crystallizes as a homopentamer with one heme b per protein subunit (14). A hexameric form of the protein from Azospira oryzae has also been crystallographically characterized (15). Further solution studies confirmed the A. oryzae protein to be pentameric, consistent with the DA-Cld structure. Moreover, the structure of Cld from candidatus Nitrospira defluvii was recently reported and also crystallized in the pentameric state (16). The possible functional significance of the homopentameric arrangement is unknown, although inspection of the crystal structure of DA-Cld suggests that oligomerization state is unlikely to influence pathways of chlorite entry or product egress. Each monomer consists of a β-sheet core flanked by α-helices in a fold unusual among known heme enzymes (Figure 1A). The area around the proximal histidine heme ligand consists of several α-helices, while the distal pocket on the opposite face of the porphyrin is lined by β-sheets (Figure 1B). The chemical functionalities of the distal pocket are generally critical to the function of heme proteins. In DA-Cld, there is a lone charged/polar residue in this pocket, Arg183, surrounded by a hydrophobic triad of Phe, Leu, and Thr (the hydroxyl group of the side-chain is oriented away from the pocket). Together, these residues provide a sterically restrictive distal volume above the porphyrin plane, which is undoubtedly important for the enzyme’s function. The distal pocket is sequestered from solvent by Arg183. In the DA-Cld crystal structure there is a nitrito ligand bound through an oxygen atom to the iron that forms a hydrogen bond to Arg183. In the absence of the ligand, this Arg183 side-chain conformer, which forms no hydrogen or ionic bonds with the protein, is likely to adopt a different conformation and/or be more mobile. As such, it is likely to be a major determinant in substrate entry and product exit under turnover conditions (14). This hypothesis is supported by work on N. defluvii Cld where mutations of the equivalent Arg183 to alanine or lysine decreased binding affinities for cyanide and lowered kcat/KM for chlorite (16).
Figure 1. Overview of Dechloromonas aromatica Cld structure.


(A) The monomer of DA-Cld is shown in green cartoon. (B) Stereo view of the heme-containing nitrite-bound active site with residues of possible catalytic importance highlighted. Relevant residues are represented as sticks, numbered according to the mature DA-Cld sequence and color-coded (carbons) by their proposed function in DA-Cld (See also Table 1). This figure was generated using PyMOL (http://www.pymol.org/).
On the proximal side, the porphyrin is ligated to a histidine residue (170) that is within hydrogen bonding distance to a glutamic acid (220, Figure 1B). Resonance Raman evidence suggests that the His/Glu interaction is weak, leading to a weak Fe-His bond where the imidazole side chain is uncharged/neutral (13). Chemical and spectroscopic evidence suggest that a ferric-hydroxide species forms on the distal side at relatively low pH, due to a combination of the weak Fe-His bond and a strong hydrogen bond donor in the distal pocket. That donor, likely the distal pocket Arg, may be pH-titratable, with the greatest catalytic activity observed when it is in its positively charged form. Also on the proximal side and spanning the heme periphery are a series of three Trp residues (155, 156, 227) and an intervening His (224), any of which might be involved in an observed migration of radical character away from Compound I and onto an amino acid side chain (14). Formation of such a Compound ES species (Fe(IV)=O/Trp·) appears to be a step along the porphyrin degradation pathway, leading to a bleached heme chromophore and non-functional enzyme (13).
3. Protein sequence diversity within the Cld family proteins
In contrast to DA-Cld, other CFP have yet to be fully characterized, and their biological functions are not entirely clear. The presence of Cld protein sequences in diverse species spanning 10 phyla suggests an ancient origin for the family. Among the Proteobacteria (Figure 2, yellow), the sequences obtained from verified perchlorate respirers (PRB) (Figure 2, indicated by a-c) along with the non-Proteobacterial Nitrospira defluvii (9), a species known to decompose chlorite into Cl− and O2 (Figure 2, d), emerge in a single cluster that branches off from the remaining members of the phylum. These remaining members presumably use the cld gene product for some purpose other than chlorite disposal. Extensive chlorite contamination is not expected in their environments, nor has chlorite decomposition by these species been documented. An analysis of 16S RNA and cld gene phylogenies from perchlorate respirers indicated that the two trees were nonidentical, suggesting the acquisition of cld in some of these species occurred via horizontal gene transfer (7, 9). In addition, none of the Proteobacterial species appears to have two cld gene copies. The first bona fide chlorite dismutase could have been a single gene product moonlighting between ancestral and acquired functions. Interestingly, those Proteobacterial cld genes with demonstrated chlorite dismutase activity have proteins preceded by a SecB-dependent signal peptide, as detected by SignalP, while the remainder lack any identifiable secretion signal.
Figure 2. Phylogenetic tree of Cld family protein sequences from diverse hosts.
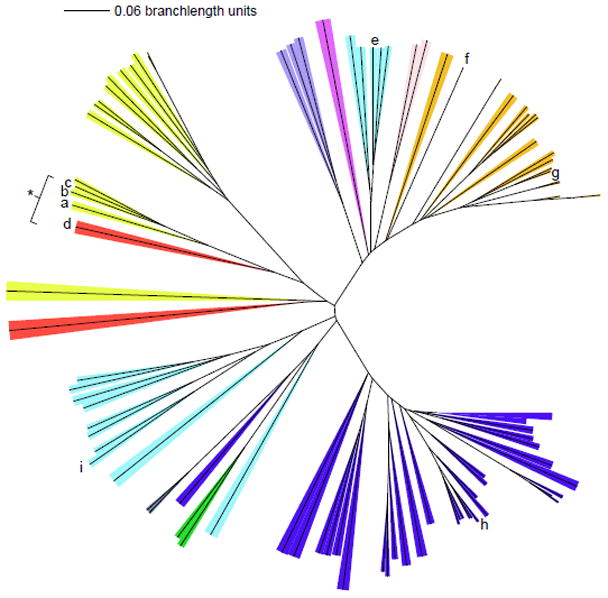
The phylum/kingdom affiliation of each species is indicated by color: Proteobacteria (yellow), Firmicutes (orange), Nitrospirae (red), Actinobacteria (blue), Archaea (light blue), Deinococcus-Thermus (grey), Chloroflexi (green), Planctomycetes (dark purple), Veruccomicrobia (light purple), and Acidobacteria (pink). The Halobacteriacea, pictured near the bottom of the tree, form their own group distinct from the other archea. Species known to carry out chlorite detoxification are indicated with a bracket/asterisk. CFPs originating from species mentioned in the text are as follows: (a) Dechloromonas aromatica; (b) Dechloromonas agitata; (c) Ideonella dechloratans; (d) Nitrospira defluvii; (e) Halobacterium sp. NRC-1; (f) Thermus thermophilus HB8; (g) Geobacillus stearothermophilus; (h) Mycobacterium tuberculosis; (i) Thermoplasma acidophilum. A three-iteration PSI-BLAST search was performed using DA-Cld as the bait sequence. The top 500 result sequences were aligned by ClustalX, and a phylogenetic tree was constructed. Representative sequences from each phylum were chosen for the above display. Settings used for the tree building were: random number = 111 and bootstrap maximum = 1000. The iTOL (Interactive Tree of Life) program was used for branch coloring and figure generation (http://itol.embl.de/). Bootstrap values for the major branching points and a table listing species of origin and accession numbers are included as Supplementary data.
Amino acid sequence conservation among the Proteobacterial sequences from PRB is extremely high, though it is less so when non-respirers from the phylum are included in the comparison. Mapping the conserved residues onto a monomer of the nitrite-bound DA-Cld crystal structure illustrates that in each case the degree of conservation is strong in the vicinity of the heme (Figure 3; see also Figure 1B). Possible residues of functional importance in the Proteobacterial PRB, including the distal pocket Arg (183), the hydrophobic Leu/Thr/Phe triad (185/198/200), and a set of three Trp (155, 156, 227) and one His (224) residues potentially involved in radical formation, are all strictly conserved (Table 1). The non-Proteobacterial N. defluvii, which contains a highly active Cld enzyme, is a nitrifying (i.e., nitrite oxidizing) member of phylum Nitrospirae (17). It furthermore is not a known perchlorate reducer, and the role of chlorite decomposition in the organism is not well understood. Equivalent active site residues are conserved between N. defluvii and the Proteobacterial PRBs, except for two of the potential radical sites: Trp (227) is a Phe in this organism, and His (224) is Asn (Figure S1). Strict conservation holds for three of the distal pocket residues in the Proteobacterial non-respirers, with the fourth, Thr (198 in DA-Cld), being an Asn in some sequences (Figure S2). On the proximal side, as in N. defluvii, the Trp and His (227, 224 respectively in DA-Cld) are not conserved in the Proteobacterial non-respirers (Table 1, Figure S2). A Thr (231) residue that coordinates a Ca2+ cation at the subunit-subunit interface of DA-Cld is strictly conserved across all the PRB but appears to be an Arg in the non-respirers (Table 1, Figure S2). In sum, though the non-respirers are not expected to have been subjected to chlorite-based selection pressure, they all possess a CFP with an active site architecture containing many structural elements postulated to be essential for chlorite detoxification. It is therefore possible that these CFPs could indeed be capable of catalyzing chlorite dismutation to some extent (Scheme 1).
Figure 3. Stereo view of DA-Cld monomer showing residues conserved among all phyla.

DA-Cld monomer shown as a faded grey cartoon. Strictly and strongly conserved residues are shown as sticks and colored by atom (carbon orange and cyan respectively). Heme is drawn in grey stick, and the iron as an orange sphere. Note: all phylogenically conserved residues fall within the C-terminal domain. This figure was generated using PyMOL (http://www.pymol.org/).
Table 1.
Conservation of residues of likely functional relevance among Cld protein family membersa
| Amino acid | Proposed rolea | Chlorite decomposing bacteriab | Proteobacteria | Firmicutes | Euryarchaeota | Actinobacteria |
|---|---|---|---|---|---|---|
| H170 | Proximal heme ligand | conserved | conserved | conserved | conserved | conserved |
| E220 | H-bonding to axial His | conserved | conserved | conserved | Lys, Arg, Met, Leu, Glu | variable |
| R183 | distal polar residue | conserved | conserved | Gln | Ser | Arg/Lys, Ala Glnc |
| L185 | distal hydrophobic triad | conserved | conserved | Ile | Thr | Leu, Iso, or Thr |
| T198 | distal hydrophobic triad | conserved | mostly conservedc | Val; Thr | Val | Thr, Val, or Leu |
| F200 | distal hydrophobic triad | conserved | conserved | Leu(Phe) | Tyr | variable |
| W155 | radical pathway | conserved | conserved | conserved | conserved | conserved |
| W156 | radical pathway | conserved | conserved | Tyr | Tyr | Trp, Tyr |
| H224 | radical pathway | strongly conservedd | Val, Iso or Asn | Arg | Trp | variable |
| W227 | radical pathway | mostly conserved | Glu or Phe | Glu | Lys, Leu, Ser, Arg, Asn | variable |
| D192 | Ca2+ligand | conserved | Gly or Ser | Gly | Gly/Ala (Glu) | Asp or Gly |
| T231 | Ca2+ligand | conserved | Arg | Phe | Ile, Leu, Val | variable |
Residues of likely functional relevance have been identified in structural and functional studies of DA-Cld. The residue numbering shown here refers to the heterologously expressed and structurally characterized DA-Cld. Colors of the amino acids named correlate with colors used in Figure 1.
Includes sequences from all experimentally verified Proteobacterial PRB and N. defluvii.
Underlined residue is at the exact position as the indicated DA-Cld residue. Conserved residues immediately up and down stream are indicated.
”Strongly conserved” indicates that 2 or fewer species had a different residue at the given position. The sequence alignments used in generating this table are given in Supporting information.
By contrast, when phylogenetically diverse CFPs are compared, the degree of observed sequence conservation is low (Figures 3, S3). As expected, the heme-coordinating His residue (170 in DA-Cld) is strictly conserved. A strictly conserved Pro residue (148 in DA-Cld) is located in the β-strand adjacent to the two strands that form the roof of the distal pocket, and likely plays a structural role in the protein fold. Additionally, two of the three active site Trp residues observed in DA-Cld are strictly (155) and strongly (156) conserved, respectively. The position of the strictly conserved Trp (155) appears to be important, as the indole ring is sandwiched between the alkyl chains of a strictly conserved Arg residue (163 in DA-Cld) and a strongly conserved Lys (151 in DA-Cld). Additionally, two hydrophobic residues near the distal heme face (Phe and Val) and an acidic residue (Glu in most phyla, but Asp in Proteobacteria and bona fide Clds) are all strongly conserved (196, 181, 195 in DA-Cld respectively). The latter forms a hydrogen bond to the strongly conserved Trp. Finally, a strongly conserved Leu (191 in DA-Cld) lies at the center of the monomer-monomer interface. It is notable that several proximal and distal pocket residues that are known or likely to be functionally significant for DA-Cld are not well conserved across the Cld family. This again suggests that the Cld proteins from NPRB have one or more biochemical functions that are distinct from the chlorite-directed reaction of DA-Cld. Primary sequence- and structure-based predictions for active site amino acid occupancies are further examined in 4.1.2. Identifying probable functions will require a closer investigation of each of the major phylogenetic groupings of CFPs identified above.
4. Possible roles for Clds: clues from synteny and structure
4.1 Gene organization
Gene organization often gives clues to function in prokaryotes. The placement of the cld gene in PRB (Figure 2, indicated by a-c) was previously examined, and shown to occur upstream or downstream of an operon of genes encoding a (per)chlorate reductase in one of multiple orientations. In the cases described, the cld gene was preceded by its own promoter and upregulated under perchlorate-reducing conditions in the perchlorate respirer D. agitata or constitutively expressed in two chlorate respirers (18, 19).
In the non-perchlorate-respiring organisms, at least three widely conserved synteny patterns are observed. In the majority of the Actinobacteria including all of the mycobacteria, the cld gene is found adjacent to a heme biosynthesis cluster and is typically located immediately downstream of hemG (encoding protoporphyrinogen oxidase) or hemH (ferrochelatase), with which it is co-operonic (20). The cld gene is likewise located within a set of hem genes in several species of Deinococci and Chloroflexi (Figure 2, grey and green respectively). In three cases within the Actinobacteria, the cld gene forms a fusion with a hemH gene. The physical linkage of two proteins into one “Rosetta Stone” sequence can imply a functional linkage (21, 22). The cld-hemH fusion therefore implies a role for cld in heme or iron metabolism, or possibly an indirect role via the sensing or metabolism of an allied species such as O2. Recent work with the cld gene from M. tuberculosis (Figure 2; indicated by h) confirmed that it indeed has a functional link to the final steps in porphyrin biosynthesis and that the gene product has catalase activity, although the precise biochemical role of the protein remains to be defined (20).
Among the archaea (Figure 2, light blue), a similar series of fusions was observed. In all of the Halobacteriaceae for which full genome sequences currently exist (8 halophilic archaeal species including Haloferax volcanii, Halobacterium sp. NRC-1 (Figure 2; indicated by e), Haloarcula marismortui, and Natronomonas pharaonis) a chlorite dismutase is found as the C-terminal domain fused to an N-terminal polyketide biosynthesis monooxygenase domain. The latter family includes functionally diverse members involved in a variety of biosynthetic and degradative processes. These include enzymes catalyzing steps in antibiotic biosynthesis as well as, interestingly, the Staphylococcus aureus heme oxygenases IsdG and IsdI, (PDB codes 1XBW and 1SQE). These proteins are involved in heme degradation, working according to a different mechanism from canonical heme oxygenases (23, 24). The linkage to IsdG/I-like domains again suggests a possible role for CFPs in heme metabolism in these organisms, possibly as heme carriers or chaperones. Shaanan et al. proposed that the O2 generated by the Cld domain could be consumed by the linked domain, though O2 generation has not yet been experimentally demonstrated for these proteins (25). Unlike all other CFPs, alignment of the halophiles Cld domains with several different Cld family members fails to identify a strictly conserved, putative heme-binding His (see 4.2.1). Notably, the halophilic archaea emerge as their own branch of the phylogenetic tree, separate from the archaea from other phyla (Figure 2; light blue, lower left). Further instances of gene fusions or widely conserved patterns of synteny were not obvious among archaea outside the halophiles (i.e., the Euryarchaeota).
Finally, among the Firmicutes (Figure 2; orange), the cld gene is almost always located adjacent to the gene for the fermentative enzyme acetyl-CoA:orthophosphate acetyltransferase phosphotransacetylase (pta or eutD). In the staphylococci, a gene cluster including a mevalonate kinase and mevalonate decarboxylase is nearby. In several bacilli (Figure 2; e.g. Geobacillus stearothermophilus indicated by g), a cluster encoding the biosynthesis of bacilysin, a bacterial antibiotic precursor, is located next to the cld gene. The cluster includes the bacA–D genes and the adjacent ywfGH genes, which together catalyze the conversion of prephenate to tetrahydroxotyrosine (26). In some annotations, the cld gene is referred to as ywfI, though it is separated from the ywfGH genes by >100 bases and does not appear to be part of the same operon. The ywfI gene was previously shown to be upregulated during the aerobic to anaerobic transition in Bacillus subtilis (27), under the control of the Fnr (fumarate and nitrate reduction) redox regulator and the resDE two component regulator. The gene product has been proposed to serve an analogous role to that in the Actinobacteria, in heme biosynthesis (20).
4.2 Structure [The associated Protein Data Bank codes are given in Table S1]
4.2.1 Known structures of Cld family proteins
Three structures for Clds with known O2-evolving functions are now available, from D. aromatica (14), A. oryzae (15), and N. defluvii (16). The crystal structure of the N. defluvii Cld shows that it shares an overall monomer structure (root mean squared deviation (RMSD) = 1.00 Å), common active site architecture, and most of its active site residues (see section 3.) (Figure S1) with the more extensively characterized DA-Cld (16). The A. oryzae monomer structure is very similar to DA-Cld (RMSD = 0.62 Å), as it is 98% identical at the primary sequence level (15). A major difference lies in the placement of a 13-residue loop (218–230) containing several active site residues in the DA-Cld structure (Glu220, His224, Trp227, Figure 1B) that could not be modeled in the A. oryzae electron density.
There are three crystal structures of heme-less CFPs, all generated by Structural Genomics Consortia, for species from three different phyla that occupy two of the major branches of the phylogenetic tree (Firmicutes, Deinococcus-Thermus {Ebihara et al (2005)}, and Euryarchaeota (Figure 2; indicated by g, f and i respectively)). These structures and their primary sequences show a high degree of similarity to DA-Cld. In each case, like DA-Cld, the protein crystallized as a pentamer, though without any associated heme.
The probable location of the heme can be discerned via the strictly conserved His residue that serves as the proximal ligand, and by inspection of the distinctive arrangement of α-helices and β-sheets (Figure 4). Based on the structures, side chains lining the probable site of heme binding, are identified in Figure 5; these are comparable to proposed key residues in DA-Cld shown in Figure 1B and listed in Table 1. Distal and proximal residues of functional importance are more difficult to identify in the heme-less crystal structures as although the main-chain overlays well with the DA-Cld structure observed side chain rotamers probably change upon heme binding,. The apo-structures from Geobacillus stearothermophilus (phylum Firmicutes, Figure 5A) and the halophilic bacterium Thermus thermophilus HB8 (phylum Deinococcus-Thermus, Figure 5B) are essentially identical in the vicinity of the heme (28). The Deinococcus-Thermus cld lies among the Firmicutes sequences on the phylogenetic tree, suggesting horizontal gene transfer; indeed, it is 53% amino acid sequence identical to the G. stearothermophilus protein. Using the G. stearothermophilus CFP numbering, the conserved axial His residue (171) defines the expected proximal pocket. The residue corresponding to a possible acidic hydrogen bonding partner to the axial His is unclear. Glu220, the sequence equivalent that performs this role in DA-Cld (Glu220) is pointing away from His171 and would need to rotate to form a hydrogen bond. There is also an Asp (219) residue N-terminal to the Glu which could be a potential hydrogen bonding partner to the axial His. This is conserved in the Firmicutes, but very variable across phyla, so it is a less likely partner than the much more conserved Glu. In the Thermoplasma acidophilum CFP (domain archaea, phylum Euryarchaeota, Figure 5C), only the conserved Glu195 is present in the proximal pocket, and it would also need to rotate into hydrogen bonding contact with the proximal His (146). In all three heme-less CFP structures, the two strictly conserved Proteobacterial CFP Trp residues that lie near the heme propionic acid side-chains are a Trp156 and Tyr157 (G. stearothermophilus numbering). The G. stearothermophilus and T. thermophilus HB8 distal pockets are lined exclusively with hydrophobic residues (Ile186, Val199, Leu201), with a strictly conserved Gln (184) at the position of the DA-Cld Arg183.. The T. acidophilum CFP distal pocket (Figure 5C) exhibits a substantially more polar environment. The active site contains three polar residues (Thr163, Ser161, Tyr178) which are strictly conserved among all Clds from Euryarchaeota (Table 1) (29). Unlike Thr198 in DA-Cld (Figure 1B), the hydroxyl group of Thr163 points into the distal pocket. Ser (161) occupies the position of the catalytically important Arg183 in DA-Cld. Thus it appears that the identity of this distal pocket residue is a key marker within phylogenetic clusters (Arg in Proteobacteria, Gln in Firmicutes, Ser in Euryarchaeota) and could be an indicator of differing CFP function (Table 1).
Figure 4. Conserved regions of secondary structure from which active site residues are derived.


(A) Structure alignment of CFP members with DA-Cld as identified via Dali. Aligned regions of secondary structure are labeled (H = helix; L = loop; E = β-sheet). To illustrate the conservation of core secondary structure elements, the alignment excludes insertions between aligned structures. Individual secondary structure elements that constitute the heme distal and proximal pockets are indicated by labeled arrows. Residues from DA-Cld are shown explicitly above their respective locations in discrete secondary structure elements (i.e. α-helix or β-sheet). (B) Secondary structure elements mapped onto the structure of DA-Cld. All structurally characterized enzymes within this family generate a heme-binding pocket from five separate secondary structure elements (purple). In the E.coli CFP, S. oneidensis CFP, and T. cucumeris CFP crystal structures, an aspartate residue is located in the distal pocket (PDB codes are given in Table S1). It invariably originates from a similarly positioned loop region (grey arrows). Color coding and labeling of these elements is according to (A). Images in this figure were generated using PyMOL (http://www.pymol.org/)
Figure 5. Active site stereo views of crystallographically characterized apo-CFPs illustrating possible heme environments.
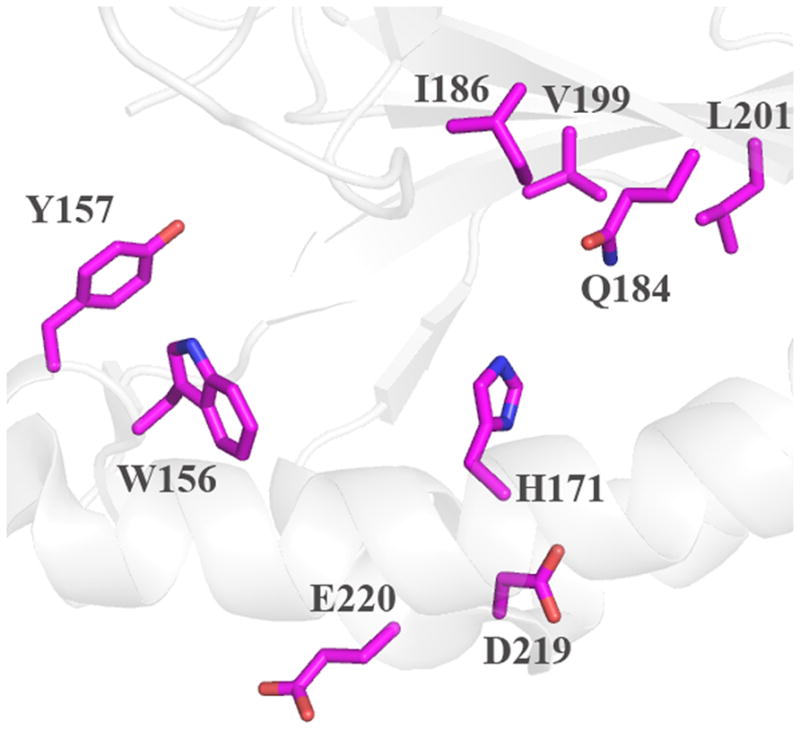
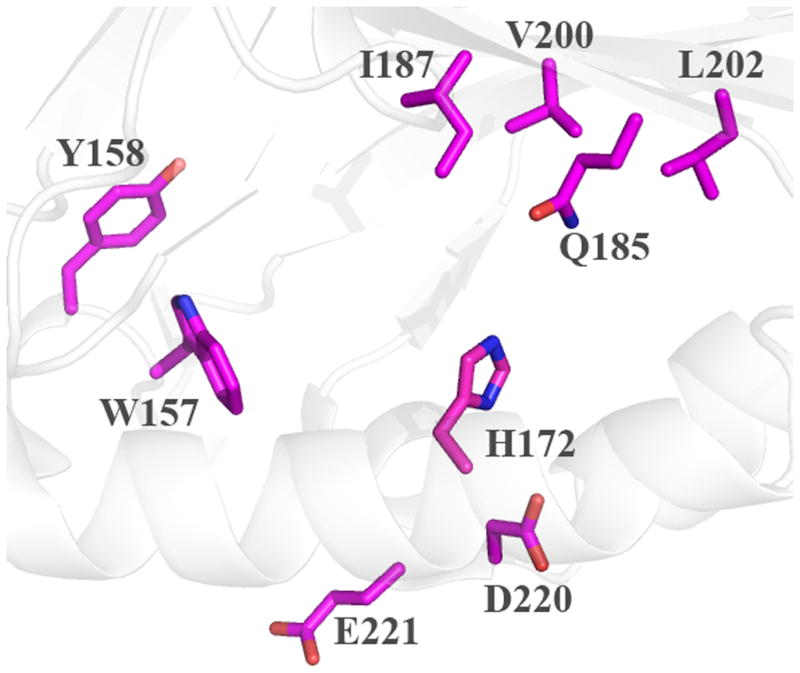
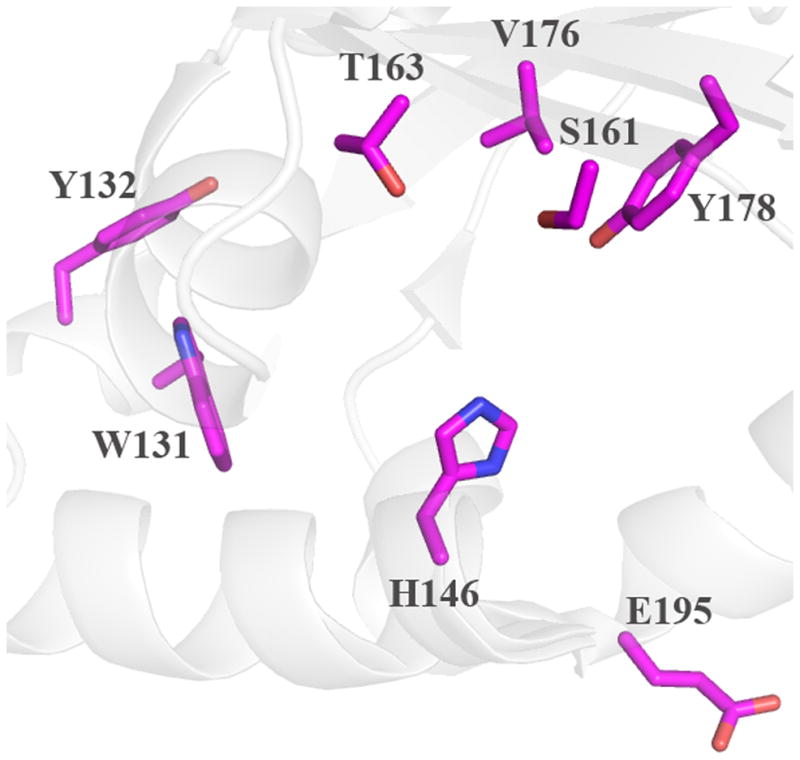
Cartoon diagram representations of protein monomers (carbon light grey) are shown, with side-chains of active site residues shown as sticks. Residues lining the expected heme pocket that fall within strictly conserved secondary structure elements are colored by atom (carbon magenta). Note that the same residues were also identified from primary sequence alignments with DA-Cld (Table 1). (A) G. stearothermophilus (Firmicutes); (B) T. thermophilus (Deinococcus-Thermus). (C) T. acidophilum (Euryarchaeota). This figure was generated using PyMOL (http://www.pymol.org/).
4.2.2 Predicted structures and possible functions of Cld family proteins (COG3253S)
Crystal structures are not available for representative members of most phyla of the phylogenetic tree, particularly the large Actinobacterial branch.. For the known crystal structures, primary sequence alignments do provide a reasonable indication of the residues around the heme, although ambiguities in the heme-less structures remain. For example, identification of a hydrogen bonding partner (if present) for the proximal His is uncertain. (A hydrogen bonding partner is not evident in the structures, and in the sequence alignment, DA-Cld Glu220 aligns with Val221 in T. thermophilus and Arg193 in T. acidophilum; Figures S2, S3.) Alignment of conserved secondary structures for all structurally characterized CFPs (and their structural relatives, 4.2.3) shows that all of the residues known to line the heme pockets derive from the same set of conserved helix/sheet substructures (Figure 4). Mapping the actual positions assumed by sequentially conserved residues onto the secondary structural elements of the known crystal structures can help narrow down and give confidence to the likely residues defining the heme environment for structurally uncharacterized proteins of low sequence identity (30). Such structure-based sequence alignments indicate a conserved Ala at the position of Arg183 in the Actinobacteria, although an Arg or Lys residue is typically located one residue upstream and Asn downstream, both of which might be distal pocket candidates (Figure S5, Table 1). A triad of potential distal hydrophobic residues is also positionally conserved in these species, though the identities of the amino acids are not. As sequence identity with the known crystal structures lessens, using the structures in the alignment becomes less effective, and reliance on the bootstrapping used to create the sequence alignments becomes dominant in the comparisons (Figure S6).
In terms of possible functions among the CFPs that are not known to have chlorite decomposition activity, the Proteobacterial proteins clearly bear the closest active site resemblance to the true Clds (Table 1). These proteins share a substantial number of strictly conserved residues with DA-Cld, particularly around the heme pocket, which include an Arg that aligns with Arg183 in DA-Cld. The Actinobacteria have an Ala at this position, but could have either an Arg/Lys or Asn if the alignment is shifted by a residue (Figure S3, Table 1). The structurally characterized apo-CFPs and the remaining species on the phylogenetic tree are all expected to have Gln or Ser at this position (Figure 5, S3, S4). Given the lack of any residue capable of acid/base chemistry or even of strongly polarizing a bound ligand, it appears unlikely that these proteins would be involved in either efficient redox chemistry or the decomposition of chlorite. Indeed, only very weak peroxidase and/or catalase activities have been observed for CFPs studied thus far (20, 28). These proteins could have other types of roles, as sensor/regulators, chaperones, or trafficking agents, possibly involving hydrogen-bonding from the distal pocket residue. Given the expected diversity of active site structures, it seems reasonable that different family members may serve different biochemical and biological functions.
Functionally essential distal pocket residues help to define the distinct properties of heme proteins. Heme-dependent peroxidases, catalases, gas sensors (O2/CO), gas transporters (O2/NO), and anion reductases all have distinct distal architectures. Typical peroxidases, for example, contain a distal His/Arg pair involved in peroxide activation/cleavage (31, 32), though some (including DyP, 4.2.3) have a functionally equivalent Asp/Arg (33). An additional aromatic residue (Phe or Trp) may be present, serving a steric role in positioning oxidizable substrates. Even though DA-Cld lacks the Asp or His of many peroxidases, it can nonetheless catalyze peroxidase chemistry, though far less efficiently than a typical peroxidase and with maximal efficiency at a much higher pH (DuBois et al., unpublished data). Bacterial catalases including the bifunctional catalase peroxidase (KatG) depend on a Trp/Arg/His triad (34–36). Canonical O2 transporters like hemoglobin possess an essential distal His that is optimally positioned to form a hydrogen bond to the bound O2 (37–39). The well-characterized gas sensing proteins CooA (CO), soluble guanylate cyclase (NO), and FixL (O2) likewise have distinct heme environments responsible for the selective recognition of their cognate ligand. Like myoglobin, the NO-binding domain of guanylate cyclase and FixL have distal residues (Tyr and Arg, respectively) that hydrogen bond to the coordinated gas molecule (40–42). CooA is 6-coordinate with an unusual His/Pro ligation, where Pro is displaced by CO (43–45). Nitrite reductase has a pair of distal His residues that are involved in protonation of the substrate (46). The distal pocket of true chlorite dismutases (those that decompose ClO2−) most closely resembles FixL s, with its single Arg residue. However, the Arg183 side chain in DA-Cld is not positioned to alternate between hydrogen bonding to the coordinated heme ligand and a propionic side chain. Moreover, the ceiling of the distal pocket in all Clds consists of β-sheet, while sensors like FixL utilize α-helices. Binding of a ligand facilitates movement of a distal pocket residue via the new hydrogen-bonding contacts that form (38, 47). In turn, the α−helices in the distal pocket are repositioned to assist in structurally propagating the message that the heme ligand is bound. There is no structural evidence in the Cld family for such behavior.
4.2.3 Structural comparisons to proteins beyond the CFPs: a CDE structural superfamily
A search of the Protein Data Bank for structural homologs via the Dali Server revealed further interesting connections between the Cld family and other proteins (48, 49). This search identified EfeB (COG2837), a structurally characterized dimeric hemoprotein from Escherichia coli, as the top structural match from outside the Cld-containing COG3253 (50, 51). With EfeU (an integral membrane Fe permease homologous to the yeast Ftr1p) and EfeO (a cupredoxin), the protein forms part of an Fe- and pH-regulated, three-gene operon involved in Fe(II) import under acidic conditions (52, 53). The protein reportedly exhibits peroxidase activity under acidic conditions. Unlike the CFPs from PRB, it is exported to the periplasm not by the Sec- but by the twin arginine or Tat-dependent pathway (54). Recent work by Wandersman et al. showed that an efeB knockout impaired E. coli’s ability to acquire iron from heme. The group further proposed that the protein product has deferrochelatase activity, or the ability to remove ferrous iron from heme, leaving protoporphyrin IX (PPIX) intact (55), though rigorous biochemical confirmation of that function has not yet been documented. Such an enzymatic activity would be important for reclaiming heme iron from organisms lacking a functional heme oxygenase, or from (facultative) anaerobes under anaerobic conditions. Whether and how such an activity would interface with the presumptive Fe(II) import function of EfeUO or merely respond to the same low pH/low Fe conditions is not known. A second distantly related paralogous protein also exists in E. coli (YfeX). This has no secretion peptide and is cytoplasmic. The yfeX and efeB genes were able to complement each other, and either was sufficient for a heme-uptake-proficient strain of E. coli to retrieve iron from heme (53).
The Dali search also identified the C-terminal domain of DyP, a dye-decolorizing peroxidase from Thanatephorus cucumeris, as well as TyrA, a bacterial prephenate dehydrogenase believed to be involved in melanin biosynthesis that has been structurally characterized for Shewanella oneidensis (56). DyP and its homologs (e.g., Termitomyces albuminous peroxidase (TAP) and cpop21, a hypothetical peroxidase from Polyporaceae sp.), EfeB and TyrA have been placed in the “DyP-type peroxidase family,” a diverse grouping of mostly dimeric fungal, bacterial, and archaeal hemoproteins that share common structural and catalytic properties (33, 57). They fall outside of the conventional three classes of non-animal heme peroxidases suggested by Welinder (58) due to their mixed bacterial and fungal origins and their unusual primary and three-dimensional structures, and they are understood to have acquired their heme-binding and peroxidase functions by convergent evolution with respect to the other peroxidases. These proteins have been of biotechnological interest due to their ability to degrade robust dyes, though their biological roles within their hosts are unclear.
The DyP-family proteins as well as EfeB are typically monomers or homodimers, but the monomers have many structural features in common with CFPs. The heme binding motif consists of αhelices proximal to the porphyrin plane that contain the strictly conserved His ligand, and a series of βstrands spanning the distal pocket in a manner highly conserved with DA-Cld and other structurally characterized CFPs. (33, 50, 51, 55) (Figure 4). This architecture stands in contrast to the well-characterized peroxidases, which consist primarily of α-helices. A superimposition of the CFP monomer structures with EfeB, TyrA and DyP monomer structures clearly indicates the shared elements of a common fold among the three families (Figure S7). This shared fold extends not only to the heme binding pocket, but also to the structurally homologous N-terminal portion that is devoid of heme (4.2.4), though with multiple sequence insertions and loop embellishments among the various available structures (Figure S5, S7). DA-Cld serves as one of the most compact examples among the known structures, having very little loop secondary structure.
Pairwise sequence comparisons indicate 8, 25, and 17% identity between DA-Cld and T. cucumeris DyP, E. coli EfeB, or S. oneidensis TyrA respectively (corresponding to E values of 0.39, 2.1, and 0.85). The former is close to the %-identity expected between two randomly chosen sequences (~6%) (59). PSI-BLAST searches using the T. cucumeris DyP as bait identified proteins annotated as EfeB-like as likely homologs. The reverse was true if the E. coli EfeB was used as bait. Phylogenetic analysis of diverse CFP, DyP, and EfeB sequences, however, indicates three distinct clades (Figure 6). It may therefore be most appropriate to describe the CFPs, DyPs, and EfeB/COG2837 proteins as separate subfamilies within a structural superfamily, which we suggest naming the “CDE structural superfamily.” Like the CFPs, the DyPs are diverse, with a variety of fungal and bacterial members. The EfeB/COG2837 proteins are much less so, being typically observed in a few genera of both Gram(+) and Gram(−) bacteria.
Figure 6. Phylogenetic tree illustrating relationships between CFPs, DyPs, and COG2837 proteins.
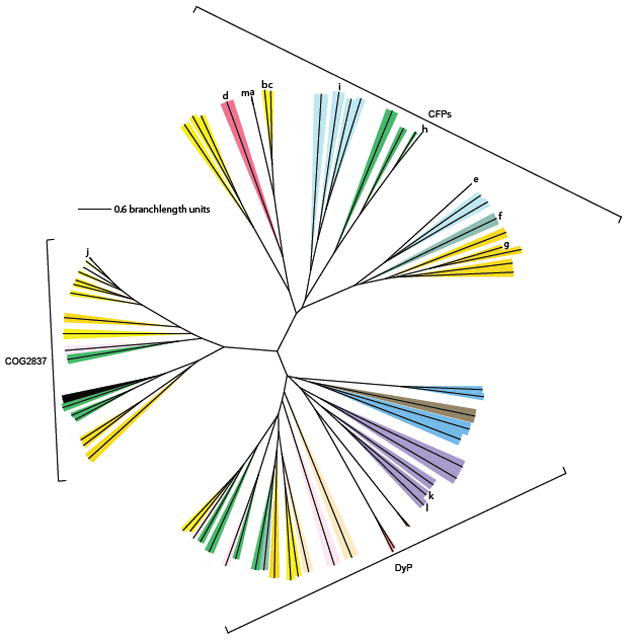
Representative CFP sequences were selected from each major phylum in Figure 2 and plotted here, along with representative DyP family and COG2837 proteins. The phylum/kingdom affiliation of each species is indicated by color: Proteobacteria (yellow), Acidobacteria (light pink), Nitrospirae (dark pink), Archaea (light blue), Cyanobacteria (royal blue), Deinococcus-Thermus (teal), Firmicutes (orange), Actinobacteria (green), Ascomycota (salmon), Plantae (black), Bacteroidetes (tan), Basidiomycota (violet), and Planctomycetes (brown). Proteins originating from species mentioned in the text are as follows: a-i see legend for Figure 2; (j) Escherichia coli EfeB; (k) Thanatephorus cucumeris DyP; (l) Shewanella oneidensis TyrA. The following was performed using bait sequences EfeB from E. coli and DyP from T. cucumeris: a three-iteration PSI-BLAST search yielded top 500 result sequences which were aligned by ClustalX. For this tree, EfeB homologs with a PSI-BLAST E-value above 3×10−68 and DyP sequences with a PSI-BLAST E-value above 2×10−22 were eliminated, leaving approximately 250 sequences from each family, a subset of which are shown on the tree. Bootstrap values for the major branching points and a table listing species of origin and accession numbers are included as Supplementary data.
The active sites of DyP-family and EfeB/COG2837 proteins all possess an Fe-His-Asp/Glu triad in the proximal pocket and an Asp-Arg pair in the distal pocket, as well as one or more hydrophobic residues in the distal pocket and on the heme periphery (Figure 7). Overlays of the DyP, TyrA, and EfeB active sites with DA-Cld show a highly similar proximal pocket with the strictly conserved histidine being hydrogen bonded to either an aspartate or glutamate residue (Figure 8A). The distal pocket of the EfeB/DyP/TyrA proteins differs primarily through the addition of an Asp that complements the distal Arg in these proteins and the Proteobacteria CFPs (including the Clds). The Asp-Arg pair is understood to be functionally equivalent to the His-Arg pair found in plant and fungal peroxidases, which together serve acid/base and polarizing roles toward the H2O2 substrate. The substitution of Asp for His in DyP is believed to be responsible for the lower-pH optimum observed for many DyP enzymes (60). In a deferrochelation mechanism, the Asp in EfeB could serve a role in ushering Fe(II) away from PPIX, while the Arg could serve to position the heme/PPIX via the cofactor’s propionic acid side-chains. All four contributing residues from the B1 and B2 β-strands of CFPs also form part of the distal pocket of the EfeB/DyP/TyrA proteins. However, there are key structural differences in the architecture of these two strands in the EfeB/DyP/TyrA proteins compared to the CFPs that enable the accommodation of the additional Asp residue in the distal pocket (Figure 8B). The B2 β-strand is shifted relative to the heme, altering the structural positions of the two B2 residues that form part of the distal pocket. The Phe200 in DA-Cld is sequentially conserved in all four enzyme structures, and although the Cα main-chain position is spatially different, the ring position still lies in the same region of the distal pocket in all four enzymes. Unlike the side-chain of Phe200, the Thr198 of DA-Cld occupies a different part of the pocket to the sequentially equivalent leucine residues of the EfeB/DyP/TyrA proteins (Leu331, 354, 255 respectively; Figure 7). The position of the distal arginine on the B1 β-strand is conserved between the EfeB/DyP/TyrA and Cld enzymes, but a bulge in the B1 β-strand of EfeB/DyP/TyrA places the other distal pocket residue from the B1 strand in a different area (DA-Cld Leu185;.EfeB Gly314; DyP Ser331; TyrA Ser244, Figures 7, 8B). The different positioning of this B1 residue, coupled with the repositioning of the B2 residue two positions N-terminal to the Phe, opens up a space such that the strictly conserved Asp residue in EfeB/DyP/TyrA can become part of the distal pocket. The distal Asp is contributed by the L1 region in EfeB/DyP/TyrA that contains no distal pocket residues in the known CFP structures (Figure 4). The position of the Asp carboxylate group of EfeB/DyP/TyrA is occupied by the Leu185 side-chain from the B1 β-strand in DA-Cld. Leu, Ile and Thr are sequentially predicted in other CFPs, and representative structures (although some are heme-less) are available in each case (Leu, DA-Cld, A. oryzae Cld, N. defluvii Cld; Ile, G. stearothermophilus CFP, T. thermophilus CFP; Thr, T. acidophilum CFP; Table S1). These structures show no evidence that an L1 residue will form part of the distal pocket. It is clear that the accommodation of the distal Asp through the structural alterations observed in the B1 and B2 β-strands of EfeB/DyP/TyrA compared to the known CFP structures will be a key distinction separating the majority, if not all, of the CFPs from the EfeB/DyP protein families. With the presence of the L1 Asp, it is not surprising that these structurally related proteins are functionally non-identical to the CFPs.
Figure 7. Top structural homologs to DA-Cld outside the CFPs as identified by Dali.



(A) EfeB from Escherichia coli; (B) DyP from Thanatephorus cucumeris; (C) TyrA from Shewanella oneidensis. PDB codes are given in Table S1. Cartoon diagram representations of protein monomers (carbon grey) are shown, with side-chains of proposed key active site residues shown as sticks. On the monomers, residues that form the enzyme active site and originate from conserved secondary structure elements as seen in Figure 4 are colored by atom (carbon magenta). An Asp residue that is strictly conserved in EfeB and DyP family proteins is absent in CFPs (carbon grey). This figure was generated using PyMOL (http://www.pymol.org/).
Figure 8. Stereo view overlay of Cld and EfeB/DyP family heme environments.


(A) Key proximal pocket residues. (B) Distal residues. Drawn as stick colored by atom. Carbon coloring DA-Cld, green; EfeB, light blue; DyP, orange; TyrA, pink. Residues in DA-Cld are labeled in black, while those of DyP are labeled in orange. DyP and TyrA distal pockets also contain an additional residue with no correlate in the other structures (S331 and S244 respectively). The heme of DA-Cld is drawn in grey sticks and the Fe center represented as an orange sphere. This figure was generated using PyMOL (http://www.pymol.org/).
Another significant difference between the EfeB/DyP/TyrA structures and the three Proteobacterial Cld structures is the location of the heme pyrrole rings within the distal pocket. Although the B and D pyrrole rings (vinyl- and propionate-bearing, respectively) are in the same position, the A and C rings (vinyl- and propionate-bearing respectively) are swapped. This results in the porphyrin being flipped between the two families (Figure 9A). In the three CFP structures, both propionates are extended away from the porphyrin, making hydrogen bonds with two main-chain amides (Asn117 and Tyr118 in DA-Cld, Figure 9B). However, in the EfeB/DyP/TyrA structures, propionate D, which is spatially equivalent to the Clds propionate D, adopts a different conformation such that the acid group lies on the distal side of the heme, and hydrogen bonds to the distal pocket Arg (Figure 9A, 9C). In DyP/TyrA, the distal Arg is also hydrogen bonded to the distal pocket Asp, giving an interacting triad of Asp/Arg/propionate D. In the EfeB/DyP/TyrA structures, the propionate of ring C interacts with an Asn side-chain nitrogen that lies at the kink in the proximal His-containing helix A2 (DyP, Asn313; TyrA, Asn229; EfeB, Asn299 (Figure 9C). The C propionate in TyrA is solvent-accessible at the surface, but in DyP and EfeB it is buried and interacts with arginine side chains from different parts of the structure (Arg315 and Arg261 respectively). The DyP propionate additionally hydrogen bonds with a Ser (174).
Figure 9. Hemes of DA-Cld and DyP.



(A) Stereo view comparing heme orientation in DA-Cld and DyP. (B) Interactions of the propionates of DA-Cld. (C) Interactions of the propionates of DyP. Active site residues and hemes are shown as sticks and colored by atom with DA-Cld (carbon green) and DyP (carbon orange). Regions associated with DyP are labeled in orange font. Pyrrole rings of the heme b cofactor are labeled and shown in bolded font. The hemes in DA-Cld and Dyp are related by a 180° flip along an axis through pyrrole positions D and B. This figure was generated using PyMOL (http://www.pymol.org/).
4.2.4 Gene duplication within the monomer and lack of heme binding in the N-terminal domain
Across the structural superfamily, the center of each monomer has a pseudo two-fold rotational axis, linking two highly similar domains that likely arose from gene duplication (Figure 10A). In one domain heme (or demetallated protoporphyrin IX, PPIX) is bound. The other is porphyrin-free. At first glance, it is not obvious why only one domain binds heme/PPIX while the other domain does not. Superimposition of the two DA-Cld domains upon one another shows the two to be remarkably similar in structure. However, in the N-terminal heme-less domain the helices and β-sheet are more compact, and this is seen in all the known structures of CFPs and DyP/TyrA/EfeB (Figure 10B). This compactness is not a consequence of the loss of heme, as the apo-CFP C-terminal domains show no evidence of helix movement akin to that of the heme-less N-terminal domains of either themselves or those of the heme-containing crystal structures. Furthermore, none of the known structures has a His located equivalently to the strictly conserved proximal His ligand of the C-terminal domain. Closer inspection of the superimposed monomers reveals an extra helix formed from the protein C-terminus that is associated with the non-heme binding domain (Figure 10). Structurally this would impede access and sterically clash with a heme positioned equivalently to that observed in the C-terminal domains. This C-terminal α-helix is observed in all the superfamily structures except for the heme-free domain of EfeB, where the C-terminus is a disordered loop. Although EfeB lacks a structured C-terminus, it still lacks a proximal His ligand and has the same compact structure observed in all the other N-terminal domains. Overall, it appears highly unlikely that any of the N-terminal domains are competent to bind heme once fully folded.
Figure 10. Structural homology between the domains of the structural superfamily.


(A) Cartoon of the DA-Cld monomer with the N-terminal domain colored in yellow, and the C-terminal heme-binding domain colored in green. The C-terminal helix is colored in red. Heme is drawn explicitly in stick colored by atom, and iron is drawn as an orange sphere. (B) N-terminal and C-terminal domains overlaid. The alignments is done with the program SUPERPOSE (30). Colors as in panel A. This figure was generated using PyMOL (http://www.pymol.org/).
5. Conclusions
The chlorite dismutase family consists of a few Proteobacterial members capable of catalyzing the remarkably specific and efficient generation of O2 and Cl− from ClO2−, and an overwhelming majority of non-Proteobacterial members that have biochemical functions that have yet to be fully described. Our sequence and structural analyses suggest active site structural diversity within the Cld family of proteins, particularly involving polar/charged residues at positions likely important for function. We have also identified elements of a common core fold with the DyP family of peroxidases, including structural superfamily members TyrA and EfeB, and suggest grouping these into the CDE superfamily. The DyP/EfeB structural homologs contain heme oriented differently compared to the CFPs, with the porphyrin flipped such that the pyrrole rings A and C exchange places in the two protein groups. The N- and C-terminal domains of the core fold likely arose through gene duplication, but heme is not observed bound in any of the N-terminal domains. The compactness of the secondary structures, lack of a suitably placed His for proximal ligation, and the presence of an additional α-helix in most of the crystal structures suggests that the N-terminal domain is incompatible with heme-binding. It is clear that the CFP heme-binding scaffold is widespread, and the few members whose function has been investigated suggest a versatile platform supporting diverse chemistries. As CFPs are found in pathogens such as Mycobacterium tuberculosis, it is possible that when more fully characterized these may become attractive drug targets. Further biochemical and in vivo studies are clearly warranted.
Supplementary Material
Acknowledgments
The NIH (1R01GM090260) is gratefully acknowledged for supporting this work. We thank Garrett Moraski for helpful conversations.
Abbreviations
- COG
Cluster of orthologs
- DyP
dye-decolorizing peroxidase
- Cld
chlorite dismutase
- NPRB
non-perchlorate respiring bacteria
- PRB
perchlorate respiring bacteria
- CFP
Cld family protein
- DA-Cld
Dechloromonas aromatica chlorite dismutase
- PPIX
protoporphyrin IX
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Brandon Goblirsch, Email: gobli033@umn.edu.
Richard C. Kurker, Email: rkurker@nd.edu.
Bennett R. Streit, Email: bstreit@nd.edu.
Carrie M. Wilmot, Email: wilmo004@umn.edu.
Jennifer L. DuBois, Email: jdubois@nd.edu.
References
- 1.Clark JJ. Toxicology of perchlorate. In: Urbansky ET, editor. Perchlorate in the Environment. Kluwer Academic/Plenum; New York: 2000. [Google Scholar]
- 2.Hauntman DP, et al. Determination of perchlorate in drinking water using ion chromotography, revision 1 Method 314, Doc No EPA/815/B-99/003. US Environmental Protection Agency; 1999. [Google Scholar]
- 3.Urbansky ET. Perchlorate as an environmental contaminant. Environ Sci Pollut Res Int. 2002;9:187–192. doi: 10.1007/BF02987487. [DOI] [PubMed] [Google Scholar]
- 4.Christen K. EPA perchlorate decision takes many by surprise. Environ Sci Technol. 2003;37:347A–348A. doi: 10.1021/es032584l. [DOI] [PubMed] [Google Scholar]
- 5.Xu JL, Song YU, Min BK, Steinberg L, Logan BE. Microbial degradation of perchlorate: Principles and applications. Environ Eng Sci. 2003;20:405–422. [Google Scholar]
- 6.Coates JD, Achenbach LA. Microbial perchlorate reduction: Rocket-fuelled metabolism. Nature Rev Microbiol. 2004;2:569–580. doi: 10.1038/nrmicro926. [DOI] [PubMed] [Google Scholar]
- 7.Bender KS, O’Connor SA, Chakraborty R, Coates JD, Achenbach LA. Sequencing and transcriptional analysis of the chlorite dismutase gene of Dechloromonas agitata and its use as a metabolic probe. Appl Environ Microbiol. 2002;68:4820–4826. doi: 10.1128/AEM.68.10.4820-4826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bender KS, Rice MR, Fugate WH, Coates JD, Achenbach LA. Metabolic primers for detection of (Per) chlorate-reducing bacteria in the environment and phylogenetic analysis of cld gene sequences. Appl Environ Microbiol. 2004;70:5651–5658. doi: 10.1128/AEM.70.9.5651-5658.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maixner F, Wagner M, Luecker S, Pelletier E, Schmitz-Esser S, Hace K, Spieck E, Konrat R, Le Paslier D, Daims H. Environmental genomics reveals a functional chlorite dismutase in the nitrite-oxidizing bacterium ‘Candidatus Nitrospira defluvii’. Environ Microbiol. 2008;10:3043–3056. doi: 10.1111/j.1462-2920.2008.01646.x. [DOI] [PubMed] [Google Scholar]
- 10.Streit BR, Blanc B, Lukat-Rodgers GS, Rodgers KR, DuBois JL. How active-site protonation state influences the reactivity and ligation of the heme in chlorite dismutase. J Am Chem Soc. 2010;132:5711–5724. doi: 10.1021/ja9082182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee AQ, Streit BR, Zdilla M, Abu-Omar MA, DuBois JL. Chlorite dismutase is a heme enzyme that is highly selective for dioxygen formation. Proc Natl Acad Sci, USA. 2008;105:15654–15659. doi: 10.1073/pnas.0804279105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DuBois JL, Abu-Omar MA, Derat E. Insight into the catalytic cycle of heme-dependent chlorite dismutase: radical mechanism for O-O bond formation. Angew Chem Int Ed (submitted) [Google Scholar]
- 13.Streit BR, DuBois JL. Chemical and steady state kinetic analyses of a heterologously expressed heme dependent chlorite dismutase. Biochemistry. 2008;47:5271–5280. doi: 10.1021/bi800163x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goblirsch B, Streit BR, DuBois JL, Wilmot CM. Structural features promoting dioxygen production by Dechloromonas aromatica chlorite dismutase. J Biol Inorg Chem. 2010;15:879–888. doi: 10.1007/s00775-010-0651-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Geus DC, Thomassen EA, Hagedoorn PL, Pannu NS, van Duijn E, Abrahams JP. Crystal structure of chlorite dismutase, a detoxifying enzyme producing molecular oxygen. J Mol Biol. 2009;387:192–206. doi: 10.1016/j.jmb.2009.01.036. [DOI] [PubMed] [Google Scholar]
- 16.Kostan J, Sjöblom B, Maixner F, Mlynek G, Furtmüller PG, Obinger C, Wagner M, Daims H, Djinovic-Carugo K. Structural and functional characterisation of the chlorite dismutase from the nitrite-oxidizing bacterium “Candidatus Nitrospira defluvii”: Identification of a catalytically important amino acid residue. J Struct Biol. 2010;172:331–342. doi: 10.1016/j.jsb.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 17.Luecker S, Wagner M, Maixner F, et al. A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc Natl Acad Sci, USA. 2010;107:13479–13484. doi: 10.1073/pnas.1003860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bender KS, Shang C, Chakraborty R, Belchik SM, Coates JD, Achenbach LA. Identification, characterization, and classification of genes encoding perchlorate reductase. J Bacteriol. 2005;187:5090–5096. doi: 10.1128/JB.187.15.5090-5096.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu JL, Trimble JJ, Steinberg L, Logan BE. Chlorate and nitrate reduction pathways are separately induced in the perchlorate-respiring bacterium Dechlorosoma sp KJ and the chlorate-respiring bacterium Pseudomonas sp PDA. 2004;38:673–680. doi: 10.1016/j.watres.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 20.Dailey TA, Boynton TO, Albetel AN, Gerdes S, Johnson MK, Dailey HA. Discovery and characterization of HemQ, an essential heme biosynthetic pathway component. J Biol Chem. 2010;285:25978–25986. doi: 10.1074/jbc.M110.142604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enright JA. Protein interaction maps for complete genomes based on gene fusion events. Nature. 1999;402:86–90. doi: 10.1038/47056. [DOI] [PubMed] [Google Scholar]
- 22.Marcotte EM. Detecting protein function and protein–protein interactions from genome sequences. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 23.Reniere ML, Ukpabi GN, Harry SR, Stec DF, Krull R, Wright DW, Bachmann BO, Murphy ME, Skaar EP. The IsdG-family of haem oxygenases degrades heme to a novel chromophore. Molec Microbiol. 2010;75:1529–1538. doi: 10.1111/j.1365-2958.2010.07076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee WC, Reniere ML, Skaar EP, Murphy MEP. Ruffling of metalloporphyrins bound to IsdG and IsdI, two heme-degrading enzymes in Staphylococcus aureus. J Biol Chem. 2008;283:30957–30963. doi: 10.1074/jbc.M709486200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bab-Dinitz E, Shmuely H, Maupin-Furlow J, Eichler J, Shaanan B. Haloferax volcanii PitA: an example of functional interaction between the Pfam chlorite dismutase and antibiotic biosynthesis monooxygenase families? Bioinformatics. 2006;22:671–675. doi: 10.1093/bioinformatics/btk043. [DOI] [PubMed] [Google Scholar]
- 26.Mahlstedt SA, Walsh CT. Investigation of anticapsin biosynthesis reveals a four-enzyme pathway to tetrahydrotyrosine in Bacillus subtilis. Biochemistry. 2010;49:912–923. doi: 10.1021/bi9021186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marino M, Hoffmann T, Schmid R, Mobitz H, Jahn D. Changes in protein synthesis during the adaptation of Bacillus subtilis to anaerobic growth conditions. Microbiol. 2000;46:97–105. doi: 10.1099/00221287-146-1-97. [DOI] [PubMed] [Google Scholar]
- 28.Ebihara A, Okamoto A, Kousumi Y, Yamamoto H, Masui R, Ueyama N, Yokoyama S, Kuramitsu S. Structure-based functional identification of a novel heme-binding protein from Thermus thermophilus HB8. J Struct Funct Gen. 2005;6:21–32. doi: 10.1007/s10969-005-1103-x. [DOI] [PubMed] [Google Scholar]
- 29.Chang C, Xu X, Savchenko A, Edwards A, Joachimiak A. Midwest Center for Structural Genomics (MCSG) unpublished data. [Google Scholar]
- 30.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr Sect D-Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 31.Finzel BC, Poulos TL, Kraut J. Crystal-structure of yeast cytochrome-c peroxidase refined at 1.7Å resolution. J Biol Chem. 1984;259:3027–3036. [PubMed] [Google Scholar]
- 32.Poulos TL, Fenna RE. Peroxidases - structure, function, and engineering. Metal ions in biological systems. 1994;30:25–75. [Google Scholar]
- 33.Sugano Y, Muramatsu R, Ichiyanagi A, Sato T, Shoda M. DyP, a unique dye-decolorizing peroxidase, represents a novel heme peroxidase family. J Biol Chem. 2007;282:36652–36658. doi: 10.1074/jbc.M706996200. [DOI] [PubMed] [Google Scholar]
- 34.Smulevich G, Jakopitsch C, Droghetti E, Obinger C. Probing the structure and bifunctionality of catalase-peroxidase (KatG) J Inorg Biochem. 2006;100:568–585. doi: 10.1016/j.jinorgbio.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 35.Carpena X, Loprasert S, Mongkolsuk S, Switala J, Loewen PC, Fita I. Catalase-peroxidase KatG of Burkholderia pseudomallei at 1.7Å resolution. J Mol Biol. 2003;327:475–489. doi: 10.1016/s0022-2836(03)00122-0. [DOI] [PubMed] [Google Scholar]
- 36.Bertrand T, Eady NAJ, Jones JN, Nagy JM, Jamart-Gregoire B, Raven EL, Brown KA. Crystal structure of Mycobacterium tuberculosis catalase-peroxidase. J Biol Chem. 2004;279:38991–38999. doi: 10.1074/jbc.M402382200. [DOI] [PubMed] [Google Scholar]
- 37.Phillips SEV. Structure of oxymyoglobin. Nature. 1978;273:247–248. doi: 10.1038/273247a0. [DOI] [PubMed] [Google Scholar]
- 38.Shaanan B. The iron oxygen bond in human oxyhaemoglobin. Nature. 1982;296:683–684. doi: 10.1038/296683a0. [DOI] [PubMed] [Google Scholar]
- 39.Shaanan B. Structure of human oxyhaemoglobin at 2·1 resolution. J Mol Biol. 1983;171:31–59. doi: 10.1016/s0022-2836(83)80313-1. [DOI] [PubMed] [Google Scholar]
- 40.Miyatake H, Mukai M, Park S-Y, Adachi S, Tamura K, Nakamura H, Nakamura K, Tsuchiya T, Iizuka T, Shiro Y. Sensory mechanism of oxygen sensor FixL from Rhizobium meliloti: crystallographic, mutagenesis and resonance Raman spectroscopic studies. J Mol Biol. 2000;301:415–431. doi: 10.1006/jmbi.2000.3954. [DOI] [PubMed] [Google Scholar]
- 41.Gong W, Hao B, Mansy SS, Gonzalez G, Gilles-Gonzalez MA, Chan MK. Structure of a biological oxygen sensor: a new mechanism for heme-driven signal transduction. Proc Natl Acad Sci USA. 1998;95:15177–15182. doi: 10.1073/pnas.95.26.15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nioche P, Berka V, Vipond J, Minton N, Tsai AL, Raman CS. Femtomolar sensitivity of a NO sensor from Clostridium botulinum. Science. 2004;306:1550–1553. doi: 10.1126/science.1103596. [DOI] [PubMed] [Google Scholar]
- 43.Lanzilotta WN, Schuller DJ, Thorsteinsson MV, Kerby RL, Roberts GP, Poulos TL. Structure of the CO sensing transcription activator CooA. Nat Struct Biol. 2000;7:876–880. doi: 10.1038/82820. [DOI] [PubMed] [Google Scholar]
- 44.Aono S, Nakajima H. Structure of the CO sensing transcription activator CooA. Coord Chem Rev. 1999;192:267–282. [Google Scholar]
- 45.Ibrahim M, Kerby RL, Puranik M, Wasbotten IH, Youn H, Roberts GP, Spiro TG. Heme displacement mechanism of CooA activation. J Biol Chem. 2006;281:29165–29713. doi: 10.1074/jbc.M605568200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams PA, Fulop V, Garman EF, Saunders NF, Ferguson SJ, Hajdu J. Haem-ligand switching during catalysis in crystals of a nitrogen-cycle enzyme. Nature. 1997;389:406–412. doi: 10.1038/38775. [DOI] [PubMed] [Google Scholar]
- 47.Lukat-Rodgers GS, Rexine JL, Rodgers KR. Heme speciation in alkaline ferric FixL and possible tyrosine involvement in the signal transduction pathway for regulation of nitrogen fixation. Biochemistry (N Y) 1998;37:13543–13552. doi: 10.1021/bi981439v. [DOI] [PubMed] [Google Scholar]
- 48.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cartron ML, Mitchell SA, Woodhall MR, Andrews SC, Watson KA. Preliminary X-ray diffraction analysis of YcdB from Escherichia coli: a novel haem-containing and Tat-secreted periplasmic protein with a potential role in iron transport. Acta Cryst F. 2007;63:37–41. doi: 10.1107/S174430910604509X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bamford VA, Rajasekaran MB, Cartron ML, Mitchell SA, Andrews SC. EfeB, a component of the EfeUOB bacterial iron transport system, shows novel removal of iron from heme, unpublished data. Protein Data Bank; [Google Scholar]
- 52.Cao J, Woodhall MR, Alvarez J, Cartron ML, Andrews SC. EfeUOB (YcdNOB) is a tripartite, acid-induced and CpxAR-regulated, low-pH Fe2+ transporter that is cryptic in Escherichia coli K-12 but functional in E-coli O157:H7. Mol Microbiol. 2007;65:857–875. doi: 10.1111/j.1365-2958.2007.05802.x. [DOI] [PubMed] [Google Scholar]
- 53.Rajasekaran MB, Nilapwar S, Andrews SC, Watson KA. EfeO-cupredoxins: major new members of the cupredoxin superfamily with roles in bacterial iron transport. Biometals. 2010;23:1–17. doi: 10.1007/s10534-009-9262-z. [DOI] [PubMed] [Google Scholar]
- 54.Sturm A, Schierhorn A, Lindenstrauss U, Lilie H, Brüser T. YcdB from Escherichia coli Reveals a Novel Class of Tat-dependently Translocated Hemoproteins. J Biol Chem. 2006;281:13972–13978. doi: 10.1074/jbc.M511891200. [DOI] [PubMed] [Google Scholar]
- 55.Létoffe S, Heuck G, Delepelaire P, Lange N, Wandersman C. Bacteria capture iron from heme by keeping tetrapyrrol skeleton intact. Proc Natl Acad Sci USA. 2009;106:11719–11724. doi: 10.1073/pnas.0903842106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zubieta C, Joseph R, Krishna SS, et al. Identification and structural characterization of heme binding in a novel dye-decolorizing peroxidase, TyrA. Proteins. 2007;69:234–243. doi: 10.1002/prot.21673. [DOI] [PubMed] [Google Scholar]
- 57.Sugano Y. DyP-type peroxidases comprise a novel heme peroxidase family. Cell Mol Life Sci. 2009;66:1387–1403. doi: 10.1007/s00018-008-8651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Welinder KG. Plant peroxidases - their primary, secondary and tertiary structures, and relation to cytochrome-c peroxidase. Eur J Biochem. 1985;151:497–504. doi: 10.1111/j.1432-1033.1985.tb09129.x. [DOI] [PubMed] [Google Scholar]
- 59.Pearson WR. Empirical statistical estimates for sequence similarity searches. J Mol Biol. 1998;276:71–84. doi: 10.1006/jmbi.1997.1525. [DOI] [PubMed] [Google Scholar]
- 60.Salinero KK, Keller K, Feil WS, Feil H, Trong S, Di Bartolo G, Lapidus A. Metabolic analysis of the soil microbe Dechloromonas aromatica str. RCB: indications of a surprisingly complex life-style and cryptic anaerobic pathways for aromatic degradation. BMC Genomics. 2009;10:351–374. doi: 10.1186/1471-2164-10-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.