

Abstract
A specific polymorphism in the hemochromatosis (HFE) gene, H63D, is over-represented in neurodegenerative disorders such as amyotrophic lateral sclerosis and Alzheimer disease. Mutations of HFE are best known as being associated with cellular iron overload, but the mechanism by which HFE H63D might increase the risk of neuron degeneration is unclear. Here, using an inducible expression cell model developed from a human neuronal cell line SH-SY5Y, we reported that the presence of the HFE H63D protein activated the unfolded protein response (UPR). This response was followed by a persistent endoplasmic reticulum (ER) stress, as the signals of UPR sensors attenuated and followed by up-regulation of caspase-3 cleavage and activity. Our in vitro findings were recapitulated in a transgenic mouse model carrying Hfe H67D, the mouse equivalent of the human H63D mutation. In this model, UPR activation was detected in the lumbar spinal cord at 6 months then declined at 12 months in association with increased caspase-3 cleavage. Moreover, upon the prolonged ER stress, the number of cells expressing HFE H63D in early apoptosis was increased moderately. Cell proliferation was decreased without increased cell death. Additionally, despite increased iron level in cells carrying HFE H63D, it appeared that ER stress was not responsive to the change of cellular iron status. Overall, our studies indicate that the HFE H63D mutant protein is associated with prolonged ER stress and chronically increased neuronal vulnerability.
Keywords: Cell Death, ER Stress, Iron, Neurodegeneration, Neurological Diseases
Introduction
A neurodegenerative disease is a condition in which neuronal cells of the brain and spinal cord are selectively lost. Despite many years of intensive research, the precise mechanisms involved in neuron degeneration remain unknown. A common event in many neurodegenerative diseases is the accumulation and deposits of inclusion bodies that contain abnormal aggregated proteins, suggesting the function of endoplasmic reticulum (ER),2 to sense and enhance the degradation of protein aggregates is impaired severely in the affected neurons (1).
Accumulation of misfolded protein in the ER provokes the early ER stress response, called unfolded protein response (UPR). UPR is a short term and protective homeostatic mechanism. It initiates both apoptotic and adaptive pathways. It is characterized by a rapid increase of UPR sensors, attenuation of protein translation, induction of ER chaperones, and degradation of misfolded proteins. If homeostasis cannot be reestablished, however, UPR declines, and prolonged ER stress activates apoptotic cell death pathways (2). ER stress has been implicated in some neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), Parkinson disease, and Alzheimer disease, as well as other disorders (3). In some cellular models, animal models, or in patients of these diseases, increased levels of Bip/GRP78 and other UPR markers have been reported (4–7). However, it is debated whether the observed ER stress is largely neuroprotective or whether it directly contributes to the disease process. In an in vivo analysis of ALS-linked mutant SOD1 mice, it has been shown that vulnerable motor neurons are selectively prone to ER stress. As a typical prolonged ER stress activation pattern, UPR is activated, peaks, and then declines before the denervation of the motor neurons in the lumbar spinal cord. This selective vulnerability underlies early disease manifestations, whereas subsequent ER stress in resistant motor neurons underlies late disease manifestation (8). These findings strongly argue that ER stress might be the driving pathogenic mechanism in neurodegenerative diseases. On the other hand, in the mouse model of Parkinson disease, activation of one of the ER stress effecter XBP1 significantly suppresses the degeneration of dopaminergic neurons (9).
Because neurodegenerative diseases have various etiologic factors and biochemical and pathological changes and mechanisms in common (10), it is not a surprise to find that two common polymorphisms in the HFE gene, C282Y and H63D, are associated increasingly with various neurodegenerative disorders. The C282Y polymorphism has been linked to Parkinson disease (11, 12) and multiple sclerosis (13). H63D is reportedly over-represented in both Alzheimer disease (14) and ALS (15–19). The presence of the HFE variant reduces the age of onset of Alzheimer disease and is associated with greater disease severity (14). However, there are also studies that did not find an increased frequency (14, 20, 21). This discrepancy may reflect a gene-environment interaction.
HFE is a major histocompatibility complex class 1 protein and mutations in the protein are associated with cellular iron overload (22). HFE protein has a wide tissue expression (23). Mice devoid of HFE show some motor impairment (24). Mice carrying Hfe H67D allele, the mouse equivalent of the human polymorphism H63D, demonstrated increased iron loading in the liver and brain (25, 26). At the cellular level, when stably expressed in a human neuronal cell line lacking endogenous HFE, both C282Y and H63D HFE are associated with higher labile iron pool and increased oxidative stress, as well as changes in glutamate regulation, chemokine secretion (27), and Tau phosphorylation (28). However, in the stable expression cells, these cellular changes may reflect the late and accumulated effects of the mutant HFE proteins.
In this report, we sought to elucidate the early cellular consequences associated with the HFE mutation, specifically H63D. We developed inducible neuronal cell lines in which the expression of HFE WT or H63D protein is under the control of tetracycline and assessed the cellular impact during the induction of their expression. Our data demonstrated that the presence of HFE H63D mutant protein initially evoked UPR, then a prolonged ER stress, along with activation of caspase and a subsequent reduction of cell proliferation. This ER stress pattern associated with HFE H63D was recapitulated in a knock-in mouse model carrying Hfe H67D. Although increasing evidence has suggested iron overload might be involved in many neurodegenerative diseases (29), and cells carrying HFE H63D have elevated iron level, it appeared that ER stress was not responsive to the change of cellular iron status alone. Our results suggest HFE H63D is associated with prolonged ER stress, which may contribute to decreased neuronal viability and aggravation of the pathogenic processes in neurodegenerative diseases.
EXPERIMENTAL PROCEDURES
Inducible HFE Expression Cell Line Construction
Human neuroblastoma SH-SY5Y cells were used as the host cells. FLAG-tagged HFE WT and H63D were cloned into the expression vector pcDNA5/FRT/TO (27). Inducible HFE cell lines were made using the Flp-In T-REX system (Invitrogen) by three integration steps as described in the product manual. In brief, the first step was to generate Flp-In host cell line. In this step, the Flp recognition target (FRT) was integrated by transfection of the FRT/lacZeo plasmid after digestion by ScaI to make single linear insertion. The use of the lacZ-Zeocin fusion gene allowed selection for Zeocin-resistant cell clones, and the LacZ portion was used to characterize the integration site by scoring the β-galactosidase activity. In the second step, to generate the Flp-In T-REX host cell line, an expression vector (pcDNA6/TR) encoding the tetracycline repressor (TetR) was introduced using blasticidin resistance. In the third step, the FLAG-tagged HFE was introduced by Flp recombinase activity from the pcDNA5/FRT/TO vector. This construct contained an FRT site in front of the hygromycin ORF that generated hygromycin resistance only if a site-directed integration occurred at the FRT site downstream of the ATG of the lacZ-Zeocin cassette. By this recombination, the FLAG-tagged HFE was integrated downstream of the FRT integration site. FLAG-tagged HFE was integrated downstream of the FRT integration site. As HFE was driven by the tetracycline operator-controlled CMV promoter (PCMV/2TetO2), tetracycline regulation was obtained. For each HFE gene, WT or H63D, six inducible clonal lines were established and assayed. Representative results were shown in the report.
Cell Culture
Human neuroblastoma SH-SY5Y cells were maintained in DMEM/F12 medium (Invitrogen) supplemented with 10% FBS (Gemini Bio-products), blastidin, hygromycin B (Roche Applied Science), penicillin-streptomycin, and nonessential amino acids (Sigma). Cells were split 1 day before the treatment with 100% EtOH (control) or 1 μg/ml tetracycline in EtOH (Invitrogen) for various times as indicated in the figure legends. For chemical treatment, 24 h after induction, cells were incubated with either EtOH or tetracycline in EtOH in the presence of tauroursodeoxycholic acid (TUDCA; Calbiochem), FeCl3-NTA, DFO, dimethyl sulfoxide, or tunicamycin (all from Sigma) for additional 30 h before harvest. All experiments were performed at 37 °C in 5% CO2 atmosphere culture conditions.
Transgenic Mice Construction and Genotyping
The Hfe H67D knock-in mice were generated as previously reported (25). In brief, the mutation H67D (199C→G) in exon 2 was introduced by site-direct mutagenesis to the murine Hfe gene fragment on the pBS vector. The H67D point mutation destroyed a BspHI restriction site. The HFE fragment was introduced between the TK and neor genes of the pPNT-loxP2 vector to generate the targeting vector. The targeting vector was linearized with NotI and introduced into the 129/Sv-derived ES cell line RW4 (Incyte Genomics Systems) by electroporation. After 24 h, the cells were placed under selection with G418 (Gibco-BRL) and ganciclovir (Syntex Chemicals) for 6 days. Targeted ES cell colonies resistant to double selection were isolated and were injected into C57BL/6J blastocysts, and chimeric males were backcrossed for germ line transmission to C57BL/6J females. The F1 mice were crossed with mice expressing Cre enzyme to remove the neor gene. The resultant neo-excised heterozygous mice were mated to produce homozygous mutant mice. Genotyping was performed by PCR analysis of DNA obtained by tail biopsies at 30 days. To amplify the murine Hfe gene, including the H67D mutation, a forward primer (5′-AGGACTCACTCTCTGGCAGCAGGAGGTAACCA-3′) and a reverse primer (5′-TTTCTTTTACAAAGCTATATCCCCAGGGT-3′) were used, resulting in amplification of an ∼500-bp fragment. Digestion with BspHI revealed the 260- and 240-bp digested fragments for the wild-type allele and the uncleaved 500-bp PCR fragment for the mutant allele. All of these mice were maintained under normal housing conditions in accordance with the IUCAC policy for animal use at Penn State University, which is in agreement with the NIH Guide for the Care and Use of Laboratory Animals.
Antibodies and Immunoblotting
The following antibodies were used: mouse monoclonal anti-FLAG (Sigma), anti-Bip (BD Biosciences), anti-XBP-1S (BioLegend and Santa Cruz Biotechnology), anti-TfR (Invitrogen); rabbit polyclonal anti-β-actin (Sigma), disulfide isomerase (PDI), inositol-requiring enzyme-1α (IRE1α) and caspase-3 (all from Cell Signaling). The HRP-conjugated secondary antibodies were purchased from Pharmacia. Immunoblots were developed using the enhanced chemiluminescence method (Pharmacia). For densitometry analyses, films were scanned and plotted using NIH ImageJ software. All the experiments were performed and quantified at least three times to ensure accuracy.
Immunocytochemistry and Confocal Microscopy
Cultured cells were fixed in 4% paraformaldehyde in PBS, rinsed, permeabilized in 0.5% Triton X-100 in PBS for 10 min, and then blocked with 5% normal goat serum in PBS containing 0.1% Triton X-100 for 1 h. Cells were then incubated with mouse anti-FLAG antibody in blocking buffer, washed, and probed with secondary Alexa 555 goat anti-mouse antibody (Invitrogen) and DAPI for 1 h. After wash, slides were mounted. Fluorescence images were acquired by confocal microscopy using a Leica LCS laser scanning microscope with a 63× objective lens.
Cytofluorometric Assay of Apoptotic Cells by Annexin V/Propidium Iodide Staining
Cultured cells were harvested and washed in PBS and then incubated with annexin V-FITC and PI (Invitrogen) for 15 min at room temperature, prior to flow cytometry analysis using FACSCalibur (BD Biosciences). More than 10,000 events were acquired and analyzed in each sample. Quadrant statistics was performed using FACSCompTM software (BD Biosciences).
Biochemical Assays
Caspase-3 activity was determined by the caspase-3 fluorometric assay kit (R&D Systems). Loss of plasma membrane integrity as an indicator of cell death was determined by measuring lactate dehydrogenase (LDH) activity released from damaged cells by the Cytotoxicity Detection kit (Roche Applied Science). Cell proliferation was assessed with the Colorimetric MTS Assay (Promega).
Statistical Analysis
All of the quantitative data, including the quantification of Western blot were subjected to statistical analysis using the Student's t test performed by GraphPad Prism software. Error bars in all figures represent standard deviation.
RESULTS
HFE H63D Is Associated with Activation of ER Stress
To reveal the early cellular consequences of expressing HFE H63D, we developed an inducible cell model using the Flp-In T-Rex system, in which FLAG-tagged HFE H63D or WT protein expression is under the control of tetracycline in a human neuronal cell line SH-SY5Y. This cell line was chosen initially because it is used widely as a neural cellular model and has no endogenous HFE expression (27). It has been reported that the FLAG tag does not interfere with the HFE protein function, in the terms of protein localization, complex formation, and cellular iron regulation (27, 30). We screened and established the Flp-In T-Rex host cell line that only contained a single FRT site (Fig. 1A). Thus, the resulting inducible HFE expression cell lines contained a single copy of the HFE gene integrated downstream of the FRT site by recombination. As this single copy of HFE was driven by the tetracycline operator-controlled CMV promoter (PCMV/2TetO2), its expression in individual cells was turned on at the homogenous level by tetracycline (Fig. 1, B and C). In the blots with a long time exposure, a leak expression was detected at a low level (Fig. 1C, dark exposure). Furthermore, this inducible system allow us to capture the transit responses associated with HFE H63D protein expression and follow the shift of these responses after the induction, which is impossible in the stable expression cell lines.
FIGURE 1.

Construction and expression of inducible HFE-expressing cells. A, Southern blot analysis of Flp-In T-Rex host cell lines. DNA of cell clones was digested with HindIII and analyzed by Southern blot using a probe specific for FRT/lacZeo. Clone 11, as marked by an asterisk, contained a single FRT site and was used as the host cell to construct inducible HFE-expressing cells. B, confocal immunofluorescence microscopy of SH-SY5Y cells with induced FLAG-tagged HFE WT or H63D expression for 2 days. Fixed cells were stained with anti-FLAG (red) and DAPI (blue). Scale bar, 40 μm. C, Western blot of SH-SY5Y cells with inducible FLAG-tagged HFE WT or H63D expression. Cells were incubated without (−) or with (+) 1 μg/ml tetracycline for 2 days and then harvested. Total protein was isolated and resolved by SDS-PAGE, followed by immunoblot for FLAG and actin. A long time exposure (dark) of FLAG blot was presented to show the leak expression of HFE-FLAG.
After a 2-day induction, the expression of a key ER stress marker chaperone protein Bip/GRP78 was increased >2-fold with HFE H63D expression, whereas WT HFE protein even at a higher expression level did not have this effect (Fig. 2). As shown in Fig. 1B and previous reports, the localization of exogenously expressed mutant HFE H63D protein is like WT HFE. There is no retention in the ER (30). Thus, we can exclude the possibility that the activation of Bip in the presence of HFE H63D expression was caused by the nonspecific effects of exogenous protein overexpression. Beyond the increase in the steady-state level of Bip, the levels of the proximal ER stress sensor serine/threonine kinase IRE1α and its downstream effector, spliced XBP-1 (XBP-1s) protein, were elevated in HFE H63D-expressing cells after a 2-day induction (Fig. 2A), further indicating the presence of ER stress in the cells expressing HFE H63D.
FIGURE 2.

Increased of expression of URP markers in HFE H63D expressing cells. A representative Western blot (A) and quantification of the Bip blots (B) were performed to monitor expression level of Bip, IRE1α, and XBP-1s in the human neuroblastoma SH-SY5Y cells where the expression of FLAG-tagged HFE WT or H63D was under the control of tetracycline. Cells were incubated in the absence (−) or presence (+) of 1 μg/ml tetracycline for 2 days and then harvested. Total protein was isolated, and then equal amount of protein was resolved by SDS-PAGE, followed by immunoblotting. Expression level of Bip was normalized to actin and calculated as the ratio to the control condition (−tet) (***, p < 0.001, n = 3).
To further examine whether this increased the expression of Bip and other UPR makers associated with HFE H63D mutant protein represented ER stress activation, we tested its response to the ER stress inhibitor TUDCA (31). First, we evaluated the efficacy of TUDCA in the HFE WT-expressing cells. In these cells, Bip expression was increased by an ER stress inducer tunicamycin (TM) (Fig. 3, A, lane 3, and B, tet+TM). The TM-mediated increase in Bip was blocked by TUDCA (Fig. 3, A, lanes 4 and 5, and B, compare tet+TM with tet+TM+TA). The Bip expression that followed induction of HFE H63D also responded to TUDCA inhibition (Fig. 3, C, lanes 2 and 3, and D, compare tet with tet+TA). As a strong ER stress inducer, TM causes protein synthesis attenuation (32). Consistent with this mechanism, in both HFE WT and H63D expressing cells, TM treatment reduced FLAG-tagged HFE expression by >10-fold (Fig. 3, A, lanes 2 and 3, and C, lanes 4 and 5), suggesting there was a general protein translation suppression. TUDCA treatment reversed the TM reduction of FLAG-HFE expression (Fig. 3A, lanes 3–5). The expression level of FLAG-HFE was not affected by TUDCA in the HFE H63D-expressing cells without TM treatment, indicating that HFE H63D did not lead to the suppression of general protein synthesis (Fig. 3C, lanes 2 and 3). Therefore, the expression of HFE H63D induced ER stress but did not cause protein translation inhibition as TM.
FIGURE 3.

HFE H63D-mediated Bip activation is sensitive to ER stress inhibitors. Representative Western blots are shown in A and C. The results of three trials are shown in graphic form in B and D (HFE WT in A and B; H63D in C and D). Cells were induced with tetracycline for 1 day and then exposed to one of the following treatments. Dimethyl sulfoxide (DMSO; vehicle control for TM), 1 μg/ml tunicamycin in dimethyl sulfoxide (TM), or TUDCA in culture medium at 2 mm (TA2) or at 10 mm (TA10) in the presence of tetracycline (tet) for 30 h. Equal amounts of proteins from cell lysates were subject to immunoblot to monitor the expression levels of FLAG-HFE, Bip, and actin. Bip expression was normalized by actin and calculated as the ratio to the control conditions: (+tet) in WT and (−tet) in H63D. The blots in each panel came from the same gel. Dividing lines were used to show the different parts of the same gel. A and B, WT HFE expressing cells: lane 1 (tet), expression of WT HFE protein; lane 2 (tet+DMSO), vehicle control for TM; lane 3 (tet+TM), TM treatment increased Bip; lane 4 and 5 (tet+TM+TA2 and +TA10): reverse Bip increase by ER stress inhibitor TA in a concentration-dependent manner. C and D, HFE H63D expressing cells: lane 1 (−tet), uninduced cell control; lane 2 (tet), induced HFE H63D expression increased Bip; lane 3 (tet+TA10), increased Bip with H63D induction is reduced by TA. In addition to the effects on Bip, decreased expression of HFE-FLAG can be observed under TM treatment: A, lane 3 (tet+TM) and C, lane 5 (tet+TM), which could be reversed by TA, A, lane 5 (tet+TM+TA10). **, p < 0.05; ***, p < 0.001 (n = 3).
Prolonged ER Stress and Caspase Activation in Presence of HFE H63D
ER stress initially activates the short term UPR. Subsequently, survival can be favored when cells adapt to the stress with the persistent up-regulation of ER chaperones, including Bip. Alternatively, if ER stress is prolonged, the caspase-mediated apoptotic pathway is activated (32). Therefore, we next examined the pattern and consequence of HFE H63D-associated ER stress using a time course study. Starting from the first day until the third day of induction, the expression levels of major UPR markers, Bip, PDI, and IRE1α, were increased, indicating that UPR was stimulated. This response was followed by a down-regulation of these markers from day 4, suggesting a shift from the short term protective UPR to persistent ER stress (Fig. 4, C and D). There was a gradual increase of these ER stress proteins in uninduced HFE H63D cells over the time course, more apparently on day 4, probably due to the leaking expression of HFE H63D (Fig. 1C). On the fourth day, up-regulated caspase-3 cleavage and a concomitant 30% increase of caspase-3 activity were present. These results indicated that at this time point, caspase was activated in association with the diminished UPR response, as shown by the attenuated IRE1α expression (Fig. 5). Induction of WT HFE protein did not alter the expression levels of these UPR markers nor induce caspase activation over the same time course (Figs. 4, A and B, and 5).
FIGURE 4.

Time course of increased ER stress in HFE H63D expressing cells. Mutant HFE H63D protein initially triggers the UPR, followed by down-regulation of UPR sensors with persistent ER stress. Western blot analyses were performed to monitor the expression level of major UPR sensors in the inducible FLAG-HFE WT (A) or H63D (C) -expressing cells during a 4-day time course. Cells were incubated in the absence (−) or presence (+) of 1 μg/ml tetracycline (tet) and harvested every day for 4 days. Equal amount of protein lysate was resolved by SDS-PAGE, followed by immunoblot for FLAG tag, Bip, PDI, IRE1α, and actin. In the quantification of immunoblots (B and D), expression levels of Bip, PDI, and IRE1 were normalized by actin and calculated as the ratio to the uninduced condition (−tet). In HFE WT-expressing cells (B), no significant change of expression of Bip, PDI, and IRE1 was detected. In HFE H63D-expressing cells (D), the increase of expression levels of the UPR sensor proteins over the first 3 days is significant (p < 0.005 at least, n = 3). d, day. Dotted lines, fold change 1.0, indicating no changes compared to the controls.
FIGURE 5.

HFE H63D induces caspase activation with prolonged ER stress in the late time point. A, Western blotting was performed to monitor expression level of IRE1α and caspase-3 p18 in HFE WT- or H63D-expressing cells. A representative blot is shown. Cells were incubated with (+) or without (−) tetracycline (tet) for 4 days and then harvested. Equal amount of protein lysate was subject to Western blot. B, caspase-3 activity was measured in cell lysate from HFE WT or H63D cells under the treatment with (+tet) or without (−tet) tetracycline for 4 days. Activity (A505) was normalized by protein concentration (mg/ml) of the lysate (**, p < 0.005, n = 4). RFU, arbitrary fluorescence units.
To determine whether the cell culture findings occurred in vivo, we examined the Hfe H67D knock-in mouse model (25). We performed longitudinal and sequential analyses on WT and Hfe H67D mice, from the brain, cervical, to lumbar spinal cord at the ages of 3, 6, and 12 months. At 3 months old, no change in Bip expression was detected in these regions of HfeH67D/H67D and WT mice (Fig. 6E). At 6 months old, the Bip expression level and spliced XBP-1 were selectively increased in the lumbar spinal cord homogenates (Fig. 6, A and F), but not in the brain or cervical lysates from HfeH67D/H67D mice (Fig. 6, B and F, and data not shown), compared with the WT littermates. In 12-month-old HfeH67D/H67D mice, Bip expression declined in lumbar spinal cord, whereas p18, the active form of caspase-3 was increased (Fig. 6, C, G, and H). These changes were limited to the lumbar spinal cord, as no changes in Bip expression were detected in the brain or cervical in HfeH67D/H67D mice (Fig. 6, D and G). There is a parallel change in XBP-1s with Bip level in the lumbar spinal cord at all the ages examined (supplemental Fig. 1). The heterozygous HfeWT/H67D mice had similar Bip and caspase-3 p18 levels as the homozygous HfeH67D/H67D mice (data not shown). These in vivo data showed that UPR was specifically activated in the lumbar spinal cord in mice carrying the Hfe H67D mutation at 6 months old and then a prolonged ER stress associated with a mild activation of caspase-3 were present at 12 months old. Interestingly, these changes seemed not progress to the regions beyond the lumbar spinal cord.
FIGURE 6.

Increased ER stress and activated caspase in Hfe H67D mice. Shown is a representative Western blot to demonstrate protein expression level change in the homogenates of lumbar spinal cord (L-SC; A and C), whole brain (B and D) from 6-month-old (A and B) and 12-month-old (C and D) Hfe WT or H67D mice. Equal amounts of protein homogenates were resolved by SDS-PAGE followed by Western blot. The blots in each panel came from the same gel. Dividing lines were used to show the different parts of the same gel. Quantification of the Bip blots on the brain, cervical and lumbar spinal cord from 3-month-old (E), 6-month-old (F), or 12-month-old mice (G) is shown in graphic form. The level of active form of caspase-3 p18 in the lumbar spinal cord at 6 and 12 months is quantified and shown in H. The expression levels of Bip and caspase-3 p18 fragment were normalized by actin and calculated as the ratio to the WT controls (**, p < 0.005; ***, p < 0.001, n = 6–8). C-SC, cervical spinal cord; L-SC, lumbar spinal cord.
Taken together, consistent data from both cell model and animal model demonstrate that in the presence of H63D HFE mutant protein, UPR is stimulated, followed by persistent ER stress and mild activation of caspase. These results also strongly justify that the inducible cell model faithfully recapitulates the cellular effects in vitro.
Increased Neuronal Vulnerability in HFE H63D-expressing Cells
Given the results that the expression of HFE H63D protein was associated with prolonged ER stress and caspase activation, we next examined its impact on neuronal cell viability.
To determine whether the moderate activation of caspase-3 was associated with apoptosis, we used annexin V (AV)/PI staining followed by flow cytometry. AV+/PI− cells represented the cell population at an early apoptotic stage, whereas AV+/PI+ cells are at end stage apoptosis and cell death. There was no significant difference in the percentage of AV+/PI− or AV+/PI+ cells 2 days after induction of either WT or HFE H63D (Fig. 7, A and B). On the fourth day following induction, a 23.5 ± 6.5% increase of AV+/PI− cells was detected in HFE H63D cells as compared with uninduced cells (Fig. 7C). However, there was no increase of AV+/PI+ cells associated with HFE H63D expression (Fig. 7D), indicating no increase in cell death.
FIGURE 7.

Evidence of increased cells in early apoptosis associated with HFE H63D expression. Cells were induced with (+tet) or without (−tet) tetracycline for 2 days (A and B) or 4 days (C and D) and then harvested and subjected to apoptosis assay using an annexin V-FITC/PI stain and flow cytometry. AV+/PI− cells represent the cell population at early apoptotic stage (A and C), whereas AV+/PI+ cells are at end stage apoptosis (B and D). The analysis was performed in duplicate or triplicate and repeated by three independent experiments for each time point. Cells induced in the same time point were analyzed in one experiment with the same setting of data acquisition and analysis. The percentage of cells of each population was obtained by quadrant statistics on gated events. Representative analysis is shown (**, p < 0.005, n = 6). tet, tetracycline.
We further measured cell death by the LDH release. There is a gradually increased LDH release in both induced and non-induced cells over the time, reflecting the basal level of cell death during the extended time in the culture. However, we did not observe a significant increase of the LDH release in either WT HFE or H63D expressing cells with the comparison to the non-HFE expressing controls (Fig. 8, A and B). Also, the H63D-expressing cells released LDH to a level comparable with that of WT, indicating the mutant protein did not increase cell death (Fig. 8C).
FIGURE 8.

HFE H63D is not associated with increased LDH release. Neuronal cell death was determined by measuring LDH activity released from damaged cells. Equal number of HFE WT (A) or H63D (B) cells were plated and incubated in uninduced (−tet) or induced (+tet) condition. LDH activity was measured in the cell-free culture supernatant every day for 4 days. The LDH activity in the medium in all conditions was normalized by that of uninduced controls in day 1. C, the impact of HFE WT or H63D was compared as the fold change of normalized LDH activity from the induced cells over those without induction (+tet/−tet). tet, tetracycline.
We next assessed the number of viable cells by an MTS-based assay. After 4 days, WT HFE induction promoted the number of proliferating cell by 22% (Fig. 9A). In contrast, mutant HFE H63D expressing cells had a gradually lowered amount of proliferating cells than the non-induced controls. On the fourth day of induction, HFE H63D-expressing cells displayed a 30% reduction in cell proliferation compared with non-induced cells (Fig. 9B). In comparison with WT, mutant H63D HFE-expressing cells had a reduced number of cells in proliferation throughout the time course. By 4 days, this reduction was as great as 45% (Fig. 9C).
FIGURE 9.

Decreased cell proliferation in HFE H63D cells. The number of viable cells was measured by an MTS-based assay in HFE WT (A) or H63D (B) cells. An equal number of HFE WT or H63D cells were plated and incubated in uninduced (−tet) or induced (+tet) condition. Cell proliferation was determined by the ratio of the number of proliferating cells between induced condition (+tet) and uninduced (−tet) conditions every day for 4 days. It was normalized to the uninduced controls in day 1. C, the impact of HFE WT or H63D on cell proliferation rates was determined by calculating the fold change in cell proliferation with tetracycline over those without induction (+tet/−tet) (*, p < 0.05; **, p < 0.005; ***, p < 0.001; n = 3). tet, tetracycline.
All of these data converge to suggest that the ER stress converted to a proapoptotic response in HFE H63D-expressing cells. A moderate increase of cells entering early apoptotic stage was present with a mild level of caspase activation. Neuronal cell lines carrying this mutant protein had a reduced proliferation, but no increase of cell death was detected.
Effect of Cellular Iron Level on ER Stress
Because of the association between HFE H63D expression and increased intracellular iron status, we tested whether there is a correlation between cellular iron level and ER stress response in these cells. To address this question, we modulated the cellular iron level and monitored Bip expression. Transferrin receptor (TfR) was used as the indicator of intracellular iron load; its expression is reciprocally regulated by cellular iron level (34). The addition of iron by FeCl3-NTA or exposure to an iron chelator DFO decreased or increased TfR expression as expected. However, neither of these reagents had a detectable effect on Bip expression in WT- or H63D-expressing cells (Fig. 10, A and B), suggesting that change of iron homeostasis alone is not sufficient to stimulate ER stress responses.
FIGURE 10.
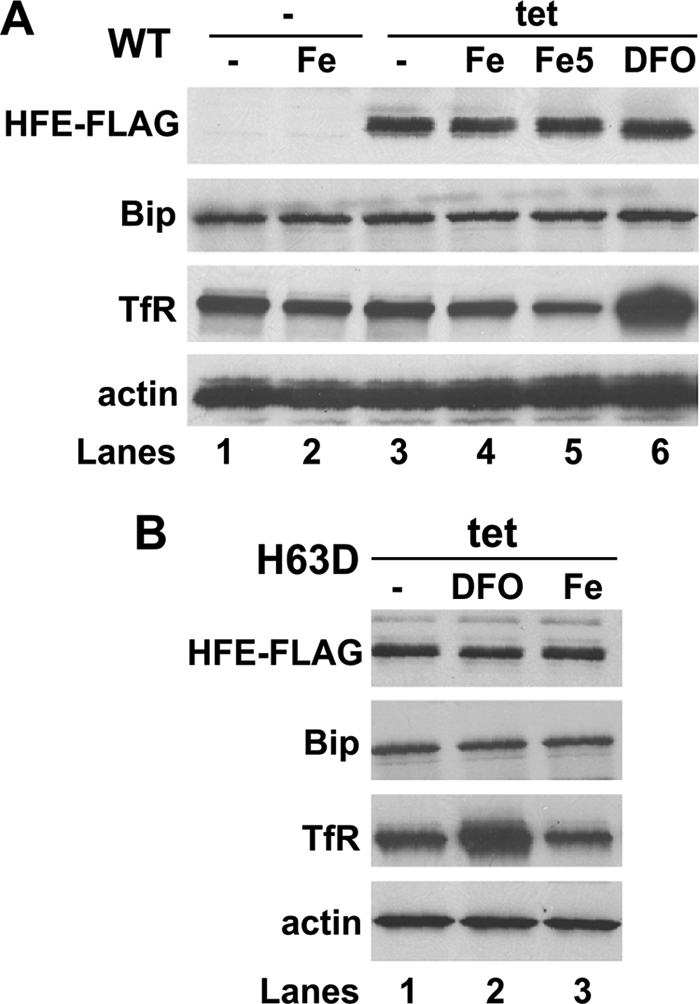
Effect of cellular iron level on ER stress. After incubating with (+) or without (−) 1 μg/ml tetracycline (tet) for 24 h, HFE WT (A) or H63D (B) cells were treated with iron reagent FeCl3-NTA at 100 μm (Fe) or 500 μm (Fe5) or iron chelator DFO at 20 μm for additional 30 h and then harvested. Equal amount of protein lysates were subjected to Western blot to monitor the expression level of FLAG tag, Bip, TfR, and actin.
DISCUSSION
In this study, we demonstrated that the presence of a mutant form of HFE protein in a neuroblastoma cell line, H63D was associated with increased ER stress. The protective UPR was initially activated, followed by a decline of UPR marker expression. This pattern is indicative of a proapoptotic rather than a prosurvival adaptive response. In the prosurvival adaptive pathway, elevated expression of ER stress markers such as IRE1α and Bip would have been extended significantly beyond the period of stress (32). Moreover, if the cell were adapting, there would have been no caspase or apoptosis activation as we observed. The prolonged ER stress and subsequently mild caspase activation in HFE H63D expressing cells was associated with decreased cell proliferation. Intriguingly, although there was a moderate increase of cells entering the early apoptotic stage, there was lack of cell death. This finding is similar to other reports in which caspase activation has been observed but not accompanied with cell death. For example, in an in vivo model of ischemic tolerance, widespread caspase-3 cleavage was observed without cell death (35) and in the adult huntingtin-deficient neurons, caspase-3 activation does not lead to cell degeneration (36).
We were able to expand our findings from the cell culture model to a transgenic mouse model. Selective activation of UPR was found in mice carrying the Hfe H67D mutation in the lumbar spinal cord at 6 months of age and then declined at 12 months in association with increased caspase-3 cleavage. Taken together, we conclude that mutant HFE H63D protein is associated with the prolonged ER stress and increased neuronal vulnerability.
ER Stress and Neuron Degeneration
ER stress has been reported in various neurodegenerative diseases. However, the exact contributions to and casual effects of ER stress in neuron degeneration are not clear (3). ER stress markers have been observed in degenerating tissues, and it has been proposed that an overloaded ER promotes cell death (37, 38). In ALS, for example, studies in fALS mutant SOD1 mice demonstrate that UPR activated, peaked, and declined selectively in vulnerable motor neuron prior to its denervation, suggesting ER stress might be the early cause for motor neuron degeneration (8). On the other hand, there are reports suggesting that ER stress could protect against or delay the onset of neurodegenerative diseases. For example, mild ER stress caused by a mutation in an ER chaperone protects photoreceptor neurons from various death stimuli in adult Drosophila (39). The active form of one ER stress marker XBP1 protein has protective effects against cell death induced by 1-methyl-4-phenylpyridinium and proteasome inhibitors. Moreover, The exogenous expression of the active-form XBP1 protein by adenoviral vectors significantly suppresses the degeneration of dopaminergic neurons in the mouse model of Parkinson Disease (9).
In HFE H63D-expressing cells, prolonged ER stress converted to a mild activation of caspase and apoptosis without dramatic cell death. These features resemble the chronic damage of the affected neurons in neurodegenerative diseases. We speculate the following model for the association of HFE H63D with neuron degeneration: on adaptation to changing conditions, normal cells, including neurons, elaborate UPR to overcome the cellular stresses. In the case of HFE H63D polymorphism, sustained ER stress attenuates the activation of this cytoprotective response to combat the misfolded proteins, such as TDP-43, SOD1, Aβ, and Tau. Dysfunction of ER in turn causes more protein aggregation, increased neuronal vulnerability, and aggravation of the disease. Thus, we propose that aggregation of these proteins might be accelerated by the presence of HFE H63D, which over the course of many years would set a permissive environment to promote nerve cell degeneration, although it does not cause neuron degeneration by itself. This model is supported by in vivo study. Heterozygous and homozygous for the HFE H67D knock-in appear to grow normally and are fertile, producing homozygous offspring in the number expected (25). No gross behavior defects are detected in these mice. Interestingly, a mouse line that carries HFE H67D with an ALS-linked SOD1 mutation has a decreased life span compared with the mouse carrying SOD1 mutant alone. The age at the onset of disease seems not different between these two mouse lines, suggesting acceleration of disease progression (26). This observation is consistent with our proposition that HFE H63D does not cause neurodegenerative diseases, but rather, as a genetic risk factor it may increase susceptibility to the disease by altering cell function critically and chronically so that pathogenic factors find the environment appropriate for inducing disease.
As these studies converge, we propose the following concept for the role of ER stress in neurodegeneration; increased level of ER stress markers as presented in UPR or mild ER stress provides protective effect on neuron viability. Once the levels of these effectors decline, prolonged ER stress may either promote or cause neuron degeneration. This concept implicates that artificially maintaining the high level of ER stress effectors, such as Bip, IRE1α, or XBP1 may be especially therapeutically valuable in patients with neurodegenerative diseases and the HFE H63D genotype.
Function of HFE and Iron Regulation
HFE is thought to play a key role in the body iron regulation, through the interaction with the transferrin receptor on the plasma membrane (22). The exact function of this protein is still not known. Both C282Y and H63D mutations alter the HFE repressor function for transferrin uptake and could result in increased cellular uptake of iron and iron deposition (27, 30). The HFE protein shares homology with major histocompatibility complex class I-like family molecules but appears not to be involved in antigen presentation (41). HFE mutations result in higher level of oxidative stress in the cells (27). The effects of the HFE mutant protein is thought to stem from mis-regulation of iron in cells. In our study, however, HFE H63D activates ER stress, although altered intracellular iron load by chemical reagents does not have the same effect. One explanation is that the function of the HFE protein is not limited to iron regulation. Alternatively, iron regulation may involve more delicate and refined sensing and feedback mechanisms than previously thought. Different reagents and subtle changes in the concentration or localization of the intracellular iron may have different outcomes on cellular pathways. It is worth noting that the mutant HFE H63D protein does not aggregate in the ER (30); thus, our data does not argue that HFE H63D protein itself directly causes ER stress as a mis-folded protein. Instead, it may induce ER stress by perturbation of protein folding machinery, insult in the ER calcium homeostasis or disturbance in lipid metabolism (42). Indeed, it has been reported that intracellular calcium level is increased in cells stably expressing HFE H63D (43). The C282Y mutant of HFE fails to be transported beyond the ER, and its aggregation triggers the UPR protein (31, 44). Both HFE mutations lead to increased ER stress, but the mechanisms that underlie this activation differs. On the other hand, two recent papers show that ER stress modulates iron metabolism through induction of the hormone hepcidin, which controls plasma iron levels and perhaps innate immunity (45, 46). HFE is necessary for regulation of hepcidin expression because HFE mutants lower hepcidin level. Hepatocyte-specific expression of HFE by recombinant adeno-associated virus in WT and HFE knock-out mice both increased HFE and hepcidin mRNA and lowered hepatic iron level (40). Our future studies will explore the correlation among HFE, ER stress, hepcidin, and iron level to reveal the delicate relationship between iron regulation and vita cellular pathways.
In summary, our studies indicate that the HFE H63D mutant protein is associated with a prolonged ER stress and increased neuronal vulnerability. The data support our hypothesis that the presence of H63D mutant protein helps to establish a permissive cellular milieu, involving ER stress, which promotes neurodegeneration. This observation has therapeutic implications because it suggests that humans with neurodegenerative disorders who have H63D mutations may have etiopathogenetic features that differ from those in patients not possessing this mutation. Thus, drug design studies and clinical treatment trials should consider stratifying subjects according to the presence or absence of the HFE mutation.
This work was supported by the Paul and Harriett Campbell Fund for ALS Research, the Zimmerman Family Love Fund, and the Judith & Jean Pape Adams Charitable Foundation.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- ALS
- amyotrophic lateral sclerosis
- FRT
- Flp recognition target
- PI
- propidium iodide
- LDH
- lactate dehydrogenase
- IRE1α
- inositol-requiring enzyme-1α
- TM
- tunicamycin
- PDI
- disulfide isomerase
- AV
- annexin V
- DFO
- desferroxamine
- NTA
- nitrilotriacetate
- MTS
- 3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium.
REFERENCES
- 1. Soto C. (2003) Nat. Rev. Neurosci. 4, 49–60 [DOI] [PubMed] [Google Scholar]
- 2. Lin J. H., Li H., Yasumura D., Cohen H. R., Zhang C., Panning B., Shokat K. M., Lavail M. M., Walter P. (2007) Science 318, 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lindholm D., Wootz H., Korhonen L. (2006) Cell Death Differ. 13, 385–392 [DOI] [PubMed] [Google Scholar]
- 4. Atkin J. D., Farg M. A., Walker A. K., McLean C., Tomas D., Horne M. K. (2008) Neurobiol. Dis. 30, 400–407 [DOI] [PubMed] [Google Scholar]
- 5. Ryu E. J., Harding H. P., Angelastro J. M., Vitolo O. V., Ron D., Greene L. A. (2002) J. Neurosci. 22, 10690–10698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoozemans J. J., Veerhuis R., Van Haastert E. S., Rozemuller J. M., Baas F., Eikelenboom P., Scheper W. (2005) Acta Neuropathol. 110, 165–172 [DOI] [PubMed] [Google Scholar]
- 7. Atkin J. D., Farg M. A., Turner B. J., Tomas D., Lysaght J. A., Nunan J., Rembach A., Nagley P., Beart P. M., Cheema S. S., Horne M. K. (2006) J. Biol. Chem. 281, 30152–30165 [DOI] [PubMed] [Google Scholar]
- 8. Saxena S., Cabuy E., Caroni P. (2009) Nat. Neurosci. 12, 627–636 [DOI] [PubMed] [Google Scholar]
- 9. Sado M., Yamasaki Y., Iwanaga T., Onaka Y., Ibuki T., Nishihara S., Mizuguchi H., Momota H., Kishibuchi R., Hashimoto T., Wada D., Kitagawa H., Watanabe T. K. (2009) Brain Res. 1257, 16–24 [DOI] [PubMed] [Google Scholar]
- 10. Price D. L. (1999) Nature 399, A3–5 [DOI] [PubMed] [Google Scholar]
- 11. Dekker M. C., Giesbergen P. C., Njajou O. T., van Swieten J. C., Hofman A., Breteler M. M., van Duijn C. M. (2003) Neurosci. Lett. 348, 117–119 [DOI] [PubMed] [Google Scholar]
- 12. Guerreiro R. J., Bras J. M., Santana I., Januario C., Santiago B., Morgadinho A. S., Ribeiro M. H., Hardy J., Singleton A., Oliveira C. (2006) BMC Neurol. 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ristić S., Lovrecić L., Brajenović-Milić B., Starcević-Cizmarević N., Jazbec S. S., Sepcić J., Kapović M., Peterlin B. (2005) Neurosci. Lett. 383, 301–304 [DOI] [PubMed] [Google Scholar]
- 14. Connor J. R., Lee S. Y. (2006) J. Alzheimers Dis. 10, 267–276 [DOI] [PubMed] [Google Scholar]
- 15. Goodall E. F., Greenway M. J., van Marion I., Carroll C. B., Hardiman O., Morrison K. E. (2005) Neurology 65, 934–937 [DOI] [PubMed] [Google Scholar]
- 16. Restagno G., Lombardo F., Ghiglione P., Calvo A., Cocco E., Sbaiz L., Mutani R., Chiò A. (2007) J. Neurol. Neurosurg. Psychiatry 78, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sutedja N. A., Sinke R. J., Van Vught P. W., Van der Linden M. W., Wokke J. H., Van Duijn C. M., Njajou O. T., Van der Schouw Y. T., Veldink J. H., Van den Berg L. H. (2007) Arch. Neurol. 64, 63–67 [DOI] [PubMed] [Google Scholar]
- 18. Wang X. S., Lee S., Simmons Z., Boyer P., Scott K., Liu W., Connor J. (2004) J. Neurol. Sci. 227, 27–33 [DOI] [PubMed] [Google Scholar]
- 19. Langou K., Moumen A., Pellegrino C., Aebischer J., Medina I., Aebischer P., Raoul C. (2010) J. Neurochem. 114, 795–809 [DOI] [PubMed] [Google Scholar]
- 20. Yen A. A., Simpson E. P., Henkel J. S., Beers D. R., Appel S. H. (2004) Neurology 62, 1611–1612 [DOI] [PubMed] [Google Scholar]
- 21. Berlin D., Chong G., Chertkow H., Bergman H., Phillips N. A., Schipper H. M. (2004) Neurobiol. Aging 25, 465–474 [DOI] [PubMed] [Google Scholar]
- 22. Lebrón J. A., West A. P., Jr., Bjorkman P. J. (1999) J. Mol. Biol. 294, 239–245 [DOI] [PubMed] [Google Scholar]
- 23. Holmström P., Dzikaite V., Hultcrantz R., Melefors O., Eckes K., Stål P., Kinnman N., Smedsrød B., Gåfvels M., Eggertsen G. (2003) J. Hepatol. 39, 308–314 [DOI] [PubMed] [Google Scholar]
- 24. Golub M. S., Germann S. L., Araiza R. S., Reader J. R., Griffey S. M., Lloyd K. C. (2005) Nutr. Neurosci. 8, 239–244 [DOI] [PubMed] [Google Scholar]
- 25. Tomatsu S., Orii K. O., Fleming R. E., Holden C. C., Waheed A., Britton R. S., Gutierrez M. A., Velez-Castrillon S., Bacon B. R., Sly W. S. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 15788–15793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nandar W., Neely E., Simmons Z., Connor J. (2010) Amyotrophic Lateral Sclerosis 11, S1, 54. [DOI] [PubMed] [Google Scholar]
- 27. Lee S. Y., Patton S. M., Henderson R. J., Connor J. R. (2007) FASEB J. 21, 564–576 [DOI] [PubMed] [Google Scholar]
- 28. Hall E. C., 2nd, Lee S. Y., Mairuae N., Simmons Z., Connor J. R. (2009) Neurobiol. Aging [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 29. Zecca L., Youdim M. B., Riederer P., Connor J. R., Crichton R. R. (2004) Nat. Rev. Neurosci. 5, 863–873 [DOI] [PubMed] [Google Scholar]
- 30. Feder J. N., Penny D. M., Irrinki A., Lee V. K., Lebrón J. A., Watson N., Tsuchihashi Z., Sigal E., Bjorkman P. J., Schatzman R. C. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 1472–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Almeida S. F., Picarote G., Fleming J. V., Carmo-Fonseca M., Azevedo J. E., de Sousa M. (2007) J. Biol. Chem. 282, 27905–27912 [DOI] [PubMed] [Google Scholar]
- 32. Rutkowski D. T., Arnold S. M., Miller C. N., Wu J., Li J., Gunnison K. M., Mori K., Sadighi Akha A. A., Raden D., Kaufman R. J. (2006) PLoS Biol. 4, e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Powell L., Jazwinska E., Halliday J. (2000) Iron Metabolism in Health and Disease, Sauders, London [Google Scholar]
- 34. Oates P. S., Morgan E. H. (1997) Am. J. Physiol. 273, G636–646 [DOI] [PubMed] [Google Scholar]
- 35. McLaughlin B., Hartnett K. A., Erhardt J. A., Legos J. J., White R. F., Barone F. C., Aizenman E. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y., Leavitt B. R., van Raamsdonk J. M., Dragatsis I., Goldowitz D., MacDonald M. E., Hayden M. R., Friedlander R. M. (2006) EMBO J. 25, 5896–5906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marciniak S. J., Ron D. (2006) Physiol. Rev. 86, 1133–1149 [DOI] [PubMed] [Google Scholar]
- 38. Yang Y., Hentati A., Deng H. X., Dabbagh O., Sasaki T., Hirano M., Hung W. Y., Ouahchi K., Yan J., Azim A. C., Cole N., Gascon G., Yagmour A., Ben-Hamida M., Pericak-Vance M., Hentati F., Siddique T. (2001) Nat. Genet. 29, 160–165 [DOI] [PubMed] [Google Scholar]
- 39. Mendes C. S., Levet C., Chatelain G., Dourlen P., Fouillet A., Dichtel-Danjoy M. L., Gambis A., Ryoo H. D., Steller H., Mollereau B. (2009) EMBO J. 28, 1296–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gao J., Chen J., De Domenico I., Koeller D. M., Harding C. O., Fleming R. E., Koeberl D. D., Enns C. A. (2010) Blood 115, 3374–3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bahram S., Gilfillan S., Kühn L. C., Moret R., Schulze J. B., Lebeau A., Schümann K. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 13312–13317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schröder M. (2008) Cell Mol. Life Sci. 65, 862–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mitchell R. M., Lee S. Y., Simmons Z., Connor J. R. (2009) Neurobiol. Aging, in press [DOI] [PubMed] [Google Scholar]
- 44. de Almeida S. F., Fleming J. V., Azevedo J. E., Carmo-Fonseca M., de Sousa M. (2007) J. Immunol. 178, 3612–3619 [DOI] [PubMed] [Google Scholar]
- 45. Vecchi C., Montosi G., Zhang K., Lamberti I., Duncan S. A., Kaufman R. J., Pietrangelo A. (2009) Science 325, 877–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oliveira S. J., Pinto J. P., Picarote G., Costa V. M., Carvalho F., Rangel M., de Sousa M., de Almeida S. F. (2009) PLoS One 4, e6618. [DOI] [PMC free article] [PubMed] [Google Scholar]