

Abstract
The L-type Ca2+ channel Cav1.2 forms macromolecular signaling complexes that comprise the β2 adrenergic receptor, trimeric Gs protein, adenylyl cyclase, and cAMP-dependent protein kinase (PKA1) for efficient signaling in heart and brain. The protein phosphatases PP2A and PP2B are part of this complex. PP2A counteracts increase in Cav1.2 channel activity by PKA and other protein kinases, whereas PP2B can either augment or decrease Cav1.2 currents in cardiomyocytes depending on the precise experimental conditions. We found that PP2A binds to two regions in the C-terminus of the central, pore-forming α1 subunit of Cav1.2: one region spans residues 1795-1818 and the other residues 1965-1971. PP2B binds immediately downstream of residue 1971. Injection of a peptide that contained residues 1965-1971 and displaced PP2A but not PP2B from endogenous Cav1.2 increased basal and isoproterenol-stimulated L-type Ca2+ currents in acutely isolated cardiomyocytes. Together with our biochemical data, these physiological results indicate that anchoring of PP2A at this site of Cav1.2 in the heart negatively regulates cardiac L-type currents, likely by counterbalancing basal and stimulated phosphorylation that is mediated by PKA and possibly other kinases.
Ca2+ influx through L-type channels controls membrane excitability (1), synaptic plasticity (2-5), and gene expression (6, 7) in neurons and triggers myocardial contraction in the heart. L-type Ca2+ channels are the main targets of so-called organic calcium channel blockers, which include dihydropyridines, phenylalkylamines, and benzothiazepines. Voltage-gated Ca2+ channels consist of a central ion-conducting pore, the α1 subunit, and auxiliary α2-δ, and β subunits (8). Cav1.2 containing the central α11.2 is the main L-type channel in the cardiovascular system, heart, and brain (8).
Cav1.2 is a point of convergence of multiple regulatory pathways. For instance, β-adrenergic stimulation upregulates our heart beat in part via phosphorylation of Cav1.2 by PKA (9, 10). Cav1.2 phosphorylation and dephosphorylation are highly dynamic with phosphatases reversing the stimulatory effect of PKA and perhaps other kinases rather quickly (11, 12). We found earlier that PP2A and PP2B (calcineurin) are constitutively bound to Cav1.2 (13, 14). Channel-associated PP2A reverses PKA-mediated phosphorylation of serine 1928 (13). Serine 1928 is one of two identified PKA sites in α11.2 the other being the most recently identified serine 1700 (15, 16). Although phosphorylation of serine 1928 is not necessary for regulation of Cav1.2 possibly because other phosphorylations can suffice in its absence (8, 15-18), it is highly regulated and continues to serve as indicator for PKA-mediated phosphorylation of α11.2.
We now narrow down the exact binding sites for PP2A to two short regions (residues 1795-1818 and 1965-1971) that independently bind PP2A. PP2B binds immediately downstream of residues 1965-1971 without competition between these two phosphatases for binding to this rather narrow region. A peptide that disrupts binding of PP2A but not PP2B to this site increases L-type-mediated Ca2+ currents in cardiomyocytes, likely by preventing the inhibitory effect of PP2A under basal and ISO1-stimulated conditions.
EXPERIMENTAL PROCEDURES
Materials, antibodies, peptides
ECL™ and ECL-Plus™ detection kits, and glutathione Sepharose were purchased from Amersham Pharmacia Biotech (Piscataway, NJ). The monoclonal mouse anti-GST antibody was purchased from NeuroMAB (Davis, CA), the monoclonal mouse anti-PP2A/C antibody 1D6 (19) from Upstate Biotechnology (Lake Placid, NY), the monoclonal rat antibody 6F9 from Dr. G. Walter (20), and the monoclonal mouse anti-PP2B antibody (21, 22) from BD Transduction Laboratories. The anti-α11.2 antibody had been produced against a segment of the cytosolic loop between domain II and III of α11.2, as described (23). Peptides for displacement studies were custom synthesized by CHI Scientific (Maynard, Massachusetts). Other chemicals were of standard biochemical quality and from usual commercial suppliers.
Peptide array overlay assay
The peptide spot array spanning residues 1784-2067 of rabbit cardiac α11.2 (for sequence see gene bank accession number CAA33546) was synthesized on a PVDF membrane as published (24). The first spot contains a 15-mer peptide covering residues 1784-1798 of α11.2. Peptides in each subsequent spot were shifted by one residue from the previous spot. The PVDF membrane was blocked with 10% milk powder in TBS (10 mM Tris-Cl, pH 7.4, 150 mM NaCl) before incubation with recombinant PP2A/C subunit expressed in E. coli (see below) in the same solution, washed, and probed with the anti-PP2A/C antibody.
In vitro binding assays of 6xHisPP2A and PP2B to GST-fusion proteins
GST-CT-8 encoding residues 1909-2029 of rabbit heart α11.2 (13) served as a template for construction of GST-fusion proteins covering residues 1909-1946 (CT-8-1), 1909-1971 (CT-8-2), 1943-2029 (CT-8-3), and 1969-2029 (CT-8-4) and for a point mutation on the otherwise full length GST-CT-8 construct to change Ala1959 to Pro (GST-CT-8-P), as described (14). GST-CT-B containing residues 1694-1817 of rat α11.2 cDNA (25) (nearly identical to residues 1726-1849 of the above rabbit α11.2) was as given earlier (13). These GST-fusion proteins as well as the 6xHis-PP2A/C construct (26) were expressed in Nova Blue (Novagen, Madison, WI) and BL21 Star (Invitrogen, San Diego, CA) E. coli strains and purified and used for in vitro pull-down interaction studies as detailed previously (13, 14, 26). PP2B was expressed in E. coli and purified for pull-down experiments as outlined earlier (14, 27). For quantification of pull-down experiments, film exposures of immunoblots were digitalized with an Epson Perfection 4180 Photo flatbed scanner and scanned immunosignals quantified by densitometry in Adobe Photoshop. Multiple exposures with increasing time length were taken to ensure that signals were in the linear range (for more details see (13, 28)). In Fig. 3-6 and Supplemental Fig. 1, immunosignals were normalized relative to the signals from the corresponding positive control samples obtained with GST-CT-8, CT-8-3, or CT-B in the absence of peptide. Means and SEMs were calculated and analyzed with Prism 3.0 (GraphPad Software, Inc.). Differences were accepted as significant at p<0.05 by paired t-test.
Figure 3.
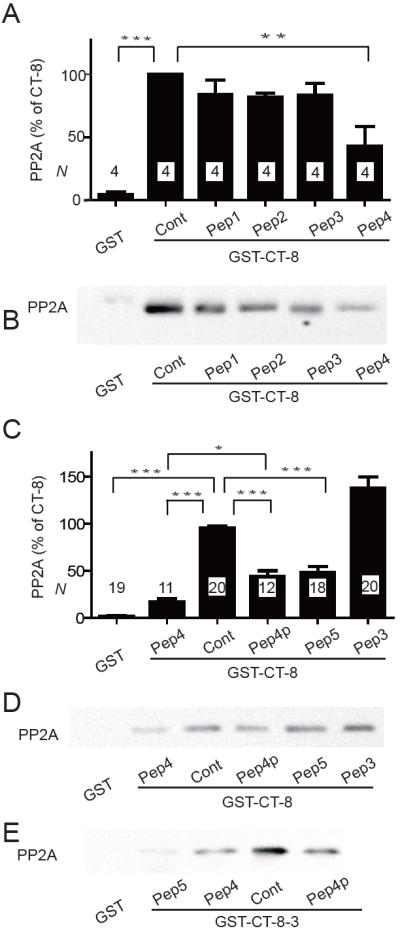
Peptides containing α11.2 residues 1965-1971 (LSPLLQR) compete with GST-CT-8 for PP2A/C binding. Graphed are means±SEM from N samples in 4 (A) and 10 (C) independent experiments and representative immunoblots (B,D,E) of pull-down experiments of purified poly-His-PP2A/C with GST-CT-8 (A-D) or GST-CT-8-3 (E) with the indicated peptides (10 μM) present during PP2A binding (see Fig. 1 for definition of Peptides 1-5). GST served as negative control and GST-CT-8 or GST-CT-8-3 without any peptide as positive control (Cont; * p<0.05; **p<0.01; ***p<0.001).
Figure 6.
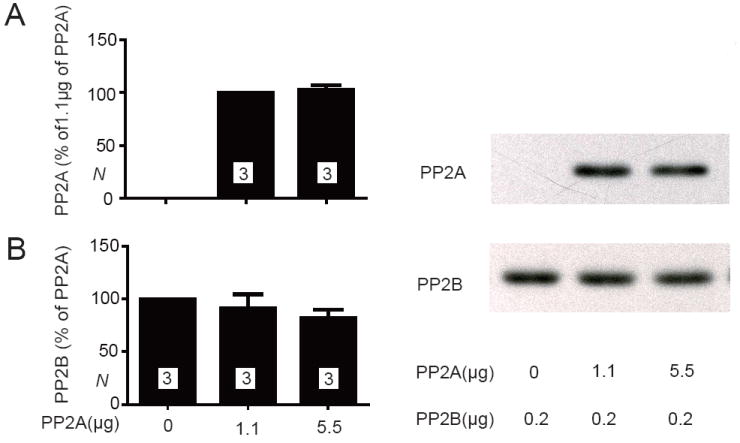
PP2A/C and PP2B do not compete for CT-8 binding. Glutathione Sepharose was sequentially incubated with GST-CT-8, then 0, 1.1, or 5.5 μg PP2A, and finally 0.2 μg PP2B. Representative immunoblots (left panels) illustrate the relative amount of PP2A (A) and PP2B (B) present in the same pull-down samples. Means±SEM from 3 independent experiments are shown in the right panels.
Immunoprecipitation
Mouse (C57black/6) hearts and brains were homogenized in 2.5 ml ice-cold solubilization buffer (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 10 mM EDTA, 10 mM EGTA, plus protease inhibitors: 1 μg/ml pepstatin A, 10 μg/ml leupeptin, 20 μg/ml aprotinin, 8 μg/ml each calpain inhibitors I and II) before centrifugation at ~250,000 × g for 30 min. 200 μl aliquots of the supernatants were incubated with 2 μg of the anti-α11.2 antibody or of non-specific control IgG and 10 μl of prewashed Protein-A Sepharose slurry (50% resin, 50% buffer) for 4-6 h and washed three times with 1% Triton X-100 in TBS (10 mM Tris-HCl pH 7.4, 150 mM NaCl) containing 0.1% SDS and once with 10mM Tris-HCl pH 7.4. Proteins were extracted by SDS sample buffer, separated by SDS-PAGE, transferred to PVDF membrane and detected as above.
PP2A and PP2B activity assays
PP2A catalytic activity was assessed using the DuoSet IC (R+D Systems) malachite green/molybdate-based PP2A activity assay (29, 30) in accordance with the manufacturer’s instructions. Murine cardiac tissue was homogenized in lysis buffer 8 (50 mM HEPES, 0.1 mM EGTA, 0.1 mM EDTA, 120 mM NaCl, 0.5% NP-40, pH7.5, 25 μg/mL leupeptin, 25 μg/mL pepstatin, 2 μg/mL aprotinin, 1 mM PMSF) and non-soluble material removed by centrifugation at ~170,000 × g for 30 min. PP2A was immobilized on microtiter plate wells from lysate (500 μg of total protein per well) using an anti-PP2A/C antibody (Millipore 05-421), washed, and pre-incubated with 20 μM Peptide 1, 4, or 5 at room temperature for 15 min before addition of 200 μM final concentration of a synthetic phosphopeptide substrate (DLDVPIPGRFDRRVS(PO3)VAAE). After 30 min at 37°C, malachite green and molybdate were added to bind free phosphate and initiate the colorimetric reaction (29, 30). Light absorbance was measured at 630 nm. The amount of released phosphate was calculated from standard curves generated in parallel using orthophosphate. To exclude that another phosphatase present in the lysate bound to the wells and contributed to the activity or that phosphate contaminations contributed to the final read out, lysate was either omitted or PP2A was blocked in some assays with 10 nM okadaic acid (IC 50 of 0.2-2.5 nM for PP2A (31)), which is specific for PP2A at this concentration (32).
PP2B/calcineurin activity was determined using the malachite green/molybdate-based Calcineurin Assay Kit from Calbiochem following manufacturer’s instructions. Purified calmodulin (CaM, 250 nM final concentration) was incubated with 40 U recombinant PP2B and 20 μM of Peptide 1, 4, or 5 at room temperature for 15 min. A synthetic phosphopeptide substrate (DVPIPGNFDNNVS(PO3)VAAE) was then added (150 μM final concentration). After 40 min at 37°C, malachite green and molybdate were added to determine how much phosphate had been released as described for PP2A. For negative control to exclude contamination by external phosphate either PP2B or calmodulin were omitted.
Electrophysiology
For recording ICa(L), freshly prepared adult rabbit ventricular myocytes were pre-incubated in Na+- and Ca2+-free Tyrode solution (LiCl 140 mM, MgCl2 1 mM, HEPES-free acid 10 mM, EGTA 1 mM, glucose 10 mM; pH = 7.4 with LiOH) for 15-20 min, and then plated on laminin-treated glass coverslips in Ca2+-free solution containing CsCl 4 mM, MgCl2 1 mM, HEPES-free acid 10 mM, tetraethylammonium (TEA)-Cl 140 mM, glucose 10 mM (pH = 7.4 with TEA-OH). For experiments, 2 mM CaCl2 was added to the TEA bath solution. Predepletion of cytosolic Na+ and use of Na+-free solutions allowed recording of ICa(L) free of interference by Na/Ca exchange or voltage-gated Na channels. In most experiments, cells were preincubated with thapsigargin (1 μM, 15 min) to block Ca2+ loading of the sarcoplasmic reticulum, and niflumic acid (30 μM) to block Cl- currents.
Ca2+ current was recorded at room temperature (22-24°C) in the whole cell perforated patch configuration using ß-escin (33). Pipettes were tip-filled with solution containing n-methylglucamine (NMG) 100 mM, TEA-Cl 20 mM, glutamic acid (free acid) 80 mM, HEPES-free acid 10 mM, MgCl2 5.66 mM, ATP (Tris salt) 5 mM GTP (Li+ salt) 0.3 mM EGTA 5 mM, and CaCl2 1.09 mM, resulting in a calculated free [Ca2+] = 50 nM (MaxChelator). pH was adjusted with HCl to 7.2. Use of EGTA buffering was intended to prevent global but not local [Ca2+] changes. This same solution with ß-escin (pre-dissolved in H2O; final concentration 50 μM in pipette) and either Peptide 1, 4, or 5 (for nomenclature, see Results and Figure 1B; final [peptide] 50 μM) or no peptide added was used to backfill pipettes. The liquid junction potential of the pipette solution, 9 mV, pipette negative, was corrected.
Figure 1.
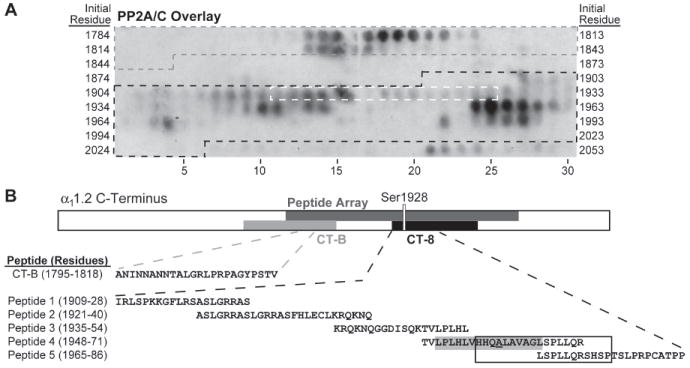
PP2A/C strongly binds to α11.2 residues 1795-1818 and 1956-1975 in peptide array overlay. (A) A peptide array spanning residues 1784-2067 of rabbit heart α11.2 was incubated with the catalytic C subunit of PP2A (PP2A/C) and probed with anti-PP2A/C antibodies. Peptides encompassing residues from CT-B (grey), CT-8 (black), and Ser1928 (white) are indicated by dashed outlines on the overlay image. (B) Topology of the linearized α11.2 C-terminus depicting the region covered by the peptide array (dark grey), GST fusion proteins CT-B (grey) and CT-8 (black), the latter including Ser1928. Below are tabulated the sequence of the residues that define the PP2A binding region in CT-B according to the overlay and the CT-8 derived peptides 1-5 used for the displacement studies below. The boxed residues in Peptide 4 and 5 are those that are encompassed by the PP2A/C binding-positive peptides on the array in the CT-8 region. The putative α-helix is shaded grey with Ala1959 underlined in Peptide 4.
After cells were sealed it took usually 15-20 min for the formation of ß-escin pores, which are of sufficient size to provide electrical access as well as access of molecules of ~10 kDa including our peptides (34). We confirmed that fluorescent dextran (MR ~10 kDa) and hence our peptides could pass through the ß-escin pores (not illustrated). Cells were continuously held under voltage clamp, with series resistance (typically 8-10 megΩ) compensated by ~75%. 200 msec depolarizations to 0 mV from -90 mV were applied each 10 sec, under control condition ([ISO] = 0) then at 10, 55 and 300 nM ISO. 300 nM ISO was prepared fresh for each session from the same sample and diluted successively. Each [ISO] was maintained ≥3 min, to apparent steady state. At each [ISO] a current-voltage relationship was also recorded, using 200 msec depolarizations from -90 mV to between -40 and +50 mV. Cells were held at -90 mV for 1-2 sec between steps to assure full recovery of channels from inactivation. Control experiments showed that 1 mM CdCl2 blocked the current identified as ICa(L) (not illustrated). Upon washout of ISO at the end of an experiment, ICa(L) returned on average to 83% of its initial value.
The transient parts of currents evoked by repeated depolarization to 0 were measured. Each individual trace was first corrected for leak conductance (initial holding current / initial holding voltage). Then we found the difference between peak current (identified directly) and final asymptotic current (obtained by fitting the post-peak decay phase of the current to
where I is the observed current, A is the identified peak and the fit parameters were k (0 < k <1, a weighting factor), τ1, τ2 (time constants) and C was a constant. As ICa(L) inactivates almost completely, constant C was usually near 0 (this procedure limits errors due to any uncorrected leakage or junctional potential-related fluxes). Current-voltage data were treated similarly, with all currents normalized to cell membrane capacitance.
Peak current-voltage data were fit to a Boltzmann function
where I is the observed current and Vm is the test depolarization potential, while the fit parameters are Gmax (maximum conductance), V50 (half-activation potential), KV (voltage dependence slope factor), Erev (net current reversal potential) and C (offset and residual leak correction constant).
We statistically analyzed the influence of ISO and the choice of peptide on Gmax and V50 using two-way analysis of variance (ANOVA; GraphPad Prism software v5.02, www.graphpad.com).
RESULTS
Peptide array overlay with PP2A/C
Pull-down experiments with GST fusion proteins previously identified two different regions in the C-terminus of α11.2 that mediate direct binding of the catalytic subunit of PP2A (PP2A/C) (13). One region lies within residues 1726-1849 and the other within residues 1909-2029 of the rabbit heart α11.2 as defined by pull-down with GST-CT-B and -CT-8 fusion proteins, respectively (13). To identify the binding sites more precisely we used a solid phase peptide library. Overlay with the catalytic subunit of PP2A (PP2A/C) expressed in E. coli as poly-His fusion protein revealed two regions with especially strong immunoreactivity: one region encompasses residues 1795-1818 of α11.2 (ANINNANNTALGRLPRPAGYPSTV; residue numbering as for the original α11.2 sequence from rabbit heart; gene bank accession number CAA33546). This segment is within the original CT-B segment (Fig. 1). The other region comprises residues 1956-1975 (HHQALAVAGLSPLLQRSHSP) within the original CT-8 segment (Fig. 1). Experiments described below suggest that all other regions on the peptide array that show detectable immunoreactivity are not strong constitutive PP2A/C interaction sites.
PP2A/C pull-down experiments with GST fusion proteins
We recently provided evidence that residues 1943-1971 within CT-8 bind yet another phosphatase, the Ca2+- and calmodulin-activated PP2B (calcineurin) (14). To define the PP2A binding site within this region relative to PP2B we used shorter fragments of GST-CT-8 (Fig. 2A). The Robson-Garnier algorithm predicts that this region forms an α-helix (14). In fact, substituting Ala1959 with Pro, a helix breaker, abrogates PP2B binding (14). All fusion proteins were first purified on glutathione Sepharose, eluted, and dialyzed to remove glutathione for immobilization of comparable amounts of protein on glutathione Sepharose and subsequent PP2A/C pull-down experiments. Several fusion proteins were partially cleaved resulting in a substantial amount of GST with only minimal or no additional channel segments (Fig. 2B, E). To allow comparison between the different fusion proteins equal amounts of the corresponding full length forms (arrowheads in Fig. 2B; see also Fig. 2E) were loaded onto the resin. To determine the portion of PP2A/C binding to CT-8 fusion proteins that was specific, pure GST was loaded in parallel roughly matching the amount of the former. Background was typically less than 20% and was subtracted from total binding signals for CT-8 pull-down samples. PP2A/C bound consistently and with high specificity to full length CT-8 as well as the proline mutant CT-8-P (Fig. 2C,D). This equivalent interaction of CT-8 and CT-8-P with PP2A/C is in striking contrast to PP2B binding, which was abrogated by the proline mutation (14). PP2A/C did not specifically bind to CT-8-1 or CT-8-4, N-terminal and C-terminal truncations of the original CT-8 fusion protein that lacked the putative α-helix (Fig. 2C,D). PP2A/C binding to CT-8-2 and CT-8-3 (both contained the putative α-helical region, residues 1943-1971), were more variable, but showed significant specific binding (p<0.05 compared to GST control) albeit weaker than to the full length CT-8.
Figure 2.

PP2A/C binds to the central region spanning residues 1943-1971 in CT-8. (A) Schematic of the parental CT-8 segment (bottom) and its derivatives. A predicted α-helix is indicated in the center of CT-8 and all derivatives that contain this segment. CT-8P encodes CT-8 with the Ala1959Pro mutation as depicted above the presumably interrupted α-helix. (B) SDS-PAGE of the various purified GST-CT-8 fusion proteins including GST itself as detected byCoomassie staining. Arrowheads indicate expected positions of the corresponding full length polypeptides. (C-E) Pull-down of purified poly-His-PP2A/C with the fusion proteins as detected by immunoblotting with anti-PP2A/C (D). Results from 18 different experiments were quantified. Means±SEM from N samples for each condition are graphed in C (* p<0.05; ***p<0.001). Reprobing with anti-GST was routinely performed to confirm that similar amounts of GST fusion proteins were present (E).
CT-8-1 encodes residues 1909-1946 and includes the first regions that showed weak binding in the peptide overlay assay (starting with residue 1910; Fig. 1). Because CT-8-1 did not show any specific binding in the pull-down test, the modest binding to the 1910 region in the Fig. 1 overlay assay likely indicates only weak association. Because this region contains Ser1928, which is phosphorylated by PKA and dephosphorylated by PP2A, this binding could indicate a catalytic site interaction (13), which would typically be brief in nature. Similarly, the weak signals in the PP2A/C overlay for peptides that cover residues 1988-2005 is also likely either non-specific or limited in nature as CT-8-4 encoding residues 1969-2029 did not show any specific binding in the pull-down experiments.
Peptide competition experiments with PP2A/C and CT-8
We used synthetic peptides in solution to further define the PP2A/C binding site in CT-8. Peptides were 20-24 residues long and covered various overlapping regions between residues 1909 and 1986 (Peptide 1 - Peptide 5; Fig. 1B). In the first set of experiments with Peptides 1 through 4, only Peptide 4 significantly inhibited pull-down of PP2A/C by GST-CT-8 (Fig. 3A,B). Peptide 4 covers the predicted α-helix in this region and most of the residues encompassed by the second strong binding region of the peptide array overlay experiment. We subsequently tested Peptide 4 with Pro substituting the Ala at position 1959 (Peptide 4P) and Peptide 5, which includes the last 7 residues of Peptide 4 plus 15 residues towards the C-terminus. Peptide 4P and Peptide 5 also disrupted PP2A/C binding to CT-8 but not as strongly as Peptide 4 (Fig. 3C,D). Similar results were obtained in two experiments using GST-CT-8-3 for PP2A pull-down (Fig. 3E).
Peptides 1 and 2 cover the region starting with residue 1910 that showed some weak binding in the peptide array overlay assay. The finding that these peptides did not diminish at all pull-down of PP2A/C by GST-CT-8 is further evidence for the above conclusion that this site interacts weakly if at all with PP2A. Subsequent attempts to further narrow down the precise binding site for PP2A within the sequence covered by Peptide 4 and 5 with 10-mer peptides failed (Supplemental Figure 1A,B; Peptides 6-10). The most likely explanation is that the affinity of the shorter peptides for PP2A is too low for effective competition with the CT-8 polypeptide because none of these peptides might have a sufficient number of contact sites for PP2A even though we increased peptide concentration from 10 μM to 20 μM (Supplemental Figure 1) and in one experiment to 40 μM (data not shown).
Peptide competition experiments with PP2A/C and CT-B
To test whether the interactions of PP2A with CT-8 and CT-B are independent of each other, we performed peptide competition PP2A pull-down experiments with GST-CT-B. None of the CT-8-derived peptides reduced PP2A pull-down by GST-CT-B, including Peptides 4, 4P, and 5 (Fig. 4). This result suggests that PP2A binds with different sites to CT-B and CT-8. Attempts to define the CT-B binding site with similar competition studies using two partially overlapping peptides derived from the peptide overlay experiment failed as one covering residues 1794-1808 was insoluble and the other one covering residues 1804-1818 had no effect (data not shown).
Figure 4.
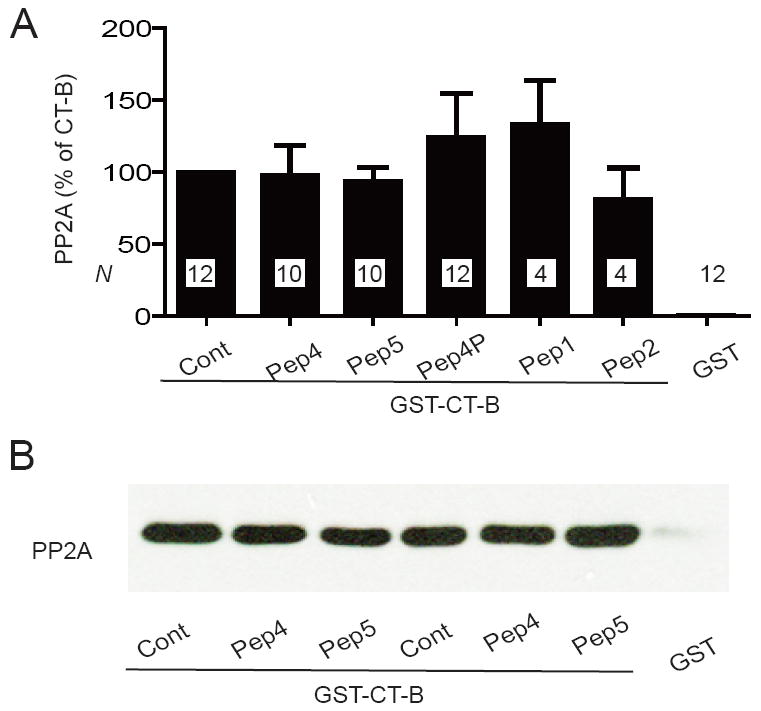
Peptides derived from CT-8 do not compete with GST-CT-B for PP2A/C binding. Graphed are means±SEM from N samples in 6 independent experiments (A) and representative immunoblots (B) of pull-down experiments of purified poly-His-PP2A/C with GST-CT-B with the indicated peptides (10 μM) present during PP2A binding. GST was the negative control and GST-CT-B without any peptide the positive control (Cont).
Peptide competition experiments with PP2B and CT-8
Because the Ala1959Pro mutation in the putative α-helix formed by residues 1943-1971 abrogated PP2B binding to GST-CT-8 (14), it appeared likely that PP2B binds at or near this region. In peptide competition studies analogous to those for PP2A above, only Peptide 5 but not Peptide 4 affected pull-down of PP2B by GST-CT-8 (Fig. 5). Peptide 4 covers nearly all of the putative α-helix including Ala1959, which is not part of Peptide 5 (Fig. 1B). It is possible that the Ala1959Pro mutation disrupts not only the conformation of the putative α-helix in its immediate vicinity but also downstream of it. Such a disruption could extend to residues covered by Peptide 5. As the N-terminal 7 residues of Peptide 5 are also present on Peptide 4, yet Peptide 4 did not disrupt PP2B binding to CT-8, those residues appear not to be sufficient for PP2B binding if they can participate at all in the interaction with PP2B. In any case, it appears that PP2B does not directly bind to the immediate vicinity of Ala1959 but rather somewhat downstream of it. We tested shorter peptides covering Peptide 4 and 5, but similar to our PP2A results none affected PP2B binding (Supplemental Fig. 1C).
Figure 5.
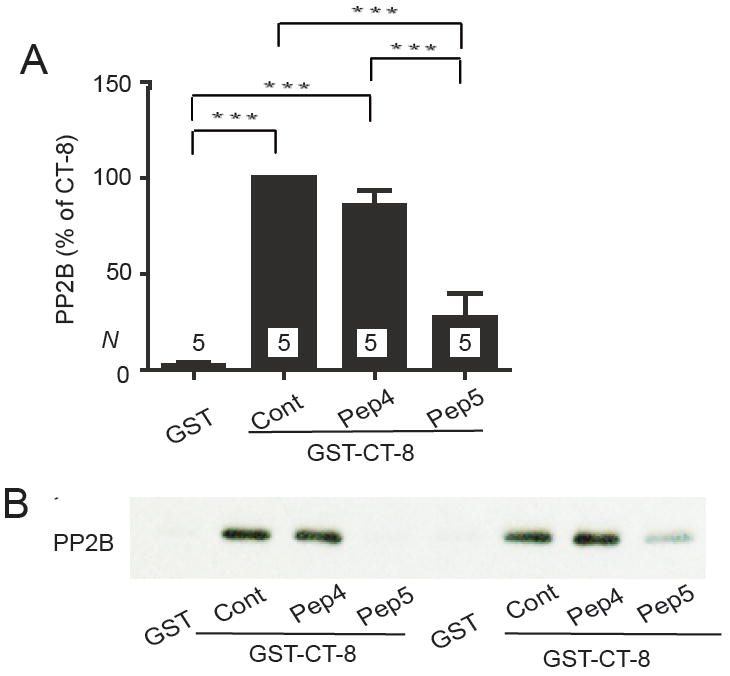
Peptide 5 but not Peptide 4 competes with GST-CT-8 for PP2B binding. Graphed are means±SEM from N samples in 3 independent experiments (A) and example immunoblot (shown in duplicate) of pull-down experiments of purified poly-His-PP2B with GST-CT-8 (B) with Peptide 4 or 5 (10 μM) present as indicated during PP2B binding. GST was the negative control and GST-CT-8 without any peptide the positive control (Cont; *** p<0.001).
Competition experiments between PP2A and PP2B
As the above peptide studies indicate that PP2A and PP2B bind to residues very close to each other in CT-8 we wondered whether these two phosphatases would compete for binding to this region. GST-CT-8 was immobilized on glutathione Sepharose and incubated with either 0, 1.1, or 5.5 μg affinity-purified PP2A and subsequently with 0.2 μg affinity-purified PP2B. Immunoblotting showed that the amount of PP2A bound to the GST-CT-8 - charged resin was indistinguishable for the 1.1 vs. 5.5 μg samples (Fig. 6A). This observation indicates that both amounts allowed saturation of the immobilized CT-8 with PP2A. Subsequent incubation with PP2B resulted in comparable amounts of PP2B pulled down by the GST-CT-8 resin whether this resin was preincubated with PP2A or not (Fig. 6B). These data suggest that PP2A and PP2B can simultaneously bind to CT-8.
Peptide 5 but not Peptide 4 displaces PP2A but not PP2B from native Cav1.2 complexes
The above in vitro binding assays define the precise PP2A and PP2B binding sites on α11.2. However, they do not show whether these peptides are effective in promoting dissociation of PP2A or PP2B from native Cav1.2 complexes. Dissociation of both phosphatases must be very slow as a substantial amount of them remains bound during the co-immunoprecipitation procedure, which takes at least 4 h after detergent extraction and dilution of tissue content. Peptides are thought to promote dissociation of otherwise relatively stable protein complexes by gaining access to interaction sites on the complementary binding partner upon partial unbinding. The latter would not lead to full dissociation itself unless interfering compounds prevent full rebinding. It is thus quite possible that some peptides compete during binding reactions but do not promote dissociation of preformed complexes.
Endogenous PP2A and PP2B co-immunoprecipitated with Cav1.2 (Fig. 7). PP2A holoenzymes typically consist the catalytic C subunit, the scaffolding A subunit and one of numerous B subunits, which mediate targeting of PP2A holoenzymes (35). We determined peptide effects on co-immunoprecipitations of PP2A/C as well as PP2A/A. All coprecipitations with Cav1.2 were specific for PP2A/C, PP2A/A, and PP2B as control IgG did not pull down any of these proteins (Fig. 7B, D, F). Inclusion of Peptide 5 but not of Peptide 4 during the immunoprecipitation step reduced co-precipitation of PP2A/A and PP2A/C with Cav1.2 by ~50%. Neither Peptide 5 nor Peptide 4 affected co-precipitation of PP2B. These results indicate that only Peptide 5 but not Peptide 4 effectively dislocates PP2A from native Cav1.2 complexes even though both peptides compete during PP2A-GST-CT-8 association reactions. Despite its effect on PP2A, Peptide 5 did not effectively dislodge PP2B from the endogenous Cav1.2 complex.
Figure 7.
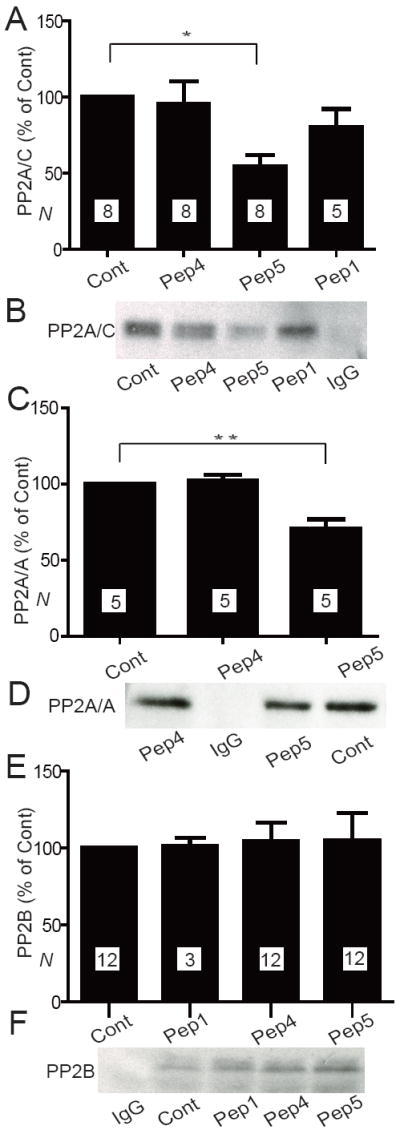
Peptide 5 displaces PP2A but not PP2B from native Cav1.2 complexes. Immunoprecipitations of Cav1.2 were performed in the absence (Cont) or presence of 20 μM Peptide 1, 4, or 5 and PP2A/C (A, B), PP2A/A (C, D), or PP2B (E, F) detected by immunoblotting. Graphed are means±SEM from N samples in 8 (A), 2 (C), and 6 (D) independent experiments. Control precipitations with non-specific IgG indicate the specificity of PPA/C (B), PP2A/A (D), and PP2B (F) co-precipitations with Cav1.2.
Perfusion of cardiomyocytes with Peptide 5 but not Peptide 4 increases L-type Ca2+ currents
To determine the functional role of the PP2A anchoring site on Cav1.2, adult rabbit ventricular myocytes were freshly prepared and ICa(L) recorded in a perforated patch configuration. The saponin-related mild detergent ß-escin was used (33) because it affords access of peptides and polypeptides to the cell interior (34) (see Methods). Inclusion of Peptide 5, which displaces PP2A but not PP2B from endogenous Cav1.2 as determined by the above immunoprecipitation experiments, resulted in a larger ICa(L) compared to Peptide 1 or no peptide (Fig. 8A,C). We used Peptide 1 as control because it has a charge content similar to that of Peptide 4 and 5 but does not affect binding of PP2A or PP2B to CT-8 nor co-immunoprecipitation of PP2A or PP2B with native Cav1.2. ANOVA showed that the choice of peptide (None or 1 vs. 4 vs. 5) affected ICa(L) at all ISO concentrations, appearing in Fig. 8C as the strong increase with Peptide 5. Independently of the choice of peptide, ISO increased ICa(L), as expected. The effect of Peptide 5 to increase basal Cav1.2 activity might reflect basal activity of a kinase, possibly PKA, that is unmasked by reducing local PP2A activity via disruption of phosphatase binding to the CT-8 region. The enhanced β-adrenergic stimulation with Peptide 5 was seen over the full range of ISO concentrations as illustrated in example traces for [ISO] = 300 nM (Fig. 8B) and as determined via Gmax derived from I/V relations (Fig. 8C). These findings indicate that endogenous PP2A is associated with Cav1.2 in cardiac myocytes to limit the magnitude of PKA-mediated enhancement of ICa(L). Half-activation potential V50 derived from the I/V relations shifted significantly negative on application of ISO as expected (12). This shift tended to be greater with Peptide 5 (maximal shift -16.0±1.9 mV SEM vs -12.9±1.9 mV with Peptide 1 or no peptide but the difference was statistically not significant; not illustrated). In agreement with the lack of Peptide 4 on PP2A co-immunoprecipitation with Cav1.2 from native tissue, Peptide 4 did not affect ICa(L) (Figure 8A,C).
Figure 8.
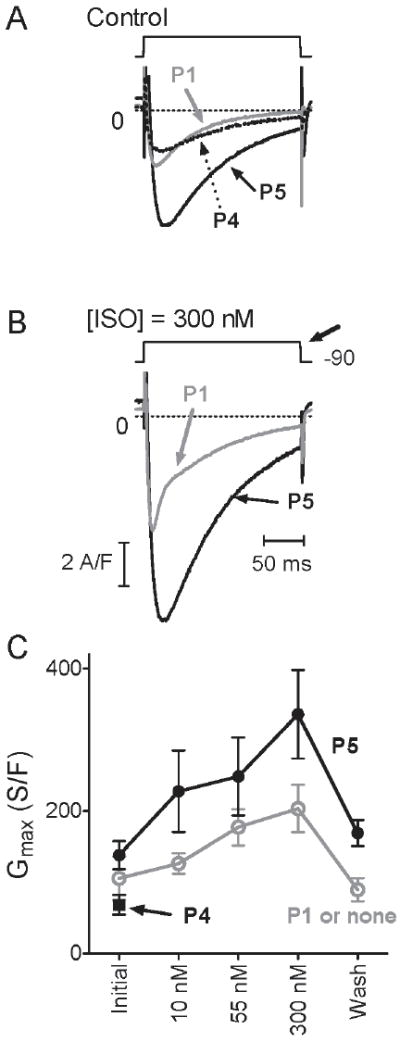
Peptide 5 increases ICa(L) density under control and ISO treatment conditions compared to Peptide 1 or no peptide. (A) Representative current traces upon depolarization from -90 mV with Peptide 1 (grey), Peptide 4 (dotted) or Peptide 5 (black) in the recording electrode ([ISO] = 0). (B) traces from same respective cells with [ISO] = 300 nM. Traces were obtained from the respective current-voltage relations, at the depolarization giving maximal peak current (bold arrow, B). The voltage for maximum current (typically -20 mV without ISO), which would be relevant for cardiac cell contraction, shifted negative in response to ISO (see Results). Traces were smoothed minimally, and vertically adjusted slightly, to plot asymptotic values at 0 (dashed lines). (C) Graphed are means±SEM of maximum conductance Gmax obtained by fit (see Methods) from peak current-voltage relations. Each data point in C represents 5-8 measurements, except wash (N≥4) from in total 3 cells with no peptide, 4 with Peptide 1, 9 with Peptide 4 and 7 with Peptide 5. In several cells, data were not recorded at all [ISO]. We combined the 3 experiments with no peptide and the 4 with Peptide 1 for our analyses as no differences between these two conditions were obvious (* p > 0.05; Mann-Whitney (U) tests).
Peptide 4 and 5 do not directly affect PP2A or PP2B catalytic activity
To ensure that in the above recordings Peptide 5 acted by displacing PP2A from Cav1.2 and not by inhibiting general phosphatase activity, we determined catalytic activity of purified PP2A and PP2B in the presence and absence of these peptides, with negative results (Fig. 9).
Figure 9.
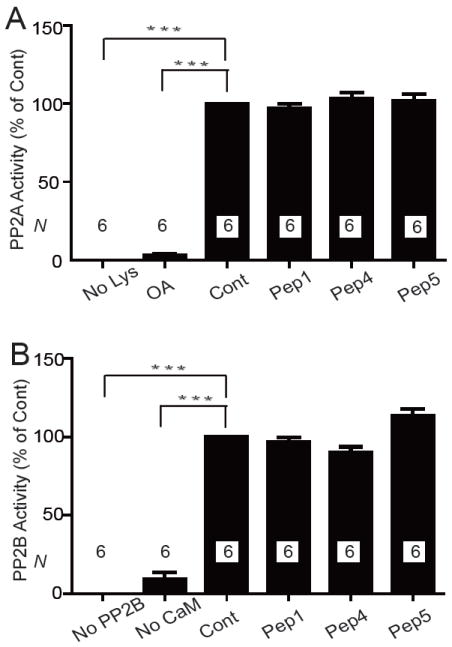
Peptides 1, 4, and 5 do not affect phosphatase activity of PP2A or PP2B. PP2A (immunoisolated from heart) and recombinant purified PP2B were pre-incubated with 20 μM Peptide 1, 4, or 5, 10 nM okadaic acid (OA), which is a specific PP2A blocker at this concentration, or without any additives (positive control for full phosphatase activity; Cont). Respective phosphopeptide substrates were added for PP2A and PP2B assays and subsequently phosphate release determined. Lysate (No Lys; PP2A), PP2B (No PP2B), or calmodulin (No CaM) were omitted in some assays to test for phosphate contaminations from other ingredients. Shown are means±SEM of phosphatase activity determined in 2 independent experiments with triplicate samples each (N=6) for each phosphatase.
DISCUSSION
Formation of inside out patches from rabbit ventricular myocytes causes run down of L-type currents that is blocked by okadaic acid (11). This observation provided early functional evidence that a phosphatase is anchored in close proximity to the channel that counteracts upregulation of Cav1.2 by phosphorylation. We found that PP2A is associated with Cav1.2 (36) and subsequently identified two attachment sites for PP2A within the C-terminus of the central α11.2 subunit (13). In the present study pull-down experiments with CT-8-derived truncated fusion proteins indicate that the segment between residues 1943 and 1971 of α11.2 is required for specific PP2A/C binding to this region. Our peptide overlay indicates that 15-mer peptides starting with residues 1956-1959 possess strong PP2A/C binding potential, with some peptides showing weaker binding. The core sequence common to all four strongly binding peptides is 1959ALAVAGLSPLLQ1970. Furthermore, Peptide 4 and Peptide 5 compete with CT-8 for PP2A/C binding. Both peptides contain the C-terminal 6 residues of that core sequence plus an additional Arg C-terminal to those residues (1965LSPLLQR1971). Finally, the Ala1959Pro mutation in CT-8 does not abrogate PP2A/C binding to CT-8 nor does this substitution in Peptide 4 prevent the resulting Peptide 4P from competing with CT-8 for PP2A/C pull down. The simplest explanation of all these results is that the main interaction sites for PP2A/C in this region of α11.2 is 1965LSPLLQ1970. This conclusion is in agreement with the finding that CT-8-3, which covers residues 1943-2029, but not CT-8-4, which covers residues 1969-2029, pulls down PP2A as the latter is lacking residues 1965-1968 from the core interaction site. Accordingly, residues 1965-1968 are critical for binding.
PP2A typically exists as a trimer consisting of a catalytic C subunit, a structural A subunit, and a B-type subunit (35). The > 15 identified B-type subunits are subcategorized into three types (B, B′, and B″) (35, 37, 38). They generally mediate targeting of the PP2A holoenzyme to its substrates, which involves at least in some cases a stable interaction. It is thus remarkable that PP2A/C itself exhibits strong constitutive binding to two different sites on α11.2 that possess different structural features. PP2A also forms a constitutive interaction with the type 2 ryanodine receptor in heart, which in this case is mediated via the B″ subunit PR130 (39). Cav1.2 and the ryanodine receptor are precisely juxtaposed in cardiomyocytes allowing Ca2+ influx through Cav1.2 to rapidly and effectively induce Ca2+ release from the sarcoplasmic reticulum. The finding that both protein complexes individually anchor PP2A indicates that PP2A has to be precisely targeted to the individual proteins for effective dephosphorylation.
Whereas PP2A/C bound equally well to CT-8 and CT-8-P, PP2B only bound to CT-8 (14). Furthermore, Peptides 4, 4P, and 5 all interfered with PP2A binding but only Peptide 5 inhibited PP2B binding to CT-8. The most parsimonious explanation for these results is that PP2B has its main attachment site immediately downstream of Arg1971. As discussed above, residues immediately upstream of Arg1971 (1965LSPLLQ1970) are most likely the main attachment site for PP2A. Because PP2A and PP2B can simultaneously bind to the region surrounding Arg1971, these upstream residues are obviously not critical for PP2B binding. Our earlier finding that CT-8-P has impaired PP2B binding is thus likely due to downstream secondary structural changes induced by the proline substitution, an alteration that could encompass residues C-terminal to Arg1971. We conclude that although PP2A and PP2B bind to the same region of α11.2, binding is through interaction with different residues, which are in close proximity to each other.
Earlier evidence suggests that PP2A in general (12) and specifically when bound to the CT-8 region of α11.2 downstream of Ser1928 (13) reduces Cav1.2 currents in the HEK-derived tsA-201 cells by counteracting PKA-mediated stimulation of these currents (13). Further, channel-associated PP2A reverses PKA-mediated phosphorylation of Ser1928 (13, 36), which is still considered a valid read-out of Cav1.2 phosphorylation by PKA even though it is not necessary for Cav1.2 regulation (8, 17, 18). In cardiomyocytes, three different modes of Cav1.2 activity have been defined (40). Channel openings are absent in mode 0, short in mode 1, and long in mode 2 with only relatively brief intermittent closures in this mode (40). β-adrenergic signaling via PKA induces transition from mode 0 to mode 1 or 2 (10, 41). Okadaic acid, which at sub-μM concentrations is more selective for PP2A than PP1, impairs the reversal of mode 2 and 1 in cardiac and smooth muscle cells, whereas application of PP2A to excised inside-out membrane patches promotes this reversal (32, 42, 43). Collectively these findings indicate that PP2A reduces Cav1.2 activity at least in part by reversing PKA-mediated stimulation of Cav1.2. Our findings that Peptide 5 displaces PP2A from native Cav1.2 and increases basal and ISO-enhanced ICa(L) now indicate that PP2A must be bound to the region containing residues 1965-1970 for effectively regulating Cav1.2 activity.
In contrast to PP2A inhibitors, several ventricular myocyte studies report that PP2B inhibition has either no detectable effect on ICa(L) in rat, mouse, or rabbit (44-47), decreases this current in rat (14, 48), or, in one study, increases ICa(L) in mouse (49)). The basis for these discrepant findings is unclear, but raises the possibility that in ventricular myocytes PP2B does not regulate channel activity as robustly as PP2A with respect to the basal activity state of ICa(L) or PKA-dependent activation of ICa(L). Dissociation of PP2B from Cav1.2 should thus either have a limited effect on ICa(L) or even decrease it in ventriculocytes. The finding that perfusion of Peptide 5 increased ICa(L) in our rabbit vetriculocytes and because Peptide 5 dislodged only PP2A but not PP2B from Cav1.2 thus suggests that the Peptide 5 - induced increase in ICa(L) is largely due to displacement of PP2A from α11.2 residues 1965-1971.
However, in neurons and HEK293 cells ectopically expressing Cav1.2, PP2B consistently acts to decrease L-type currents (50-52). Thus our previous findings (14), which are based on multiple lines of evidence leaving little doubt that PP2B stimulates ICa(L) in cardiomyocytes, could reflect that PP2B acts under certain conditions in cardiomyocytes not directly on Cav1.2 but rather on other regulators of this channels, perhaps PP2A itself. For instance, the B-type subunit B’δ (B56δ) is phosphorylated by PKA to increase the phosphatase activity of the corresponding PP2A holoenzyme (53). PP2B could act in heart by reversing this or other analogously acting PP2A phosphorylations to decrease PP2A activity.
The A kinase anchor protein AKAP150 (corresponding to AKAP79 in humans and AKAP75 in cows) is the main AKAP at postsynaptic sites in brain (54, 55). Cav1.2 is clustered at these sites (56, 57) and AKAP150 co-immunoprecipitates with Cav1.2 from brain and heart (8, 58, 59). AKAP150 recruits PKA and PP2B to Cav1.2 in neurons (52, 58, 60, 61). It also links PP2B and the adenylyl cyclases 5 and 6, but not PKA, to Cav1.2 in heart (59), where AKAP15/18 links PKA to Cav1.2 (16, 62). Because AKAP150 binds not only to the C-terminus of α11.2 but also to its N-terminus and its intracellular loop between domains I and II (58), AKAP150 and with it PP2B can bind simultaneously with AKAP15/18 to Cav1.2. In fact AKAP150 is strictly required for co-immunoprecipitation of PP2B with Cav1.2 (59). The observation that Peptide 5 does not displace PP2B from native Cav1.2 complexes could thus be due to the apparently stronger interaction of PP2B with Cav1.2 via AKAP150 than the direct PP2B binding to α11.2. Accordingly, AKAP150 is necessary for stable PP2B association with Cav1.2 although it is not clear whether it is sufficient. The lack of effect of Peptide 5 on PP2B co-immunoprecipitation with Cav1.2 indicates at the first glance that the corresponding interaction is not required and the AKAP150-mediated interaction is sufficient but the lack of Peptide 5 effect could be explained by a lack of access to this interaction site in the native complex.
Peptide 5 augments ICa(L) not only under basal conditions but also enhances in absolute magnitude the increase in ICa(L) by β-adrenergic stimulation with ISO. Accordingly Peptide 5 could act at least in part by enhancing the PKA-mediated increase in Cav1.2 activity. Our results also indicate that PP2A effectively reverses the PKA-stimulated activity of cardiac Cav1.2 only if bound to residues 1965-1970 and surrounding residues in α11.2. An indirect implication of our results is that the phosphatase activity at Cav1.2 may be relatively high, keeping basal ICa(L) activation low, limiting the extent of PKA-dependent current enhancement and potentially accelerating the reversal of current activation at the end of a bout of sympathetic activation.
In conclusion, there are at least two different attachment sites in the Cav1.2 channel complex for PP2A as defined by our CT-B and CT-8 constructs. The latter site is clearly important for negative regulation of Cav1.2 by PP2A but the former site awaits yet more precise definition and functional studies.
Supplementary Material
Acknowledgments
The authors thank Dr. S. S. Taylor (HHMI, UC San Diego) for providing the solid phase peptide library and Dr. G. Walter (UC San Diego) for an aliquot of the 6F9 antibody against PP2A/A.
Footnotes
This work was supported by grant No [2007] 3020 from the China Scholarship Council (to H.X.), the National Institute of Health research grants R01-NS035563 and R01-AG017502 (to J.W.H.), R01-GM056900 (to M.C.H.), R01-HL075173 (to J.A.H.), R37-HL30077 (to D.M.B.), and the American Heart Association grants 0535235N (to D.D.H.) and (0640084N) (to J.A.H.).
Abbreviations used are: GST, glutathione-S transferase; ISO, isoproterenol; PAGE, polyacrylamide gel electrophoresis; PKA, cAMP-dependent protein kinase; PP, protein phosphatase; PVDF, polyvinyldifluoride; SDS, sodium dodecylsulphate.
SUPPORTING INFORMATION
Supplemental Figure 1 shows that peptide 6-10, which are 10 residues long and cover Peptide 4 and 5, do not inhibit PP2A and PP2B binding to GST-CT-8.
This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Marrion NV, Tavalin ST. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- 2.Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- 3.Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:148–152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- 4.Christie BR, Schexnayder LK, Johnston D. Contribution of voltage-gated Ca2+ channels to homosynaptic long-term depression in the CA1 region in vitro. J Neurophysiol. 1997;77:1651–1655. doi: 10.1152/jn.1997.77.3.1651. [DOI] [PubMed] [Google Scholar]
- 5.Wang HX, Gerkin RC, Nauen DW, Bi GQ. Coactivation and timing-dependent integration of synaptic potentiation and depression. Nat Neurosci. 2005;8:187–193. doi: 10.1038/nn1387. [DOI] [PubMed] [Google Scholar]
- 6.Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 7.Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 8.Dai S, Hall DD, Hell JW. Supramolecular Assemblies and Localized Regulation of Voltage-gated Ion Channels. Physiol Rev. 2009;89:411–452. doi: 10.1152/physrev.00029.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reuter H. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 1983;301:569–574. doi: 10.1038/301569a0. [DOI] [PubMed] [Google Scholar]
- 10.Bean BP, Nowycky MC, Tsien RW. β-Adrenergic modulation of calcium channels in frog ventricular heart cells. Nature. 1984;307:371–375. doi: 10.1038/307371a0. [DOI] [PubMed] [Google Scholar]
- 11.Ono K, Fozzard HA. Phosphorylation restores activity of L-type calcium channels after rundown in inside-out patches from rabbit cardiac cells. J Physiol. 1992;454:673–688. doi: 10.1113/jphysiol.1992.sp019286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sculptoreanu A, Rotman E, Takahashi M, Scheuer T, Catterall WA. Voltage-dependent potentiation of the activity of cardiac L-type calcium channel alpha 1 subunits due to phosphorylation by cAMP-dependent protein kinase. Proc Natl Acad Sci. 1993;90:10135–10139. doi: 10.1073/pnas.90.21.10135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall DD, Feekes JA, Arachchige Don AS, Shi M, Hamid J, Chen L, Strack S, Zamponi GW, Horne MC, Hell JW. Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochem. 2006;45:3448–3459. doi: 10.1021/bi051593z. [DOI] [PubMed] [Google Scholar]
- 14.Tandan S, Wang Y, Wang TT, Jiang N, Hall DD, Hell JW, Luo X, Rothermel BA, Hill JA. Physical and functional interaction between calcineurin and the cardiac L-type Ca2+ channel. Circ Res. 2009;105:51–60. doi: 10.1161/CIRCRESAHA.109.199828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hell JW. {beta}-Adrenergic Regulation of the L-Type Ca2+ Channel CaV1.2 by PKA Rekindles Excitement. Sci Signal. 2010;3:pe33. doi: 10.1126/scisignal.3141pe33. [DOI] [PubMed] [Google Scholar]
- 16.Fuller MD, Emrick MA, Sadilek M, Scheuer T, Catterall WA. Molecular Mechanism of Calcium Channel Regulation in the Fight-or-Flight Response. Sci Signal. 2010;3:ra70. doi: 10.1126/scisignal.2001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganesan AN, Maack C, Johns DC, Sidor A, O’Rourke B. Beta-adrenergic stimulation of L-type Ca2+ channels in cardiac myocytes requires the distal carboxyl terminus of alpha1C but not serine 1928. Circ Res. 2006;98:e11–18. doi: 10.1161/01.RES.0000202692.23001.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lemke T, Welling A, Christel CJ, Blaich A, Bernhard D, Lenhardt P, Hofmann F, Moosmang S. Unchanged beta-adrenergic stimulation of cardiac L-type calcium channels in Ca v 1.2 phosphorylation site S1928A mutant mice. J Biol Chem. 2008;283:34738–34744. doi: 10.1074/jbc.M804981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang CS, Vitto MJ, Busby SA, Garcia BA, Kesler CT, Gioeli D, Shabanowitz J, Hunt DF, Rundell K, Brautigan DL, Paschal BM. Simian virus 40 small t antigen mediates conformation-dependent transfer of protein phosphatase 2A onto the androgen receptor. Mol Cell Biol. 2005;25:1298–1308. doi: 10.1128/MCB.25.4.1298-1308.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kremmer E, Ohst K, Kiefer J, Brewis N, Walter G. Separation of PP2A core enzyme and holoenzyme with monoclonal antibodies against the regulatory A subunit: abundant expression of both forms in cells. Mol Cell Biol. 1997;17:1692–1701. doi: 10.1128/mcb.17.3.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang H, Venema VJ, Wang X, Ju H, Venema RC, Marrero MB. Regulation of angiotensin II-induced phosphorylation of STAT3 in vascular smooth muscle cells. J Biol Chem. 1999;274:19846–19851. doi: 10.1074/jbc.274.28.19846. [DOI] [PubMed] [Google Scholar]
- 22.Jicha GA, Weaver C, Lane E, Vianna C, Kress Y, Rockwood J, Davies P. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer’s disease. J Neurosci. 1999;19:7486–7494. doi: 10.1523/JNEUROSCI.19-17-07486.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davare MA, Dong F, Rubin CS, Hell JW. The A-kinase anchor protein MAP2B and cAMP-dependent protein kinase are associated with class C L-type calcium channels in neurons. J Biol Chem. 1999;274:30280–30287. doi: 10.1074/jbc.274.42.30280. [DOI] [PubMed] [Google Scholar]
- 24.Burns-Hamuro LL, Ma Y, Kammerer S, Reineke U, Self C, Cook C, Olson GL, Cantor CR, Braun A, Taylor SS. Designing isoform-specific peptide disruptors of protein kinase A localization. Proc Natl Acad Sci. 2003;100:4072–4077. doi: 10.1073/pnas.2628038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snutch TP, Tomlinson WJ, Leonard JP, Gilbert MM. Distinct calcium channels are generated by alternative splicing and are differentially expressed in the mammalian CNS. Neuron. 1991;7:45–57. doi: 10.1016/0896-6273(91)90073-9. [DOI] [PubMed] [Google Scholar]
- 26.Bennin DA, Don AS, Brake T, McKenzie JL, Rosenbaum H, Ortiz L, DePaoli-Roach AA, Horne MC. Cyclin G2 associates with protein phosphatase 2A catalytic and regulatory B’ subunits in active complexes and induces nuclear aberrations and a G1/S phase cell cycle arrest. J Biol Chem. 2002;277:27449–27467. doi: 10.1074/jbc.M111693200. [DOI] [PubMed] [Google Scholar]
- 27.Mondragon A, Griffith EC, Sun L, Xiong F, Armstrong C, Liu JO. Overexpression and purification of human calcineurin alpha from Escherichia coli and assessment of catalytic functions of residues surrounding the binuclear metal center. Biochem. 1997;36:4934–4942. doi: 10.1021/bi9631935. [DOI] [PubMed] [Google Scholar]
- 28.Davare MA, Hell JW. Increased phosphorylation of the neuronal L-type Ca(2+) channel Ca(v)1.2 during aging. Proc Natl Acad Sci. 2003;100:16018–16023. doi: 10.1073/pnas.2236970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geladopoulos TP, Sotiroudis TG, Evangelopoulos AE. A malachite green colorimetric assay for protein phosphatase activity. Anal Biochem. 1991;192:112–116. doi: 10.1016/0003-2697(91)90194-x. [DOI] [PubMed] [Google Scholar]
- 30.Harder KW, Owen P, Wong LK, Aebersold R, Clark-Lewis I, Jirik FR. Characterization and kinetic analysis of the intracellular domain of human protein tyrosine phosphatase beta (HPTP beta) using synthetic phosphopeptides. Biochem J. 1994;298:395–401. doi: 10.1042/bj2980395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fathi AR, Krautheim A, Lucke S, Becker K, Juergen Steinfelder H. Nonradioactive technique to measure protein phosphatase 2A-like activity and its inhibition by drugs in cell extracts. Anal Biochem. 2002;310:208–214. doi: 10.1016/s0003-2697(02)00377-9. [DOI] [PubMed] [Google Scholar]
- 32.Wiechen K, Yue DT, Herzig S. Two distinct functional effects of protein phosphatase inhibitors on guinea-pig cardiac L-type Ca2+ channels. J Physiol (Lond) 1995;484:583–592. doi: 10.1113/jphysiol.1995.sp020688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res. 2006;98:403–411. doi: 10.1161/01.RES.0000202707.79018.0a. [DOI] [PubMed] [Google Scholar]
- 34.Fan JS, Palade P. Perforated patch recording with beta-escin. Pflugers Arch. 1998;436:1021–1023. doi: 10.1007/pl00008086. [DOI] [PubMed] [Google Scholar]
- 35.Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Davare MA, Horne MC, Hell JW. Protein Phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J Biol Chem. 2000;275:39710–39717. doi: 10.1074/jbc.M005462200. [DOI] [PubMed] [Google Scholar]
- 37.McCright B, Rivers AM, Audlin S, Virshup DM. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem. 1996;271:22081–22089. doi: 10.1074/jbc.271.36.22081. [DOI] [PubMed] [Google Scholar]
- 38.Price NE, Mumby MC. Brain protein serine/threonine phosphatases. Curr Opin Neurobiol. 1999;9:336–342. doi: 10.1016/s0959-4388(99)80049-x. [DOI] [PubMed] [Google Scholar]
- 39.Marx SO, Reiken S, Hisamatsu Y, Gaburjakova M, Gaburjakova J, Yang YM, Rosemblit N, Marks AR. Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J Cell Biol. 2001;153:699–708. doi: 10.1083/jcb.153.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hess P, Lansman JB, Tsien RW. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature. 1984;311:538–544. doi: 10.1038/311538a0. [DOI] [PubMed] [Google Scholar]
- 41.Trautwein W, Hescheler J. Regulation of cardiac L-type calcium current by phosphorylation and G proteins. Annu Rev Physiol. 1990;52:257–274. doi: 10.1146/annurev.ph.52.030190.001353. [DOI] [PubMed] [Google Scholar]
- 42.Ono K, Fozzard HA. Two phosphatase sites on the Ca2+ channel affecting different kinetic functions. J Physiol. 1993;470:73–84. doi: 10.1113/jphysiol.1993.sp019848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Groschner K, Schuhmann K, Mieskes G, Baumgartner W, Romanin C. A type 2A phosphatase-sensitive phosphorylation site controls modal gating of L-type Ca2+ channels in human vascular smooth-muscle cells. Biochem J. 1996;318:513–517. doi: 10.1042/bj3180513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.duBell WH, Wright PA, Lederer WJ, Rogers TB. Effect of the immunosupressant FK506 on excitation-contraction coupling and outward K+ currents in rat ventricular myocytes. J Physiol. 1997;501:509–516. doi: 10.1111/j.1469-7793.1997.509bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCall E, Li L, Satoh H, Shannon TR, Blatter LA, Bers DM. Effects of FK-506 on contraction and Ca2+ transients in rat cardiac myocytes. Circ Res. 1996;79:1110–1121. doi: 10.1161/01.res.79.6.1110. [DOI] [PubMed] [Google Scholar]
- 46.Yatani A, Honda R, Tymitz KM, Lalli MJ, Molkentin JD. Enhanced Ca2+ channel currents in cardiac hypertrophy induced by activation of calcineurin-dependent pathway. J Mol Cell Cardiol. 2001;33:249–259. doi: 10.1006/jmcc.2000.1296. [DOI] [PubMed] [Google Scholar]
- 47.Su Z, Sugishita K, Li F, Ritter M, Barry WH. Effects of FK506 on [Ca2+]i differ in mouse and rabbit ventricular myocytes. J Pharmacol Exp Therap. 2003;304:334–341. doi: 10.1124/jpet.102.041210. [DOI] [PubMed] [Google Scholar]
- 48.Fauconnier J, Lacampagne A, Rauzier JM, Fontanaud P, Frapier JM, Sejersted OM, Vassort G, Richard S. Frequency-dependent and proarrhythmogenic effects of FK-506 in rat ventricular cells. Am J Physiol Heart Circ Physiol. 2005;288:H778–786. doi: 10.1152/ajpheart.00542.2004. [DOI] [PubMed] [Google Scholar]
- 49.Santana LF, Chase EG, Votaw VS, Nelson MT, Greven R. Functional coupling of calcineurin and protein kinase A in mouse ventricular myocytes. J Physiol. 2002;544:57–69. doi: 10.1113/jphysiol.2002.020552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burley JR, Sihra TS. A modulatory role for protein phosphatase 2B (calcineurin) in the regulation of Ca2+ entry. Eur J Neurosci. 2000;12:2881–2891. doi: 10.1046/j.1460-9568.2000.00178.x. [DOI] [PubMed] [Google Scholar]
- 51.Lukyanetz EA, Piper TP, Sihra TS. Calcineurin involvement in the regulation of high-threshold Ca2+ channels in NG108-15 (rodent neuroblastoma x glioma hybrid) cells. J Physiol. 1998;510:371–385. doi: 10.1111/j.1469-7793.1998.371bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oliveria SF, Dell’acqua ML, Sather WA. AKAP79/150 Anchoring of Calcineurin Controls Neuronal L-Type Ca(2+) Channel Activity and Nuclear Signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahn JH, McAvoy T, Rakhilin SV, Nishi A, Greengard P, Nairn AC. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56delta subunit. Proc Natl Acad Sci. 2007;104:2979–2984. doi: 10.1073/pnas.0611532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 2007;26:4879–4890. doi: 10.1038/sj.emboj.7601884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weisenhaus M, Allen ML, Yang L, Lu Y, Nichols CB, Su T, Hell JW, McKnight GS. Mutations in AKAP5 disrupt dendritic signaling complexes and lead to electrophysiological and behavioral phenotypes in mice. PLoS One. 2010;5:e10325. doi: 10.1371/journal.pone.0010325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- 57.Obermair GJ, Szabo Z, Bourinet E, Flucher BE. Differential targeting of the L-type Ca2+ channel alpha1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur J Neurosci. 2004;19:2109–2122. doi: 10.1111/j.0953-816X.2004.03272.x. [DOI] [PubMed] [Google Scholar]
- 58.Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochem. 2007;46:1635–1646. doi: 10.1021/bi062217x. [DOI] [PubMed] [Google Scholar]
- 59.Nichols CB, Rossow CF, Navedo MF, Westenbroek RE, Catterall WA, Santana LF, McKnight GS. Sympathetic Stimulation of Adult Cardiomyocytes Requires Association of AKAP5 With a Subpopulation of L-Type Calcium Channels. Circ Res. 2010;107:747–756. doi: 10.1161/CIRCRESAHA.109.216127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, Scott JD. Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science. 1995:108–111. doi: 10.1126/science.7528941. [DOI] [PubMed] [Google Scholar]
- 61.Oliveria SF, Gomez LL, Dell’Acqua ML. Imaging kinase--AKAP79--phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol. 2003;160:101–112. doi: 10.1083/jcb.200209127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA. Beta-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci. 2003;100:13093–13098. doi: 10.1073/pnas.2135335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.