

Abstract
Selective alkylation of a chosen sequence of DNA typically relies on ligand-directed delivery of a compound that expresses an intrinsic reactivity. A significant and biologically relevant enhancement in specificity is theoretically possible if such an intrinsic reactivity could be replaced by a latent activity induced solely by the target of interest, but examples of this are rare and not easily emulated. A simple strategy for target-promoted alkylation is now illustrated by an intramolecular adduct formed by an oligonucleotide–quinone methide conjugate. This adduct persists in the absence of a complementary sequence of DNA for at least 8 days, yet remarkably is able to alkylate target DNA upon duplex hybridization. Neither formation of the intramolecular self-adduct nor transfer of the quinone methide to its target is significantly quenched by 450-fold excess 2-mercaptoethanol. Similarly, noncomplementary DNA is neither subject to alkylation by the self-adduct nor able to effect its consumption. Reversible trapping of the nascent quinone methide through an intramolecular reaction thus appears efficient enough to inhibit competing intermolecular reaction. Only complementary base pairing induces a conformational change necessary to promote intermolecular transfer of the quinone methide. Generalization of this approach based on reversible intramolecular trapping of a reactive intermediate by a ligand with multiple recognition subdomains has the potential for wide-ranging applications in targeting nucleic acids and proteins.
Selective covalent modification of biological macromolecules has been a topic of longstanding interest due in part to the breadth and diversity of its applications to basic, applied, and medical research. The intrinsic chemical reactivity of many reagents may be used to distinguish between general types of solvent accessible amino acid chains or nucleobases, but rarely can they identify individual sites of interest. For this additional selectivity, intrinsic chemistry must be combined with a site-directing ligand to form an affinity reagent (1, 2). This latter approach has been applied with great success when studying proteins and nucleic acids in vitro. Applications to more complex systems are often stymied by unavoidable side reactions caused by an abundance of nonspecific competitors that overwhelms the potential kinetic advantage of affinity-based localization. A still higher level of specificity can be obtained from mechanism-based reagents for targets with catalytic activity (3). This strategy relies on the binding and catalytic properties of a target to localize and activate a reagent's inducible reactivity. Paradoxically, the ultimate specificity may be attained by avoiding covalent reactivity altogether and instead designing a species with a slow rate of dissociation from its target that approaches the physiological equivalent of covalent modification. This objective is now frequently achieved with transition-state analogues and often serves as an initial step in drug development (4).
Reliance on covalent reactivity remains widespread out of necessity when targets, in particular DNA, lack a predictable catalytic activity. Such suboptimal approaches are perhaps most pertinent to treatment of life-threatening diseases such as cancer for which few alternatives are available. Highly toxic and nonspecific alkylating agents of DNA such as the N-mustards chlorambucil and cyclophosphamide are still frequently prescribed against various forms of cancer (5). Only few covalently reactive drugs such as bleomycin and mitomycin gain an additional degree of specificity by their need for cellular induction and their ability to form preassociation complexes with duplex DNA (6, 7). Still, sequence specificity remains minimal.
Unlike enzymes, DNA-dependent activation of a bound substrate or ligand is extremely rare and consequently highly prized (8). The most common manner in which DNA can induce or accelerate reactions is through its ability to act as a template and localize reactants. For example, a single strand of DNA can selectively bind and orient two smaller complementary strands to direct their respective reactive appendages (9–12). Additionally, single- and double-stranded DNA as well as RNA are capable of catalyzing polymerization or ligation of complementary sequences through duplex and triplex recognition and assembly (13–17).
Rate acceleration afforded by a limited number of examples seems to exceed that easily explained by the template effects of DNA alone. In these cases, the conformation and surface properties of nucleic acids likely contribute to the observed rate enhancements of reaction (18–25). Probably the best studied example of this type of DNA-promoted process involves nucleobase reaction with cyclopropylpyrroloindole derivatives such as those found in natural products CC-1065 and the duocarmycins as well as a related synthetic analog bizelsin, currently undergoing clinical investigation (26–29). Unfortunately, the prospects of translating this process more generally to other molecular structures is not obvious. In contrast, the approach described in this article has the potential for broad application because it is based on (i) reversible formation of an intramolecular adduct between an electrophilic appendage and its attached target-directing ligand and (ii) intermolecular transfer of the appendage to the target in a manner dependent on its recognition (Fig. 1).
Fig. 1.

A conformational switch induced by target recognition may promote selective delivery of a reactive intermediate (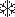 ).
).
Materials and Methods
All organic solvents and reagents obtained commercially were used without purification unless noted otherwise. Oligonucleotides were purified by polyacrylamide gel electrophoresis and 32P-labeled at their 5′ terminus using standard protocols. Oligonucleotide conjugates, self-adducts, and interstrand alkylation products were also separated by reverse-phase (C-18, Varian Microsorb-MV, 300 Å pore, 250 mm) HPLC using a gradient (30 min, 1 ml/min) of 10–55% acetonitrile in aqueous triethylammonium acetate (50 mM, pH 5.0) as controlled by a Jasco PU-980 system (Jasco, Easton, MD). Samples (20–150 pmol DNA) were manually collected during elution from the HPLC and analyzed by electrospray ionization mass spectroscopy (ESI-MS, Thermo Finnigan LCQ) (30) using direct injection with a voltage of –3.6 kV and a capillary temperature of 170°C.
Oligonucleotide Conjugates. Oligonucleotide conjugates were formed by combining the appropriate activated succimidyl ester (1 mg, preparation described in the supporting information, which is published on the PNAS web site) in CH3CN/DMF (2:1, 400 μl) with the appropriate 5′-aminohexyloligonucleotide (20 nmol) in Mops buffer (250 mM, pH 7.5, 400 μl) and incubating the mixture for 24 h at ambient temperature. The desired conjugates were purified by HPLC, dialyzed against water and lyophilized. ESI-MS (m/z) calculated for the conjugate of OD1-NH2 and its quinone methide precursor (Fig. 2, OD1-QMP) (M-3H+)3– was 1,701, and ESI-MS (m/z) found was 1,701. ESI-MS (m/z) calculated for (M-4H+)4– was 1,276, and ESI-MS (m/z) found was 1,276. ESI-MS (m/z) calculated for the quinone methide precursor conjugate of T10-NH2 (T10-QMP) (M-2H +)2– 1,746, and ESI-MS (m/z) found was 1,746. ESI-MS (m/z) calculated for the conjugate of diphenylacetate and OD2-NH2 (Fig. 2, diphenyl-OD2) (M-4H+)4– 1,618, and ESI-MS (m/z) found was 1,618.
Fig. 2.

Fluoride-dependent generation of a quinone methide (QM), its subsequent alkylation by a nucleophile (nuc), and the oligonucleotides used to investigate target promoted alkylation.
Self-Adduct of the Oligonucleotide Conjugate (OD1-QMP). Reaction of OD1-QMP (2.2 μM) was initiated by desilylation with KF (1.0 M) in Mes (20 mM, pH 7.0, 1.0 ml) and maintained at 25°C. Formation of the self-adduct was monitored by reverse-phase (C-18) HPLC analysis using m-cresol (1.0 μM) as an internal standard. Conversion of OD1-QMP to its self-adduct was complete within 24 h, purified by HPLC, and confirmed by ESI-MS (see Results and Discussion).
Nucleophilic Quenching of the Quinone Methide Generated by T10-QMP. Reaction of T10-QMP (2.5 μM) was initiated by desilylation with KF (1.0 M) in Mes (20 mM, pH 7.0) and maintained at 25°C in the alternative presence and absence of 2-mercaptoethanol (1.1 mM). Reaction progress was monitored by reverse-phase (C-18) HPLC analysis using m-cresol (1.0 μM) as an internal standard. Product formation was complete within 24 h. In the absence of the thiol, only the water adduct of the quinone methide intermediate was observed and confirmation by ESI-MS [m/z calculated for (M-2H+)2–, 1,668; found, 1,668. m/z calculated for (M-3H+)3–, 1,111; found, 1,112). In the presence of the thiol, both the thiol adduct (82%) and water (18%) adduct of the quinone methide intermediate were observed [ESI-MS for the thiol adduct (m/z): calculated for (M-2H+)2–, 1,698; found, 1,698; calculated for (M-3H+)3– 1,131; found, 1,132.). Yields were based on the integrated absorption at 260 nm measured during chromatographic analysis using the conditions described above.
Alkylation of Oligonucleotide Targets by the OD1-QM Self-Adduct. The self-adduct was generated in situ by incubating OD1-QMP (5.0 μM) with KF (1.0 M) in Mes (20 mM, pH 7.0, 1.0 ml) for 8 days at 25°C unless indicated otherwise. The desired conversion was confirmed by HPLC analysis. Target alkylation was then initiated by adding diphenyl-OD2 (2.5 μM) to the self-adduct (2.5 μM) in 20 mM Mes pH 7 (25°C), and the reaction progress was monitored by HPLC analysis. Alternatively, for electrophoretic analysis, the OD1-QM self-adduct (2.2 μM) was generated equivalently. Reaction was initiated by addition of the self-adduct (1.1 μM) to 5′-[32P]OD2 (90 nCi, 1.0 μM; 1 Ci = 37 GBq) and maintained at ambient temperature for 8 days. Products were separated by denaturing gel electrophoresis (20% acrylamide/7 M urea) and quantified by phosphoimagery using imagequant software.
Results and Discussion
Formation of an Oligonucleotide–Quinone Methide Self-Adduct. A recent appreciation for the reversible nature of quinone methide alkylation (31, 32) suggested that its transient adducts with DNA might function as effective delivery agents for this highly reactive intermediate just as a malondialdehyde adduct of deoxyguanosine (dG) efficiently transports malondialdehyde in vivo (33, 34). Our laboratory had traditionally relied on a fluoride dependent deprotection of O-silylated precursors (QMP) and their site-directed conjugates for inducible alkylation by an ortho-benzoquinone methide intermediate (QM) (Fig. 2) (32, 35–39). However, a self-adduct formed within the oligonucleotide conjugate by its QM has now demonstrated a capacity for target-promoted QM transfer as described below.
The possibility of target promoted reaction based on QM chemistry originated from two essential observations: (i) formation of an oligonucleotide-QM self-adduct; and (ii) alkylation of a complementary strand of DNA by this self-adduct. A combination of HPLC and ESI-MS analysis provided the first evidence that OD1-QMP formed a self-adduct efficiently. Treatment of OD1-QMP with KF rapidly removed the t-butyldimethylsilyl (TBDMS) group and generated the unprotected “acetate derivative” as evident from its m/z of 1,663 ([M-3H+]3– calculated, 1,663) (Fig. 3A). This derivative in turn spontaneously, but more slowly, eliminated acetate to form a new product with a m/z of 1,643 that is consistent with a QM self-adduct ([M-3H+]3– calculated, 1,643). This product was formed in >90% yield from the parent OD1-QMP assuming no significant change in extinction coefficients.
Fig. 3.

(A) C-18 HPLC chromatogram and ESI-MS analysis of fluoride-dependent loss of the t-butyldimethylsilyl group (TBDMS) from OD1-QMP and subsequent formation of its self-adduct. (B) Alkylation of diphenyl-OD2 by the self-adduct of OD1-QM formed from prior incubation with fluoride (40 h).
Addition of Water to the Quinone Methide Is Not Competitive with Intramolecular Reaction. The absence of any detectable water adduct formed in competition with the self-adduct was initially surprising. Despite the slow rate of water addition to a simple quinone methide, (31) its concentration is typically orders of magnitude greater than other potential nucleophiles. Additionally, reaction with water is irreversible and might have been expected to accumulate slowly over time as the OD1-QMP self-adduct continued to generate its QM intermediate reversibly (32). Still, no m/z of 1,649 ([M-3H+]3– calculated for the water adduct) was observed at any point through the HPLC chromatogram of a sample incubated for 24 h (Fig. 3). Further incubation for an additional 7 days failed to generate the water adduct, and instead, the self-adduct remained evident by chromatography. Finally, dehydration of the potential water adduct was not expected during ESI-MS analysis but, if it had, the same mass as the self-adduct would have been observed. Consequently, a positive control for the formation and detection of a water adduct was necessary.
The only nucleotide that does not readily form an adduct with QM is thymidine (32, 37), and therefore a QMP conjugate containing only thymidine (T10-QMP) was synthesized. Incubation of this conjugate with KF for 24 h as described above yielded a single product in 98% as detected by HPLC. ESI-MS of this product was consistent with the water adduct ([M-2H+]2– of 1,668, calculated 1,668, and [M-3H+]3– of 1,112, calculated 1,111) and no self-adduct was evident. Thus, the lack of a water adduct for OD1-QMP reflects its lack of formation rather than its inability to be detected.
Target DNA Is Alkylated by a Complementary Oligonucleotide–Quinone Methide Self-Adduct. HPLC and ESI-MS also contributed to the second critical observation, alkylation of a DNA strand by its complementary OD1-QM self-adduct. Because the target oligonucleotide, OD2, coeluted with the self-adduct when using reverse-phase (C-18) chromatography, the 5′ terminus of the target was extended with a diphenylacetylated aminolinker (diphenyl-OD2) (Fig. 2). This modification is distal to the possible sites of reaction and allowed for resolution of all reactants by chromatography. Within 1 day, a single new species was evident from a stoichiometric mixture of the self-adduct and diphenyl-OD2 (Fig. 3B). This product alone continued to accumulate while the self-adduct and diphenyl-OD2 decreased over 4 days. The m/z signals detected for the product (1,628 and 1,900) are consistent with those calculated for an alkylated duplex formed by intermolecular QM transfer ([M-7H+]7–, calculated, 1,629; [M-6H+]6–, calculated, 1,900) (see supporting information). Again, there was no indication that water was able to quench the OD1-QM self-adduct.
Stability of the Oligonucleotide–Quinone Methide Self-Adduct in Aqueous Solution. Persistence of the OD1-QM self-adduct in the absence of external nucleophiles was next monitored by its enduring ability to alkylate 5′-[32P]OD2. This alkylation proceeded in a maximum yield of almost 45% when both OD2 and OD1-QMP were present during induction by fluoride (Fig. 4). Complementary analysis by HPLC revealed consistent results by indicating that OD1-QMP partitioned almost equally between self-adduct formation and target alkylation when annealed to diphenyl-OD2 before fluoride addition. As illustrated by Fig. 3, the self-adduct was the only product generated in the absence of diphenyl-OD2, and this self-adduct retained its ability to alkylate DNA upon subsequent addition of diphenyl-OD2. The capacity for target alkylation slowly decreased following a first order decay, but still >20% of OD2 could be alkylated after the self-adduct had previously aged under neutral and ambient conditions for 8 days. Thus, only ≈50% of the self-adduct's ability to alkylate its target was lost over 8 days. In contrast, consumption of the self-adduct was much more rapid in the presence of its target OD2.
Fig. 4.

Stability and persistence of the OD1-QMP self-adduct as suggested by its extended ability to alkylate OD2. Desilylation of OD1-QMP was initiated by adding KF (1.0 M) in Mes (pH 7.0, 20 mM) under ambient conditions. At the indicated time, 5′-[32P]OD2 (1.1 μM, 1.1 equivalents) was added to the solution and maintained under ambient conditions for 8 days. The yield of alkylation (%) was calculated relative to the total radiolabel after electrophoretic separation. Two independent determinations were performed and fit (solid line) to a first-order decay.
A Proposed Mechanism for Target Promoted Alkylation. The apparent stability of the self-adduct in the absence of a complementary strand is not incompatible with its reactivity in the presence of such a strand. The self-adduct likely remains in equilibrium with its high energy QM intermediate because the most reactive nucleophiles of DNA add reversibly to the QM (Fig. 5). For example, the N1 of dA is both a strong nucleophile and an efficient leaving group (32). Intramolecular addition to reform the self-adduct remains favorable even in the presence of water, and intermolecular reaction only becomes competitive after hybridization to a complementary strand.
Fig. 5.

Target recognition may be used to suppress intramolecular reaction that limits base pairing and promote selective intra- and intermolecular reaction that supports full base pairing. (a, intramolecular trapping; b, intermolecular trapping.)
Target recognition and duplex formation is likely retained by the nucleotides that are not significantly constrained by the intramolecular QM linkage (Fig. 5). Most importantly, barriers to forming the remaining base pairs are removed once the self-adduct equilibrates back to its QM intermediate. The energy gained from the additional base pairing then creates a thermodynamic barrier preventing a reformation of any intrastrand adduct that might again limit base pairing (Fig. 5). For example, adducts of dA N1 and dC N3 form readily in the absence of duplex DNA but are mutually exclusive of Watson–Crick base pairing (32). The nascent quinone methide is consequently left to partition between intra- and interstrand reactions that do not corrupt the integrity of duplex DNA. Hence, base pairing energy may be used to drive partitioning of QM alkylation from an intramolecular to intermolecular process.
The exact nucleobase(s) involved in formation of the self-adducts have not yet been identified. Preliminary results suggest that the self-adduct of OD1-QM may be a collection of species differing in their nucleobase attachment. For example, alternative substitution of all dC or dA residues with dT in OD1-QMP still supports formation of self-adduct formation and alkylation of the appropriate complementary sequences (unpublished observations). The products of interstrand alkylation by the self-adduct are also likely to be heterogeneous. Hydroxyl radical footprinting of products formed by interstrand reaction in the original QMP system suggested alkylation occurred in both the single- and double-strand stranded regions proximal to the QM conjugate (40), and a similar profile is expected for the products of OD2 alkylation.
Noncomplementary DNA Does Not Compete for Alkylation by the Oligonucleotide–Quinone Methide Self-Adduct. The mechanism of target promoted alkylation proposed in Fig. 5 suggests that alkylation is limited to complementary regions of DNA that can divert reclosure of the self-adduct. This finding was confirmed by adding a noncomplementary strand (OD3, see Fig. 2) in 10-fold excess (10 μM) over the self-adduct of OD1-QM and its complement OD2. Electrophoretic analysis of the alkylation products indicates that OD3 had no appreciable effect on the yield of OD2 alkylation (Fig. 6, lane 2 vs. lane 3). A similar lack of competition was observed in the presence of native or denatured calf thymus DNA (Fig. 6, lanes 4 and 5). Additionally, no alkylation of 5′-[32P]OD3 was detected by the self-adduct of OD1-QM in the absence OD2 (see supporting information). The self-adduct thus acts to transfer its QM to a complementary sequence selectively. Neither water nor nucleophilic sites of noncomplementary DNA effectively quenched the transient QM formed by the self-adduct.
Fig. 6.

Selective alkylation of OD2 by the self-adduct of OD1-QM persisting in solution for 8 days. 5′-[32P]OD2 (1.0 μM, lane 1) was incubated with the self-adduct of OD1-QM (1.1 μM) for 8 days in the absence of competitor DNA (lane 2) and alternative presence of excess noncomplementary strand OD3 (10 μM, lane 3), native calf thymus DNA (10 μM, lane 4), and heat-denatured calf thymus DNA (10 μM, lane 5).
Even Excess 2-Mercaptoethanol Does Not Compete for Alkylation by the Oligonucleotide–Quinone Methide Self-Adduct. The inability of solvent to trap the QM intermediate during equilibration with its intramolecular self-adduct may be rationalized by the relatively low nucleophilicity of water. Similarly, the inability of noncomplementary DNA to trap the QM may be rationalized by electrostatic repulsion of the two nucleotide strands. A lack of quenching by 450 equivalents of 2-mercaptoethanol is considerably more remarkable. Alkylation of one equivalent of 5′-[32P]OD2 (1 μM) by 1.1 equivalents of the OD1-QM self-adduct was not significantly effected by simultaneous addition of 2-mercaptoethanol (0, 45, and 450 μM) (Fig. 7, lanes 2, 3, and 4, respectively).
Fig. 7.

Alkylation of OD2 by the self-adduct of OD1-QM in the presence of a strong nucleophile. 5′-[32P]OD2 (1.0 μM, lane 1) was incubated with the self-adduct of OD1-QM (1.1 μM) for 8 days in the absence of 2-mercaptoethanol (lane 2) and presence of excess 2-mercaptoethanol (45 μM, lane 3; 450 μM, lane 4).
Thiols represent the strongest and most abundant nucleophiles in cells, and yet even this model thiol was not competitive with intrastrand and intraduplex reaction of the nascent QM. Formation and preincubation of the self-adduct with 450 equivalents of the thiol for 8 days before adding 5′-[32P]OD2 reduced the yield of target alkylation only from 21% to 16% (Fig. 8 lane 2 vs. lane 3). Little of this decrease can be ascribed to inhibiting the initial formation of the OD1-self-adduct because a similar decrease in target alkylation was detected after generating the self-adduct in the absence of 2-mercaptoethanol (2 day) before its preincubation in the presence of thiol (6 days) (Fig. 8, lane 4). These results are made all of the more astonishing after considering that the rate constant for addition of 2-mercaptoethanol to the simple ortho QM under neutral conditions is >4 orders of magnitude greater than that of water (31) and the closest nucleophile within the oligonucleotide conjugate (dA N1) is 26 atoms from the reactive exo-methylene position of the attached QM.
Fig. 8.

OD1-QM self-adduct formation and persistence in the presence of a thiol nucleophile as detected by target alkylation. 5′-[32P]OD2 (1.0 μM, lane 1) was added to initiate its alkylation (20 mM Mes, pH 7, ambient temperature, 8 days) with the self-adduct of OD1-QM (1.1 μM) generated under various conditions. The self-adduct was formed by preincubating OD1-QMP with 1.0 M KF in 20 mM Mes, pH 7, for 8 days under ambient conditions in the absence (lane 2) and presence of 450 equivalents of 2-mercaptoethanol (lane 3). The self-adduct was also generated equivalently in the absence of 2-mercaptoethanol (2 days) and then incubated in its presence (450 equivalents) for an additional 6 days (lane 4).
Consistent with the electrophoretic analysis above, no adducts containing 2-mercaptoethanol were detected by HPLC analysis after incubation of this thiol, diphenyl-OD2, and the self-adduct of OD1-QM. To demonstrate the chemical competence of thiol addition to a nascent oligonucleotide QM conjugate, the non-nucleophilic thymidine system (T10-QMP) was again examined because it does not support formation of an intrastrand self-adduct. Treatment of this QMP with fluoride and 450 equivalents of 2-mercaptoethanol under aqueous conditions did indeed yield the thiol adduct (82%) and the water adduct (18%) as separated by HPLC and detected by A260 and ESI-MS (see supporting information).
Intramolecular trapping of the QM intermediate is consequently more efficient for an attached oligonucleotide of mixed sequence (OD1) than intermolecular trapping by a strong thiol nucleophile. This result is best understood as a kinetic phenomenon, because once the thiol adduct forms, it should remain stable, as suggested by the constant ratio of water vs. thiol addition during consumption of T10-QMP (24 h). In this example, the QM was too unstable to allow thiol expulsion, although a more stable quinone methide derived from anthraquinone had previously been shown to support reversible thiol addition (41). These complementary observations illustrate how structure and electronics may be used in the design of future QMP derivatives to control their kinetic and functional group specificity.
Target Promoted Alkylation of DNA. Alkylation of a chosen sequence of DNA by a QM self-adduct introduces a new approach to site-induced reaction. The self-adduct expresses minimal reaction over 8 days with competing external nucleophiles and yet maintains an ability to alkylate a complementary strand. This is most likely accomplished by the thermodynamics of target recognition that shifts the equilibrium between QM addition and elimination from an intrastrand to interstrand process. The principles governing this conversion need not be limited to targets of single-stranded DNA or even nucleic acids in the future. Rather, this strategy should be readily generalized to many types of reagents and targets and may offer a bridge between the broad concepts of affinity reagents and mechanism based inactivators. Requirements for such a system include (i) an efficient but reversible method for intramolecular trapping of a reactive intermediate within a probe, (ii) significant recognition of a chosen target by the self-adduct, and (iii) more complete recognition of the target after the self-adduct regenerates the reactive intermediate that establishes (iv) a thermodynamic driving force promoting the ultimate intermolecular coupling between the target and probe.
Supplementary Material
Acknowledgments
We thank Dr. Brian Balgley for assistance with ESI-MS analysis. This work was supported by National Institutes of Health Grant CA-81571.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: QMP, quinone methide precursor; QM, ortho-benzoquinone methide; ESI, electrospray ionization.
References
- 1.Knorre, D. G. & Vlassov, V. V. (1989) Affinity Modification of Biopolymers (CRC Press, Boca Raton, FL).
- 2.Knorre, D. G., Vlassov, V. V. & Zarytova, V. F. (1994) Design and Targeted Reactions of Oligonucleotide Derivatives (CRC Press, Boca Raton, FL).
- 3.Silverman, R. B. (1988) Mechanism Based Enzyme Inactivation: Chemistry and Enzymology (CRC Press, Boca Raton, FL).
- 4.Wolfenden, R. & Radzicka, A. (1991) Curr. Opin. Struct. Biol. 1, 780–787. [Google Scholar]
- 5.Chabner, B. A., Ryan, D. P., Paz-Ares, L., Garcia-Carbonero, R. & Calabresi, P. (2001) in Goodman and Gilman's The Pharmacological Basis of Therapeutics, eds. Hardman, J. G., Limbird, L. E. & Gilman, A. G. (McGraw–Hill, New York), 10th Ed., pp. 1389–1459.
- 6.Tomasz, M. (1995) Chem. Biol. 2, 575–579. [DOI] [PubMed] [Google Scholar]
- 7.Stubbe, J., Kozarich, J. W., Wu, W. & Vanderwall, D. E. (1996) Acc. Chem. Res. 29, 322–330. [Google Scholar]
- 8.Wolkenberg, S. E. & Boger, D. L. (2002) Chem. Rev. 102, 2477–2495. [DOI] [PubMed] [Google Scholar]
- 9.Ma, Z. & Taylor, J.-S. (2000) Proc. Natl. Acad. Sci. USA 97, 11159–11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sando, S. & Kool, E. T. (2002) J. Am. Chem. Soc. 124, 2096–2097. [DOI] [PubMed] [Google Scholar]
- 11.Calderone, C. T., Puckett, J. W., Gartner, Z. J. & Liu, D. R. (2002) Angew. Chem. Int. Ed. 41, 4104–4108. [DOI] [PubMed] [Google Scholar]
- 12.Gartner, Z. J., Grubina, R., Calderone, C. T. & Liu, D. R. (2003) Angew. Chem. Int. Ed. 42, 1370–1375. [DOI] [PubMed] [Google Scholar]
- 13.Luebke, K. J. & Dervan, P. B. (1989) J. Am. Chem. Soc. 111, 8733–8735. [Google Scholar]
- 14.Rohatgi, R., Bartel, D. P. & Szostak, J. W. (1996) J. Am. Chem. Soc. 118, 3332–3339. [DOI] [PubMed] [Google Scholar]
- 15.Li, T., Weinstein, D. A. & Nicolaou, K. C. (1997) Chem. Biol. 4, 209–214. [DOI] [PubMed] [Google Scholar]
- 16.Li, X., Zhan, Z.-Y., Knipe, R. & Lynn, D. G. (2002) J. Am. Chem. Soc. 124, 746–747. [DOI] [PubMed] [Google Scholar]
- 17.Sando, S. & Kool, E. T. (2002) J. Am. Chem. Soc. 124, 9686–9687. [DOI] [PubMed] [Google Scholar]
- 18.Geacintov, N. E., Yoshida, H., Ibanez, V. & Harvey, R. G. (1982) Biochemistry 21, 1864–1869. [DOI] [PubMed] [Google Scholar]
- 19.Fernando, H., Huang, C.-R., Milliman, A., Shu, L. & LeBreton, P. R. (1996) Chem. Res. Toxicol. 9, 1391–1402. [DOI] [PubMed] [Google Scholar]
- 20.Lamm, G., Wang, L. & Pack, G. R. (1996) J. Am. Chem. Soc. 118, 3326–3331. [Google Scholar]
- 21.Shaw, J.-P., Milligan, J. F., Krawczyk, S. H. & Matteucci, M. (1991) J. Am. Chem. Soc. 113, 7765–7766. [Google Scholar]
- 22.Wilson, C. & Szostak, J. W. (1995) Nature 374, 777–782. [DOI] [PubMed] [Google Scholar]
- 23.Nagatsugi, F., Kawasaki, T., Usui, D., Maeda, M. & Sasaki, S. (1999) J. Am. Chem. Soc. 121, 6753–6754. [Google Scholar]
- 24.Lee, A. H. F., Chan, A. S. C. & Li, T. (2002) Chem. Commun., 2112–2113. [DOI] [PubMed]
- 25.Gao, X., Stassinopoulos, A., Ji, J., Kwon, Y., Bare, S. & Goldberg, I. H. (2002) Biochemistry 41, 5131–5143. [DOI] [PubMed] [Google Scholar]
- 26.Lin, C. H., Beale, J. M. & Hurley, L. H. (1991) Biochemistry 30, 3597–3602. [DOI] [PubMed] [Google Scholar]
- 27.Warpehoski, M. A. & Harper, D. E. (1995) J. Am. Chem. Soc. 117, 2951–2952. [Google Scholar]
- 28.Boger, D. L. & Garbaccio, R. M. (1999) Acc. Chem. Res. 32, 1043–1052. [Google Scholar]
- 29.Pitot, H. C., Reid, J. M., Sloan, J. A., Ames, M. M., Adjei, A. A., Rubin, J., Bagniewski, P. G., Atherton, P., Rayson, D., Goldberg, R. M. & Erlichman, C. (2002) Clin. Cancer Res. 8, 712–717. [PubMed] [Google Scholar]
- 30.Harsch, A., Sayer, J. M., Jerina, D. M. & Vouros, P. (2000) Chem. Res. Toxicol. 13, 1342–1348. [DOI] [PubMed] [Google Scholar]
- 31.Modica, E., Zanaletti, R., Freccero, M. & Mella, M. (2001) J. Org. Chem. 66, 41–52. [DOI] [PubMed] [Google Scholar]
- 32.Veldhuyzen, W. F., Shallop, A. J., Jones, R. A. & Rokita, S. E. (2001) J. Am. Chem. Soc. 123, 11126–11132. [DOI] [PubMed] [Google Scholar]
- 33.Dedon, P. C., Plastaras, J. P., Rouzer, C. A. & Marnett, L. J. (1998) Proc. Natl. Acad. Sci. USA 95, 11113–11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plastaras, J. P., Riggins, J. N., Otteneder, M. & Marnett, L. J. (2000) Chem. Res. Toxicol. 13, 1235–1242. [DOI] [PubMed] [Google Scholar]
- 35.Li, T. & Rokita, S. E. (1991) J. Am. Chem. Soc. 113, 7771–7773. [Google Scholar]
- 36.Zeng, Q. & Rokita, S. E. (1996) J. Org. Chem. 61, 9080–9081. [Google Scholar]
- 37.Pande, P., Shearer, J., Yang, J., Greenberg, W. A. & Rokita, S. E. (1999) J. Am. Chem. Soc. 121, 6773–6779. [Google Scholar]
- 38.Veldhuyzen, W., Lam, Y.-F. & Rokita, S. E. (2001) Chem. Res. Toxicol. 14, 1345–1351. [DOI] [PubMed] [Google Scholar]
- 39.Zhou, Q., Pande, P., Johnson, A. E. & Rokita, S. E. (2001) Bioorg. Med. Chem. 9, 2347–2354. [DOI] [PubMed] [Google Scholar]
- 40.Li, T. (1992) Ph.D. dissertation (State University of New York, Stony Brook).
- 41.Angle, S. R. & Yang, W. (1992) Tetrahedron Lett. 33, 6089–6092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.