

Abstract
Biological responses to oxygen availability play important roles in development, physiological homeostasis, and many disease processes. In mammalian cells, this adaptation is mediated in part by a conserved pathway centered on the hypoxia-inducible factor (HIF). HIF is a heterodimeric protein complex composed of two members of the basic helix–loop–helix Per-ARNT-Sim (PAS) (ARNT, aryl hydrocarbon receptor nuclear translocator) domain family of transcriptional activators, HIFα and ARNT. Although this complex involves protein–protein interactions mediated by basic helix–loop–helix and PAS domains in both proteins, the role played by the PAS domains is poorly understood. To address this issue, we have studied the structure and interactions of the C-terminal PAS domain of human HIF-2α by NMR spectroscopy. We demonstrate that HIF-2α PAS-B binds the analogous ARNT domain in vitro, showing that residues involved in this interaction are located on the solvent-exposed side of the HIF-2α central β-sheet. Mutating residues at this surface not only disrupts the interaction between isolated PAS domains in vitro but also interferes with the ability of full-length HIF to respond to hypoxia in living cells. Extending our findings to other PAS domains, we find that this β-sheet interface is widely used for both intra- and intermolecular interactions, suggesting a basis of specificity and regulation of many types of PAS-containing signaling proteins.
Cellular responses to oxygen availability are essential for the development and homeostasis of mammalian cells, demonstrated most critically by the link between the cellular adaptation to reduced tissue oxygenation and disease progression (1, 2). In mammalian cells, these responses are mediated in part by the hypoxia-inducible factor (HIF), a heterodimeric transcription factor composed of HIFα and aryl hydrocarbon receptor nuclear translocator (ARNT, also known as HIFβ) (3). HIF activity is tightly controlled under normoxic conditions by multiple O2-dependent hydroxylation events of the HIFα subunit, which coordinately promote the ubiquitin-mediated destruction of this protein (4) and impair its ability to interact with transcriptional coactivators (5, 6) (Fig. 1a). These controls are relieved during hypoxia, allowing HIF to activate the transcription of genes that facilitate metabolic adaptation to low oxygen levels and increase local oxygen supply by angiogenesis (7).
Fig. 1.

Oxygen-dependent regulation and domain architecture of HIF proteins. (a) HIF regulation is tightly linked to intracellular oxygen levels. Under normoxic conditions, HIFα is posttranslationally hydroxylated, promoting its degradation [modification of the oxygen-dependent degradation domain (ODD)] and interfering with its ability to interact with CBP/p300 coactivators (modification of the transcriptional activation domains NTAD and CTAD). These modifications are not made under hypoxic conditions, allowing HIFα to accumulate and enter the nucleus where it associates with ARNT and binds to HREs upstream of hypoxia-activated genes. The red box highlights the HIFα and ARNT PAS-B domains. (b) Domain topology of HIFα subunits, including a bHLH domain, two PAS domains, and C-terminal regulatory domains. A sequence alignment of the HIFα PAS-B orthologs is shown, with bold letters indicating the mutated residues described in the text. HIF-2α PAS-B secondary structure elements are indicated with a gray background.
All three isoforms of HIFα [HIF-1α, -2α (EPAS1), and -3α] (8, 9) and ARNT belong to the basic helix–loop–helix (bHLH)–Per-ARNT-Sim (PAS) family of eukaryotic transcription factors, which contain bHLH and PAS domains (Fig. 1). The bHLH domains of these proteins serve as dimerization elements, helping determine the specificity of complex formation while providing a DNA-binding interface composed of the basic regions from each monomer (10). PAS domains are widespread components of signal transduction proteins, currently identified in >2,000 proteins from organisms in all three kingdoms of life. These domains, shown to be protein–protein interaction elements in several systems (11), also appear to contribute to the dimerization process and thus increase the specificity of bHLH-PAS transcription factor formation (12, 13). In the case of the HIFα/ARNT complex, coimmunoprecipitation and gel mobility-shift experiments using truncated forms of HIFα and ARNT suggest that although the bHLH domains alone are able to dimerize, the PAS domains are required to build a stable heterodimer capable of robust DNA binding (14, 15). These data suggest a model of the complex where the bHLH, PAS-A, and PAS-B domains of ARNT interact with their counterparts in HIFα (Fig. 1a). However, most of this model remains speculative in light of the sparse data describing how PAS domains bind to each other, or more generally, to any protein partner.
To provide insight into this general topic of PAS domain signaling, particularly its importance in the hypoxia response pathway, we have studied the structure and interactions of the C-terminal PAS domain of human HIF-2α (HIF-2α PAS-B) by NMR spectroscopy. We report that HIF-2α PAS-B adopts a structure similar to other members of this family, with a central β-sheet flanked on one face by several α-helices. We further show that HIF-2α PAS-B binds directly to the human ARNT PAS-B domain in vitro, identifying the interface as a group of residues located in the central strands of the β-sheet. With structure-based mutations of this interface in the PAS-B domains of HIF-1α and -2α, we demonstrate that such changes interfere with the binding of isolated PAS-B domains in vitro but more importantly disrupt the ability of full-length HIF proteins to respond to hypoxia in living cells. These observations led us to compare PAS domains from multiple systems, showing that the β-sheet interface participates in a wide range of inter- and intramolecular interactions and suggesting a way that specificity and regulation may be achieved among these versatile domains.
Materials and Methods
Protein Expression and Purification. DNA-encoding fragments of human HIF-2α PAS-B (residues 240–350) and ARNT PAS-B (residues 356–470) were subcloned into the pGβ1-parallel and pHis-parallel expression vectors, respectively (16, 17). Escherichia coli BL21(DE3) cells transformed with these plasmids were grown in M9 media containing 1 g/liter 15NH4Cl for U-15N samples (supplemented with 3 g/liter 13C6 glucose for U-15N/13C labeled samples). These cultures were grown at 37°C to an A600 of 0.6–1.0, then induced overnight at 20°C by the addition of 0.5 mM isopropyl β-d-thiogalactoside.
The purification of HIF-2α PAS-B has been detailed (18). NMR samples typically contained 0.9 mM protein in 50 mM Tris buffer (pH 7.3), 15 mM NaCl, 5 mM DTT, 5 mM NaN3 and a protease inhibitor mixture (Sigma) in 90% H2O/10% D2O, unless otherwise noted. ARNT PAS-B was expressed and purified as described in Supporting Methods, which is published as supporting information on the PNAS web site.
Parallel studies on human HIF-1α PAS-B used a construct containing residues 238–349, chosen by homology with HIF-2α PAS-B. Expression and purification of HIF-1α PAS-B were done as described for HIF-2α PAS-B.
NMR Spectroscopy. All NMR data were recorded at 30°C with Varian Inova 500 and 600 MHz spectrometers by using nmrpipe for data processing (19) and nmrview for analysis (20). Chemical-shift assignments were made by using standard methods (21) as detailed in Supporting Methods.
Deuterium exchange reactions were started by resuspending lyophilized 15N-labeled HIF-2α PAS-B in 99% D2O (uncorrected pH 7.3). These samples were then placed into a prewarmed magnet (T = 30°C), and 15N/1H heteronuclear sequential quantum correlation (HSQC) spectra were sequentially acquired approximately every 15 min. Observed 2H exchange rates were converted into protection factors by using standard methods (22).
Structure Determination. Interproton distance constraints were obtained from 3D 15N edited NOESY (τm = 150 ms), 15N, 13C edited NOESY (τm = 100 ms), and 2D NOESY (τm = 120 ms) spectra. Hydrogen bond constraints (1.3 Å < dNH-O < 2.5 Å, 2.3 Å < dN-O < 3.5 Å) were set for backbone amide protons protected for >30 min from exchange with D2O solvent (30°C, pH 7.3). Constraints for the ϕ and ψ dihedral angles were generated by chemical-shift analyses by using talos (23), with two times the standard deviation of talos predictions as the bounds (minimum ± 30°). For 19 residues without talos predictions, ϕ dihedral angle constraints were obtained from an analysis of a 3D HNHA spectrum. Finally, 78 15N-1H residual dipolar coupling constraints were obtained from a sample partially aligned in 5% (wt/vol) DMPC/DHPC ratio of 3:1 (Avanti Polar Lipids) and 5 mM cetyltrimethylammonium bromide at 35°C.
Initial structures were determined without manual assignments by using aria1.2 (24, 25) and subsequently refined with a mix of automated and manual assignment of NOESY spectra. Of 1,000 structures, the 20 lowest-energy structures were analyzed with molmol (26) and procheck-nmr (27).
From this ensemble, the structure closest to the mean was superimposed against other PAS domains with the deepview Swiss Protein Data Bank program (28) with the automatic fit option. The calculated rms deviations ranged between 1.4 and 1.65 Å for HERG (Research Collaboratory for Structural Bioinformatics Protein Data Bank ID 1BYW), hPASK (1LL8), RmFixL (1D06), and Phy3 (1G28). The HIF-2α PAS-B structure was also used to generate a model of the HIF-1α PAS-B structure (74% sequence identity) by using modeller (29).
HIFα and ARNT PAS-B Titration. Titrations were conducted by the stepwise addition of natural abundance ARNT PAS-B (up to 800 μM) to a sample of 200-μM HIF-2α at 35°C. The peak heights of HIF-2α PAS-B signals that do not show ARNT-dependent chemical shift changes (38 residues) were fit to Eq. 1 to obtain the corresponding Kd:
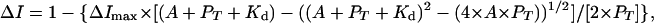 |
[1] |
where ΔI is the observed change in peak height at ARNT concentration A, ΔImax is the change in peak height at saturation, and PT is the total HIFα concentration. Eq. 1 is similar to the equation used to extract Kd from chemical-shift changes observed in titrations of complexes undergoing fast exchange (30), and we apply it here only to sites without chemical-shift changes (fast exchange) to ensure that the observed peak line widths are a population-weighted average of the free- and bound-state line widths (31). The binding of HIF-2α PAS-B mutants to ARNT PAS-B was assessed by adding 900 μM natural abundant ARNT PAS-B to 250 μM HIFα PAS B at 25°C.
Mutagenesis. Point mutants of full-length HIF-1α and -2α were created from wild-type DNA and primers including the desired mutation(s). PAS-B domains containing these mutations were obtained by PCR amplification of the corresponding full-length sequence and subcloned into the pGβ1-parallel vector. Transformation, protein induction, and purification were performed as described above.
Transfections. Cells were plated onto 48-well plates (3.5 × 104 cells per well) in 200 μl of HyQ DME/F-12 1:1 media (HyClone) supplemented with 5% FBS 24 h before transfection. Cells were transfected with 10 ng of each HIFα construct and 20 ng of the 3HRE-tk-luc (HRE, hypoxia-responsive element) luciferase reporter construct (8) by using the Lipofectamine PLUS reagent (Invitrogen). After 3 h, the media were changed and, after an additional 2 h, cells were incubated for 15 h under normoxic or hypoxic (1.0% O2) conditions. Luciferase activity was measured as described (32).
Results
Solution Structure of HIF-2α PAS-B. We determined the solution structure of HIF-2α PAS-B by using standard double- and triple-resonance NMR experiments conducted on uniformly 15N and 15N 13C labeled protein samples (Fig. 2). This structure is based on >2,500 geometric constraints obtained from measurements of interproton distances, dihedral angles, and 15N-1H residual dipolar couplings of a partially oriented sample (Table 1). All of these data are well satisfied by the high-precision ensemble of the 20 lowest-energy structures subsequently used for further analysis.
Fig. 2.

Solution structure of HIF-2α PAS-B. (a) Superimposition of 20 lowestenergy structures for HIF-2α PAS-B, calculated as indicated in the text. (b) Ribbon diagram of the structure closest to the mean of the ensemble shown in a. Circles indicate the approximate locations of the ligand-binding sites of several PAS domains (17, 33–36).
Table 1. Statistics for HIF-2α PAS-B solution structure determination.
| List of constraints | |
| NOE distance restraints | |
| Unambiguous | 2,767 |
| Ambiguous | 496 |
| Hydrogen bond restraints | 60 |
| Dihedral angle restraints | 96 |
| 15N-1H residual dipolar couplings | 78 |
| Stereospecific assignments (Val γ, Leu δ) | 12 |
| Structural analysis | |
| Mean rms deviation from experimental restraints | |
| NOE, Å | 0.022 +/- 0.002 |
| Dihedral angles, deg | 1.04 +/- 0.16 |
| Average number of: | |
| NOE violations >0.5 Å | 0 |
| NOE violations >0.3 Å | 1.9 +/- 1.2 |
| Dihedral violations >5° | 1.6 +/- 1.1 |
| Mean rms from idealized covalent geometry | |
| Bonds, Å | 0.0045 |
| Angles, deg. | 0.65 |
| Impropers, deg. | 1.69 |
| Geometric analysis of residues 6-91 and 98-112 | |
| rms deviation to mean | 0.53 +/- 0.07 Å (backbone) |
| 1.08 +/- 0.10 Å (all heavy) | |
| Ramachandran analysis (procheck) | 81.0% most-favored |
| 16.4% additionally allowed | |
| 1.6% generously allowed | |
| 1.0% disfavored |
HIF-2α PAS-B adopts a typical α/β PAS domain fold, characterized by several α-helices flanking a five-stranded antiparallel β sheet. The similarity of this structure to other PAS domains is demonstrated by the low-backbone rms deviation values (1.4–1.65 Å) of pairwise comparisons between representative PAS structures and HIF-2α PAS-B. Although several other PAS domains bind cofactors within their hydrophobic cores to regulate protein–protein interactions in response to various physical stimuli (11), a combination of NMR, mass spectrometry, and visible spectroscopy shows that HIF-2α PAS-B does not copurify with any such compound (data not shown). Further, no preformed cavities are present in the protein core, even at sites occupied by ligands in some other PAS domains (17, 33–36) (Fig. 2b).
Identification of ARNT PAS-B-Binding Surface on HIF-2α PAS-B. The PAS domains in bHLH-PAS transcription factors are thought to cooperate with the bHLH domains to facilitate dimerization (12, 13), which implies that the HIFα and ARNT PAS domains bind to each other (Fig. 1a). To experimentally demonstrate this interaction, we titrated unlabeled ARNT PAS-B into 15N-labeled HIF-2α PAS-B and monitored changes in the HIF-2α 15N/1H HSQC spectrum (Fig. 3a). Peaks in these spectra showed both chemical-shift changes and line broadening on addition of ARNT PAS-B, consistent with binding on the intermediate and fast exchange time scales. In contrast, we found that HIF-2α PAS-B signals were not affected by the addition of a PAS domain from PAS kinase, a protein not involved in the hypoxia response (17) (data not shown), suggesting that the changes observed on addition of ARNT PAS-B reflect a specific HIF-2α/ARNT interaction.
Fig. 3.

Characterization of the HIF-2α/ARNT PAS-B-binding interaction. (a) Titration of unlabeled ARNT PAS-B (black, 0 μM; light blue, 200 μM; blue, 600 μM; red, 800 μM ARNT) into a 200 μM 15N-labeled HIF-2α PAS-B solution. Arrow shows direction of peak shifts with increasing amounts of ARNT. Residues with peak broadening beyond detection during the titration are indicated with *.(b) Normalized peak heights of HIF-2α PAS-B (38 resonances) plotted against increasing amounts of ARNT PAS-B. The concentration dependence of the observed reduction in peak heights can be fit to a 1:1 binding event with a Kd of ≈30 μM (dotted line).
The ARNT-induced changes in the HIF-2α line widths demonstrate two important effects. First, we observed a general increase in line width for HIF-2α peaks during the titration, which we attribute to the slower tumbling of the larger 27-kDa heterodimeric complex compared with an isolated HIF-2α PAS-B domain. By monitoring this broadening via the decrease in peak heights as ARNT PAS-B was added, we observed a titration consistent with a 1:1 binding event with a 30 μM Kd (Fig. 3b). This effect saturated at a 1:3 (HIF/ARNT) ratio, establishing that it is not caused by nonspecific increases in sample viscosity or aggregation. Second, we observed that a subset of residues preferentially broadened on the addition of substoichiometric amounts of ARNT PAS-B. Such differential effects have been observed in several complexes (17, 37, 38) and arise from exchange broadening at sites experiencing significant chemical-shift changes on complex formation. Mapping sites that exhibit either this differential broadening or significant ARNT-induced chemical shift changes onto the HIF-2α PAS-B structure shows that they cluster on the face of the central β-sheet (Fig. 4). This provides a chiefly hydrophobic surface for ARNT binding that is conserved among the HIF isoforms (Fig. 1), suggesting that the PAS-B domains of all three interact similarly with ARNT.
Fig. 4.

Identification of the ARNT PAS-B-binding site on the surface of HIF-2α PAS-B. (a) Chemical-shift changes from 15N/1H HSQC spectra of HIF-2α PAS-B in the presence of 800 μM ARNT are plotted as a function of residue number. The red line indicates chemical-shift changes >0.16 ppm. Residues with peak broadening beyond detection are shown as an arbitrary chemical-shift change of 0.5 ppm. Secondary structure elements are indicated with a gray background. (b) Surface representations of HIF-2α PAS-B showing the location of the ARNT PAS-B-binding site. Colors indicate residues with large chemical-shift changes (>0.4 ppm) or broadening beyond detection (red), residues with a significant chemical-shift changes (>0.16 ppm) (orange), and the site of the mutated residues that disrupt the HIF/ARNT PAS-B interactions (yellow). Figs. 4 and 6 were made with pymol (www.pymol.org).
Evidence for the importance of this interface in the HIF/ARNT PAS-B dimer was obtained from studies of PAS-B domains containing point mutations. Based on our structure, we altered three residues with solvent-exposed side chains within the Hβ and Iβ strands (Q322E, M338E, and Y342T) (Fig. 4b). 15N/1H HSQC spectra of this triple mutant (trHIF-2α PAS-B) retain the chemical-shift dispersion and general pattern of the wild-type protein, confirming that the protein structure remains intact (Fig. 5a). The ARNT-binding capability of this mutant was assessed by comparing 15N/1H HSQC spectra before and after addition of unlabeled ARNT PAS-B. As demonstrated by the minimal ARNT-induced changes in peak locations and intensities, the interaction of the triple mutant HIF-2α PAS-B with ARNT has been very significantly weakened. These data establish that subtle changes on the surface of the Hβ and Iβ strands of HIF-2α PAS-B can disrupt the HIF–ARNT PAS-B interaction.
Fig. 5.

Point mutations in the HIFα PAS-B central β-sheet disrupt the binding of ARNT PAS-B. (a) Superimposed 15N/1H HSQC spectra of 250 μM 15N labeled HIF-2α PAS-B (Left) or triple mutant (Q322E/M338E/Y342T) (Right). Spectra in the presence of 900 μM unlabeled ARNT PAS-B are shown with red contours; those without ARNT are shown in black contours. Similar data for HIF-1α PAS-B are provided in Supporting Methods. (b) PAS-B domain interaction is important to form a biologically active HIF/ARNT complex. A construct expressing a luciferase reporter under the control of an HRE promoter was transfected into Ka-13 (columns 1–5) or CHO (column 6) cells along with various HIFα constructs. Values represent the average luciferase activity of three samples, with bars indicating standard error. Luciferase expression was induced by cotransfection of HIF-1α (column 2) or HIF-2α (column 4), particularly under hypoxic conditions. Cotransfection of trHIF-1α (column 3) or trHIF-2α (column 5), full-length HIFα proteins containing the three PAS-B mutations, shows a significant drop in luciferase activity compared with wild-type HIFα.
Comparison of HIF-1α and -2α PAS-B. Sequence alignments of HIF-1α and -2α indicate that the PAS-B domains of these proteins are extremely similar (74% identity; Fig. 1b). Nevertheless, the HIF-1α homolog of our HIF-2α PAS-B construct was too poorly behaved in solution to be amenable to structure determination. However, by combining the high sequence identity between these PAS-B domains and the HIF-2α PAS-B solution structure, we generated a homology model of HIF-1α PAS-B (29). This guided our mutation of three solvent-exposed residues in the HIF-1α PAS-B β-sheet (Q320E/V336E/Y340T) at sites analogous to those changed in HIF-2α PAS-B. Interestingly, this HIF-1α PAS-B triple mutant was significantly better behaved in solution than the wild-type domain. To determine whether HIF-1α PAS-B bound ARNT PAS-B in a similar fashion as HIF-2α PAS-B, we recorded 15N/1H HSQC of wild-type and triple-mutant HIF-1α PAS-B in the presence and absence of ARNT PAS-B (Fig. 7, which is published as supporting information on the PNAS web site). In the case of wild-type HIF-1α PAS-B, the combination of specific peak shifting and line broadening on the addition of ARNT PAS-B indicated binding. In contrast, this interaction was disrupted in the triple mutant (Fig. 7), demonstrating that the interface identified in HIF-2α is as crucial for the ARNT-binding function of HIF-1α.
Functional Importance of PAS-B in Full-Length HIFα. Our demonstration of specific interactions between the isolated HIFα and ARNT PAS-B domains suggested that they function as dimerization elements in the HIF heterodimer. To address this question in living cells with full-length HIFα, we used a luciferase-based assay that uses Chinese hamster ovary (CHO) cells lacking functional HIFα (CHO-Ka13) (39). CHO-Ka13 cells were transiently transfected with full-length HIFα constructs under the control of the HIF-1 promoter (40) and encoding either wild-type or a mutant sequence containing the three PAS-B domain mutations. Low levels of DNA were transfected to roughly match the HIF activity in wild-type CHO cells. The abilities of these HIFα constructs to activate transcription were determined by measuring the expression of luciferase under the control of a minimal HRE promoter (8).
After expressing either wild-type HIF-1α or -2α, the hypoxia response of the transfected CHO-Ka13 cells mimicked that of wild-type CHO cells, confirming that HIFα activity can be rescued by expression from these plasmids (Fig. 5b). Cells transfected with HIF-2α constructs either lacking the entire PAS-B domain (Δ240–350) or containing a mutation known to unfold PAS-B (C339P; unfolded as established by NMR) showed no significant luciferase activity under any conditions (data not shown). Although these results suggest that HIFα PAS-B plays a crucial role in forming a biologically active HIFα/ARNT heterodimer, the question remained whether the subtler PAS-B modifications that disrupted in vitro binding would have a similar effect. Therefore, we transfected CHO-Ka13 cells with full-length HIF-1α and -2α constructs containing the three PAS-B mutations (trHIFα). For both mutant proteins, the HRE-driven luciferase response is decreased compared with that observed in cells containing vectors expressing wild-type HIFα. In the case of trHIF-1α, the response is virtually eliminated (Fig. 5b, column 3), whereas a less pronounced but significant reduction is observed for trHIF-2α (Fig. 5b, column 5). These results are surprising in light of the three apparently redundant sets of protein–protein interactions implied by the model of the HIF heterodimer (Fig. 1a). Our data show that the subtle disruption of the PAS-B interaction within the full length HIFα protein is sufficient to prevent the formation of the HIF transcription factor, thereby abolishing the hypoxia response.
Discussion
Despite the intense interest in hypoxia signaling, previous understanding of the roles of various protein–protein interactions in the HIF heterodimer remained unclear. Much of the data underlying prior work in this area were based on deletions generated at a time when the lack of structural data hampered the design of constructs cleanly corresponding to PAS domain boundaries. Unfortunately, this approach has complicated the interpretation of these findings with respect to the functional importance of individual PAS domains. In this context, our results provide the structural insight to clarify how PAS-B domains contribute to the stability of HIF and other bHLH-PAS transcription factor complexes. We demonstrate that the PAS-B domains of HIFα and ARNT directly associate in vitro, using a group of predominantly hydrophobic residues located on the solvent-exposed face of the HIFα β-sheet. Critically, mutations that disrupt the interaction between isolated PAS-B domains in vitro also interfere with the ability of the full-length heterodimer to activate transcription in living cells. These results establish the importance of the β-sheet interaction surface of HIFα PAS-B domains within the complexes that are central to the hypoxia response pathway.
More broadly, the β-sheet interface we identified appears to be important for a wide array of inter- and intramolecular interactions within several PAS domains (Fig. 6). Evidence of this commonality is provided by PAS domains from two blue-light photoreceptors, phototropin1 (AsLOV2) (36) and photoactive yellow protein (33). Both of these domains are flanked by additional α-helical elements that fold back on the PAS domain itself, associating with the same β-sheet region used by HIFα to bind ARNT (Fig. 6). An additional example is provided by the human ether-a-go-go-related gene (HERG) potassium channel, which contains a PAS domain that controls the poststimulation deactivation kinetics of this channel, possibly by interacting with the S4-S5 linker (41). Point mutations in the PAS domain that impair channel function have been identified by both alanine scanning mutagenesis (42) and sequencing of HERG genes from individuals affected by long QT syndrome (41), an inherited cardiac disorder associated with defects in HERG and other ion channels. Biochemical studies of channels containing one of several conservative mutations in solvent-exposed residues (F29L, F29A, N33T, Y43A, and R56Q; Fig. 6) demonstrate that the region analogous to the HIF/ARNT interface plays an important role in regulating HERG channel kinetics. In summary, these comparisons indicate that the exposed face of the central β-sheet in PAS domains is well suited for making many functionally important types of associations.
Fig. 6.

Versatility of protein interactions involving PAS domain β-sheets. HIF2α is shown in the same orientation as Fig. 4b and colored by residues experiencing significant 15N/1H chemical shifts on complex formation (red) and those used to generate the complex-disrupting trHIF-2α (blue). Phototropin (AsLOV2) (36) and photoactive yellow protein (33) highlight the α-helices external to the PAS core (magenta) and any atoms located within 5 Å of those helices (pink). HERG (42) shows functionally important, solvent-exposed residues (dark blue) and residues present in a surface hydrophobic patch suggested to be important for channel function (light blue) (42).
This comparison also suggests a route that might be exploited to regulate the formation of the HIFα–ARNT complex by using small organic compounds. Although neither a natural ligand nor a ligand-binding cavity has been identified for the HIFα PAS-B domains, this should not be interpreted to mean this domain cannot bind any small compounds. Indeed, it has been demonstrated that certain artificial chemicals can specifically bind into the well packed hydrophobic core of a PAS domain from PAS kinase (Fig. 2b) (17), likely producing significant conformational changes therein. Parallel studies of two PAS domains with natural cofactors, AsLOV2 and photoactive yellow protein, have shown that relatively small structural changes in these internally bound compounds are sufficient to displace α-helices bound onto the same β-sheet interface we identified in HIFα (36, 43). Combining these observations and an earlier model proposed for PAS domain signaling (44) raises the possibility that binding compounds in the HIFα PAS-B core could generate structural changes that disrupt the interactions needed to form the HIF heterodimer, analogous to the HIFα mutations demonstrated here. Given the central role of HIF in the hypoxia response and the importance of this pathway in cancer progression, this approach may serve as the basis for a novel therapeutic strategy (1, 2).
Supplementary Material
Acknowledgments
We thank M. Rhima for technical assistance; P. Ratcliffe (Oxford University, Oxford) for the gift of the Ka13 cell line; and D. Russell, J. Garcia, and S. McKnight for comments on the manuscript. This work was supported by grants from the National Institutes of Health (CA90601 to K.H.G. and CA95471 to K.H.G. and R.K.B.), the University of Texas Southwestern Endowed Scholars Program (to K.H.G. and R.K.B.), and the Robert A. Welch Foundation (I-1424 to K.H.G.). P.B.C. was supported by a National Institutes of Health Training Grant GM08297 to the University of Texas Southwestern Graduate Program in Molecular Biophysics. R.K.B. is supported by a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HIF, hypoxia-inducible factor; PAS, Per-ARNT-Sim; HIF-2α PAS-B, C-terminal PAS domain of human HIF-2α; ARNT, aryl hydrocarbon receptor nuclear translocator; bHLH, basic helix–loop–helix; HSQC, heteronuclear sequential quantum correlation; CHO, Chinese hamster ovary; HRE, hypoxia responsive element.
Data deposition: The atomic coordinates for the HIF-2α PAS-B domain have been deposited in the Protein Data Bank (PDB ID 1P97).
References
- 1.Semenza, G. L. (2000) Genes Dev. 14, 1983–1991. [PubMed] [Google Scholar]
- 2.Ratcliffe, P. J., Pugh, C. W. & Maxwell, P. H. (2000) Nature 6, 1315–1340. [DOI] [PubMed] [Google Scholar]
- 3.Wang, G., Jiang, B., Rue, E. & Semenza, G. (1995) Proc. Natl. Acad. Sci. USA 92, 5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Min, J. H., Yang, H., Ivan, M., Gertler, F., Kaelin, W. G., Jr., & Pavletich, N. P. (2002) Science 296, 1886–1889. [DOI] [PubMed] [Google Scholar]
- 5.Dames, S. A., Martinez-Yamout, M., De Guzman, R. N., Dyson, H. J. & Wright, P. E. (2002) Proc. Natl. Acad. Sci. USA 99, 5271–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freedman, S. J., Sun, Z. Y., Poy, F., Kung, A. L., Livingston, D. M., Wagner, G. & Eck, M. J. (2002) Proc. Natl. Acad. Sci. USA 99, 5367–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semenza, G. L. (1999) Annu. Rev. Cell Dev. Biol. 15, 551–578. [DOI] [PubMed] [Google Scholar]
- 8.Tian, H., McKnight, S. & Russell, D. (1997) Genes Dev. 11, 72–82. [DOI] [PubMed] [Google Scholar]
- 9.Gu, Y.-Z., Moran, S. M., Hogenesch, J. B., Wartman, L. & Bradfield, C. A. (1998) Genes Dev. 7, 205–213. [PMC free article] [PubMed] [Google Scholar]
- 10.Massari, M. E. & Murre, C. (2000) Mol. Cell. Biol. 20, 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor, B. L. & Zhulin, I. B. (1999) Microbiol. Mol. Biol. Rev. 63, 479–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pongratz, I., Antonsson, C., Whitelaw, M. L. & Poellinger, L. (1998) Mol. Cell. Biol. 18, 4079–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hogenesch, J. B., Chan, W. K., Jackiw, V. H., Brown, R. C., Gu, Y.-Z., Pray-Grant, M., Perdew, G. H. & Bradfield, C. A. (1997) J. Biol. Chem. 272, 8581–8593. [DOI] [PubMed] [Google Scholar]
- 14.Jiang, B.-H., Rue, E., Wang, G. L., Roe, R. & Semenza, G. L. (1996) J. Biol. Chem. 271, 17771–17778. [DOI] [PubMed] [Google Scholar]
- 15.Gradin, K., McGuire, J., Wenger, R., Kvietikova, I., Whitelaw, M., Toftgard, R., Tora, L., Gassmann, M. & Poellinger, L. (1996) Mol. Cell. Biol. 16, 5221–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheffield, P., Garrard, S. & Derewenda, Z. (1999) Protein Expr. Purif. 15, 34–39. [DOI] [PubMed] [Google Scholar]
- 17.Amezcua, C. A., Harper, S. M., Rutter, J. & Gardner, K. H. (2002) Structure (Cambridge, U.K.) 10, 1349–1361. [DOI] [PubMed] [Google Scholar]
- 18.Erbel, P. J. A., Barr, K., Gao, N., Gerwig, G. J., Rick, P. D. & Gardner, K. H. (2003) J. Bacteriol. 185, 1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J. & Bax, A. (1995) J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 20.Johnson, B. A. & Blevins, R. A. (1994) J. Biomol. NMR 4, 603–614. [DOI] [PubMed] [Google Scholar]
- 21.Sattler, M., Schleucher, J. & Griesinger, C. (1999) Prog. NMR Spectrosc. 34, 93–158. [Google Scholar]
- 22.Bai, Y., Milne, J. S., Mayne, L. & Englander, S. W. (1993) Proteins 17, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cornilescu, G., Delaglio, F. & Bax, A. (1999) J. Biomol. NMR 13, 289–302. [DOI] [PubMed] [Google Scholar]
- 24.Brünger, A. T., Adams, P. D., Clore, G. M., Delano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M. & Pannu, N. S. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 25.Nilges, M. & O'Donoghue, S. I. (1998) Prog. NMR Spectrosc. 32, 107–139. [Google Scholar]
- 26.Koradi, R., Billeter, M. & Wüthrich, K. (1996) J. Mol. Graphics 14, 51–55. [DOI] [PubMed] [Google Scholar]
- 27.Laskowski, R. A., Rullman, J. A. C., MacArthur, M. W., Kaptein, R. & Thornton, J. M. (1996) J. Biomol. NMR 8, 477–486. [DOI] [PubMed] [Google Scholar]
- 28.Guex, N., and Peitsch, M. C. (1997) Electrophoresis 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- 29.Marti-Renom, M. A., Stuart, A., Fiser, A., Sanchez, R., Melo, F. & Sali, A. (2000) Annu. Rev. Biophys. Biomol. Struct. 29, 291–325. [DOI] [PubMed] [Google Scholar]
- 30.Dalvit, C., Flocco, M., Knapp, S., Mostardini, M., Perego, R., Stockman, B. J., Veronesi, M. & Varasi, M. (2002) J. Am. Chem. Soc. 124, 7702–7709. [DOI] [PubMed] [Google Scholar]
- 31.Palmer, A. G., III, Kroenke, C. D. & Loria, J. P. (2001) Methods Enzymol. 339, 204–238. [DOI] [PubMed] [Google Scholar]
- 32.Bruick, R. K. (2000) Proc. Natl. Acad. Sci. USA 97, 9082–9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borgstahl, G. E. O., Williams, D. R. & Getzoff, E. D. (1995) Biochemistry 34, 6278–6287. [DOI] [PubMed] [Google Scholar]
- 34.Crosson, S. & Moffat, K. (2001) Proc. Natl. Acad. Sci. USA 98, 2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gong, W., Hao, B., Mansy, S. S., Gonzalez, G., Gilles-Gonzalez, M. A. & Chan, M. K. (1998) Proc. Natl. Acad. Sci. USA 95, 15177–15182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harper, S. M., Neil, L. C. & Gardner, K. H. (2003) Science 301, 1541–1544. [DOI] [PubMed] [Google Scholar]
- 37.Sette, M., van Tilborg, P., Spurio, R., Kaptein, R., Paci, M., Gualerzi, C. O. & Boelens, R. (1997) EMBO J. 16, 1436–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsuo, H., Walters, K. J., Teruya, K., Tanaka, T., Gassner, G. T., Lippard, S. J., Kyogoku, Y. & Wagner, G. (1999) J. Am. Chem. Soc. 121, 9903–9904. [Google Scholar]
- 39.Wood, S. M., Wiesener, M. S., Yeates, K. M., Okada, N., Pugh, C. W., Maxwell, P. H. & Ratcliffe, P. J. (1998) J. Biol. Chem. 273, 8360–8368. [DOI] [PubMed] [Google Scholar]
- 40.Minet, E., Ernest, I., Michel, G., Roland, I., Remacle, J., Raes, M. & Michiels, C. (1999) Biochem. Biophys. Res. Commun. 261, 534–540. [DOI] [PubMed] [Google Scholar]
- 41.Chen, J., Zou, A., Splawski, I., Keating, M. T. & Sanguinetti, M. C. (1999) J. Biol. Chem. 274, 10113–10118. [DOI] [PubMed] [Google Scholar]
- 42.Morais Cabral, J. H., Lee, A., Cohen, S. L., Chait, B. T., Li, M. & Mackinnon, R. (1998) Cell 95, 649–655. [DOI] [PubMed] [Google Scholar]
- 43.Hoff, W. D., Xie, A., Van Stokkum, I. H. M., Tang, X. J., Gural, J., Kroon, A. R. & Hellingwerf, K. J. (1999) Biochemistry 38, 1009–1017. [DOI] [PubMed] [Google Scholar]
- 44.Cusanovich, M. A. & Meyer, T. E. (2003) Biochemistry 42, 4759–4770. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.