

Abstract
Protein tyrosine phosphatases (PTPs) are a diverse family of enzymes encoded by 107 genes in the human genome. Together with protein tyrosine kinases (PTKs), PTPs regulate various cellular activities essential for the initiation and maintenance of malignant phenotypes. While PTK inhibitors are now used routinely for cancer treatment, the PTP inhibitor development field is still in the discovery phase. In this article, the suitability of targeting PTPs for novel anticancer drug discovery is discussed. Examples are presented for PTPs that have been targeted for anticancer drug discovery as well as potential new PTP targets for novel anticancer drug discovery.
Keywords: Protein tyrosine phosphatase inhibitor, Shp2, PTP1B, Cdc14, PRL-3, LMW-PTP, Cdc25, Eya
INTRODUCTION
Discovered three decades ago, protein tyrosine phosphorylation is now well recognized as a major regulatory mechanism of cell signaling and activities [1]. Aberrant tyrosine phosphorylation is either the primary cause of human cancer or is required for maintaining the oncogenic state that include abnormal survival, replication, growth, metastasis, angiogenesis, resistance to growth inhibition, evasion of immune surveillance, and stress support of human cancer [2, 3]. Protein tyrosine phosphorylation is controlled reciprocally by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). Many PTKs have been identified as human oncogenes and are validated targets for cancer therapy. Small molecule PTK inhibitors such as Bcr-Abl inhibitors imatinib and dasatinib, epidermal growth factor receptor (EGFR) inhibitors gefitinib and erlotinib are in clinical use for human cancer treatment. Whereas the PTK-targeted therapy field has advanced to the United States Food and Drug Administration (FDA)-approved drugs, PTP inhibitor development is in the early discovery phase. In this article, we discuss evidence that specific PTPs are targets for development of novel anticancer drug and summarize selected PTP drug discovery efforts. Readers are referred to several previous review articles of PTPs and PTP inhibitors for additional information [4-9]. Table I lists PTP inhibitors that have been tested in vivo. For comprehensive description of PTP classification, the articles by Alonso and colleagues [10] and Andersen and colleagues [11] are recommended.
Table 1.
PTP inhibitors that have been evaluated in vivo
| Inhibitor | PTP | Animal Studies | Human Trials | Firm | Reference |
|---|---|---|---|---|---|
| BN82002 Cmp 26* |
Cdc25 | Inhibit Mia PaCa-2 pancreatic tumor xenograft growth. |
IPSEN | [156] | |
| BN82685 Cmp 27 |
Cdc25 | Inhibit Mia PaCa-2 pancreatic tumor xenograft growth. |
IPSEN | [157] | |
| IRC-083864 Debio-0931 Cmp 25 |
Cdc25 | Inhibit Mia PaCa-2 pancreatic tumor and LNCaP prostate tumor xenograft growth. |
Phase I (planned) | IPSEN/ Debiopharm |
[155] |
| Cmp 17 | PTP1B | Lower blood glucose level. Delay onset of ErbB2-induced mammary tumor. |
Merck Frosst | [102] | |
| Ertiprotafib Cmp 18 |
PTP1B | Lower fasting blood glucose and insulin. Improve glycemic excursion. Improve lipid profiles. |
Phase II in type II diabetes (Discontinued because of insufficient efficacy and concerns about side-effects due to inhibition of other targets) |
Wyeth Research |
[107] |
| MSI-1436 Trodusquemine Cmp 19 |
PTP1B | Suppress appetite Induce fat-specific weight loss Improve insulin and leptin levels |
Phase I | Genaera | [108] |
| Cmp 13 | Glepp1 | Inhibit thioglycolate-induced peritonitis. Inhibit dermatitis. Inhibit ulcerative colitis. |
Merck Serono |
[90] |
Cmp: compound
PROTEIN TYROSINE PHOSPHATASES AS ANTICANCER DRUG TARGETS
At least three criteria need to be considered when selecting a PTP as the direct target for novel anticancer drug discovery:
Does the PTP play a positive role in the pathogenesis of human cancer such that inhibition of its activity suppresses malignant phenotypes? This is fulfilled if the PTP is an oncogene that causes human cancer or is a non-oncogene addiction gene required for maintenance of malignant phenotypes.
Alternatively, does the PTP contribute to resistance to an anticancer therapy used in the standard care of cancer patients such that inhibition of its activity sensitizes the anticancer therapy?
Does the potential PTP drug target have appropriate surface properties (surface topology, charge distribution, lipophilic potential) at its active site and the surrounding area such that high affinity and specific binding of a small molecule inhibitor may be achieved?
Although it is not impossible, small molecule activators of proteins are much more difficult to obtain than small molecule inhibitors. Drug discovery efforts generally aim at development of inhibitors. Since aberrant activation of many PTKs is associated with various types of human cancer and PTPs catalyze the reverse reaction of PTKs, PTPs were initially thought as negative regulators of cancer phenotypes [4, 12, 13]. Thus, inhibition of PTPs would be predicted to facilitate, rather than suppress, oncogenesis. This concept has proven to be inaccurate. Biological systems are much more complex than the simple in vitro chemical reaction (Fig. (1)). While many specific tyrosine phosphorylation sites on proteins serve as positive signals to propagate activating responses, some of these tyrosine phosphorylation sites also trigger negative-feedback mechanism to terminate the activation signal. Moreover, certain tyrosine phosphorylation sites have suppressive effect on enzyme activities. For example, phosphorylation of human c-Src at Tyr-530 by Csk tyrosine kinase inhibits the c-Src tyrosine kinase activity. Dual phosphorylation of Cdk1 at Thr-14 and Tyr-15 blocks its kinase activity. Dephosphorylation of these residues leads to enzyme activation. In fact, increasing evidence suggests that cell signaling requires coordinate action of both PTK and PTP activities [5]. Therefore, PTPs could cooperate with PTKs, in addition to antagonizing them, in promoting cancer growth and progression.
Fig. (1).

Positive and negative roles of tyrosine phosphorylation in cell signaling. In this illustration, three tyrosine residues (Y1, Y2, Y3) on a protein may be subject to phosphorylation by a PTK. Phosphorylation of Y1 increases the activity of the protein. Phosphorylation of Y2 inhibits the activity of the protein. Phosphorylation of Y3 induces feedback inhibition such as recruitment of E3 ligase that causes degradation of the protein or GTPase Activator Protein (GAP) that turns off G-proteins. While dephosphorylation of Y1 by PTP1 inactivate the protein, dephosphorylation of Y2 and Y3 by PTP2 and PTP3 are necessary for sustained activity of the protein. Thus, PTP1 is a negative regulator whereas PTP2 and PTP3 are positive regulators that coordinately control the activity of the protein.
Another dogma contributing to the slow start of PTP drug discovery efforts was that PTKs are highly regulated and specific, whereas a few constitutive, non-specific PTPs passively counteract the function of PTKs [14]. It is now known that there are at least 107 PTP genes in the human genome, providing highly regulated and specific function in various types of human cells [10, 13]. Human PTPs are grouped into three classes of Cys-based PTPs and a fourth family of Asp-based PTPs. Although designated as PTPs, besides phosphotyrosine-specific phosphatases, PTPs include dual specificity phosphatases (DSPs) that dephosphorylate protein tyrosine and serine/threonine residues and phosphatases that their known physiological substrates are phosphothreonine residues, phospholipids, and mRNA. Among Class I phosphotyrosine-specific classical PTPs, the transmembrane PTPα (encoded by the PTPRA gene) is an activator of c-Src. The non-receptor PTP Shp2 (PTPN11) is a positive regulator of growth factor signaling. Gain-of-function Shp2 mutants have been established as oncogenes. Both positive and negative effects of PTP1B on tumorigenesis have been reported. Cell cycle requires at least three groups of PTPs to modulate Cdks and their substrates: Class III PTPs Cdc25s and Class I dual-specific PTPs Cdc14s, and Kap (CDKN3). Cdc25s dephosphorylate the dual Thr-Tyr phosphorylation sites at the N-terminal region of Cdks to activate these kinases to drive the cell cycle progression. Cdc14s regulate mitosis exit and centrosome separation. Cdc25 and Cdc14, therefore, are potential targets for inhibition of cell proliferation. Kap dephosphorylates the activating Thr-160 of Cdk2 and thus is a Cdk inactivator [15]. While the role of Kap in tumorigenesis is controversial [16, 17], CDKN3 mRNA is frequently elevated in human cancer. Furthermore, inactivation of Cdk2 is required for mitotic exit in some organisms [18]. The Class I dual specific PTP PRL-3 (PTP4A3) promotes cancer metastasis [19]. The low molecular weight PTP (LMW-PTP, ACP1) is the sole member of the Class II PTP. Overexpression of LMW-PTP is sufficient to transform MCF-10A mammary epithelial cells [20] and NIH3T3 fibroblast cells [21]. Recent studies have shown that phosphorylation of Tyr-142 at the C-terminal region of γ-H2A.X histone prevents DNA-damage repair and induces apoptosis [22, 23]. The Asp-based PTPs Eya1 and Eya3 are responsible for dephosphorylation of Tyr-142 on γ-H2A.X [23, 24]. Conceivably, blocking Eya1/Eya3 PTP activity could be used to enhance the therapeutic efficacy of DNA damage-based cancer therapy. Thus, candidates of anticancer drug targets are found in every family of PTPs.
In a study employing RNA interference screen to identify anti-apoptosis genes in Hela cells, 28 of the 107 human PTPs genes were found to be positive regulators of cell survival, whereas only 4 PTPs were identified as cell death phosphatases [25]. Thus, the number of anti-apoptotic PTPs is 7-times of the number of pro-apoptotic PTPs in Hela cells. The overall number of PTP genes contributing to the survival of human cancer cells is likely to be higher because Hela cells are likely to express only a portion of the human phosphatome and because different requirements for survival in various types of cancer cells of diverse genetic origin. Therefore, there are several known and likely many yet to be identified PTPs that satisfy Criteria A and B described above.
Needless to say, some of PTPs are established or potential tumor suppressors [4, 13]. Mutational and promoter methylation analyses have provided links between several PTP genes to various types of human cancer. These include PTEN, PTPRF, PTPRG, PTPRJ, PTPRO, PTPRT, PTPN3, PTPN6, PTPN13, PTPN14, and DUSP6 [5, 13]. Among these PTPs, Pten is clearly established as a tumor suppressor in various types of human cancer through extensive investigation that includes mouse models of tumorigenesis [4, 5]. Notably, physiological substrates of Pten are phosphoinositide 3-phosphates, not proteins. There is substantial evidence that DEP1 (PTPRJ) is a tumor suppressor [4, 13]. Roles of other PTPs as tumor suppressors are less well-established. If some of them are confirmed as tumor suppressors and if the loss-of-function is due to epigenetic silencing, indirect re-activation through epigenetic approaches such as the use of demethylating agents can be explored to restore expression of these PTP tumor suppressors as an anticancer strategy.
The potential tumor suppressor function of some PTPs raises the concern about potential cross-inhibition of these PTP tumor suppressors by poorly selective PTP inhibitors. This concern can be addressed by answering three questions. First, does the PTP target have distinct surface properties surrounding the active site to allow selective binding of small molecule inhibitors? Second, if cross-inhibition of a small molecule PTP inhibitor cannot be completely avoided, does inhibition of the targeting PTP have a dominant effect over inhibition of the cross-inhibited PTP? Third, is cross-inhibition of a putative PTP tumor suppressor in normal cells sufficient to turn normal cells cancerous? It should be noted that, since tumor suppressors are defective or absent in cancer cells, a drug that cross-inhibits a tumor suppressor in cancer cells under this condition has no functional significance.
The first question brings up Criteria C described above. Crystal structures of catalytic domains of Class I Cys-based PTPs share a similar Cα-backbone trace [11, 26]. Despite the overall conserved ternary structure, comparison of the surface surrounding these PTP catalytic sites revealed diverse topology, electrostatic potential, and lipophilic potential characteristics [10, 26, 27]. Barr et al. mapped conserved residues of all experimental structures of PTPs onto the surface of PTP1B and the D1 domain of RPTPμ [26]. They found only a few conserved surface patches surrounding the active site. These distinct surface properties exist even between close members of PTPs, such as Shp1 and Shp2 [26, 27]. Thus, PTPs have different properties on the surface surrounding the catalytic site that allow development of specific inhibitors. In support of this notion, selective PTP inhibitors have been reported [28, 29].
A challenge in targeting the PTP catalytic site is the relatively shallow pocket of the catalytic site. Nevertheless, after the discovery that PTP1B has a secondary substrate binding pocket [30, 31], high affinity bidentate inhibitors with Ki < 10 nM have been developed [32] (see Fig. (2)). In a recent large-scale structure analysis of catalytic domains of classical PTPs, it was revealed that many PTPs have a secondary pocket proximal to the catalytic site [26]. Therefore, the limitation of the pocket depth of the catalytic site of PTPs can be overcome by utilizing bidentate inhibitors that bind simultaneously to the catalytic and the secondary pockets.
Fig. (2).
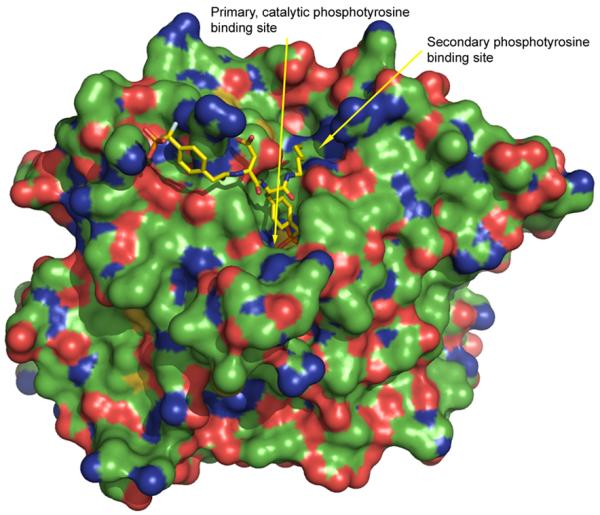
Illustration of the secondary phosphotyrosine binding site of PTP1B (from pdb code 1PXH), colored by element: carbon, green; oxygen, red; nitrogen blue, sulfur, yellow.
Selective PTP inhibitors that display >10-fold selectivity against similar PTPs and >100-fold against other PTPs are obtainable. However, a monospecific PTP with >100-fold selectivity against all other PTPs has yet to be identified. Therefore, an issue needed to be considered in the PTP anticancer drug discovery is whether inhibition of the targeting PTP has a dominant effect over inhibition of the cross-inhibited PTP, particularly if the cross-inhibited PTP is a putative tumor suppressor. In general, a PTP is considered as a target for anticancer drug discovery because it is required for survival, proliferation, or metastasis of cancer cells or it contributes to drug resistance. Inhibition of the single PTP target alone in cancer cells by molecular biology approaches is sufficient to suppress malignant phenotypes. Thus, inhibition of the targeting PTP is predicted to have a dominant effect on suppression of cancer cells. Although further study is required to address this issue, we are not aware of any evidence that any PTP inhibitor can promote cancer cell survival and growth.
Similarly, the synthetic effects of inactivation of a tumor-promoting PTP and a tumor-suppressor PTP in normal cells remain to be studied. Asides from genetic/epigenetic links, the role of a PTP as a tumor suppressor is generally assessed by the expression of a functional PTP in cancer cells. There is paucity of evidence that inactivation of a putative PTP tumor suppressor is sufficient to cause cancer, except the phosphoinositide phosphatase Pten. For instance, PTPRJ (DEP1 gene)-null mice do not develop spontaneous tumor [33]. Therefore, although pre-clinical and clinical evaluations will be required, it is predicted that a selective PTP inhibitor, even if it weakly cross-inhibits a putative PTP tumor suppressor, is unlikely to cause therapy-induced tumor and therefore it is acceptable as an anticancer drug candidate in this regard.
Another issue is the potential toxicity of inhibiting the targeted PTPs in normal cells. Although this needs to be tested in each case through clinical trials, it is believed that therapeutic windows exist for exploration of selective toxicity to cancer cells. PTPs selected as drug targets usually are aberrantly active in the cancer cells, which may confer specific dependency of cancer cells to the PTPs. For instance, it has been reported that Shp2 knockdown specifically inhibits primary chronic myeloid leukemia (CML) cells but not normal CD34+ cells [34]. Furthermore, for certain terminal diseases, short term, low grade toxicity with drugs that have proven benefits to the disease management may be acceptable.
In the following sections, we describe Shp2 as a target for novel anticancer drug discovery and summarize other established and potential PTP targets for anticancer drug discovery.
SHP2 (PTPN11) AS AN ANTICANCER DRUG TARGET
Shp2 is a Class I non-receptor classical Cys-based PTP containing tandem Src homology-2 (SH2) domains [35]. The presence of phosphotyrosine-binding SH2 domains provides an immediately recognizable link to PTK signaling when it was cloned by several laboratories in 1992-1993. Shp2 is a homolog of Drosophila corkscrew (csw) gene product. Shortly after mammalian Shp2 was cloned, several laboratories tested effects of catalytic-Cys mutated Shp2 on insulin- or epidermal growth factor (EGF)-stimulated Ras and Erk1/Erk2 (Erk1/2) activation. These experiments consistently showed that a catalytic-inactive Shp2 displayed a dominant-negative effect on insulin- or EGF-induced Ras/Erk1/2 activation [36, 37]. These and other experiments documented Shp2 as a positive mediator of mitogenic signaling of growth factor receptor tyrosine kinases and showed that the PTP activity is essential.
Structure
Human Shp2 contains two SH2 domains in the N-terminal region, a single PTP domain following the tandem SH2 domains, and a C-terminal region with Tyr phosphorylation sites. Two Shp2 [593 amino acid (aa) and 597 amino acid residues] exist in humans and rodents that differ in a stretch of 4 amino acid residues in the catalytic domain. These two Shp2 are derived from a single gene and are believed to be functionally identical. The tandem SH2 domains are designated as N-SH2 (aa 2-104) and C-SH2 (aa 112-215) domain, respectively. Shp2 SH2 domains preferred a consensus binding motif of (T/V/I/y)-X-pY-(A/s/t/v)-X-(I/V/L) [38, 39]. Cys-459 in the catalytic domain (aa 220-525) is the catalytic Cys. The mechanism of tyrosine dephosphorylation is illustrated in Fig. (3). The C-terminal region has two tyrosine residues (Tyr-546 and Tyr-584) that may be phosphorylated after growth factor stimulation, especially PDGF and FGF. These two tyrosine residues are located within Grb SH2 domain binding motif (pYxNx) and are reported to mediate Grb2 binding.
Fig. (3).

General mechanism of tyrosine dephosphorylation using Shp2 residue numbering. The nucleophylic cysteine residue Cys459, part of the signature PTP loop sequence (residues 457-467), VHCSAGIGRTG, attacks the phosphorus atom to break the PO bond in an SN2-like fashion. The aspartic acid residue Asp 425, that forms part of the WPD loop, acts as the proton donor to protonate the tyrosine phenol. This generates a labile thiophosphate intermediate that is subsequently hydrolysed to liberate the free cysteine residue. The aspartic acid plays a role as a general base, by abstracting a proton from the water molecule. The phosphotyrosine binding site of PTPs is generally populated with positively charged residues to stabilize the signature, in particular the arginine residue Arg465 of the P loop is important in stabilizing the negative charge of the pentavalent phosphorus formed upon initial addition of the cysteine to the phosphotyrosine group.
The crystal structure [40] of human Shp2 apo-protein (aa 1-525, without the C-terminal region) shows that the phosphotyrosine binding sites of SH2 domains face outward away from the catalytic domain while the backside of N-SH2 domain (primarily via the D’E loop) wedges into the catalytic pocket (Fig. (4)). Thus, unliganded Shp2 is auto-inhibited by its N-SH2 domain and has a low basal PTP activity. Binding of Shp2 SH2 domains to specific phosphotyrosine containing peptides induces a conformational change that relieves the intramolecular autoinhibition. This is predicted from crystal structures [40, 41] and based on experimental evidence [42]. The tandem SH2 domains are necessary for high affinity binding to bisphosphoryl tyrosine-based activation motifs (BTAMs) [42-44].
Fig. (4).
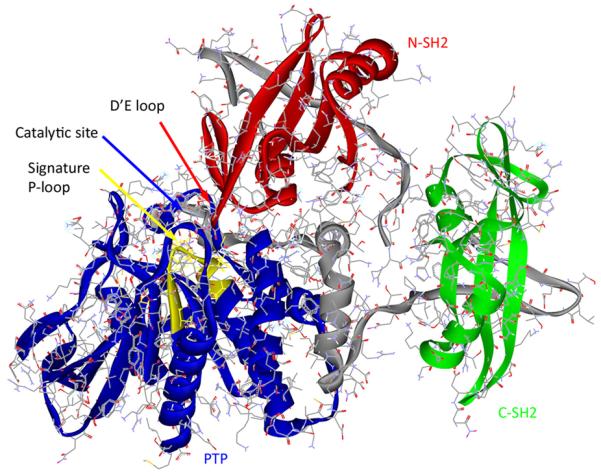
X-ray crystal structure of Shp2 (pdb code 2SHP) showing the N-SH2 (red), C-SH2 (green) and PTP (blue) domains. The two linking sequences that join these three domains are shown in gray. The signature P-loop motif (VHCSAGIGRTG) is illustrated in yellow.
Binding of Shp2 to specific BTAMs such as those located on Gab family of docking proteins are required for its biological function. Deletion of N-SH2 domain or both SH2 domains results in fully activated Shp2. However, these deletion mutants are not biologically functional. Similarly, point-mutations that disrupt the N-SH2 domain or C-SH2 domain binding activity render the mutant Shp2 biologically non-functional. In contrast to the catalytic Cys-mutant, these SH2 domain-defective Shp2 mutants do not have a dominant effect over endogenous wildtype Shp2.
Besides the crystal structure of full length Shp2 (lacking the C-terminal region) [40], a crystal structure of the Shp2 PTP domain in complex with malic acid was published recently [26]. In these crystal structures, the catalytic WPD loop is in an inactive “open” conformation [26] (Fig. (5)). In contrast, the WPD loop of the closely related Shp1 PTP is in an active “closed” conformation in the absence of a substrate in crystal structures [26, 27]. The open conformation of the Shp2 WPD loop in crystals may be due to a lower energy state of this conformation in the absence of substrate or because of spatial constraint in these crystals. Enzyme kinetic studies have shown that the Shp1 catalytic domain is a much more active enzyme than the Shp2 catalytic domain in vitro [45, 46], suggesting that the Shp2 catalytic domain is in a less optimal, perhaps “open” conformation in aqueous solution similar to that observed in the crystal structures. The distinct WPD loop conformation of these two PTPs contributes to the different surface charge distribution at areas surrounding the catalytic site [26]. This may provide a ground for PTP inhibitor selectivity. Similar to PTP1B, the Shp2 PTP domain has a secondary substrate binding pocket proximal to the catalytic site [26]. Thus, it may be possible to obtain highly potent, bidentate Shp2 PTP inhibitors that dock into these two sites simultaneously in analogy to PTP1B inhibitors [47].
Fig. (5).
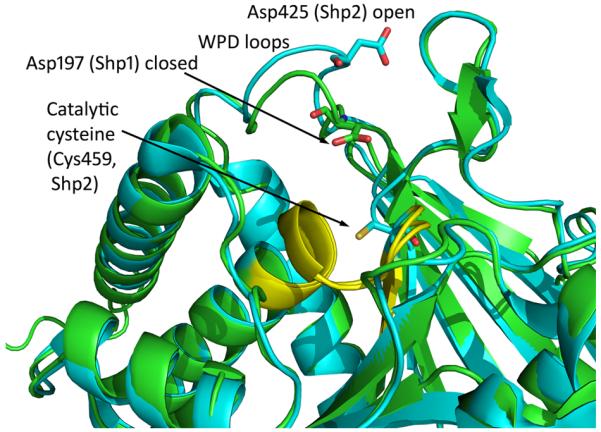
Overlay of the X-ray crystal structures of the PTP domain of Shp1 (pdb 1GWZ, colored green) and Shp2 (pdb 3B7O) colored blue, clearly indicating the closer proximity of the WPD aspartic acid residue to the catalytic cysteine residue for Shp1 as a consequence f the closed WPD loop conformation.
Pathways and substrates
The most consistently reported Shp2-regulated signaling pathway is the Ras-Erk1/2 mitogen-activated (MAP) kinase pathway. Increased peak signal strength, prolonged activation period, or both have been observed. This includes Ras-Erk1/2 activation by various growth factor receptor tyrosine kinases such as EGFR, ErbB2, c-Met, PDGFR, insulin, FGFR1, cytokine receptors such as GM-CSF and IL-6, G-protein-coupled receptors such as lysophosphatidic acid, tyrosine kinase oncogenes such as Bcr-Abl, and oncogenic bacterial protein CagA. Shp2 positively regulates the Ras-Erk1/2 MAP kinase pathway is supported by evidence obtained using dominant-negative Shp2 mutants, Shp2 knockdown with RNA interference, Shp2-defective mouse embryo fibroblast cells, Shp2 knockout mice, gain-of-function Shp2 mutants in cell cultures and in animals. Shp2-mediated Ras-Erk1/2 MAP kinase pathway is involved in regulation of cell survival, proliferation, differentiation, adhesion, and migration depending on cell contexts.
Shp2 binds directly to PDGFRβ. Only one Shp2 binding site on PDGFRβ cytoplasmic region is known. It is believed that Shp2 binds to a PDGFR dimer to achieve sufficient affinity but this has not been proven. In most growth factor- and cytokine-simulated cells, including PDGF-stimulated cells, Shp2 binds to PH-domain containing docking proteins Gab1 and/or Gab2 through interaction between the Shp2 SH2 domains and a BTAM located at the C-terminal region of Gab proteins [42]. Disruption of Gab1 and Shp2 binding impairs EGF-stimulated Erk1/2 activation [42]. Conversely, expression of a Gab1-Shp2 fusion protein results in constitutive Erk1/2 activation [42, 48]. In addition to Gab family of proteins, Shp2 binds to fibroblast growth receptor substrate 2α (FRS2α) in FGF-stimulated cells and to insulin receptor substrate-1 (IRS-1) in insulin-stimulated cells [49-51]. Shp2-FRS2α interaction is involved in FGF-stimulated Erk1/2 activation while the role of Shp2-IRS-1 interaction in Erk1/2 activation is less clear.
How Shp2 activates the Ras-Erk1/2 pathway is not completely resolved although both receptor tyrosine kinase specific and shared mechanisms have been reported (Fig. (6)). It has been found that Shp2 activates Src family kinases (SFKs) by dephosphorylating Csk docking proteins paxillin [52] or Cbp/PAG [53]. Dissociation of Csk from Src leads to Src activation. However, the link from an activated Src to Ras activation remains a subject of investigation. Shp2 is recruited to paxillin by Gab1. It is unclear how Shp2 is recruited to Cbp/PAG.
Fig. (6).

Signaling pathways from the EGF receptor to Erk1/2 and Src in adherent cells. Representative mechanisms are shown to illustrate that Shp2 activates Src family kinases by preventing recruitment of Csk to Src complexes and activates Ras-Erk1/2 by regulating RasGAP and Src.
It was reported that Shp2 dephosphorylates p120RasGAP binding sites on EGFR [54] or ErbB2 [55] to prevent membrane localization of p120RasGAP via these ErbB proteins. This is analogous to corkscrew binding to Torso in the Drophophila PDGFR pathway [56] and is receptor-specific. It was also reported that p120RasGAP binds to tyrosine-phosphorylated Gab1 and Shp2 prevents such binding by dephosphorylation the tyrosine phosphorylation site thereby activate Ras [57]. Shp2 was reported to dephosphorylate Sprouty in FGF-stimulated cells that results in dissociation of Grb2 from Sprouty [58].
Several transmembrane glycoproteins bind Shp2. These include Shp substrate 1 (SHPS-1)/Signal regulatory protein (SIRP) [59], PECAM-1/CD31 [60], myelin protein zero-like-1 (MPZL1/PZR) [61] and several immunoreceptors that contain immunoreceptor tyrosine-based inhibitor motif (ITIM) such as FcγRIIB [62]. In contrast to specific binding of non-transmembrane docking proteins to Shp2, these transmembrane proteins bind both Shp1 and Shp2 and, in some cases, the phosphoinositide 5-phosphatase SHIP.
Shp2 is a regulator of cell adhesion and migration. These functions are mediated by dephosphorylation of focal adhesion kinase (FAK), activation of Src kinase, and the Ras-Erk1/2 pathway in addition to transcription-mediated events. Shp2 was reported to bind β4 integrin in Met-activated cells, which also activates c-Src [63].
While Shp2 is a positive regulator of cell growth and migration, it is a negative regulator of interferon signaling. Interferons induce STAT1 or STAT1/STAT2 tyrosine phosphorylation. STAT1 is a Shp2 PTP substrate [64]. Increased IFN-γ-induced responses are observed in mouse embryo fibroblasts lacking Shp2 [64, 65].
Roles in cancer
Shp2-regulated Ras-Erk1/2 MAP kinase pathway, SFKs, and FAK are involved in tumor growth and metastasis. Overexpression of Shp2 has been observed in infiltrating breast carcinoma [66] and in primary leukemia cells [67]. It was reported that inhibition of Shp2 with a catalytic-inactive Shp2 mutant (Shp2C459S) abolished anchorage-independent growth of breast cancer cell lines and knockdown of Shp2 in BT20 and BT474 breast cancer cells resulted in a normal breast epithelial morphology [68]. Activating mutations of FGFR3 are associated with bladder carcinoma and multiple myeloma. It was reported that transformation of NIH3T3 cells by a constitutively active FGFR3 mutant is inhibited by a Shp2 C459S mutant [69]. The aberrant EGFRvIII (deletion exon 2-7) mutant is detected in 40-50% of human glioblastoma. EGFRvIII is also detected in about 5% of human lung squamous cell carcinoma [70]. Soft agar colony formation and tumor growth in nude mice of EGFRvIII-expressing glioblastoma cells are inhibited by a Shp2C459S mutant [71], indicating that Shp2 PTP activity contributes to EGFvIII-induced cell transformation and tumor growth. The Shp2 docking protein Gab2 is a Bcr-Abl substrate. In fact, it was originally cloned as a Shp2-associated tyrosine-phosphorylated protein from Bcr-Abl-transformed cells. It has been reported that proliferation of Bcr-Abl+ CML cell lines and primary CML CD34+ cells, but not normal CD34+ cells, is inhibited by Shp2 shRNAs [34]. Anaplastic large cell lymphoma is associated with NPM-ALK fusion gene. Shp2 was found to bind NPM-ALK oncogene and regulated anaplastic lymphoma cell growth [72]. Virulence factor CagA-positive H. pylori are carcinogenic bacteria that cause gastric cancer. After injecting into human gastric epithelial cells, the membrane-associated CagA becomes tyrosine phosphorylated and recruits Shp2 to induce gastric epithelial cell transformation [73]. Together, these findings suggest that Shp2 is required for cell transformation and malignant growth of several oncogenic tyrosine kinases and the bacterial oncogenic protein CagA. The list of tyrosine kinase oncogenes known to be dependent on Shp2 for their malignant activities is likely to increase in the coming years.
While the wildtype Shp2 may function as a non-oncogene addiction gene, gain-of-function Shp2 mutants have been established as oncogenes. Mutations in the PTPN11 gene that encodes Shp2 were originally discovered in the childhood development disorder Noonan syndrome as germline mutations [74]. Subsequently, somatic mutations in PTPN11 genes were identified in 35% of juvenile myelomonocytic leukemia (JMML), 10% of childhood myelodysplasic syndrome (MDS), 7% of B-cell acute lymphoblastic leukemia (B-ALL), 4% of childhood acute myelogenous leukemia (AML), and in some cases of colon cancer, lung cancer, melanoma, and neuroblastoma [75-77]. JMML is an aggressive myeloproliferative disorder (MPD)/MDS. JMML cells are hypersensitive to GM-CSF. Ras and neurofibromin (NF1) mutations together are associated with 50% of JMML patients. Thus, PTPN11 is the most frequently mutated genes in JMML patients. Cancer-associated Shp2 mutations occur most frequently in N-SH2 domain in residues that affect N-SH2 domain and PTP domain interaction. Cancer-associated Shp2 mutations either have been demonstrated or are predicted to be gain-of-function mutants. No loss-of-function Shp2 mutant has been found in human cancer. Several laboratories have reported leukemia-associated Shp2 mutants (E76K, D61Y, D61V) are able to confer GM-CSF and IL-3 hypersensitivity of murine hematopoietic progenitor cells from fetal liver or bone marrow and evoked GM-CSF-independent colonies [78-80]. The Shp2 PTP activity and both SH2 domains are required for the transformation activity. In Balb/c mice, transplant of bone marrow cells infected with Shp2 mutant-encoding retroviruses results in fatal JMML-like disease and lymphoproliferation [80]. In human cytokine-dependent TF-1 myeloid cells, expression of the leukemia-associated Shp2 E76K mutant results in cytokine-independent transformation of these cells [81]. Recently, Chan et al reported that conditional expression of the leukemia-associated Shp2 D61Y mutant in hematopoietic cells of knockin mice led to a fatal MPD [82]. Thus, mutant Shp2 has been established as an oncogene in leukemia.
Small molecule Inhibitors
By screening the NCI Diversity Set-1 library, NSC-87877 (1, Fig. (7)) was identified as a Shp2 PTP inhibitor with an IC50 of 0.33 μM [83]. NSC-87877 inhibits Shp1 with the same potency. It is 5- and 24-fold less potent at inhibiting PTP1B and HePTP, respectively, and displays >200-fold selectivity against DEP1, CD45, and LAR. Molecular docking analysis suggests that NSC-87877 forms hydrogen bonds with the backbone NH group of Arg-465 in the PTP signature motif and with side-chains of Lys-280 and Asn-281. Mutational experiments with the non-conserved Lys and Asn suggested that these two residues are involved in the interaction with NSC-87877. Comparison of NSC-87877 sensitivity of wildtype Shp2, Shp2E76K mutant, and Shp2 PTP domain indicated that the wildtype Shp2 is ~4.5-fold less sensitive to NSC-87877. Because the catalytic pocket of the wildtype Shp2 is blocked by N-SH2 domain, the required higher concentration of NSC-87877 necessary to achieve the same degree of inhibition of wildtype Shp2 supports the notion that NSC-87877 binds to the catalytic pocket. NSC-87877 has been reported to inhibit Shp2 in epithelial/carcinoma cells, endothelial cells, fibroblasts, muscle cells, and neuronal/glioma cells [71, 83-86]. However, NSC-87877 does not appear to be able to enter hematopoietic cells. With two arylsulfonic acid groups, we suspect that NSC-87877 is a substrate of the organic anion transporter (OATP) family of membrane transporters, although this has not been proven experimentally.
Fig. (7).

Representative of Shp2 inhibitors. (1), NSC-87877. (3) and (4) are synthesized based on screening hit (2). (5) and (6), PHPS1 and PHPS4.
Based on another hit, NSC-117199 (2a), (Shp2 IC50 = 47 μM) from a Shp2 PTP inhibitor screen of the NCI Diversity Set-1 chemical library, >200 analogs were synthesized [29]. The focused library synthesis involved exploitation of sulfonic acid moiety and the nitro group in the NSC-117199 scaffold to generate potent compounds with IC50 values of around 1 μM (e.g. 2b). The library of isatins contained polar replacements of the sulfonic acid of 2a phosphotyrosine mimics. The docking pose of isatin 2b is shown in Fig. (8). In this case the compounds docks best to the Shp2 catalytic site with the ortho-carboxylate bound close to the catalytic cystein residue. Similarly, the nitro group in NSC-117199 was replaced with more stable carboxylic acid moiety. These moieties appear to be essential for Shp2 inhibitory activity and provide a pharmacophore for further development of novel compounds for Shp2. Several compounds have IC50 to Shp2 ~ 1 μM, with various degrees of selectivity against Shp1 and PTP1B were identified. Two compounds from this series (3 and 4), have > 10-fold selectivity against Shp1 and PTP1B.
Fig. (8).
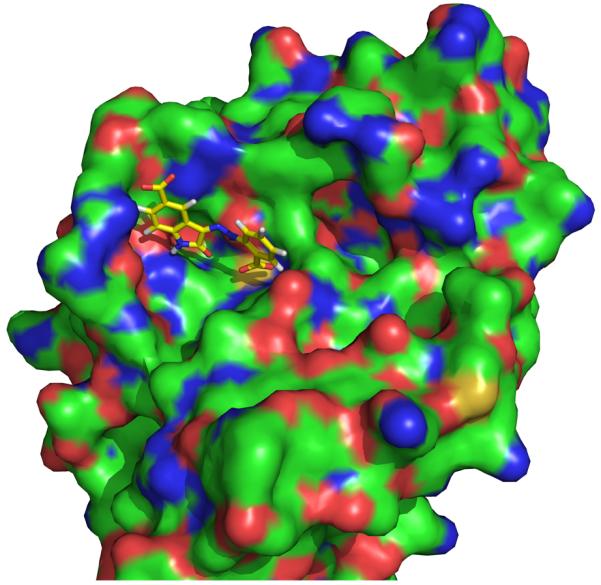
Docked structure of isatin (2b) with Shp2 (derived from structure pdb code 2SHP), colored by element: carbon, green; oxygen, red; nitrogen blue, sulfur, yellow: hydrogens removed from the protein for clarity.
Using a homology model of Shp2 PTP domain based upon a crystal structure of a PTP1B in complex with a potent small molecule inhibitor, Birchmeier and colleagues performed virtual screen of ~2.7 million compounds and identified phenylhydrazonopyrazolone sulfonate 1 (5) (PHPS1) and PHPS4 (6) as Shp2 selective inhibitors [28]. PHPS1 is less potent (Shp2 IC50: 2.1 μM) than PHPS4 (Shp2 IC50: 0.63 μM) but has greater selectivity against other PTPs. The phenyl sulfonate group of PHPS1 appears to be essential for the inhibitor activity. The computer model suggests that the phenyl sulfonate group inserts into the active site and forms hydrogen bonds with ILE-463, Gly-464, Arg-465, Ser-460, and Ala-461, whereas the pyrazolone core and two other phenyl rings make contacts with residues at the periphery of the catalytic pocket (Fig. (9)). The model is supported by mutational studies using PTP1B and Shp2 mutants. PHPS1 is able to inhibit Shp2 PTP in several cell-based assays [28].
Fig. (9).

Proposed schematic docking model of PHPS1 (5) with the catalytic site of Shp2. Hydrogen bonds between the ligand and phosphatase as predicted by MOE v2006.3 are shown as hatched lines.
By screening a natural product-based compound collection, Waldmann and colleagues identified a furanodictine class of Shp2 inhibitor (7, Shp2 IC50: 2.5 μM) that is highly selective against PTP1B (PTP1B IC50: >100 μM) [87]. With >40-fold selectivity against PTP1B, compound (7) represents the most selective Shp2 inhibitor against PTP1B identified so far. Three other less selective furanodictine compounds were also identified that inhibited both Shp2 and PTP1B with IC50 <10 μM. It is unknown whether these are competitive Shp2 inhibitors or how they interact with the Shp2 PTP domain. No cell-based experiment was reported in that study so that it is unclear whether these compounds are cell permeable or are indeed cell active in a Shp2-dependent manner.
Geronikaki et al synthesized and tested three series of 2-thiazolylimino/heteroarylimino-5arylidene-4-thiazolidinones with thiazole-, benzo[d]thiazole-, or benzo[d]isothizole rings as Shp2 inhibitors [88]. Computer docking suggests that the phenyl ring is oriented to the catalytic pocket. The most potent compounds (8 and 9, both have a Ki against full length GST-Shp2 of 11.7 μM as measured by the pNPP assay) have a 4-methoxyl-substitued on the phenyl ring. These compounds do not contain a negatively charged sulfonic acid or carboxylic acid group featured in many PTP inhibitors. However, the pKa of the thiazolidinone may be sufficiently low in these systems to render these compounds ionic under physiological conditions. Nevertheless, these compounds are > 10-times less potent than NSC-87877, compound (4), and PHPS1, which have sub-micromolar Ki to Shp2 ([28] and our unpublished data). The selectivity and cell permeability of these compounds were not reported.
Based on a putative binding pocket of Shp2 for substrate pY+5 residues, which is different between Shp1 and Shp2, Yu et al screened a database of 1.3 million compounds by docking into this secondary pocket [89]. Compound (10) (Fig. (10)) was identified that inhibits Shp2 in a PTP assay using an insulin receptor-derived decapeptide as the substrate (IC50: ~100 μM) but did not inhibit Shp1 at 100 μM. If the binding mode is confirmed, this may provide a precursor for synthesizing bidentate inhibitors in combination with inhibitors that bind to the catalytic site.
Fig. (10).

Shp2 inhibitors and other PTP inhibitors that cross-inhibit Shp2. (10), an Shp2 selective inhibitor identified from virtual screening of secondary binding site. (11), a potent PTP1B that cross inhibits Shp2. (13) and (14), orally bioavailable Glepp1 inhibitors that cross-inhibits Shp2. (15), a nuclear receptor small heterodimer partner that cross inhibits Shp2.
Inhibitors designed for PTP1B that cross-inhibit Shp2 have also been reported. These include a bidentate PTP1B inhibitor (11), (PTP1B Ki: 2.4 nM; Shp2 IC50: 10 μM) [47] and a bis(trifluoromethanesulfonamide) (12) (Shp2 IC50: 1.8 μM). Gobert et al synthesized two Glepp1 PTP inhibitors (13 and 14, IC50: 0.12 μM, 0.34 μM ) that potently cross-inhibit Shp2 (IC50: 0.39 μM, 0.65 μM) [90]. Compounds (13) and (14) are orally bioavailable and have protective effect on dextran sulfate sodium-induced ulcerative colitis in mice [90]. In an effort to develop retinoid-derived orphan nuclear receptor small heterodimer partners that induce apoptosis in leukemic cells, compound (15) was found to inhibit Shp2 (IC50: 0.45 μM) [91].
Assessment of target inhibition
PTP activity assay in vitro
PTP inhibitors are usually screened by in vitro PTP activity inhibition assay [14]. Shp2 PTP activity may be assayed in vitro using the fluorogenic 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) (available from Invitrogen). DiFMUP is non-fluorescent and is hydrolyzed to the fluorescent DiFMU (maximal excitation/emission at ~358/450 nM) by the PTP. The fluorescent signal can be read without stopping the PTP reaction. In chemical library screening, potential hits may be missed if these compounds are fluorogenic. The chromogenic p-nitrophenyl phosphate (pNPP) is another generic PTP substrate frequently used in PTP activity assay in vitro. Hydrolysis of pNPP yields the yellow colored p-nitrophenol at alkaline pH that can be measured with a spectrophotometer at a wavelength of 405 nm. A potential problem is that some of screening samples may be colored and may interfere with the assay readout. Thus, appropriate controls without the PTP should be included in a confirmation screen. Alternatively, hydrolysis of pNPP can be determined by measuring the liberated free phosphate with the Malachite green assay [42] or the use of Biomol Green reagent (available from Biomol). Free-phosphate contamination is the major interference using the Biomol Green reagent. The pNPP-based assays require stopping of reaction for color development so that only one time point can be obtained from each reaction. Commercial pNPP usually does not have sufficient purity and gives a high background signal. Thus, further purification of pNPP obtained from commercial sources may be necessary. In addition to small molecule substrates, PTP activity can be assayed using phosphopeptide substrates of the PTP and free phosphate release measured using Malachite green/Biomol Green reagents. This has the benefit of potentially identifying compounds that inhibit the PTP activity by binding to the peptide substrate binding site. In chemical library screening, secondary confirmation assays should be performed using different substrates and/or methods to evaluate whether a hit interferes with the assay measurement and is not a true inhibitor of the PTP. Ideally, PTP activity is assayed using a substrate at the concentration that equals its Km. Under this condition, a competitive inhibitor will have an IC50 = 2 x Ki.
Assessment of target inhibition in vivo
Shp2 PTP is activated in the cells after EGF stimulation. Therefore, inhibition of Shp2 by a Shp2 inhibitor may be determined directly by immunoprecipitation of Shp2 from cells after stimulation with EGF and the Shp2 PTP activity measured by an appropriate assay. This has been used in our study of NSC-87877, in which we assayed Shp2 inhibition in EGF-stimulated HEK293 and MDA-MB-468 cells [83]. This assay requires that the Shp2 inhibitor binds tightly to Shp2 so that the inhibitor-enzyme complex does not dissociate completely during immunoprecipitation. For an inhibitor such as a competitive inhibitor that does not covalently modify the enzyme, it is difficult to correlate the extent of Shp2 PTP inhibition in the cells by a sub-saturated concentration of the inhibitor with the percentage of Shp2 PTP inhibition in the immune complex PTP assay. This is because the amount of inhibitor dissociated from the immune complex during immunoprecipitation is unknown.
A Shp2 substrate in EGF-stimulated MDA-MB-468 breast cancer cells is paxillin [52]. Paxillin is tyrosine-phosphorylated under serum-starved conditions and is dephosphorylated by Shp2 after EGF-stimulation. The Shp2 inhibitor NSC-87877 has been shown to block EGF-induced paxillin tyrosine dephosphorylation in MDA-MB-468 cells [83]. Similarly, Hellmuth et al found that HGF also induced paxillin dephosphorylation in MDCK cells, which was attenuated by the Shp2 inhibitor PHPS1 [28].
Shp2 mediates Ras-Erk1/2 activation by growth factors. Shp2-mediated signaling pathways may regulate the strength of the peak activity or the length of the activation phase. Growth factor-induced Erk1/2 activation has been used as a surrogate marker for Shp2 inhibition in EGF- and HGF-stimulated cells by Shp2 inhibitors [28, 83]. In HEK293 cells, NSC-87877 inhibited EGF-stimulated Erk1/2 activation with an EC50 of 6 μM [83]. In MDCK cells, although PHPS1 did not affect HGF-stimulated phospho-Erk1/2 (pErk1/2) signal at 5 min, it inhibited HGF-stimulated pErk1/2 signal at 15-360 min [28].
Three approaches may be used to rule out the possibility that a Shp2 inhibitor non-specifically blocks Erk1/2 activation. The first is by comparison of Erk1/2 activation by a growth factor and by phorbol ester. While Shp2 mediates EGF- and HGF-induced Erk1/2 activation, the phorbol ester-induced Erk1/2 activation is independent of Shp2. Therefore, phorbol ester-induced Erk1/2 activation should not be affected by inhibition of Shp2 PTP activity. This has been used for determination of specific inhibition of the Shp2-dependent Erk1/2 activation by NSC-87877 and PHPS1 [28, 83]. The second approach utilizes active Shp2 constructs that by-pass the upstream signaling steps of receptor tyrosine kinases. Constitutive Erk1/2 activation can be initiated intracellularly using a chimeric protein of Gab1 PH domain and N-SH2 domain deletion Shp2 mutant or using a myristoylated Shp2 [48]. This approach has been used in evaluation of NSC-87877 [83]. In addition, certain cell lines that express the leukemia-associated Shp2E76K mutant also result in constitutive Erk1/2 activation [28, 81]. These cell lines may be used to assess Shp2 inhibition by monitoring pErk1/2 as reported [28]. Because Ras is downstream of Shp2, constitutively active Ras mutant-induced Erk1/2 activation by-passes the requirement of Shp2 PTP activity. Thus, cells expressing a constitutively active Ras, such as H-RasV12, is used as an approach to rule out the possibility that a Shp2 inhibitor suppresses Erk1/2 activation by affecting a signaling step downstream of Ras [28].
In addition to these biochemical analyses, cell-based biological responses have also been used to assess Shp2 PTP inhibition in the cells. HGF-induced scattering and branching morphogenesis of MDCK cells is dependent on Shp2 PTP activity [92, 93]. HGF-induced MDCK cell scattering was used as a secondary assay in the screening of Shp2 inhibitors by Hellmuth et al [28], while MDCK cell scattering induced by HGF and by a myristoylated, active Shp2 construct was used to examine PHPS1 on inhibition of Shp2 in MDCK cells. The effect of PHPS1 on MDCK cells was phenocopied by Shp2 shRNAs.
In solid tumor-derived cell lines, inhibition of Shp2 with small molecule PTP inhibitors, shRNAs or dominant negative mutants can significantly reduce cell proliferation and soft-agar colony formation, indicating that inhibition of Shp2 can suppress cancer cell proliferation and the transformation phenotype. However, the extent of inhibition varies among different cancer cell lines [28]. Parameters for prediction of sensitivity to Shp2 inhibition in carcinoma cell lines have not been established.
Gain-of-function Shp2 mutants such as Shp2E76K and Shp2Y61D can transform certain cytokine-dependent myeloid cell line [81] and can cause fatal myeloproliferative disease in conditional knockin mice [82]. It is anticipated that these cell lines and animal models will be used in the future for pre-clinical studies of Shp2 PTP inhibitors.
OTHER PTP CANDIDATES OF ANTICANCER DRUG TARGETS
PTP1B
PTP1B (encoded by the PTPN1 gene) is a Class I non-receptor classical PTP. It contains a PTP domain, two Pro-rich sequences and a hydrophobic endoplasmic reticulum anchor domain at the C-terminal region. PTP1B dephosphorylates insulin receptor, which inhibits insulin signaling and thus contributes to insulin resistance in Type II diabetes. PTP1B was reported to activate c-Src by dephosphorylation of the inhibitory pTyr-530 [94]. PTP1B-mediated Src activation is required for transformation of MCF-10A by ErbB2 (HER2/Neu) [95] and contributes to tumor growth of SW48 colon cancer cells [96]. Another PTP1B substrate is p62Dok, which binds p120RasGAP and is a negative regulator of the Ras-Erk1/2 pathway. PTP1B-deficient fibroblasts have reduced PDGF-induced Ras and Erk1/2 activation but increased p62Dok tyrosine phosphorylation and active Akt [97]. PTP1B activity also promotes cell migration [98].
PTP1B is overexpressed in breast and ovarian cancer, which correlates with ErbB2 overexpression [99, 100]. In transgenic mouse models of mammary tumorigenesis, PTP1B deficiency delays tumor onset induced by ErbB2 and resistance to lung metastasis [101, 102], whereas overexpression of PTP1B in mammary gland results in development of spontaneous mammary tumors [102]. The effects of PTP1B deficiency in these transgenic mice correlates with reduced Erk activation. These results illustrate PTP1B as a tumor promoter and suggest PTP1B as a therapeutic target in ErbB2-positive breast cancer.
However, tumor suppressor effects of PTP1B have also been observed. PTP1B deficient mice do not have spontaneous tumor incidence. In p53 null mice, PTP1B deficiency renders mice more susceptible to B-cell lymphomas [103], which correlates with decreased apoptosis. PTP1B is a negative regulator of c-Met, Bcr-Abl, and p130Cas. Thus, PTP1B has dual roles in cancer.
Since PTP1B was identified as a target for diabetes and obesity, extensive efforts have been undertaken by industrial and academic laboratories to develop PTP1B PTP inhibitors [6, 7]. The discovery of the secondary substrate binding site near the catalytic pocket of the PTP1B has facilitated the development of bidentate inhibitors [6, 31, 32, 47]. Several PTP1B inhibitors with low nanomolar Ki have been identified [6]. Among the most potent and selective PTP1B inhibitor is the bidentate difluoromethylphosphonate containing compound (11) (Fig. (10)), which has 2.4 nM Ki to PTP1B, > 10-fold selectivity against TCPTP and >600-fold selectivity against other PTPs [47]. However, compound (11) is not cell permeable. Cell permeable analogs such as compound (16) (Fig. (11)) and prodrug strategies have been used to deliver compound (11) into cells [104, 105]. Four series of difluoromethylphosphonate containing compounds were optimized at Merck Frosst Canada using bioavailability in rodents as a key parameter [106]. Compound (17) (Fig. (11)) was identified as an orally active PTP1B inhibitor (IC50: 120 nM) with good pharmacokinetic properties. The selectivity of compound (17) against other PTPs is unclear except that it does not inhibit CD45 (IC50: >50 μM). Compound (17) is able to lower blood glucose level in mice in a dose dependent manner, indicative of PTP1B inhibition in the animals [106]. In an ErbB2-induced mammary tumor mouse model, orally administration of compound (17) (30 mg/kg for 21 days) significantly delays the onset of mammary tumor development [102]. The median time to tumor onset was extended from 28 days in the vehicle-treated mice to 75 days in the drug-treated mice. It is possible that compound (17) is not specific for PTP1B and the potential cross-inhibition of other PTPs may contribute to its anti-tumor effect. However, the combination of genetic and pharmacologic evidence demonstrates PTP1B as target for intervention of ErbB2-dependent breast cancer.
Fig. (11).

Representative of PTP1B inhibitors. (16), a cell permeable analog of compound (11). (17), an orally bioavailable PTP1B inhibitor. (18), Ertiprotafib. (19) MSI-1436 (Trodusquemine)
Although not tested as anti-cancer drugs, two PTP1B inhibitors, Ertiprotanib (18) and MSI-1436 (Trodusquemine) (19) have progressed to human clinical trials as anti-diabetes and anti-obesity drugs [107, 108]. The development of Ertiprotanib was discontinued because of insufficient efficacy and concern of multiple in vivo targets. Trodusquemine is a natural product isolated from the dogfish shark squalus acanthias [109]. It suppresses appetite and causes fat-specific weight loss in diet-induced obsese mice [108]. Trodusquemine is in phase I trials.
PRL-3
The phosphatase of regenerating liver (PRL) sub-group of DSPs are ~20 kDa small PTPs with three members PRL-1 (PTP4A1), PRL-2 (PTP4A2), and PRL-3 (PTP4A3) [19, 110]. The PTP domain of PRLs is followed by a polybasic region and the CAAX prenylation motif at the C-terminus. The polybasic region facilitates interaction with membrane phospholipids and nuclear localization. PRLs can be farnesylated via the C-terminal CAAX box. Farnesylated PRLs are associated with the plasma membrane and early endosome [111]. PRL-1 has been detected in nuclei.
After the seminal discovery that PRL-3 is overexpressed in metastatic colorectal cancer [112], many laboratories have investigated the roles of PRLs in human cancer. Elevated PRL-3 expression has been detected in metastatic lesions of colorectal cancer regardless the site of metastasis [113]. High levels of PRL-3 expression in primary colorectal cancer correlated with poor prognosis [114]. Up-regulation of PRL-3 has also been reported in primary breast, gastric, liver, ovarian, nasopharyngeal carcinoma, myeloma, and various cancer cell lines [19, 115-121]. In tumor xenograft experiments, Zeng et al reported that Chinese hamster ovary (CHO) cells expressing PRL-1 and PRL-3 develop metastatic tumors in mice [122] and that the PTP activity is essential for tumor metastasis [123]. Association of PRL-1 and PRL-2 expression with human cancer has not been thoroughly examined. However, PRL-1 and PRL-2 are expressed in most tissues.
PRLs have been found to regulate RhoA and RhoC [124], down-regulate PTEN [125], control Src expression and activity, and activate Erk1/2 [126, 127]. Forte et al reported that PRL-3 dephosphorylates pThr-567 on Ezrin in HCT116 colon cancer cells [128]. In cell cultures, PRL-3 has been shown to promote cell proliferation, migration, invasion, epithelial-mesenchymal transition, and angiogenesis. Therefore, extensive data have shown that PRLs, especially PRL-3, play important roles in metastatic cancer.
The abundant evidence supporting the cancer promoting roles of PRLs has sparked interests in the search for PRL inhibitors. Solution structures of human PRL-3 [129, 130] and a crystal structure of human PRL-1 [131] have been determined. The catalytic pockets of PRLs are unusually shallow and wide, indicative of difficult drug targets. Nevertheless, Daouti et al employed a fluorescence polarization assay to screen the Roche chemical library and identified thienopyridone (20) (Fig. (12)) as a selective PRL inhibitor [132]. Thienopyridone inhibits PRL-3 with an in vitro IC50 of 0.457 μM and displays > 100-fold selectivity against other PTPs, although the mechanism of inhibition has not been defined. Thienopyridone is cell active and induces p130Cas cleavage and anoikis and inhibits anchorage-independent growth of colon cancer cells.
Fig. (12).
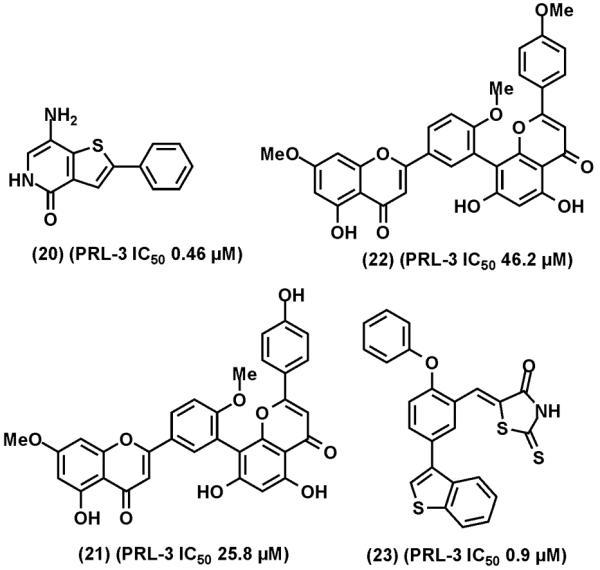
Representative of PRL-3 inhibitors. (20), thienopyridone. (21), ginkgetin. (22), sciadoptysin. (23), a rhodanine derivative.
In other efforts, natural products ginkgetin (21) and sciadoptysin (22) were identified as weak inhibitors of PRL-3 (IC50: 25.8 and 46.2 μM, respectively) [133]. Twelve lead compounds of PRL-3 inhibitors, with IC50 values in the range of 10-50 μM, were identified by virtual screening of an interbioscreen library using the solution structure of PRL-3 [134]. A panel of rhodanine derivatives were evaluated as PRL-3 inhibitors and compound (23) was found to inhibit PRL-3 with an IC50 value of 0.9 μM [135]. Furthermore, Pathak et al reported that the anti-protozoa drug pentamidine inhibits PTPs including PRL-3 [136].
Given that PRL-3 is frequently up-regulated in various types of metastatic cancer, effort has also been made to suppress PRL-3 expression. While the molecular target is unknown, curcumin, a polyphenol component of the spice turmeric, has been found to downregulate PRL-3 mRNA in a manner independent of p53 [137]. While this does not represent a traditional approach to drug discovery of molecular cancer targets, it highlights the usefulness of compounds derived from natural products.
CDC14
Cdc14 is an evolutionally conserved DSP. In budding yeast, Cdc14 is required for mitosis exist by inactivating mitotic Cdk through dephosphorylation of Cdk substrates Cdh1 and Sic1 [138]. Mammalian cells have two Cdc14, Cdc14A and Cdc14B. Cdc14s are Pro-directed phosphatases that dephosphorylate specific phosphorylation sites within the pSer/pThr-Pro motif, which are substrate sites of Pro-directed kinases Cdks or MAP kinases. Cdc14B has a nuclear targeting sequence in the N-terminal region. Besides the PTP domain, Cdc14s have non-conserved C-terminal regions. Cdc14A and Cdc14B are differently regulated in mammalian cells. Cdc14A is localized at the interphase centrosome and at the spindle midzone. Cdc14B is localized at nucleoli during interphase and at the mitotic spindle during mitosis. Overexpression of Cdc14A results in premature centrosome splitting and multi-polar spindles [139], suggesting that over-reactive Cdc14 contributes to genomic instability. Down regulation of Cdc14A leads to centrosome separation and cytokinesis defects and induces cell death [139]. Cdc14A is up-regulated in human cancer cell lines that contain p53 mutants [140]. Although the critical Cdc14 substrates that regulate centrosome separation and cytokinesis in mammalian cells remain to be identified, a few Cdc14 substrates have been reported. These include RN-tre, Cdc25A and p53 [140-142].
No Cdc14 inhibitor discovery effort has been reported. The structure of Cdc14 PTP catalytic domain is consisted of tandem A- and B-DSP domains, in which the PTP signature motif C(X)5R is located in the B-domain. [143]. The catalytic site is located in a narrow, long groove formed at the interface of the A- and B-domains. Three acidic residues are located at one end of the substrate binding groove. These interesting features of the Cdc14 catalytic site are attractive characteristics of a potentially useful drug target.
LMW-PTP
Low molecular weight PTPs (LMW-PTPs) are encoded by the ACP1 gene. ACP1 is the sole Class II Cys-based PTP gene in human [144]. LMW-PTPs are ~18-kDa and have the conserved Cys-based PTP signature motif C(X)5R but little sequence homology with other PTPs. Four isoforms are produced by alternative splicing. Only isoforms 1 and 2 (IF1 and IF2) have PTP activity. While IF1 and IF2 differ in in vitro enzyme kinetics properties [145], their functional difference in the cells is unclear. A number of substrates of LMW-PTP have been reported that include EphA2, FAK, PDGFR, insulin receptor, ZAP-70, and STAT5. It has been reported that upregulation of LMW-PTP in colon cancer and neuroblastoma correlates with unfavorable outcome [146]. Overexpression of LMW-PTP is sufficient to transform MCF10A breast epithelial cells [20]. Overexpression of LMW-PTP in NIH3T3 cells causes early onset and larger tumors in nude mice while the catalytic inactive LMW-PTP inhibit NIH3T3 tumor growth [21]. Cell transformation and tumor promoting activities of LMW-PTP are associated with dephosphorylation of the EphA2 receptor tyrosine kinase [20, 21].
Two natural products morin and ferruginol are reported to weakly inhibit LMW-PTP [147, 148]. In an effort to optimize flavonoid-based LMW-PTP inhibitors, Forghieri et al identified several compounds having sub-micromolar to low micromolar IC50 to LMW-PTP, although these compounds display similar inhibitory activity to PTP1B [149]. In another effort, Maccari et al characterized a series of compounds based on the 4-[(5-aryldene-2,4-dioxothanolidin-3-yl)methyl]benzoic acid scaffold and identify several potent LMW-PTP inhibitors having sub-micromolar IC50 values [150]. Again, these compounds cross-inhibit PTP1B.
Cdc25
Cdc25 are class III Cys-based PTPs. Three isoforms are present in mammals, Cdc25A, Cdc25B, and Cdc25C. They are dual specificity phosphatases (DSPs) that activate cyclin-dependent kinases (Cdks) by dephosphorylation of the inhibitory dual Thr-Tyr phosphorylation sites (Thr-14 and Tyr-15 on Cdk1) in the N-terminal area of Cdks [151] (Fig. (13)). Cdc25s are positive regulators of cell cycle progression and also play a central role in the checkpoint response to DNA damage. Cdc25 expression and activity are subject to transcriptional and posttranslational regulation. Cdc25 overexpression has been reported in many types of human cancer and is correlated with higher grade tumors and poor disease-free survival. Although Cdc25 overexpression is not sufficient to cause cancer, overexpression of Cdc25A reduces the latency of MMTV-H-ras induces mammary tumors in mice and gives rise to more invasive mammary tumors in MMTV-Neu mice. [152]. Homozygous Cdc25A knockout is embryonic lethal in mice. In heterozygous Cdc25A knockout mice, the latencies of MMTV-Ras and MMTV-Neu evoked mammary tumors are significantly delayed [153]. Cdc25A+/− mouse embryonic fibroblasts are resistant to transformation by co-expression of H-RasV12 and a dominant-negative p53 and are more responsive to radiation-induced cell cycle arrest. These findings suggest that Cdc25 overexpression contributes to tumorigenesis. Interestingly, Cdc25C is located in chromosome 5q, which is deleted in a sub-group of MDS patients. MDS patients with 5q deletion are responsive to lenalidomide treatment, which has been attributed to the haplodeficiency of Cdc25C [154]. It will be important to test if a Cdc25 inhibitor can sensitize non-5q deletion MDS cells to lenalidomide.
Fig. (13).
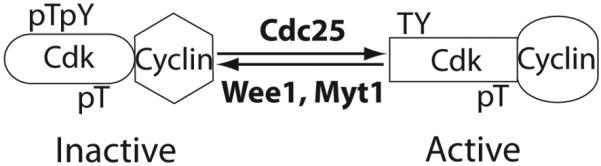
Cdc25 dual specificity phosphatases are positive regulators of Cdks. The activity of a Cdk is regulated by three phosphorylation sites Thr14, Tyr15, and Thr161 (residues numbers based on Cdk1). Thr14 and Tyr15 phosphorylation catalyzed by Wee1 and Myt1 kinases are inactivating while Thr161 phosphorylation by Cak is activating. Cdc25 phosphatases dephosphorylate Thr14 and Tyr15, resulting in Cdk activation.
Cdc25s have been targeted for novel anticancer drug discovery for more than a decade. Many Cdc25 inhibitors have been reported. These Cdc25 inhibitors belong to six classes of small molecules as summarized by Lazo and Wipf [12] that include natural products, lipophilic acids, quinones, electrophiles, phosphate mimics, peptide analogs. In contrast to PTP1B inhibitor discovery efforts in which phosphotyrosine mimetics are frequently used in the inhibitor design, redox and microreactive compounds are frequently identified among Cdc25 PTP inhibitors. Many of these reactive Cdc25 inhibitors, such as NSC-663284 (24), Fig. (14), have IC50 values in the sub-micromolar range. The most potent Cdc25 inhibitor is the bis heterocyclic quinone IRC-083864 (25) [155]. IRC-083864 inhibits Cdc25s with IC50 values in the range of ~20 nM but does not significantly inhibit other PTPs tested at 50 nM. IRC-083864 and two other less potent Cdc25 PTP inhibitors, BN82002 (26) and BN82685 (27), have been shown to inhibit tumor xenograft growth in mice [155-158].
Fig. (14).
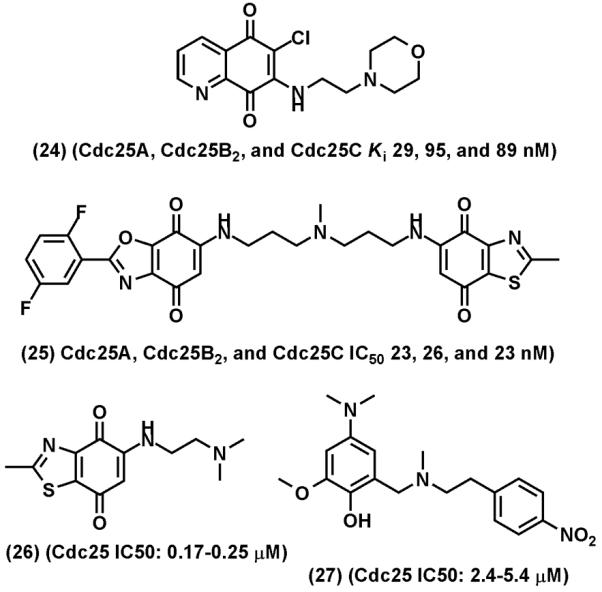
Representative of Cdc25 inhibitors. (24), NSC-663284. (25), IRC-083864, (26), BN82002, (27) BN82685.
Eya
Eyes Absents (EYAs) are Asp-based PTPs that include four members Eya1, Eya2, Eya3 and Eya4. Eya is a component of the SIX-EYA-DACH transcription network [159-161]. Eya2 is overexpressed in ovarian carcinoma and promotes tumor xenograft growth [162]. Recently, Eya has been identified as the histone H2A.X PTP, serving as the switch between DNA-damage repair and apoptosis after DNA double-strand breaks (DSBs). Tyr-142 at the C-terminus of histone H2A.X is constitutively phosphorylated under unstressed conditions by the WSTF tyrosine kinase [22, 23]. After DNA DSBs, ATM/ATR phosphorylates H2A.X at Ser-139 that generates γ-H2A.X and phosphorylates Eya3 at Ser-219 that results in localization of Eya1/Eya3 complex to γ-H2A.X and dephosphorylation of Tyr-142. Tyr-142 dephosphorylation by Eya1/Eya3 is necessary for recruitment of repairs factors via binding of DNA damage checkpoint protein MDC1 to the pSer-139 site of γ-H2A.X for repairing damaged DNA. While inhibiting MDC1 binding to pSer-139, Tyr-142 phosphorylated γ-H2A.X recruits JNK1 to initiate pro-apoptotic program. Thus, blocking Tyr-142 dephosphorylation by inhibition of Eya prevents DNA damage repair and promotes apoptosis [23, 24]. Conceivably, inhibition of Eya PTP activity may be used to enhance the therapeutic efficacy of DNA damage-based cancer therapy, such as radiation and certain chemotherapy agents.
A structure of Eya2 PTP domain shows that five conserved Asp and five conserved Glu are located on the surface around the active site [24]. The highly negative-charged active site is consistent with the finding that Eya recognizes the basic substrate histone H2A.X. While no Eya PTP inhibitor discovery effort has been reported, the recent realization of the critical role of Eya in DNA damage repair and apoptosis is likely to stimulate the quest for novel Eya PTP inhibitors.
CONCLUDING REMARK
Protein phosphatases represent 4% of the druggable genome [163]. It is now realized that specific PTPs can function as oncogenes or non-oncogene addiction genes that contribute positively to the initiation and maintenance of human cancer. Potential PTP targets for novel anticancer drug discovery are identified in all four classes of PTPs. While catalytic pockets of many PTPs are considered challenging for drug discovery efforts, appropriate surface properties surrounding PTP catalytic sites do exist that allow development of potent and selective inhibitors for many PTPs. This notion has been supported by the identification of a number of highly potent and selective inhibitors to specific PTPs.
Although progress has been made, the field of PTP inhibitor development is in its infancy. Major efforts are required by academic institutions and industries to overcome various hurdles in order to successfully develop the first PTP inhibitor as a drug. Target identification and validation in vitro and in vivo, lead discovery and optimization, identification of specific substrates in vivo and surrogate markers for assessment of target inhibition, pharmacologic profiling and selective toxicity analysis all require great efforts. The realization that PTPs do not simply counter the function of PTKs, but could cooperate with PTKs to promote oncogenesis should attract great interests in developing PTP inhibitors for cancer therapy in the coming years.
Acknowledgment
Support by National Institutes of Health grants P01CA118210, R01CA077467, and U56CA118809 is acknowledged.
ABBREVIATIONS
- AML
acute myelogenous leukemia
- BTAMs
bisphosphoryl tyrosine-based activation motifs
- Cdk
cyclin dependant kinase
- CML
chronic myeloid leukemia
- csw
corkscrew
- DiFMUP
6,8-difluoro-4-methylumbelliferyl phosphate
- DSB
double strand break
- DUSP/DSP
dual specificity phosphatase
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- Erk1/2
extracellular signal regulated kinase 1 and 2
- Eyas
eyes absents
- FAK
focal adhesion kinase
- FDA
Food and drug administration
- FGFR
fibroblast growth factor receptor
- FRS2α
Fibroblast growth factor receptor substrate 2α
- GAP
GTPase activating protein
- GM-CSF
granulocyte macrophage-colony stimulating factor
- IFN-γ
interferon gamma
- IL
interleukin
- IRS-1
insulin receptor substrate-1
- ITIM
immunoreceptor tyrosine-based inhibitor motif
- JMML
juvenile myelomonocytic leukemia
- LMW
low molecular weight
- MAP
mitogen-activated protein
- MDS
myelodysplastic syndrome
- MPD
myeloproliferative disorder
- MPZL1
myelin protein zero-like-1
- OTAP
organic anion transporter
- PDGFR
platelet-derived growth factor receptor
- PECAM-1
platelet endothelial cell adhesion molecule
- PH
pleckstrin homology
- PHPS
phenylhydrazonopyrazolone sulfonate
- pNPP
p-nitrophenyl phosphate
- PRL
phosphatase of regenerating liver
- PTEN
phosphatase and tensin homolog 10
- PTK
protein tyrosine kinase
- PTP
protein tyrosine phosphatase
- pY
phosphotyrosine
- SFK
Src family kinase
- SH2
src-homology-2
- SHPS1
Shp substrate-1
- SIRP
signal regulatory protein
- STAT
signal transduction activator of transcription
REFERENCES
- [1].Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009;21:140–6. doi: 10.1016/j.ceb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- [3].Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ostman A, Hellberg C, Bohmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6:307–20. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- [5].Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–46. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- [6].Bialy L, Waldmann H. Inhibitors of protein tyrosine phosphatases: next-generation drugs? Angew Chem Int Ed Engl. 2005;44:3814–39. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- [7].Jiang ZX, Zhang ZY. Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer Metastasis Rev. 2008;27:263–72. doi: 10.1007/s10555-008-9113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tautz L, Pellecchia M, Mustelin T. Targeting the PTPome in human disease. Expert Opin Ther Targets. 2006;10:157–77. doi: 10.1517/14728222.10.1.157. [DOI] [PubMed] [Google Scholar]
- [9].Vintonyak VV, Antonchick AP, Rauh D, Waldmann H. The therapeutic potential of phosphatase inhibitors. Curr Opin Chem Biol. 2009;13:272–83. doi: 10.1016/j.cbpa.2009.03.021. [DOI] [PubMed] [Google Scholar]
- [10].Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- [11].Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, et al. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. 2001;21:7117–36. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lazo JS, Wipf P. Is Cdc25 a druggable target? Anticancer Agents Med Chem. 2008;8:837–42. doi: 10.2174/187152008786847738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Andersen JN, Jansen PG, Echwald SM, Mortensen OH, Fukada T, Del Vecchio R, et al. A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB J. 2004;18:8–30. doi: 10.1096/fj.02-1212rev. [DOI] [PubMed] [Google Scholar]
- [14].Tautz L, Mustelin T. Strategies for developing protein tyrosine phosphatase inhibitors. Methods. 2007;42:250–60. doi: 10.1016/j.ymeth.2007.02.014. [DOI] [PubMed] [Google Scholar]
- [15].Poon RY, Hunter T. Dephosphorylation of Cdk2 Thr160 by the cyclin-dependent kinase-interacting phosphatase KAP in the absence of cyclin. Science. 1995;270:90–3. doi: 10.1126/science.270.5233.90. [DOI] [PubMed] [Google Scholar]
- [16].Lee SW, Reimer CL, Fang L, Iruela-Arispe ML, Aaronson SA. Overexpression of kinase-associated phosphatase (KAP) in breast and prostate cancer and inhibition of the transformed phenotype by antisense KAP expression. Mol Cell Biol. 2000;20:1723–32. doi: 10.1128/mcb.20.5.1723-1732.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yu Y, Jiang X, Schoch BS, Carroll RS, Black PM, Johnson MD. Aberrant splicing of cyclin-dependent kinase-associated protein phosphatase KAP increases proliferation and migration in glioblastoma. Cancer Res. 2007;67:130–8. doi: 10.1158/0008-5472.CAN-06-2478. [DOI] [PubMed] [Google Scholar]
- [18].D’Angiolella V, Costanzo V, Gottesman ME, Avvedimento EV, Gautier J, Grieco D. Role for cyclin-dependent kinase 2 in mitosis exit. Curr Biol. 2001;11:1221–6. doi: 10.1016/s0960-9822(01)00352-9. [DOI] [PubMed] [Google Scholar]
- [19].Bessette DC, Qiu D, Pallen CJ. PRL PTPs: mediators and markers of cancer progression. Cancer Metastasis Rev. 2008;27:231–52. doi: 10.1007/s10555-008-9121-3. [DOI] [PubMed] [Google Scholar]
- [20].Kikawa KD, Vidale DR, Van Etten RL, Kinch MS. Regulation of the EphA2 kinase by the low molecular weight tyrosine phosphatase induces transformation. J Biol Chem. 2002;277:39274–9. doi: 10.1074/jbc.M207127200. [DOI] [PubMed] [Google Scholar]
- [21].Chiarugi P, Taddei ML, Schiavone N, Papucci L, Giannoni E, Fiaschi T, et al. LMW-PTP is a positive regulator of tumor onset and growth. Oncogene. 2004;23:3905–14. doi: 10.1038/sj.onc.1207508. [DOI] [PubMed] [Google Scholar]
- [22].Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009;457:57–62. doi: 10.1038/nature07668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458:591–6. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Krishnan N, Jeong DG, Jung SK, Ryu SE, Xiao A, Allis CD, et al. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J Biol Chem. 2009;284:16066–70. doi: 10.1074/jbc.C900032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol. 2005;7:591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- [26].Barr AJ, Ugochukwu E, Lee WH, King ON, Filippakopoulos P, Alfano I, et al. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell. 2009;136:352–63. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yang J, Liang X, Niu T, Meng W, Zhao Z, Zhou GW. Crystal structure of the catalytic domain of protein-tyrosine phosphatase SHP-1. J Biol Chem. 1998;273:28199–207. doi: 10.1074/jbc.273.43.28199. [DOI] [PubMed] [Google Scholar]
- [28].Hellmuth K, Grosskopf S, Lum CT, Wurtele M, Roder N, von Kries JP, et al. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc Natl Acad Sci U S A. 2008;105:7275–80. doi: 10.1073/pnas.0710468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lawrence HR, Pireddu R, Chen L, Luo Y, Sung SS, Szymanski AM, et al. Inhibitors of Src homology-2 domain containing protein tyrosine phosphatase-2 (Shp2) based on oxindole scaffolds. J Med Chem. 2008;51:4948–56. doi: 10.1021/jm8002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. 2000;6:1401–12. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- [31].Puius YA, Zhao Y, Sullivan M, Lawrence DS, Almo SC, Zhang ZY. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: a paradigm for inhibitor design. Proc Natl Acad Sci U S A. 1997;94:13420–5. doi: 10.1073/pnas.94.25.13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sun JP, Fedorov AA, Lee SY, Guo XL, Shen K, Lawrence DS, et al. Crystal structure of PTP1B complexed with a potent and selective bidentate inhibitor. J Biol Chem. 2003;278:12406–14. doi: 10.1074/jbc.M212491200. [DOI] [PubMed] [Google Scholar]
- [33].Trapasso F, Drusco A, Costinean S, Alder H, Aqeilan RI, Iuliano R, et al. Genetic ablation of Ptprj, a mouse cancer susceptibility gene, results in normal growth and development and does not predispose to spontaneous tumorigenesis. DNA Cell Biol. 2006;25:376–82. doi: 10.1089/dna.2006.25.376. [DOI] [PubMed] [Google Scholar]
- [34].Scherr M, Chaturvedi A, Battmer K, Dallmann I, Schultheis B, Ganser A, et al. Enhanced sensitivity to inhibition of SHP2, STAT5, and Gab2 expression in chronic myeloid leukemia (CML) Blood. 2006;107:3279–87. doi: 10.1182/blood-2005-08-3087. [DOI] [PubMed] [Google Scholar]
- [35].Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–93. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- [36].Milarski KL, Saltiel AR. Expression of catalytically inactive Syp phosphatase in 3T3 cells blocks stimulation of mitogen-activated protein kinase by insulin. J Biol Chem. 1994;269:21239–43. [PubMed] [Google Scholar]
- [37].Bennett AM, Hausdorff SF, O’Reilly AM, Freeman RM, Neel BG. Multiple requirements for SHPTP2 in epidermal growth factor-mediated cell cycle progression. Mol Cell Biol. 1996;16:1189–202. doi: 10.1128/mcb.16.3.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Imhof D, Wavreille AS, May A, Zacharias M, Tridandapani S, Pei D. Sequence specificity of SHP-1 and SHP-2 Src homology 2 domains. Critical roles of residues beyond the pY+3 position. J Biol Chem. 2006;281:20271–82. doi: 10.1074/jbc.M601047200. [DOI] [PubMed] [Google Scholar]
- [39].Sweeney MC, Wavreille AS, Park J, Butchar JP, Tridandapani S, Pei D. Decoding protein-protein interactions through combinatorial chemistry: sequence specificity of SHP-1, SHP-2, and SHIP SH2 domains. Biochemistry. 2005;44:14932–47. doi: 10.1021/bi051408h. [DOI] [PubMed] [Google Scholar]
- [40].Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92:441–50. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- [41].Eck MJ, Pluskey S, Trub T, Harrison SC, Shoelson SE. Spatial constraints on the recognition of phosphoproteins by the tandem SH2 domains of the phosphatase SH-PTP2. Nature. 1996;379:277–80. doi: 10.1038/379277a0. [DOI] [PubMed] [Google Scholar]
- [42].Cunnick JM, Mei L, Doupnik CA, Wu J. Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J Biol Chem. 2001;276:24380–7. doi: 10.1074/jbc.M010275200. [DOI] [PubMed] [Google Scholar]
- [43].Ottinger EA, Botfield MC, Shoelson SE. Tandem SH2 domains confer high specificity in tyrosine kinase signaling. J Biol Chem. 1998;273:729–35. doi: 10.1074/jbc.273.2.729. [DOI] [PubMed] [Google Scholar]
- [44].Pluskey S, Wandless TJ, Walsh CT, Shoelson SE. Potent stimulation of SH-PTP2 phosphatase activity by simultaneous occupancy of both SH2 domains. J Biol Chem. 1995;270:2897–900. doi: 10.1074/jbc.270.7.2897. [DOI] [PubMed] [Google Scholar]
- [45].Niu T, Liang X, Yang J, Zhao Z, Zhou GW. Kinetic comparison of the catalytic domains of SHP-1 and SHP-2. J Cell Biochem. 1999;72:145–50. doi: 10.1002/(sici)1097-4644(19990101)72:1<145::aid-jcb15>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- [46].Mishra AK, Zhang A, Niu T, Yang J, Liang X, Zhao ZJ, et al. Substrate specificity of protein tyrosine phosphatase: differential behavior of SHP-1 and SHP-2 towards signal regulation protein SIRPalpha1. J Cell Biochem. 2002;84:840–6. doi: 10.1002/jcb.10090. [DOI] [PubMed] [Google Scholar]
- [47].Shen K, Keng YF, Wu L, Guo XL, Lawrence DS, Zhang ZY. Acquisition of a specific and potent PTP1B inhibitor from a novel combinatorial library and screening procedure. J Biol Chem. 2001;276:47311–9. doi: 10.1074/jbc.M106568200. [DOI] [PubMed] [Google Scholar]
- [48].Cunnick JM, Meng S, Ren Y, Desponts C, Wang HG, Djeu JY, et al. Regulation of the mitogen-activated protein kinase signaling pathway by SHP2. J Biol Chem. 2002;277:9498–504. doi: 10.1074/jbc.M110547200. [DOI] [PubMed] [Google Scholar]
- [49].Hadari YR, Kouhara H, Lax I, Schlessinger J. Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol Cell Biol. 1998;18:3966–73. doi: 10.1128/mcb.18.7.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kuhne MR, Pawson T, Lienhard GE, Feng GS. The insulin receptor substrate 1 associates with the SH2-containing phosphotyrosine phosphatase Syp. J Biol Chem. 1993;268:11479–81. [PubMed] [Google Scholar]
- [51].Myers MG, Jr., Mendez R, Shi P, Pierce JH, Rhoads R, White MF. The COOH-terminal tyrosine phosphorylation sites on IRS-1 bind SHP-2 and negatively regulate insulin signaling. J Biol Chem. 1998;273:26908–14. doi: 10.1074/jbc.273.41.26908. [DOI] [PubMed] [Google Scholar]
- [52].Ren Y, Meng S, Mei L, Zhao ZJ, Jove R, Wu J. Roles of Gab1 and SHP2 in paxillin tyrosine dephosphorylation and Src activation in response to epidermal growth factor. J Biol Chem. 2004;279:8497–505. doi: 10.1074/jbc.M312575200. [DOI] [PubMed] [Google Scholar]
- [53].Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T, et al. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol Cell. 2004;13:341–55. doi: 10.1016/s1097-2765(04)00050-4. [DOI] [PubMed] [Google Scholar]
- [54].Agazie YM, Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol Cell Biol. 2003;23:7875–86. doi: 10.1128/MCB.23.21.7875-7886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhou X, Agazie YM. Molecular mechanism for SHP2 in promoting HER2-induced signaling and transformation. J Biol Chem. 2009;284:12226–34. doi: 10.1074/jbc.M900020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cleghon V, Feldmann P, Ghiglione C, Copeland TD, Perrimon N, Hughes DA, et al. Opposing actions of CSW and RasGAP modulate the strength of Torso RTK signaling in the Drosophila terminal pathway. Mol Cell. 1998;2:719–27. doi: 10.1016/s1097-2765(00)80287-7. [DOI] [PubMed] [Google Scholar]
- [57].Montagner A, Yart A, Dance M, Perret B, Salles JP, Raynal P. A novel role for Gab1 and SHP2 in epidermal growth factor-induced Ras activation. J Biol Chem. 2005;280:5350–60. doi: 10.1074/jbc.M410012200. [DOI] [PubMed] [Google Scholar]
- [58].Hanafusa H, Torii S, Yasunaga T, Matsumoto K, Nishida E. Shp2, an SH2-containing protein-tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor Sprouty. J Biol Chem. 2004;279:22992–5. doi: 10.1074/jbc.M312498200. [DOI] [PubMed] [Google Scholar]
- [59].Fujioka Y, Matozaki T, Noguchi T, Iwamatsu A, Yamao T, Takahashi N, et al. A novel membrane glycoprotein, SHPS-1, that binds the SH2-domain-containing protein tyrosine phosphatase SHP-2 in response to mitogens and cell adhesion. Mol Cell Biol. 1996;16:6887–99. doi: 10.1128/mcb.16.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jackson DE, Ward CM, Wang R, Newman PJ. The protein-tyrosine phosphatase SHP-2 binds platelet/endothelial cell adhesion molecule-1 (PECAM-1) and forms a distinct signaling complex during platelet aggregation. Evidence for a mechanistic link between PECAM-1- and integrin-mediated cellular signaling. J Biol Chem. 1997;272:6986–93. doi: 10.1074/jbc.272.11.6986. [DOI] [PubMed] [Google Scholar]
- [61].Zhao R, Zhao ZJ. Dissecting the interaction of SHP-2 with PZR, an immunoglobulin family protein containing immunoreceptor tyrosine-based inhibitory motifs. J Biol Chem. 2000;275:5453–9. doi: 10.1074/jbc.275.8.5453. [DOI] [PubMed] [Google Scholar]
- [62].Famiglietti SJ, Nakamura K, Cambier JC. Unique features of SHIP, SHP-1 and SHP-2 binding to FcgammaRIIb revealed by surface plasmon resonance analysis. Immunol Lett. 1999;68:35–40. doi: 10.1016/s0165-2478(99)00027-9. [DOI] [PubMed] [Google Scholar]
- [63].Bertotti A, Comoglio PM, Trusolino L. Beta4 integrin activates a Shp2-Src signaling pathway that sustains HGF-induced anchorage-independent growth. J Cell Biol. 2006;175:993–1003. doi: 10.1083/jcb.200605114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wu TR, Hong YK, Wang XD, Ling MY, Dragoi AM, Chung AS, et al. SHP-2 is a dual-specificity phosphatase involved in Stat1 dephosphorylation at both tyrosine and serine residues in nuclei. J Biol Chem. 2002;277:47572–80. doi: 10.1074/jbc.M207536200. [DOI] [PubMed] [Google Scholar]
- [65].You M, Yu DH, Feng GS. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol Cell Biol. 1999;19:2416–24. doi: 10.1128/mcb.19.3.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zhou X, Coad J, Ducatman B, Agazie YM. SHP2 is up-regulated in breast cancer cells and in infiltrating ductal carcinoma of the breast, implying its involvement in breast oncogenesis. Histopathology. 2008;53:389–402. doi: 10.1111/j.1365-2559.2008.03103.x. [DOI] [PubMed] [Google Scholar]
- [67].Xu R, Yu Y, Zheng S, Zhao X, Dong Q, He Z, et al. Overexpression of Shp2 tyrosine phosphatase is implicated in leukemogenesis in adult human leukemia. Blood. 2005;106:3142–9. doi: 10.1182/blood-2004-10-4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Zhou XD, Agazie YM. Inhibition of SHP2 leads to mesenchymal to epithelial transition in breast cancer cells. Cell Death Differ. 2008;15:988–96. doi: 10.1038/cdd.2008.54. [DOI] [PubMed] [Google Scholar]
- [69].Agazie YM, Movilla N, Ischenko I, Hayman MJ. The phosphotyrosine phosphatase SHP2 is a critical mediator of transformation induced by the oncogenic fibroblast growth factor receptor 3. Oncogene. 2003;22:6909–18. doi: 10.1038/sj.onc.1206798. [DOI] [PubMed] [Google Scholar]
- [70].Ji H, Zhao X, Yuza Y, Shimamura T, Li D, Protopopov A, et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc Natl Acad Sci U S A. 2006;103:7817–22. doi: 10.1073/pnas.0510284103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhan Y, Counelis GJ, O’Rourke DM. The protein tyrosine phosphatase SHP-2 is required for EGFRvIII oncogenic transformation in human glioblastoma cells. Exp Cell Res. 2009;315:2343–57. doi: 10.1016/j.yexcr.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Voena C, Conte C, Ambrogio C, Boeri Erba E, Boccalatte F, Mohammed S, et al. The tyrosine phosphatase Shp2 interacts with NPM-ALK and regulates anaplastic lymphoma cell growth and migration. Cancer Res. 2007;67:4278–86. doi: 10.1158/0008-5472.CAN-06-4350. [DOI] [PubMed] [Google Scholar]
- [73].Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4:688–94. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- [74].Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–8. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- [75].Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–50. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- [76].Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64:8816–20. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- [77].Loh ML, Reynolds MG, Vattikuti S, Gerbing RB, Alonzo TA, Carlson E, et al. PTPN11 mutations in pediatric patients with acute myeloid leukemia: results from the Children’s Cancer Group. Leukemia. 2004;18:1831–4. doi: 10.1038/sj.leu.2403492. [DOI] [PubMed] [Google Scholar]
- [78].Chan RJ, Leedy MB, Munugalavadla V, Voorhorst CS, Li Y, Yu M, et al. Human somatic PTPN11 mutations induce hematopoietic-cell hypersensitivity to granulocyte-macrophage colony-stimulating factor. Blood. 2005;105:3737–42. doi: 10.1182/blood-2004-10-4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Schubbert S, Lieuw K, Rowe SL, Lee CM, Li X, Loh ML, et al. Functional analysis of leukemia-associated PTPN11 mutations in primary hematopoietic cells. Blood. 2005;106:311–7. doi: 10.1182/blood-2004-11-4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Mohi MG, Williams IR, Dearolf CR, Chan G, Kutok JL, Cohen S, et al. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell. 2005;7:179–91. doi: 10.1016/j.ccr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- [81].Ren Y, Chen Z, Chen L, Woods NT, Reuther GW, Cheng JQ, et al. Shp2E76K mutant confers cytokine-independent survival of TF-1 myeloid cells by up-regulating Bcl-XL. J Biol Chem. 2007;282:36463–73. doi: 10.1074/jbc.M705789200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chan G, Kalaitzidis D, Usenko T, Kutok JL, Yang W, Mohi MG, et al. Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood. 2009;113:4414–24. doi: 10.1182/blood-2008-10-182626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Chen L, Sung SS, Yip ML, Lawrence HR, Ren Y, Guida WC, et al. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol. 2006;70:562–70. doi: 10.1124/mol.106.025536. [DOI] [PubMed] [Google Scholar]
- [84].Zhao XT, Qian YK, Chan AW, Madhavan R, Peng HB. Regulation of ACh receptor clustering by the tyrosine phosphatase Shp2. Dev Neurobiol. 2007;67:1789–801. doi: 10.1002/dneu.20556. [DOI] [PubMed] [Google Scholar]
- [85].Qian YK, Chan AW, Madhavan R, Peng HB. The function of Shp2 tyrosine phosphatase in the dispersal of acetylcholine receptor clusters. BMC Neurosci. 2008;9:70. doi: 10.1186/1471-2202-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Chatterjee PK, Al Abed Y, Sherry B, Metz CN. Cholinergic agonists regulate JAK2/STAT3 signaling to suppress endothelial cell activation. Am J Physiol Cell Physiol. 2009 doi: 10.1152/ajpcell.00160.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Noren-Muller A, Reis-Correa I, Jr., Prinz H, Rosenbaum C, Saxena K, Schwalbe HJ, et al. Discovery of protein phosphatase inhibitor classes by biology-oriented synthesis. Proc Natl Acad Sci U S A. 2006;103:10606–11. doi: 10.1073/pnas.0601490103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Geronikaki A, Eleftheriou P, Vicini P, Alam I, Dixit A, Saxena AK. 2-Thiazolylimino/heteroarylimino-5-arylidene-4-thiazolidinones as new agents with SHP-2 inhibitory action. J Med Chem. 2008;51:5221–8. doi: 10.1021/jm8004306. [DOI] [PubMed] [Google Scholar]
- [89].Yu WM, Guvench O, Mackerell AD, Qu CK. Identification of small molecular weight inhibitors of Src homology 2 domain-containing tyrosine phosphatase 2 (SHP-2) via in silico database screening combined with experimental assay. J Med Chem. 2008;51:7396–404. doi: 10.1021/jm800229d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Gobert RP, van den Eijnden M, Szyndralewiez C, Jorand-Lebrun C, Swinnen D, Chen L, et al. GLEPP1/protein-tyrosine phosphatase phi inhibitors block chemotaxis in vitro and in vivo and improve murine ulcerative colitis. J Biol Chem. 2009;284:11385–95. doi: 10.1074/jbc.M807241200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Dawson MI, Xia Z, Jiang T, Ye M, Fontana JA, Farhana L, et al. Adamantyl-substituted retinoid-derived molecules that interact with the orphan nuclear receptor small heterodimer partner: effects of replacing the 1-adamantyl or hydroxyl group on inhibition of cancer cell growth, induction of cancer cell apoptosis, and inhibition of SRC homology 2 domain-containing protein tyrosine phosphatase-2 activity. J Med Chem. 2008;51:5650–62. doi: 10.1021/jm800456k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Schaeper U, Gehring NH, Fuchs KP, Sachs M, Kempkes B, Birchmeier W. Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses. J Cell Biol. 2000;149:1419–32. doi: 10.1083/jcb.149.7.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Maroun CR, Naujokas MA, Holgado-Madruga M, Wong AJ, Park M. The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol Cell Biol. 2000;20:8513–25. doi: 10.1128/mcb.20.22.8513-8525.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bjorge JD, Pang A, Fujita DJ. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J Biol Chem. 2000;275:41439–46. doi: 10.1074/jbc.M004852200. [DOI] [PubMed] [Google Scholar]
- [95].Arias-Romero LE, Saha S, Villamar-Cruz O, Yip SC, Ethier SP, Zhang ZY, et al. Activation of Src by protein tyrosine phosphatase 1B Is required for ErbB2 transformation of human breast epithelial cells. Cancer Res. 2009;69:4582–8. doi: 10.1158/0008-5472.CAN-08-4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zhu S, Bjorge JD, Fujita DJ. PTP1B contributes to the oncogenic properties of colon cancer cells through Src activation. Cancer Res. 2007;67:10129–37. doi: 10.1158/0008-5472.CAN-06-4338. [DOI] [PubMed] [Google Scholar]
- [97].Dube N, Cheng A, Tremblay ML. The role of protein tyrosine phosphatase 1B in Ras signaling. Proc Natl Acad Sci U S A. 2004;101:1834–9. doi: 10.1073/pnas.0304242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Zhang Z, Lin SY, Neel BG, Haimovich B. Phosphorylated alpha-actinin and protein-tyrosine phosphatase 1B coregulate the disassembly of the focal adhesion kinase x Src complex and promote cell migration. J Biol Chem. 2006;281:1746–54. doi: 10.1074/jbc.M509590200. [DOI] [PubMed] [Google Scholar]
- [99].Wiener JR, Hurteau JA, Kerns BJ, Whitaker RS, Conaway MR, Berchuck A, et al. Overexpression of the tyrosine phosphatase PTP1B is associated with human ovarian carcinomas. Am J Obstet Gynecol. 1994;170:1177–83. doi: 10.1016/s0002-9378(94)70118-0. [DOI] [PubMed] [Google Scholar]
- [100].Wiener JR, Kerns BJ, Harvey EL, Conaway MR, Iglehart JD, Berchuck A, et al. Overexpression of the protein tyrosine phosphatase PTP1B in human breast cancer: association with p185c-erbB-2 protein expression. J Natl Cancer Inst. 1994;86:372–8. doi: 10.1093/jnci/86.5.372. [DOI] [PubMed] [Google Scholar]
- [101].Bentires-Alj M, Neel BG. Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Res. 2007;67:2420–4. doi: 10.1158/0008-5472.CAN-06-4610. [DOI] [PubMed] [Google Scholar]
- [102].Julien SG, Dube N, Read M, Penney J, Paquet M, Han Y, et al. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat Genet. 2007;39:338–46. doi: 10.1038/ng1963. [DOI] [PubMed] [Google Scholar]
- [103].Dube N, Bourdeau A, Heinonen KM, Cheng A, Loy AL, Tremblay ML. Genetic ablation of protein tyrosine phosphatase 1B accelerates lymphomagenesis of p53-null mice through the regulation of B-cell development. Cancer Res. 2005;65:10088–95. doi: 10.1158/0008-5472.CAN-05-1353. [DOI] [PubMed] [Google Scholar]
- [104].Lee SY, Liang F, Guo XL, Xie L, Cahill SM, Blumenstein M, et al. Design, construction, and intracellular activation of an intramolecularly self-silenced signal transduction inhibitor. Angew Chem Int Ed Engl. 2005;44:4242–4. doi: 10.1002/anie.200462004. [DOI] [PubMed] [Google Scholar]
- [105].Boutselis IG, Yu X, Zhang ZY, Borch RF. Synthesis and cell-based activity of a potent and selective protein tyrosine phosphatase 1B inhibitor prodrug. J Med Chem. 2007;50:856–64. doi: 10.1021/jm061146x. [DOI] [PubMed] [Google Scholar]
- [106].Han Y, Belley M, Bayly CI, Colucci J, Dufresne C, Giroux A, et al. Discovery of [(3-bromo-7-cyano-2-naphthyl)(difluoro)methyl] phosphonic acid, a potent and orally active small molecule PTP1B inhibitor. Bioorg Med Chem Lett. 2008;18:3200–5. doi: 10.1016/j.bmcl.2008.04.064. [DOI] [PubMed] [Google Scholar]
- [107].Erbe DV, Wang S, Zhang YL, Harding K, Kung L, Tam M, et al. Ertiprotafib improves glycemic control and lowers lipids via multiple mechanisms. Mol Pharmacol. 2005;67:69–77. doi: 10.1124/mol.104.005553. [DOI] [PubMed] [Google Scholar]
- [108].Lantz KA, Hart SG, Planey SL, Roitman MF, Ruiz-White IA, Wolfe HR, et al. Inhibition of PTP1B by Trodusquemine (MSI-1436) Causes Fat-specific Weight Loss in Diet-induced Obese Mice. Obesity (Silver Spring) 2010 doi: 10.1038/oby.2009.444. [DOI] [PubMed] [Google Scholar]
- [109].Rao MN, Shinnar AE, Noecker LA, Chao TL, Feibush B, Snyder B, et al. Aminosterols from the dogfish shark Squalus acanthias. J Nat Prod. 2000;63:631–5. doi: 10.1021/np990514f. [DOI] [PubMed] [Google Scholar]
- [110].Stephens BJ, Han H, Gokhale V, Von Hoff DD. PRL phosphatases as potential molecular targets in cancer. Mol Cancer Ther. 2005;4:1653–61. doi: 10.1158/1535-7163.MCT-05-0248. [DOI] [PubMed] [Google Scholar]
- [111].Zeng Q, Si X, Horstmann H, Xu Y, Hong W, Pallen CJ. Prenylation-dependent association of protein-tyrosine phosphatases PRL-1, -2, and -3 with the plasma membrane and the early endosome. J Biol Chem. 2000;275:21444–52. doi: 10.1074/jbc.M000453200. [DOI] [PubMed] [Google Scholar]
- [112].Saha S, Bardelli A, Buckhaults P, Velculescu VE, Rago C, St Croix B, et al. A phosphatase associated with metastasis of colorectal cancer. Science. 2001;294:1343–6. doi: 10.1126/science.1065817. [DOI] [PubMed] [Google Scholar]
- [113].Bardelli A, Saha S, Sager JA, Romans KE, Xin B, Markowitz SD, et al. PRL-3 expression in metastatic cancers. Clin Cancer Res. 2003;9:5607–15. [PubMed] [Google Scholar]
- [114].Peng L, Ning J, Meng L, Shou C. The association of the expression level of protein tyrosine phosphatase PRL-3 protein with liver metastasis and prognosis of patients with colorectal cancer. J Cancer Res Clin Oncol. 2004;130:521–6. doi: 10.1007/s00432-004-0563-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Rouleau C, Roy A, St Martin T, Dufault MR, Boutin P, Liu D, et al. Protein tyrosine phosphatase PRL-3 in malignant cells and endothelial cells: expression and function. Mol Cancer Ther. 2006;5:219–29. doi: 10.1158/1535-7163.MCT-05-0289. [DOI] [PubMed] [Google Scholar]
- [116].Wang L, Peng L, Dong B, Kong L, Meng L, Yan L, et al. Overexpression of phosphatase of regenerating liver-3 in breast cancer: association with a poor clinical outcome. Ann Oncol. 2006;17:1517–22. doi: 10.1093/annonc/mdl159. [DOI] [PubMed] [Google Scholar]
- [117].Miskad UA, Semba S, Kato H, Yokozaki H. Expression of PRL-3 phosphatase in human gastric carcinomas: close correlation with invasion and metastasis. Pathobiology. 2004;71:176–84. doi: 10.1159/000078671. [DOI] [PubMed] [Google Scholar]
- [118].Zhao WB, Li Y, Liu X, Zhang LY, Wang X. Evaluation of PRL-3 expression, and its correlation with angiogenesis and invasion in hepatocellular carcinoma. Int J Mol Med. 2008;22:187–92. [PubMed] [Google Scholar]
- [119].Ren T, Jiang B, Xing X, Dong B, Peng L, Meng L, et al. Prognostic Significance of Phosphatase of Regenerating Liver-3 Expression in Ovarian Cancer. Pathol Oncol Res. 2009 doi: 10.1007/s12253-009-9153-1. [DOI] [PubMed] [Google Scholar]
- [120].Dai N, Lu AP, Shou CC, Li JY. Expression of phosphatase regenerating liver 3 is an independent prognostic indicator for gastric cancer. World J Gastroenterol. 2009;15:1499–505. doi: 10.3748/wjg.15.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Fagerli UM, Holt RU, Holien T, Vaatsveen TK, Zhan F, Egeberg KW, et al. Overexpression and involvement in migration by the metastasis-associated phosphatase PRL-3 in human myeloma cells. Blood. 2008;111:806–15. doi: 10.1182/blood-2007-07-101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Zeng Q, Dong JM, Guo K, Li J, Tan HX, Koh V, et al. PRL-3 and PRL-1 promote cell migration, invasion, and metastasis. Cancer Res. 2003;63:2716–22. [PubMed] [Google Scholar]
- [123].Guo K, Li J, Tang JP, Koh V, Gan BQ, Zeng Q. Catalytic domain of PRL-3 plays an essential role in tumor metastasis: formation of PRL-3 tumors inside the blood vessels. Cancer Biol Ther. 2004;3:945–51. doi: 10.4161/cbt.3.10.1111. [DOI] [PubMed] [Google Scholar]
- [124].Fiordalisi JJ, Keller PJ, Cox AD. PRL tyrosine phosphatases regulate rho family GTPases to promote invasion and motility. Cancer Res. 2006;66:3153–61. doi: 10.1158/0008-5472.CAN-05-3116. [DOI] [PubMed] [Google Scholar]
- [125].Wang H, Quah SY, Dong JM, Manser E, Tang JP, Zeng Q. PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 2007;67:2922–6. doi: 10.1158/0008-5472.CAN-06-3598. [DOI] [PubMed] [Google Scholar]
- [126].Achiwa H, Lazo JS. PRL-1 tyrosine phosphatase regulates c-Src levels, adherence, and invasion in human lung cancer cells. Cancer Res. 2007;67:643–50. doi: 10.1158/0008-5472.CAN-06-2436. [DOI] [PubMed] [Google Scholar]
- [127].Luo Y, Liang F, Zhang ZY. PRL1 Promotes Cell Migration and Invasion by Increasing MMP2 and MMP9 Expression through Src and ERK1/2 Pathways (dagger) Biochemistry. 2009;48:1838–46. doi: 10.1021/bi8020789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Forte E, Orsatti L, Talamo F, Barbato G, De Francesco R, Tomei L. Ezrin is a specific and direct target of protein tyrosine phosphatase PRL-3. Biochim Biophys Acta. 2008;1783:334–44. doi: 10.1016/j.bbamcr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- [129].Kozlov G, Cheng J, Ziomek E, Banville D, Gehring K, Ekiel I. Structural insights into molecular function of the metastasis-associated phosphatase PRL-3. J Biol Chem. 2004;279:11882–9. doi: 10.1074/jbc.M312905200. [DOI] [PubMed] [Google Scholar]
- [130].Kim KA, Song JS, Jee J, Sheen MR, Lee C, Lee TG, et al. Structure of human PRL-3, the phosphatase associated with cancer metastasis. FEBS Lett. 2004;565:181–7. doi: 10.1016/j.febslet.2004.03.062. [DOI] [PubMed] [Google Scholar]
- [131].Jeong DG, Kim SJ, Kim JH, Son JH, Park MR, Lim SM, et al. Trimeric structure of PRL-1 phosphatase reveals an active enzyme conformation and regulation mechanisms. J Mol Biol. 2005;345:401–13. doi: 10.1016/j.jmb.2004.10.061. [DOI] [PubMed] [Google Scholar]
- [132].Daouti S, Li WH, Qian H, Huang KS, Holmgren J, Levin W, et al. A selective phosphatase of regenerating liver phosphatase inhibitor suppresses tumor cell anchorage-independent growth by a novel mechanism involving p130Cas cleavage. Cancer Res. 2008;68:1162–9. doi: 10.1158/0008-5472.CAN-07-2349. [DOI] [PubMed] [Google Scholar]
- [133].Choi SK, Oh HM, Lee SK, Jeong DG, Ryu SE, Son KH, et al. Biflavonoids inhibited phosphatase of regenerating liver-3 (PRL-3) Nat Prod Res. 2006;20:341–6. doi: 10.1080/14786410500463312. [DOI] [PubMed] [Google Scholar]
- [134].Park H, Jung SK, Jeong DG, Ryu SE, Kim SJ. Discovery of novel PRL-3 inhibitors based on the structure-based virtual screening. Bioorg Med Chem Lett. 2008;18:2250–5. doi: 10.1016/j.bmcl.2008.03.013. [DOI] [PubMed] [Google Scholar]
- [135].Ahn JH, Kim SJ, Park WS, Cho SY, Ha JD, Kim SS, et al. Synthesis and biological evaluation of rhodanine derivatives as PRL-3 inhibitors. Bioorg Med Chem Lett. 2006;16:2996–9. doi: 10.1016/j.bmcl.2006.02.060. [DOI] [PubMed] [Google Scholar]
- [136].Pathak MK, Dhawan D, Lindner DJ, Borden EC, Farver C, Yi T. Pentamidine is an inhibitor of PRL phosphatases with anticancer activity. Mol Cancer Ther. 2002;1:1255–64. [PubMed] [Google Scholar]
- [137].Wang L, Shen Y, Song R, Sun Y, Xu J, Xu Q. An Anticancer Effect of Curcumin Mediated by Down-regulating PRL-3 Expression on Highly Metastatic Melanoma Cells. Mol Pharmacol. 2009 doi: 10.1124/mol.109.059105. [DOI] [PubMed] [Google Scholar]
- [138].Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell. 1998;2:709–18. doi: 10.1016/s1097-2765(00)80286-5. [DOI] [PubMed] [Google Scholar]
- [139].Mailand N, Lukas C, Kaiser BK, Jackson PK, Bartek J, Lukas J. Deregulated human Cdc14A phosphatase disrupts centrosome separation and chromosome segregation. Nat Cell Biol. 2002;4:317–22. doi: 10.1038/ncb777. [DOI] [PubMed] [Google Scholar]
- [140].Paulsen MT, Starks AM, Derheimer FA, Hanasoge S, Li L, Dixon JE, et al. The p53-targeting human phosphatase hCdc14A interacts with the Cdk1/cyclin B complex and is differentially expressed in human cancers. Mol Cancer. 2006;5:25. doi: 10.1186/1476-4598-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Esteban V, Vazquez-Novelle MD, Calvo E, Bueno A, Sacristan MP. Human Cdc14A reverses CDK1 phosphorylation of Cdc25A on serines 115 and 320. Cell Cycle. 2006;5:2894–8. doi: 10.4161/cc.5.24.3566. [DOI] [PubMed] [Google Scholar]
- [142].Lanzetti L, Margaria V, Melander F, Virgili L, Lee MH, Bartek J, et al. Regulation of the Rab5 GTPase-activating protein RN-tre by the dual specificity phosphatase Cdc14A in human cells. J Biol Chem. 2007;282:15258–70. doi: 10.1074/jbc.M700914200. [DOI] [PubMed] [Google Scholar]
- [143].Gray CH, Good VM, Tonks NK, Barford D. The structure of the cell cycle protein Cdc14 reveals a proline-directed protein phosphatase. EMBO J. 2003;22:3524–35. doi: 10.1093/emboj/cdg348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Souza AC, Azoubel S, Queiroz KC, Peppelenbosch MP, Ferreira CV. From immune response to cancer: a spot on the low molecular weight protein tyrosine phosphatase. Cell Mol Life Sci. 2009;66:1140–53. doi: 10.1007/s00018-008-8501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Cirri P, Fiaschi T, Chiarugi P, Camici G, Manao G, Raugei G, et al. The molecular basis of the differing kinetic behavior of the two low molecular mass phosphotyrosine protein phosphatase isoforms. J Biol Chem. 1996;271:2604–7. doi: 10.1074/jbc.271.5.2604. [DOI] [PubMed] [Google Scholar]
- [146].Malentacchi F, Marzocchini R, Gelmini S, Orlando C, Serio M, Ramponi G, et al. Up-regulated expression of low molecular weight protein tyrosine phosphatases in different human cancers. Biochem Biophys Res Commun. 2005;334:875–83. doi: 10.1016/j.bbrc.2005.06.176. [DOI] [PubMed] [Google Scholar]
- [147].Miranda MA, Okamoto AK, Ferreira CV, Silva TL, Granjeiro JM, Aoyama H. Differential effects of flavonoids on bovine kidney low molecular mass protein tyrosine phosphatase. J Enzyme Inhib Med Chem. 2006;21:419–25. doi: 10.1080/14756360500179523. [DOI] [PubMed] [Google Scholar]
- [148].Bispo de Jesus M, Zambuzzi WF, Ruela de Sousa RR, Areche C, Santos de Souza AC, Aoyama H, et al. Ferruginol suppresses survival signaling pathways in androgen-independent human prostate cancer cells. Biochimie. 2008;90:843–54. doi: 10.1016/j.biochi.2008.01.011. [DOI] [PubMed] [Google Scholar]
- [149].Forghieri M, Laggner C, Paoli P, Langer T, Manao G, Camici G, et al. Synthesis, activity and molecular modeling of a new series of chromones as low molecular weight protein tyrosine phosphatase inhibitors. Bioorg Med Chem. 2009;17:2658–72. doi: 10.1016/j.bmc.2009.02.060. [DOI] [PubMed] [Google Scholar]
- [150].Maccari R, Ottana R, Ciurleo R, Paoli P, Manao G, Camici G, et al. Structure-based optimization of benzoic acids as inhibitors of protein tyrosine phosphatase 1B and low molecular weight protein tyrosine phosphatase. ChemMedChem. 2009;4:957–62. doi: 10.1002/cmdc.200800427. [DOI] [PubMed] [Google Scholar]
- [151].Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer. 2007;7:495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- [152].Ray D, Terao Y, Fuhrken PG, Ma ZQ, DeMayo FJ, Christov K, et al. Deregulated CDC25A expression promotes mammary tumorigenesis with genomic instability. Cancer Res. 2007;67:984–91. doi: 10.1158/0008-5472.CAN-06-3927. [DOI] [PubMed] [Google Scholar]
- [153].Ray D, Terao Y, Nimbalkar D, Hirai H, Osmundson EC, Zou X, et al. Hemizygous disruption of Cdc25A inhibits cellular transformation and mammary tumorigenesis in mice. Cancer Res. 2007;67:6605–11. doi: 10.1158/0008-5472.CAN-06-4815. [DOI] [PubMed] [Google Scholar]
- [154].Wei S, Chen X, Rocha K, Epling-Burnette PK, Djeu JY, Liu Q, et al. A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. Proc Natl Acad Sci U S A. 2009;106:12974–9. doi: 10.1073/pnas.0811267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Brezak MC, Valette A, Quaranta M, Contour-Galcera MO, Jullien D, Lavergne O, et al. IRC-083864, a novel bis quinone inhibitor of CDC25 phosphatases active against human cancer cells. Int J Cancer. 2009;124:1449–56. doi: 10.1002/ijc.24080. [DOI] [PubMed] [Google Scholar]
- [156].Brezak MC, Quaranta M, Mondesert O, Galcera MO, Lavergne O, Alby F, et al. A novel synthetic inhibitor of CDC25 phosphatases: BN82002. Cancer Res. 2004;64:3320–5. doi: 10.1158/0008-5472.can-03-3984. [DOI] [PubMed] [Google Scholar]
- [157].Brezak MC, Quaranta M, Contour-Galcera MO, Lavergne O, Mondesert O, Auvray P, et al. Inhibition of human tumor cell growth in vivo by an orally bioavailable inhibitor of CDC25 phosphatases. Mol Cancer Ther. 2005;4:1378–87. doi: 10.1158/1535-7163.MCT-05-0168. [DOI] [PubMed] [Google Scholar]
- [158].Brezak MC, Kasprzyk PG, Galcera MO, Lavergne O, Prevost GP. CDC25 inhibitors as anticancer agents are moving forward. Anticancer Agents Med Chem. 2008;8:857–62. doi: 10.2174/187152008786847701. [DOI] [PubMed] [Google Scholar]
- [159].Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003;426:247–54. doi: 10.1038/nature02083. [DOI] [PubMed] [Google Scholar]
- [160].Tootle TL, Silver SJ, Davies EL, Newman V, Latek RR, Mills IA, et al. The transcription factor Eyes absent is a protein tyrosine phosphatase. Nature. 2003;426:299–302. doi: 10.1038/nature02097. [DOI] [PubMed] [Google Scholar]
- [161].Rayapureddi JP, Kattamuri C, Steinmetz BD, Frankfort BJ, Ostrin EJ, Mardon G, et al. Eyes absent represents a class of protein tyrosine phosphatases. Nature. 2003;426:295–8. doi: 10.1038/nature02093. [DOI] [PubMed] [Google Scholar]
- [162].Zhang L, Yang N, Huang J, Buckanovich RJ, Liang S, Barchetti A, et al. Transcriptional coactivator Drosophila eyes absent homologue 2 is up-regulated in epithelial ovarian cancer and promotes tumor growth. Cancer Res. 2005;65:925–32. [PubMed] [Google Scholar]
- [163].Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–30. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]