

Abstract
Estimation of gene diversity (heterozygosity) at 1,442 single-nucleotide polymorphism (SNP) loci in an ethnically diverse sample of humans revealed consistently reduced gene diversities at SNP loci causing amino acid changes, particularly those causing amino acid changes predicted to be disruptive to protein structure. The reduction of gene diversity at these SNP loci, in comparison to SNPs in the same genes not affecting protein structure, is evidence that negative natural selection (purifying selection) has reduced the population frequencies of deleterious SNP alleles. This, in turn, suggests that slightly deleterious mutations are widespread in the human population and that estimation of gene diversity even in a sample of modest size can help guide the search for disease-associated genes.
In the effort to explain the genetic contribution to complex diseases such as cancer and heart disease, large numbers of polymorphisms have been surveyed for statistical associations with disease phenotypes in human populations (1). To date, several million single-nucleotide polymorphisms (SNPs) have been reported in the public database dbSNP (2), making it desirable to devise strategies to analyze known SNPs for likely candidates for disease association (3–9). Recent interest has focused particularly on SNPs located in protein-coding genes, both because of the ease in assaying biallelic single-nucleotide variation and because the large number of SNPs in genes encoding proteins of known biological function (estimated to be between 50,000 and 250,000) are often plausible candidates for the underlying causes of disease processes (1, 10–12).
One feature that can be indicative of the deleterious consequences of a given allele is evidence of purifying selection (13). Purifying selection is the form of natural selection that acts to eliminate selectively deleterious mutations. For example, purifying selection is expected to act against mutations that have deleterious effects on protein structure by causing change to functionally important amino acid residues or on gene expression by altering regulation (14, 15). To test whether evidence of purifying selection can be obtained from population frequency data at SNP loci, we typed 1,442 SNPs at 234 protein-coding loci in a population of 102 anonymized subjects belonging to the major culturally defined ethnic groups contributing to the United States population (African American/African, non-Hispanic Caucasian, Hispanic, and Pacific Rim). SNPs were restricted to known genes and heavily biased toward exons, intron–exon borders, and regulatory regions within 5 kb of the start or end of the ORFs.
Materials and Methods
Samples. DNA from 102 unrelated individuals of self-described heritage were selected from the Coriell Institute for Medical Research (Camden, NJ; http://locus.umdnj.edu/nigms), and included four of the major culturally defined ethnic groups contributing to the United States population: 31 non-Hispanic Caucasians, 24 African/African Americans, 23 Hispanic, and 24 of Pacific Rim heritage. Note that, because these are culturally defined ethnic groups, they do not necessarily correspond to historic subdivisions of the human population. For example, the “non-Hispanic Caucasian” and “Hispanic” populations are probably both largely of European ancestry. All samples are anonymized and do not have available phenotype data, except sex and self-described ethnic heritage. An analysis of nine satellite tandem repeats (Applied Biosystems profiler, Foster City, CA) confirmed the uniqueness of each of the 102 individuals in the study population.
Resequencing. SNPs were deliberately chosen within or closely situated to genes, and the selection of genes and SNPs for analysis was drawn from publicly available databases. Sequencing primers were designed for bidirectional sequencing by using primer3 software (www.basic.nwu.edu/biotools/Primer3). Each primer was tagged with a universal sequencing primer, M13 (TGTAAAACGACGGCCAGT) for forward and M13 (CAGGAAACAGCTATGACC) for reverse. Amplicons were optimized in 8 of the 102 individuals on a gradient PTC-225 Tetrad Unit (MJ Research, Waltham, MA) by using a matrix to determine the suitable annealing temperature (between 54°C and 69°C) for the additional 94 samples, and checked by 2% agarose gel before sequence analysis. Bidirectional sequencing was performed on all 102 samples by using the BigDye Terminator (Applied Biosystems) mix 2.0, 3.0, and 3.1, according to the manufacturer's direction, but at a dilution of 1:8, and run on either ABI 3100 or 3700 machines (Applied Biosystems). Sequences of all sequencing primers are available at http://snp500cancer.nci.nih.gov.
Sequence tracings were analyzed by using sequencher 4.0.5 (Genecodes, Ann Arbor, MI). After alignment of bidirectional sequence reads, two independent reviewers analyze each contig for SNPs. The criteria for completing sequence alignment of each contig included 190 separate sequence traces at 70% assembly parameters (sequencher 4.0.5). Genotype calls were determined for each of the 102 individuals and loaded into an Oracle database. Allelic frequency data for all SNPs are available at http://snp500cancer.nci.nih.gov.
Statistical Analyses. For a given locus, gene diversity (heterozygosity) was estimated by 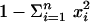 , where n is the number of alleles and xi is the population frequency of the ith allele (15). Missense SNPs (SNPs causing a change of amino acid residue) were categorized as radical if the amino acid replacement involved two amino acids with a pairwise stereochemical difference >3.0 according to Miyata et al.'s (16) scale (based on amino acid residue volume and polarity). Otherwise, missense SNPs were categorized as conservative.
, where n is the number of alleles and xi is the population frequency of the ith allele (15). Missense SNPs (SNPs causing a change of amino acid residue) were categorized as radical if the amino acid replacement involved two amino acids with a pairwise stereochemical difference >3.0 according to Miyata et al.'s (16) scale (based on amino acid residue volume and polarity). Otherwise, missense SNPs were categorized as conservative.
In comparisons with mouse sequences, the amino acid sequence of a putative mouse ortholog was obtained from the NCBI sequence databases (http://ncbi.nlm.nih.gov), in most cases from the RefSeq database (17). Amino acid sequences of human and mouse were aligned by using the clustalw program (18). There is a possibility of polymorphism at these sites in the mouse, which could not be addressed given available data. However, if there are two alleles in mouse, one of which is considerably more common than the other (as in human, see Fig. 2B), on average it is expected that the mouse RefSeq sequence will be more likely to represent the more common mouse allele. Following the principle of maximum parsimony (19), we assumed that a residue that represents the more common allele in both human and mouse has been conserved since the most recent common ancestor of the two species.
Fig. 2.

(A) Mean gene diversity at missense SNP sites causing conservative amino acid changes. Error bars indicate standard errors and red horizontal lines indicate median values. One-way analysis of variance, F2,276 = 5.38; P = 0.005. The other means were significantly different from that for SNP sites for which the most frequent allele encoded the same amino acid residue as in the mouse (Dunnett's test; family error rate of 0.05 and individual error rate of 0.0265). Medians were significantly different by the Kruskal–Wallis nonparametric analysis of variance, P = 0.001. (B) Mean allele frequency at 200 conservative missense SNP sites for which a mouse ortholog was available and one SNP encoded a residue the same as the mouse. Error bars indicate standard errors, and red horizontal lines indicate median values. Paired t test, P < 0.0001; Wilcoxon signed rank test, P < 0.0001.
Results
Striking differences with respect to gene diversity (heterozygosity) were seen between sites at which SNPs caused amino acid changes and those at which no amino acid change was caused (Fig. 1A). All sites at which SNPs did not cause an amino acid change, including sites in 5′- and 3′ noncoding regions, sites in introns, and synonymous sites in exons, had remarkably similar levels of gene diversity, ≈20% (Fig. 1 A). This corresponds to an average frequency of ≈12% for the less common allele. By contrast, mean gene diversities were on average lower at nonsynonymous SNP sites in exons. Mean gene diversity was 17.2% for missense (amino acid altering) SNPs causing a conservative amino acid change, 7.9% for missense SNPs causing a radical amino acid change, and 2.8% for SNPs introducing a stop codon (nonsense mutation) (Fig. 1 A). The difference in mean gene diversity between conservative and radical missense SNPs was statistically significant (Fig. 1 A).
Fig. 1.

(A) Mean gene diversity (heterozygosity) at SNP sites categorized by location in the gene and effect on protein coding. Numbers of SNP sites in each category are indicated. Error bars indicate standard errors and red horizontal lines indicate median values. One-way analysis of variance, F6,1435 = 3.77; P = 0.001. All means except that for nonsense SNPs were significantly different from that for missense, radical SNPs (Dunnett's test; family error rate of 0.05 and individual error rate of 0.0167). Medians were significantly different by the Kruskal–Wallis nonparametric analysis of variance, P = 0.001. (B) Mean gene diversity compared pairwise between replacement (conservative missense, radical missense, and nonsense) SNP sites and silent SNP sites in the same genes. Error bars indicate standard errors and red horizontal lines indicate median values. Numbers indicate numbers of loci. Significance levels for paired-sample t tests are shown. Significance levels for Wilcoxon paired tests of the equality of medians were as follows: conservative missense, n.s.; radical missense, P = 0.017; nonsense, n.s.)
Median gene diversities, although consistently lower than the corresponding mean values, showed the same pattern as mean gene diversities, with radical missense and nonsense SNPs showing the lowest median gene diversities (Fig. 1 A). Because the median is less sensitive to outliers than is the mean, the fact that mean values were lower than median values is evidence of a modest positive skew of gene diversity values within each category of SNP. For the seven categories of SNPs in Fig. 1 A, the skewness values were as follows: 5′ noncoding, 0.37; intron, 0.38; synonymous, 0.35; 3′ noncoding, 0.31; missense (conservative), 0.67; missense (radical), 1.85; nonsense, 0.44. For the complete data set, skewness was 0.44. A positively skewed distribution of single-locus gene diversity values has been observed in many data sets, and is theoretically predicted to occur when loci are evolving subject to genetic drift (20).
To control for differences in gene diversity across loci, we further compared mean gene diversities at nonsynonymous SNP sites in a pairwise fashion with those at silent SNP sites (i.e., those not causing amino acid changes) in the same gene (Fig. 1B). At missense SNP sites causing conservative amino acid changes, mean gene diversity was not significantly different from that at silent SNP sites in the same gene; in both cases, mean gene diversity was ≈18% (Fig. 1B). By contrast, at missense SNP sites causing radical amino acid changes, mean gene diversity (8.9%) was significantly lower than that at silent SNP sites (18.6%) in the same gene (Fig. 1B). Likewise, mean gene diversity at nonsense SNP sites (2.7%) was significantly lower than that at silent SNP sites (28.3%) in the same gene (Fig. 1B). These results are consistent with the hypothesis that purifying selection has affected allele frequencies at SNP loci in the human population, because gene diversities are lowest at sites where mutation is expected to have the greatest impact on protein structure.
To test for evidence of purifying selection on missense SNPs causing conservative amino acid changes, we compared human protein sequences with those of orthologous mouse protein sequences. A mouse ortholog was available for 279 conservative missense SNPs at 128 loci. At 200 of these SNP sites (71.7%), one of the two human alleles encoded an amino acid residue identical to that found in the mouse database sequence, whereas at 79 SNP sites (28.3%) both human alleles encoded residues different from that in mouse database sequence. Significant differences in mean gene diversity were observed among sites in which both amino acid residues were different from that seen in mouse, sites at which only the higher-frequency residue was different from that seen in mouse, and sites at which only the lower-frequency residue was the same as that seen in mouse (Fig. 2A).
Mean gene diversity was lowest (14.1%) when the higher-frequency residue was the same as that seen in mouse (Fig. 2 A). Mean gene diversities were similar when the lower-frequency allele was the same as that seen in mouse (22.7%) and when neither residue was the same as that seen in mouse (20.3%) (Fig. 2 A). This result is consistent with the hypothesis that purifying selection is acting against many mutations that introduce amino acid residues differing from those seen in the mouse because of functional constraints on evolutionarily conserved amino acid residues.
Furthermore, when one of the two alleles encoded the same residue as the mouse, while the other encoded a different residue, the mean allelic frequency of the former allele (75.3%) was over three times the mean frequency of the latter allele (24.7%) (Fig. 2B). This result provides additional evidence of purifying selection at these sites, because the higher-frequency allele tends to encode a residue conserved over mammalian evolutionary history, as is consistent with functional importance of many of these residues. It is also consistent with previous evidence that disease-associated human SNPs are likely to involve residues different from those seen in mouse orthologs (21).
Discussion
We found evidence of purifying selection in the case of nonsynonymous SNPs, many of which were found to occur at relatively high population frequencies. The frequencies of the lower-frequency allele at many of these loci were in the range of 1–10%. Wong et al. (22) similarly reported numerous nonsynonymous SNPs with similar allelic frequencies in a sample of 114 human genes. These frequencies are much higher than those reported for human genes causing severe disease phenotypes such as cystic fibrosis or Huntington chorea (23). This in turn suggests that the selection coefficients at many of these SNP sites are rather modest in comparison with those at loci associated with severe disease.
Ohta's (24–26) nearly neutral theory of molecular evolution emphasizes the evolutionary importance of slightly deleterious mutations and predicts that, because population sizes fluctuate over evolutionary time, slightly deleterious mutants can reach high frequencies as a result of genetic drift during population bottlenecks. Several lines of evidence support the hypothesis that the human population has expanded since the origin of modern humans 100,000–250,000 years ago (27, 28). The slightly deleterious alleles revealed by the present analyses may include mutants that drifted to relatively high frequency in the smaller ancestral population. In addition, the human population has historically been subdivided into a number of partially isolated subpopulations (28, 29), and drift may have acted similarly within subpopulations. The effects of drift during a population bottleneck on subsequent population gene diversity are predicted to last hundreds of thousands of years (30), and this process would explain the presence of purifying selection at sites with relatively high gene diversities. Thus, our results provide support for the nearly neutral model and for its application to human population genetics.
Our results show strong evidence of purifying selection at nonsynonymous polymorphic sites in the human genome. The effects of this selection, in terms of reduced gene diversity in comparison to silent SNPs in the same genes, are readily observable in a sample of modest size. Furthermore, because the effect of purifying selection is to reduce the frequency of a deleterious allele, our results suggest a strategy for nominating SNPs for genetic association studies of complex disease. A nonsynonymous SNP site with low gene diversity, relative to other SNP sites in the same gene that do not cause amino acid changes, might constitute a good candidate allele for disease association. This would be particularly true if the polymorphism causes a radical amino acid change or an amino acid change known to affect protein structure (4–7), and/or if the higher-frequency allele encodes a residue conserved in orthologous proteins of other mammals (8).
Consistent with the nearly neutral model, our results suggest that there are likely to be numerous slightly deleterious mutations present at relatively high frequency in human populations. If so, it seems likely that common disease phenotypes may be associated with different sets of variants in different individuals as a function both of each individual's genetic makeup as a whole and of environmental factors. This, in turn, implies that understanding the contributions of numerous genes of small phenotypic effect may be required before the genetic basis of a given disease is fully elucidated.
Acknowledgments
This project has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract NO1-CO-12400. Partial support was provided by National Institutes of Health Grant GM43940 (to A.L.H.).
Abbreviation: SNP, single nucleotide polymorphism.
References
- 1.Lohnmueller, K. E., Pearce, C. L. Pike, M., Lander, E. S. & Hirschhorn, J. N. (2003) Nat. Genet. 33, 177–182. [DOI] [PubMed] [Google Scholar]
- 2.Wheeler, D. L., Church, D. M., Lash, A. E., Leipe, D. D., Madden, T. L., Pontius, J. U., Schuler, G. D., Schriml, L. M., Tatusova, T. A., Wagner, L. & Rapp, B. A. (2002) Nucleic Acids Res. 30, 13–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sunyaev, S., Ramensky, V., Koch, I., Lathe, W., III, Kondrashov, A. & Bork, P. (2001) Hum. Mol. Genet. 10, 591–597. [DOI] [PubMed] [Google Scholar]
- 4.Wang, Z. & Moult, J. (2001) Hum. Mutat. 17, 263–270. [DOI] [PubMed] [Google Scholar]
- 5.Ng, P. C. & Henikoff, S. (2002) Genome Res. 12, 436–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramensky, V., Bork, P. & Sunyaev, S. (2002) Nucleic Acids Res. 30, 3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stitzel, N. O., Tseng, Y. Y., Pervouchine, D., Goddeau, D., Kasif, S. & Liang, J. (2003) J. Mol. Biol. 327, 1021–1030. [DOI] [PubMed] [Google Scholar]
- 8.Fleming, M. A., Potter, J. D., Ramirez, C. J., Ostrander, G. K. & Ostrander, E. A. (2003) Proc. Natl. Acad. Sci. USA 100, 1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Botstein, D. & Risch, N. (2003) Nat. Genet. Suppl. 33, 228–237. [DOI] [PubMed] [Google Scholar]
- 10.Gray, I. C., Campbell, D. A. & Spurr, N. K. (2000) Hum. Mol. Genet. 9, 2403–2408. [DOI] [PubMed] [Google Scholar]
- 11.Risch, N. J. (2000) Nature 405, 847–856. [DOI] [PubMed] [Google Scholar]
- 12.Chanock, S. (2001) Disease Markers 17, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao, Z., Fu, Y.-X., Hewett-Emmett, D. & Boerwinkle, E. (2003) Gene 312, 207–213. [DOI] [PubMed] [Google Scholar]
- 14.Kimura, M. & Ohta, T. (1974) Proc. Natl. Acad. Sci. USA 71, 2848–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nei, M. (1987) Molecular Evolutionary Genetics (Columbia Univ. Press, New York).
- 16.Miyata, T., Miyazawa, S. & Yasunaga, T. (1979) J. Mol. Evol. 12, 219–236. [DOI] [PubMed] [Google Scholar]
- 17.Pruitt, K. D. & Maglott, D. R. (2001) Nucleic Acids Res. 29, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson, J. D., Higgins, D. G. & Gibson, T. (1994) Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fitch, W. M. (1971) Syst. Zool. 20, 406–416. [Google Scholar]
- 20.Fuerst, P. A., Chakraborty, R. & Nei, M. (1977) Genetics 86, 455–483. [PMC free article] [PubMed] [Google Scholar]
- 21.Mouse Genome Sequencing Consortium (2002) Nature 420, 520–562. [DOI] [PubMed] [Google Scholar]
- 22.Wong, G. K.-S., Yang, Z., Passey, D. A., Kibukawa, M., Paddock, M., Liu, C.-R., Bolund, L. & Yu, J. (2003) Genome Res. 13, 1873–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKusick, V. A. & Francomano, C. A. (1997) Mendelian Inheritance in Man: a Catalog of Human Genes and Genetic Disorders (Johns Hopkins Univ. Press, Baltimore), 12th Ed.
- 24.Ohta, T. (1973) Nature 246, 96–98. [DOI] [PubMed] [Google Scholar]
- 25.Ohta, T. (1976) Theor. Popul. Biol. 10, 254–275. [DOI] [PubMed] [Google Scholar]
- 26.Ohta, T. (2002) Proc. Natl. Acad. Sci. USA 99, 16134–16137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harpending, H. C., Batzer, M. A., Gurven, M., Jorde, L. B., Rogers, A. R. & Sherry, S. T. (1998) Proc. Natl. Acad. Sci. USA 95, 1961–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhitovsky, L. A., Rosenberg, N. A. & Feldman, M. W. (2003) Am. J. Hum. Genet. 72, 1171–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watkins, W. S., Rogers, A. R., Ostler, C. T., Wooding, S., Bamshad, M. J., Brassington, A. M., Carroll, M. L., Nguyen, S. V., Walker, J. A., Prasad, B. V., et al. (2003) Genome Res. 13, 1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nei, M., Murayama, T. & Chakroborty, R. (1975) Evolution (Lawrence, Kans.) 29, 1–10. [Google Scholar]