

Abstract
Multi-scale lithography and cryogenic deep reactive ion etching techniques were used to create ensembles of nanoporous, picoliter volume, reaction vessels within a microfluidic system. The fabrication of these vessels is described and how this process can be used to tailor vessel porosity by controlling the width of slits that constitute the vessel pores is demonstrated. Control of pore size allows the containment of nucleic acids and enzymes that are the foundation of biochemical reaction systems, while allowing smaller reaction constituents to traverse the container membrane and continuously supply the reaction. In this work, a 5.4kB DNA plasmid was retained within the reaction vessels and labeled under microfluidic control with ethidium bromide as an initial proof-of-principle. Subsequently, a coupled enzyme reaction, in which glucose oxidase and horseradish peroxidase were contained and fed with a substrate solution of glucose and Amplex Red™ to produce fluorescent Resorufin, was carried out under microfluidic control and monitored using fluorescent microscopy. The fabrication techniques presented are broadly applicable and can be adapted to produce devices in which a variety of high aspect ratio, nanoporous silicon structures can be integrated within a microfluidic network. The devices shown here are amenable to being scaled in number and organized to implement more complex reaction systems for applications in sensing and actuation as well as fundamental studies of biological reaction systems.
Introduction
Biochemical reactions such as protein synthesis and enzymatic conversion are fundamental to the function of living systems and are vital tools in industry and research. As our understanding of these reactions, and the living cellular systems in which they are carried out, has grown, the importance of cellular organization and efficiencies achieved by operating at the cellular scale have become more apparent1. This, combined with a desire to understand “what constitutes life,” has led to efforts to create synthetic cellular-scale containers, cell mimics, in which basic biochemical processes can be sustained2-6.
Chemical reactions within the cell depend on the cellular membrane, as it 1) defines the spatial extent (volume) of the cell, and 2) regulates the transfer of reactants and products between the cellular reaction volume and its surroundings. Within the small volume of the cell, passive transport of molecules via diffusion is rapid and facilitates the efficient mixing of molecules within the cell1. Furthermore, because the cell can contain only a limited, small number of molecules, small changes in the numbers of molecules can lead to drastic changes in concentration within the cell. Such changes, can lead to significant alterations in cellular function and fate7. Beyond its passive role as a “container,” the cellular membrane plays a dynamic role in affecting internal concentrations by regulating the exchange of materials between the internal volume of the cell and the external microenvironment, both in concert with, and against electrical and chemical potentials.
A growing body of work has examined the efficacy of biomimetic systems for carrying out biological processes in micro/cellular-scale systems. Such work includes the solution synthesis of liposomes and vesicles5, 6, 8-10, serial creation of surface bound, single and networked vesicles11, microfluidic generation of cellular scale droplets formed in multiphase fluidics12, 13, and inorganic microscale containers 2, 14-18. In general, these approaches have all sought to capture the basic function of the cellular membrane, defining small volumes in which diffusive transport is efficient, and the transfer of materials between the internal reaction volume and the external environment is regulated.
Norieaux et al8 examined the use of vesicle bioreactors for carrying out cell free transcription and translation for the production of GFP, and found that reactions could be sustained for up to four days with the incorporation of an alpha-hemolysin pore that made the membrane permeable to external reactants. In similar work, Murtas et al. successfully carried out the expression of enhanced GFP in liposomes using a specific and well-defined set of minimal enzymes6. A more thorough review of work done in this area is provided by both Monnard5 and by Walde and Ichikawa10. Despite the success of these solution based approaches, the isolation of individual vesicles for monitoring individual reactions (addressability) and potential difficulties involving long term stability and storage of such vesicles in extreme conditions of tonicity (osmolarity), pH and temperature remain a challenge for the integration of these vesicle reactors into a sensing and actuation platforms.
In related work, Karlsson et al11 utilized vesicles formed on surfaces, which could be connected by small nanoscale vesicle channels, to create small reaction volumes and networks. Because each volume could be formed using a different microinjection pipette, each could be filled with individual reagents and scaled according to the desired function. The technique was highly effective, and allowed tuning of mass transport from one volume to the other by controlling the interconnecting channel width and length, establishing a predictable concentration gradient (flux) between reaction containers. The approach demonstrates improved addressability over conventional liposome and vesicle based approaches, allowing the progression of a reaction within a single volume to be monitored over time. However, the serial nature by which the reaction volumes are created coupled with their inability to be stored over long periods makes the creation and use of many identical reaction volumes difficult. Furthermore, like vesicles synthesized in solution, the approach is likely to suffer from poor stability under more extreme reaction conditions.
Advances in soft lithography have made the study of reactions in microscale droplets created within lab-on-a-chip (LOC) systems accessible. Significant contributions have been made in the development and study of droplet generating and mixing systems12, 13. The exquisite control of droplet size and mixing afforded by microfluidics has even facilitated studies of reaction kinetics in sub-cellular scale droplets19. Song et al review the topic of reactions in droplets in detail, highlighting the techniques and describing various applications for microfluidic droplet generation and mixing20. While droplet-based microfluidics provide exquisite control of single reaction volumes, the multiphase nature of these systems can make the exchange of materials between reaction containers difficult.
To develop a cellular scale reaction container that can be spatially addressed for monitoring and filling, while allowing both the storage and exchange of chemical information across the reactor membrane, a combination of conventional microfabrication and advanced lithographic techniques was used to create ensembles of nanoporous, picoliter volume reaction vessels that are integrated within a microfluidic system. Similar concepts have been used in the development of nanofabricated chemostats and cellular containers formed in silicon14, 21, 22 and silicone elastomer23. In these systems, microchannels allow for loading and restricted movement of cells, and nanoscale pores or slits allow the exchange of molecular species (nutrients, signaling molecules, etc.) with the external environment or adjacent bioreactors. In this work, methods for refining pore size at a level that allows the differentiation between large and small molecules, as opposed to cellular and molecular scale materials, is demonstrated. The reaction vessels described here are batch fabricated, allowing over a thousand identical containers to be produced in a single fabrication run, and can be stored indefinitely. Each of the reaction containers can be individually filled with different reagents at the time of use. Because the process used to fabricate the devices combines direct write electron beam lithography to define individual reaction volumes, the physical spacing of the containers and their individual properties, including total volume, pore size and shape, can be tailored for specific applications. For example, placing reaction vessels proximal to one another within a microfluidic channel, that contains only aqueous solution, can allow for reaction products to be exchanged between vessels via diffusion across membranes and convection along the microchannel. This would enable the study of reaction networks where the product of one reaction regulates or initiates a second reaction occurring downstream. The level of communication between vessels can be tailored for specific applications by controlling the proximity and porosity of containers, their positions relative to the direction of fluid flow, and the overall rate of fluid flow within the microchannel.
The fabrication techniques used to create these containers and the impact of those techniques on pore size and functionality are described here. In addition, the methods used to tailor the effective size of device pores and a demonstration of the ability to contain larger biomolecules (plasmid DNA, and enzymes) within the reaction containers while microfluidically addressing the reaction containers and exchanging small molecules are provided. Two simple reactions were demonstrated; 1) the labeling of DNA in the reaction vessel with ethidium bromide, and 2) a coupled enzyme reaction of HRP and GOX with a fluorogenic substrate to determine glucose concentrations within the microfluidic channel. While the use of inorganic membrane materials does not allow the direct incorporation of biological materials or function into the membrane itself, the long-term and overall stability, addressability, and ability to discretely tailor reaction vessel properties as a means of controlling material exchange suggest that the described platform represents an useful complementary alternative to bioreactors based on organic membrane systems.
Materials and Methods
Chemicals and Reagents
Fluorsbrite™ 100nm and 350nm latex beads used in size exclusion experiments were obtained from Polysciences Inc. (Warrington, PA) Amplex Red™ was purchased from Invitrogen. Ethidium bromide, horseradish peroxidase, glucose oxidase and other reagents were purchased from Sigma Aldrich. A Sylgard 184 Elastomer Kit was obtained from Fisher Scientific.
Plasmid DNA (5.4kB) used in the DNA containment and labeling experiments was constructed by recombining a gene for enhanced GFP (eGFP) into pDEST17 (Invitrogen) to allow expression of 6xHis-GFP from a T7 promoter. Plasmid DNA was purified from E. coli BL21 (DE3) (Invitrogen) using a Qiagen Midiprep kit (Valancia, CA) according to manufacturer’s instructions. Production of eGFP, was carried out by using the plasmid described above to transform E. coli BL21 (DE3) cells. IPTG was added to a final concentration of 1mM to induce expression of 6xHis-GFP. His-tagged protein was purified from cell lysates on a Ni-NTA column according to manufacturer’s instructions (Qiagen, no 31314). Protein concentration was determined using a Bradford assay (Pierce).
Nanoporous Reaction Vessel Fabrication
The fabrication of nanoporous reaction vessels is carried out using a combination of electron beam and optical lithography techniques to define the etch masks for the reaction vessels and surrounding microchannels. Electron beam lithography is used to define the etch mask for the reaction containers, and conventional contact photolithography is used to align and define the microchannels. Once the etch masks are defined, the vessels and microchannels are etched simultaneously using a cryogenic silicon etching process. A brief plasma enhanced chemical vapor deposition process is used to coat the reaction vessel with silicon dioxide and to modify the final pore size. (Figure 1)
Figure 1.
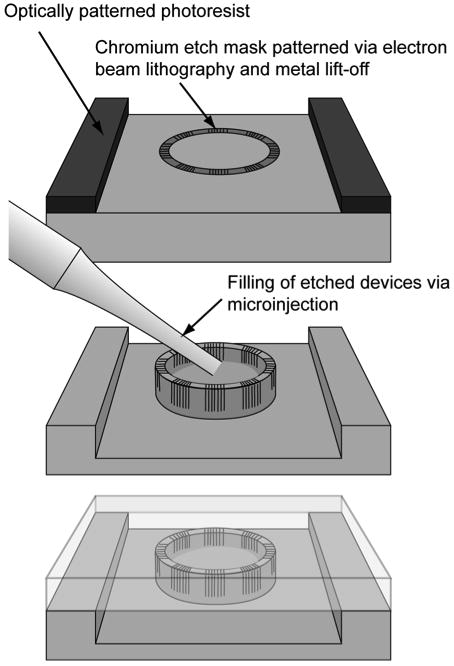
Fabrication of nanoporous reaction vessels within microfluidic channels is carried out by combining electron beam lithography and metal lift-off techniques with contact photolithography to define the reaction vessel and channel geometries (top). The pattern is transferred into the exposed silicon using cryogenic silicon etching and individual vessels are filled with reactants via microinjection (middle). Whole chips are then sealed with a silicone elastomer lid (bottom)
Fabrication was carried out using standard 4” silicon wafers with a <100> crystal orientation. To define the etch mask for the reaction vessels, wafers were spin-coated with NANO™PMMA 495 A4 electron beam resist (Microchem Corp., Newton, MA) at 2500 RPM for 45 seconds and baked on a hotplate for 10 minutes at 180°C. A JEOL JBX9300-FS electron beam lithography system operating at 100kV and 2nA was used to expose the geometry of the reaction container and optical alignment marks for subsequent processing. The dose was approximately 1250μC/cm2, but was modulated through iterative exposure and examination via scanning electron microscopy to yield the desired pore sizes. Patterns were developed for one minute in 1:3 methyl isobutyl ketone (MIBK): isopropyl alcohol (IPA), rinsed with IPA, and dried with nitrogen. The patterns were then exposed to a brief oxygen plasma (10sccm, 150mT, 100W, 6 seconds) before depositing 15 nm of chromium via electron beam evaporation. Samples were soaked in acetone, and rinsed with IPA and water, leaving behind a chromium etch mask in the areas exposed by the electron beam.
Microchannel masks were defined using conventional contact alignment optical lithography. A negative photoresist, JSR Micro NFR 016 D2 55cp (JSR Micro Inc., Sunnyvale, CA) was chosen as the etch mask for the microchannels as it was more resistant to cracking during the cryogenic etching process. Resist was spin-coated at 6000 RPM following wafer treatment with Microprime P20 Adhesion promoter. Wafers were baked on a hot-plate at 90°C for 90 seconds, exposed, and baked for an additional 90 seconds at 90°C. Samples were then developed in CD26 (Microchem Corp., Newton, MA) (<5% tetramethyl ammonium hydroxide) for 20 seconds, until the microchannels were visibly developed. After rinsing and drying with N2, samples were baked for 3 minutes at 180°C.
Once the chromium and photoresist etch masks were defined, samples were exposed to a brief oxygen plasma (10sccm O2, 150mT, 400W, 1 min) to remove any organic solvent and resist residue left on the exposed silicon. Samples were then etched in an Oxford Plasmalab 100 Inductively Coupled Plasma Reactive Ion Etching systems using a cryogenic silicon etching process. This process is carried out by exposing samples to a plasma of SF6 and O2 at -110°C. The oxygen flow rate and RF bias of the plasma are used to balance etching and sidewall passivation to allow highly anisotropic etching of the exposed silicon to yield high aspect ratio features in silicon. Reaction vessels and channels were etched to a depth of 15μm at a rate of approximately 3 μm per minute. Following the completion of etching, mask materials were removed by an exposure to a more aggressive oxygen plasma and brief soak in chromium etchant.
Following removal of the etch mask materials an Oxford Plasmalab 100 Plasma Enhanced Chemical Vapor deposition system was used to deposit silicon dioxide on the structures. The duration of the deposition was varied to control final pore dimensions and tune size selectivity of the nanoporous reaction vessels.
Device Packaging and General Imaging Techniques
Polydimethylsiloxane (PDMS) used to seal the microchannels was mixed at a 10:1 w/w ratio, degassed and cured for approximately 1 hour at 70°C. PDMS was cut into 4mm thick pieces matching the microfluidic chip size. Inlet and outlet holes were punched in the device using an 18-gauge blunt tip needle. 0.020” OD polyethylene tubing was press fit into the PDMS. The packaged chip measured approximately 4 cm long by 15 mm wide. The number and type of individual reaction vessels can be modified from one chip to the next. Each chip configuration consisted of 30 vessels, three groups of five in each of two channels, spaced at equal intervals along the channel. Each group of five devices can be imaged simultaneously in the viewing area of a Zeiss Axioscope epifluorescent microscope with a 10x objective. For the experiments described here, each of the vessels was created with the same nominal pore size, a 200nm wide pore that extended across the full width of the vessel wall. The ability to control pore size down to tens of nanometers via electron beam lithography, prior to coating with silicon dioxide, was demonstrated by the creation of test reaction vessels with 5 different size pores. Pore width was designed to be 30, 50, 80, 100, and 200nm. The four smallest sizes started at 200nm wide and tapered to a pore of width 30, 50, 80, or 100nm and length of 600nm.
Loading of Nanoporous Reaction Vessels
Prior to sealing with a PDMS “roof,” chips were treated with a 1 mg/mL solution of bovine serum albumin for ten minutes to prevent nonspecific adsorption of reaction constituents to the device walls. Individual vessels were filled using an adapted cell microinjection system. A hydraulic manipulator with manual injection pump was attached to a Burleigh Micromanipulator. Pulled micropipettes with tip diameters of two microns (Small Parts Inc., Logansport, IN) were backfilled with the desired reagent using a flexible polyimide needle, (World Precision Instruments, Sarasota, FL). Chips were placed under a stereo microscope and were manually filled by touching the filled pipette into the center of the reaction vessels. The volume of the device structure and viscosity of the filling solution dictated the amount of material injected, as materials generally wetted the device to the inner wall of the membrane, allowing repeatable filling of devices. For DNA experiments solutions contain 4% glycerol to reduce the risk of overfilling devices
Functional Demonstration of Size Exclusion and Molecular Containment
To demonstrate the functional size range of the 200nm pore devices, microchannels were loaded with solutions of 100nm and 350nm Fluorsbrite™ latex beads. Care was taken to insure that air bubbles were not trapped in the reaction vessels. DNA containment and labeling experiments were carried out by filling reaction vessels with concentrations of DNA ranging from 0.06 to 600 ng/μL in buffer. A labeling solution of ethidium bromide (EtBr) at a concentration of 100μg/mL was injected into the microchannel. Images were captured before adding EtBr, immediately after adding EtBr, and after 30 minutes under flow at a flow rate of 10 μL/hour.
Containment of Proteins and Coupled Enzyme Reactions
Flow experiments were conducted by first filling the reaction vessels with a 100 μg/mL solution of GFP. Under a steady flow of 10 μL/hour ensembles of reaction vessels were imaged. A Retiga firewire camera and QCapture software were used to capture fluorescent and brightfield images. Care was taken to ensure that exposure times, binning and other relevant camera settings were maintained to allow reasonable comparisons to be made between experiments. A shutter was used to minimize photobleaching when images were not being captured.
Coupled enzyme reactions were carried out by first filling reaction vessels with a mixture of Horseradish Peroxidase (0.25 U/mL) and glucose oxidase (0.25 U/mL) in PBS buffer. Channels were filled with a solution containing 100μM amplex red and glucose ranging in concentrations from 100μM to 100mM. Fluorescent images were recorded at different time intervals under constant flow conditions.
Results and Discussion
Device Fabrication
The hybrid lithography techniques used in the reaction vessel fabrication, combining optical and electron beam lithography, provide for a robust and flexible process by which device geometries and configurations can be readily varied for different applications. The use of electron beam lithography to define the reaction vessel etch mask allows routine definition of features below 30nm. Additionally, because the containers are defined using a direct write technology, the number, organization and physical attributes of containers can be varied across samples and from run to run. Batch fabrication of samples allowed eight chips to be completed per wafer. Electron beam lithography write times were relatively short, 45 minutes per wafer, allowing multiple samples to be produced in an afternoon. Further reductions in write times could be realized by using higher sensitivity electron beam resist, increasing the electron beam current and increasing the writing shot pitch. Completed vessels had an inner diameter of 40 microns, a 2 micron thick wall, and 56 pores (8 sets of 7 pores spaced at 45° intervals). Structures and channels were nominally 15 microns tall, with heights measured using physical profilometry. The internal volume of the structures were approximately 18 picoliters. A diameter of 40 microns was chosen because of the relative ease with which the devices could be imaged and filled using microinjection. Figure 2 illustrates the multi-scale features of the completed devices.
Figure 2.

Scanning electron micrographs of reaction vessels taken prior to filling and sealing show the integration of porous vessels within a microfluidic channel (left), the overall device size and pore locations (center) and nominal pore size (right). Images were taken at a 30° stage tilt.
While electron beam lithography could be used to create pores below 30nm (figure 3) we ultimately found that the modification of larger pores through silicon dioxide deposition was a more effective strategy for reducing and controlling pore size. Thus the containment experiments discussed in this manuscript were performed with devices having initial pore widths of 200nm and a minimal silicon dioxide coating (2min duration). One of the primary drivers for making this choice is the coupling of slit width with etch depth as shown in figure 4.
Figure 3.

Top view scanning electron micrographs of the porous membranes taken at a magnification of 35000X show the different dimension pores achieved via electron beam lithography and cryogenic silicon etching. Pore width (wp), corresponds to the dimension used in the lithography patterns. Final dimensions ranged from approximately 10 to 30nm smaller than the CAD-defined pore width.
Figure 4.

Membrane test structures with different sized pores (labeled in nm) were used to examine the effects of pore width and oxygen flow rates on pore depth. At 12 sccm of oxygen flow (top) no passivating material is visible and 200nm pores etch more than 60% of the membrane height. At higher oxygen flow rates (bottom), passivation is increased leaving passivating polymer deposited on structures and severe reductions in pore depth.
As can be seen in figures 2 and 4, even though the pores and the larger vessel structure are etched simultaneously the overall depth of the device and channel are nearly twice the depth of the pore slit. This can be attributed to aspect ratio dependent etching24 resulting from poor diffusion of reactive gases and depletion of etching reactants within the narrower slits. Test structures combining different pore widths side by side were constructed to illustrate the effects of pore width on etch depth. In addition, the effects of plasma oxygen content on sidewall profile are illustrated (figure 4). During the cryogenic etch process a silicon-oxy-fluorine polymer forms on the sidewalls of the structure while reactive fluorine ions etch exposed silicon. By balancing the lateral etch rate with the passivating polymer deposition an anisotropic etch process can be developed. At low oxygen content some undercutting can occur as shown in the 12 sccm O2 flow condition (top), while at higher oxygen content the deposition and presence of the passivating polymer becomes notable (bottom). The sensitivity of the balance in passivation and etching is notable near the slits where a small pillar-like structure is noticeable beneath the pore. We hypothesize that this results from the local consumption of etching gases in the etching of the pore slit. Locally this results in an excess of passivating gases below the slit, resulting in local variations in sidewall profile and the appearance of the pillar-like structures below the slit. It is important to note, and indicated from figure 4, that the cryogenic etching process is sensitive to oxygen content. The choice of oxygen flow rate must therefore be fine-tuned for different structure designs and geometries. Etch profiles were also affected by variations in RF power, though the effects were less dramatic (not shown).
Functional Demonstration of Size Exclusion and Molecular Containment
In separate experiments 100 and 350 nm beads were injected into microfluidic devices containing vessels with 200nm pores. 100 nm beads easily entered the vessels while 350 nm beads were excluded. Figure 5 demonstrates that vessels are defect free and have a functional pore size, and size distribution between 100nm and 350nm. In previous work we examined the use of patterned carbon nanofiber membranes for similar separations, but found that defect density resulting from the nanofiber growth resulted in “leaky” membrane structures25, 26. In addition to being free of defects, the deterministic nature of the structures used here help make modeling and simulation of these systems more tractable. This basic demonstration helps illustrate the potential utility in using the reaction vessels to exclude molecules or functionalized particles based on size. In future work, surface modification techniques may be used to create pores that enable selection based on charge or selective binding.
Figure 5.

Fluorescent polystyrene beads were introduced into microchannels at modest flow rates. (a) 100nm diameter beads traversed the reaction vessel membrane as expected, while larger, (b) 350nm beads were excluded.
In DNA labeling experiments 600ng/μL, 60ng/μL, 6ng/μL and 0.6ng/μL and 0.06ng/μL of 5.4kb plasmid DNA were loaded in reaction vessels using the Burleigh microinjection system. Images taken 30 minutes after ethidium bromide is introduced through the microfluic channel illustrate a fluorescent response proportional to DNA concentration (figure 6). Only limited degradation of the fluorescent signal was observed over time indicating that the DNA is contained within the reaction vessels over extended periods. The successful containment of DNA while allowing small molecules to traverse the reactor membrane and intercalate the DNA suggest a potential path for the development of more complex systems based on cell-free transcription and translation. DNA containment experiments were carried out in reaction vessels with a 200nm pore and minimal silicon oxide coating.
Figure 6.

Plasmid DNA was loaded into reaction vessels with a minimal silicon dioxide coating at different concentrations (600ng/μL – 0.06ng/μL) corresponding to the total mass shown. Plasmid DNA was labeled in situ with ethidium bromide. Fluorescence was evident immediately and did not diminish, indicating that plasmid DNA is retained, and small molecules are able to traverse the membrane and react with the plasmid.
A 5.4kb plasmid has an experimentally and computationally predicted radius of gyration between 80 and 240nm27, 28. This suggests an overall diameter that fluctuates near the 200nm pore slit width. Assuming a diffusion coefficient of approximately 3×10-8cm2/s for the 5.4kb plasmid, a simple numerical model of free leakage of DNA through the 200nm pores approximates a 50% reduction in concentration in less than 3 minutes, and a near zero concentration within 10 minutes (supplementary material). The persistence of fluorescence from the labeled DNA after 30 minutes and the absence of fluorescent material outside of the vessels indicates that the plasmid is not able to freely diffuse through the reaction vessel membrane. Ultimately, to control the transport of specific proteins or small molecules across and between reaction vessels, the pore size and surface charge or functionality would need to be tuned further. Towards that end we examined the use of plasma enhanced chemical vapor depostion to reduce pore size (width) and limit the flux of GFP across the vessel membranes. A comparison of GFP retention within devices having different thickness coatings of silicon dioxide was carried out. Individual device chips were subjected to different duration PECVD silicon dioxide depositions. From the scanning electron micrographs shown in figure 7 it is clear that there is a considerable reduction in pore size as more silicon dioxide is deposited on the reaction vessels. Though it would be difficult to directly measure the effective gap size through the entire thickness of the membrane using imaging techniques, changes in the retention of GFP within devices that have different thickness silicon dioxide coatings suggests that we can modify pore size to tune the flux of small molecules across the reactor membrane and to contain proteins of a particular size (figure 7).
Figure 7.

The deposition of silicon dioxide on membrane structures can be used to reduce pore width. At a deposition rate of approximately 60nm/min on flat horizontal surfaces, deposition on vertical, pore sidewalls is considerably less. Pores are shown after (a) 2, (b) 4, (c) 6, and (d) 8 minutes of PECVD silicon dioxide deposition. The retention of green fluorescent protein was observed after 5 minutes in reaction vessels coated with silicon dioxide for (e) 4 minutes (100ms digital camera exposure) and (f) 6 minutes (50ms digital camera exposure). Each of the devices shown was filled with the same concentration of GFP.
Coupled Enzyme Reaction System
The ability to controllably contain macromolecules opens the possibility for applications in both sensing and the production or conversion of biological materials. Coupled enzyme reactions were carried out in vessels subjected to a 7 minute PECVD silicon dioxide coating. The retention of GOX and HRP within the reactors is demonstrated by the continued fluorescence of the coupled reaction system under steady flow. It is conceivable that nonspecific adsorption could account for some percentage of the retained fluorescence, but one would expect that this would manifest as some initial drop in fluorescence intensity followed by a longer steady state condition. We see a proportional response of fluorescence with glucose concentration 100 seconds after the addition of the substrate. Figure 8 illustrates the measured fluorescence intensity for the coupled enzyme reaction at a fixed time and shows a predictable increase in intensity with glucose concentration. In future work we intend to characterize, in greater detail, the effects of enzyme concentration and device geometry on enzyme kinetics and compare results with predictive reaction/diffusion models for our platform.
Figure 8.
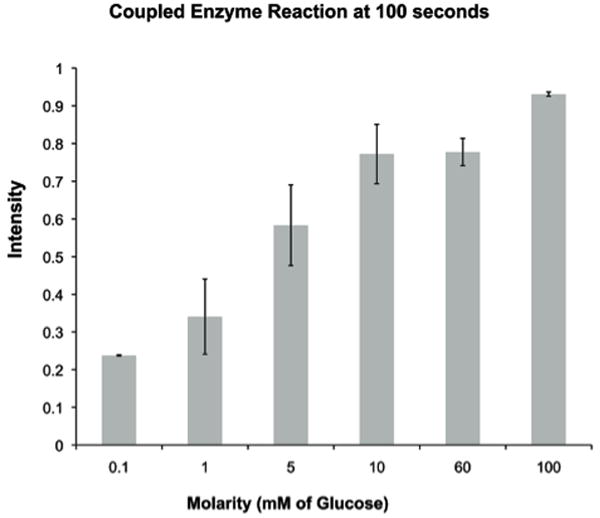
Fluorescence intensity resulting from a coupled GOX/HRP enzymatic conversion of glucose and amplex red to fluorescent resorufin at a fixed time point of 100 seconds. GOX/HRP were loaded into reaction vessels at a concentration of 0.25U/mL.
Conclusions
Lessons gleaned from nature clearly point towards the biological cell as a model system for carrying out a multitude of functions. While it is a tremendous endeavor to synthetically engineer and build all of the complexities of the cell into a synthetic system from the bottom-up, basic physical aspects of cell design can be replicated in synthetic nanoengineered systems. Here, the concepts of controlled flux and scaled transport drive the design of synthetic reaction vessels which can eventually be used to contain and create more complex networks. One can envision reaction systems akin to synthetic gene regulatory networks as described in synthetic biology pursuits29, 30. The flux of material into or between chambers could be tailored to modulate the level of feedback between systems or to allow response to changes in the local environment. Using multi-scale fabrication techniques to create ensembles of addressable, nanoporous, cellular scale reactors allows the retention of larger species that represent information or functionality within a reaction system while allowing materials and energy to freely traverse the porous membrane. Such reaction vessels may prove useful in fundamental studies of protein-based complex reaction systems and will enable the development of responsive sensors and therapeutic platforms that rely upon the on-demand conversion or production of biological materials.
Supplementary Material
Acknowledgments
This research was supported by NIH Grant EB000657. STR and MJD would like to acknowledge funding from the Center for Nanophase Materials Sciences, which is sponsored at Oak Ridge National Laboratory by the Division of Scientific User Facilities, U.S. Department of Energy. This work was performed at the Oak Ridge National Laboratory, managed by UT-Battelle, LLC, for the U.S. DOE under Contract No. DE-AC05- 00OR22725.
References
- 1.Verkman AS. Trends Biochem Sci. 2002;27:27–33. doi: 10.1016/s0968-0004(01)02003-5. [DOI] [PubMed] [Google Scholar]
- 2.Doktycz MJ, Simpson ML. Mol Syst Biol. 2007;3 doi: 10.1038/msb4100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pohorille A, Deamer D. Trends Biotechnol. 2002;20:123–128. doi: 10.1016/s0167-7799(02)01909-1. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Ruder WC, Leduc PR. Trends Biotechnol. 2008;26:14–20. doi: 10.1016/j.tibtech.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Monnard PA. J Membr Biol. 2003;191:87–97. doi: 10.1007/s00232-002-1046-0. [DOI] [PubMed] [Google Scholar]
- 6.Murtas G, Kuruma Y, Bianchini P, Diaspro A, Luisi PL. Biochem Biophys Res Commun. 2007;363:12–17. doi: 10.1016/j.bbrc.2007.07.201. [DOI] [PubMed] [Google Scholar]
- 7.Raj A, van Oudenaarden A. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noireaux V, Libchaber A. P Natl Acad Sci USA. 2004;101:17669–17674. doi: 10.1073/pnas.0408236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pileni MP. J Phys Chem-Us. 1993;97:6961–6973. [Google Scholar]
- 10.Walde P, Ichikawa S. Biomol Eng. 2001;18:143–177. doi: 10.1016/s1389-0344(01)00088-0. [DOI] [PubMed] [Google Scholar]
- 11.Karlsson A, Sott K, Markstrom M, Davidson M, Konkoli Z, Orwar O. J Phys Chem B. 2005;109:1609–1617. doi: 10.1021/jp0459716. [DOI] [PubMed] [Google Scholar]
- 12.He MY, Edgar JS, Jeffries GDM, Lorenz RM, Shelby JP, Chiu DT. Anal Chem. 2005;77:1539–1544. doi: 10.1021/ac0480850. [DOI] [PubMed] [Google Scholar]
- 13.Tice JD, Song H, Lyon AD, Ismagilov RF. Langmuir. 2003;19:9127–9133. [Google Scholar]
- 14.Desai TA, West T, Cohen M, Boiarski T, Rampersaud A. Adv Drug Deliver Rev. 2004;56:1661–1673. doi: 10.1016/j.addr.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 15.Fletcher BL, Retterer ST, McKnight TE, Melechko AV, Fowlkes JD, Simpson ML, Doktycz MJ. Acs Nano. 2008;2:247–254. doi: 10.1021/nn700212k. [DOI] [PubMed] [Google Scholar]
- 16.Leoni L, Desai TA. Adv Drug Deliver Rev. 2004;56:211–229. doi: 10.1016/j.addr.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto T, Nojima T, Fujii T. Lab Chip. 2002;2:197–202. doi: 10.1039/b205010b. [DOI] [PubMed] [Google Scholar]
- 18.Fletcher BL, Hullander ED, Melechko AV, McKnight TE, Klein KL, Hensley DK, Morrell JL, Simpson ML, Doktycz MJ. Nano Lett. 2004;4:1809–1814. [Google Scholar]
- 19.Liu Y, Jung SY, Collier CP. Anal Chem. 2009;81:4922–4928. doi: 10.1021/ac900624h. [DOI] [PubMed] [Google Scholar]
- 20.Song H, Chen DL, Ismagilov RF. Angew Chem Int Ed Engl. 2006;45:7336–7356. doi: 10.1002/anie.200601554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keymer JE, Galajda P, Lambert G, Liao D, Austin RH. Proc Natl Acad Sci U S A. 2008;105:20269–20273. doi: 10.1073/pnas.0810792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keymer JE, Galajda P, Muldoon C, Park S, Austin RH. Proc Natl Acad Sci U S A. 2006;103:17290–17295. doi: 10.1073/pnas.0607971103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hung PJ, Lee PJ, Sabounchi P, Lin R, Lee LP. Biotechnol Bioeng. 2005;89:1–8. doi: 10.1002/bit.20289. [DOI] [PubMed] [Google Scholar]
- 24.Yeom J, Wu Y, Selby JC, Shannon MA. J Vac Sci Technol B. 2005;23:2319–2329. [Google Scholar]
- 25.Fowlkes JD, Fletcher BL, Retterer ST, Melechko AV, Simpson ML, Doktycz MJ. Nanotechnology. 2008;19 doi: 10.1088/0957-4484/19/41/415301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Retterer ST, Melechko A, Hensley DK, Simpson ML, Doktycz MJ. Carbon. 2008;46:1378–1383. doi: 10.1016/j.carbon.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith DE, Perkins TT, Chu S. Macromolecules. 1996;29:1372–1373. [Google Scholar]
- 28.Langowski J, Hammermann M, Klenin K, May R, Toth K. Genetica. 1999;106:49–55. doi: 10.1023/a:1003720610089. [DOI] [PubMed] [Google Scholar]
- 29.Hasty J, McMillen D, Collins JJ. Nature. 2002;420:224–230. doi: 10.1038/nature01257. [DOI] [PubMed] [Google Scholar]
- 30.Purnick PE, Weiss R. Nat Rev Mol Cell Biol. 2009;10:410–422. doi: 10.1038/nrm2698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.