

Abstract
Iron-sulfur clusters are versatile electron transfer cofactors, ubiquitous in metalloenzymes such as hydrogenases. In the oxygen-tolerant Hydrogenase I from Aquifex aeolicus such electron “wires” form a relay to a diheme cytb, an integral part of a respiration pathway for the reduction of O2 to water. Amino acid sequence comparison with oxygen-sensitive hydrogenases showed conserved binding motifs for three iron-sulfur clusters, the nature and properties of which were unknown so far. Electron paramagnetic resonance spectra exhibited complex signals that disclose interesting features and spin-coupling patterns; by redox titrations three iron-sulfur clusters were identified in their usual redox states, a [3Fe4S] and two [4Fe4S], but also a unique high-potential (HP) state was found. On the basis of 57Fe Mössbauer spectroscopy we attribute this HP form to a superoxidized state of the [4Fe4S] center proximal to the [NiFe] site. The unique environment of this cluster, characterized by a surplus cysteine coordination, is able to tune the redox potentials and make it compliant with the [4Fe4S]3+ state. It is actually the first example of a biological [4Fe4S] center that physiologically switches between 3+, 2+, and 1+ oxidation states within a very small potential range. We suggest that the (1 + /2+) redox couple serves the classical electron transfer reaction, whereas the superoxidation step is associated with a redox switch against oxidative stress.
Keywords: electrochemistry, EPR, iron-sulfur centers, O2-sensitivity
Hydrogenases are metalloproteins occurring in the metabolic pathway of a wide variety of microbial organisms and catalyze the reversible oxidation of dihydrogen: H2⇌2H+ + 2e- (1). The growing interest in alternative sources of energy has focused scientific research on understanding and engineering these enzymes for future applications (2). One of the major limitations of hydrogenases, however, is their sensitivity towards oxygen. Recently, the discovery of hydrogenases that retain catalytic activity in oxygenic environments has potentially opened new applications as “green” vanguard catalysts, in particular as electrocatalysts on electrodes for biofuel cells (3, 4).
Aquifex aeolicus is a hyperthermophilic Knallgas bacterium with optimum growth temperature of 85 °C (5). This microorganism harbors three distinct [NiFe] hydrogenases, among which Hase I is located in the aerobic respiration pathway and attached to the membrane via a diheme cytb (6). Hase I consists of two subunits; the large subunit contains the hetero-bimetallic nickel-iron site and the small subunit the electron transfer cofactors, namely iron-sulfur clusters (6). Based on spectroelectrochemical studies, this enzyme exhibits enhanced thermostability and tolerance for inhibitors (e.g., O2 and CO) (4, 7).
Although the structures of O2-sensitive hydrogenases are well characterized (8, 9), such information is still lacking for O2-tolerant enzymes. The molecular mechanism and structural determinants for this increased oxygen tolerance remain to be understood. Recent EPR and FTIR studies have shown that the [NiFe] center of Hase I has a geometric and electronic structure similar to standard hydrogenases (6, 7). The as-isolated enzyme is in the Ni-B state, a dormant form known to carry a bridging hydroxyl ligand between the two metals. The Ni-B state is quickly reactivated and Hase I enters the catalytic cycle consisting of three intermediates (7). Reactivation occurs at potentials similar to those of standard hydrogenases, whereas catalysis sets in at approximately 100 mV more positive potentials (7).
In standard [NiFe] hydrogenases electrons are channeled to the redox partners through a “wire” consisting of one [3Fe4S] cluster placed in-between two low-potential [4Fe4S] clusters (8) (Fig. 1). In the sequence of the small subunit of Hase I, the ten cysteines and one histidine that bind these three iron-sulfur centers are fully conserved, indicating that one [3Fe4S] and two [4Fe4S] clusters are also present in Hase I (Fig. 1). The EPR signals of these clusters, however, are more complex (6) than in standard hydrogenases, showing additional species and magnetic interactions (10).
Fig. 1.

Top: Sequence alignment of O2-sensitive and O2-tolerant hydrogenases. A set of columns of the same color represents the conserved residues that bind a specific cluster, highlighted in the structure of D. vulgaris MF (1WUI pdb code). The two extra cysteines in the A. aeolicus enzyme are depicted in yellow. Bottom: Localization of the [NiFe] site and the three [FeS] clusters in the standard hydrogenases and the closest metal-to-metal distances between different cofactors. The [FeS] clusters are denoted as proximal (P), medial (M), and distal (D), based on their distance from the [NiFe] site (Fig.S1).
A closer inspection of the iron-sulfur center proximal to the [NiFe] active site and homology structure models show that the latter has an atypical sequence motif 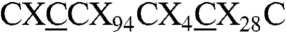 , with two extra cysteines (
, with two extra cysteines (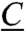 ); these may potentially participate in its coordination and thus affect its electronic and redox properties. Interestingly these extra cysteines are also present in the membrane bound hydrogenase (MBH) from Ralstonia eutropha and Hyd-1 from Escherichia coli (Fig. 1), which show similar spectroscopic features (11, 12). The presence of these residues has been shown to contribute to the oxygen stability of the R. eutropha enzyme and is therefore associated with a possible redox chemistry protecting the enzyme against oxidative stress (13). In the present work the type and redox properties of the constituent iron-sulfur clusters are investigated with the aim to (i) portray the energy coupled processes between Hase I and its redox partners and (ii) outline a relation between O2-tolerance and electronic/chemical events occurring at the iron-sulfur clusters.
); these may potentially participate in its coordination and thus affect its electronic and redox properties. Interestingly these extra cysteines are also present in the membrane bound hydrogenase (MBH) from Ralstonia eutropha and Hyd-1 from Escherichia coli (Fig. 1), which show similar spectroscopic features (11, 12). The presence of these residues has been shown to contribute to the oxygen stability of the R. eutropha enzyme and is therefore associated with a possible redox chemistry protecting the enzyme against oxidative stress (13). In the present work the type and redox properties of the constituent iron-sulfur clusters are investigated with the aim to (i) portray the energy coupled processes between Hase I and its redox partners and (ii) outline a relation between O2-tolerance and electronic/chemical events occurring at the iron-sulfur clusters.
Results
At low temperatures (< 55 K) the continuous wave (cw) X-Band EPR spectrum of the as-isolated Hase I from A. aeolicus exhibits complex spectral features in the region, where an almost isotropic signal at g = 2.02 from an oxidized [3Fe4S]+ (S = 1/2) is observed in standard hydrogenases (Fig. 2, dashed line, Table S1) (6, 14). Double integration of the iron-sulfur cluster signals accounted for 1.9 ± 0.3 spins/mol of protein, indicating the presence of two S = 1/2 centers. In the case of magnetically noninteracting species, their EPR signals appear at the same positions on a g-factor scale at different microwave frequencies. In Hase I, however, these multiline patterns are different for the spectra recorded at S-, X-, and Q-band frequencies (Fig. 2, Fig. S2). This dependence argues for spin-spin interactions between these two species with exchange and dipolar contributions (10, 15, 16). Such magnetic interactions can be observed in cw EPR spectra between paramagnetic centers localized less than 15 Å apart, suggesting that these centers in Hase I are adjacent to each other (17) (Fig. 1). In addition, the splitting patterns of these EPR spectra show that the exchange interaction lies in an intermediate range (|2J| ≈ ΔgμBB) (15, 16). Furthermore, there could be an additional interaction with the paramagnetic [NiFe] center (S = 1/2), see below. The relaxation of all components of the spectrum has very similar temperature dependence between 10 and 50 K (Fig. S3); above 50 K all signals are broadened beyond detection.
Fig. 2.

EPR spectra of the [FeS] clusters in the as-isolated state of Hase I (pH 7.4) at S-band (cw), X-band (cw), and Q-Band (pulse) frequencies at T = 10 K. The asterisk marks a cavity impurity. The dashed line denotes the position of the [3Fe4S]1+ signal. Experimental Conditions: Modulation amplitude 0.3 mT and 1 mT; mw power 6 mW and 2 mW for S- and X-band, respectively.
EPR Redox Titration (pH 7.4).
To unravel the complex features in the EPR spectra of Hase I and identify the contributions from the individual [FeS] clusters, potentiometric titrations were carried out. Selected spectra illustrating all detectable iron-sulfur centers are shown for pH 7.4 in Fig. 3. The initial working hypothesis is that three iron-sulfur cofactors are expected to yield three single-electron redox transitions.
Fig. 3.

(A, B, C, and D) cw X-Band EPR spectra of the EPR signals corresponding to the [FeS] clusters of Hase I at different potentials (9.433 GHz, 15 K) and (E) their redox titration as a function of the potential at pH 7.4. The normalized individual EPR intensities of all redox components are plotted against the redox potential and fitted to curves calculated from the Nernst equation corresponding to single-electron transitions. Mod. amplitude 1 mT, mw power 2 mW.
As shown in Fig. 3 A–D the spectral patterns of the iron-sulfur clusters are drastically changing with redox potential allowing the identification of four groups of EPR signals, corresponding to interacting paramagnetic centers. By monitoring the intensity of these complex patterns four distinct one-electron transitions related to the iron-sulfur clusters of Hase I (Fig. 3E) could be identified (Table 1). The pH-dependence of these redox transitions has also been examined (Table S2). Considering our initial hypothesis, there appears to be a contradiction with respect to the number of redox centers available and the number of observed redox transitions.
Table 1.
Apparent midpoint potentials of the iron-sulfur clusters in O2-sensitive and O2-tolerant enzymes
| Redox center | Apparent midpoint potential, mV | ||||
| A. aeolicus (this work) | R. eutropha H16 / R. metallidurans CH34 (28) | D. vulgaris MF (14) | D. gigas (19) | D. fructosovorans (18) | |
| HP | + 232 | +160/+240 | – | – | – |
| [3Fe4S]1+/0 (medial) | + 68 | +25/+100 | − 70 | −35, − 70 | + 65 |
| [4Fe4S]2+/1+ (proximal) | + 87 | −60/+50 | < -300 | − 290 | − 340 |
| [4Fe4S]2+/1+ (distal) | − 78 | −180/−80 | < -300 | − 340 | − 340 |
Values are quoted relative to the normal hydrogen electrode (NHE). The error is ± 20 mV (for A. aeolicus).
Assignment of the [FeS] Clusters.
The [3Fe4S]1+ cluster can be assigned in a straightforward way on the basis of its g-value (giso = 2.019) to the signal denoted as 2 (Fig. 3). For the remaining [FeS] centers identification is more difficult as their assignments require knowledge of the structure and interaction patterns. Therefore, an additional species that could be linked to these spectral features and used as a probe to identify the localization of these centers is required. This information can be obtained by observing the magnetic interaction with the paramagnetic [NiFe] center.
The most oxidized Ni-B state of the [NiFe] center has a rhombic EPR spectrum (gx = 2.30, gy = 2.17, and gz = 2.01) (7). In standard hydrogenases, reduction of the iron-sulfur cluster proximal to the [NiFe] site, induces a splitting of the nickel signals and was previously characterized to result from spin exchange and magnetic dipole-dipole interactions (15). In Hase I, reduction of the [4Fe4S] cluster denoted as 3 (Fig. 3 B and C) is accompanied by the formation of a “splitting” of the Ni-B signals (Fig. 4A), indicative of a magnetic interaction between these two centers (15, 17). When the sample is annealed to 80 K the uncoupled EPR spectrum of the Ni-B state is recovered, due to the faster relaxation of [FeS] clusters at such temperatures (Fig. 4B). The EPR signal 3 in Hase I is thus assigned to the [4Fe4S]1+ cluster proximal to the [NiFe] site (Fig. 1). Each principal g-value of the Ni-B spectrum is split symmetrically into two components of comparable amplitude (18), showing that the exchange interaction between the [NiFe] site and the [4Fe4S] cluster is weak (|2J| < < ΔgμBB) (15). The spectrum in Fig. 4A has been simulated with the spin Hamiltonian described in the SI Text, taking into account three interacting species (Ni-B, [4Fe4S]P, and [3Fe4S]M). The magnitude of the isotropic exchange between the [NiFe] site and the proximal tetranuclear [FeS] cluster (JNiB-P) is 36 × 10-4 cm-1, while the isotropic exchange between the proximal and medial clusters (JP-M) is 7 × 10-4 cm-1.
Fig. 4.

cw X-Band EPR spectra at +12 mV and their simulations at 15 (A) and 80 K (B) are shown for pH 7.4. The asterisks denote cavity or undetermined signals. Mod. amplitude 1 mT, mw power 2 mW (15 K) and 20 mW (80 K) at 9.433 GHz.
At potentials as low as -29 mV, the spectral features of the proximal [4Fe4S]1+ cluster decrease in intensity and more composite signals appear (signals denoted as 4, Fig. 3) that correspond to the one-electron reduction of another [FeS] center. This center has the most reducing potential and is assigned to the distal [4Fe4S]1+ cluster (18–20). On the basis of homology model structures of Hase I, the distance between proximal and distal clusters is similar to the one in standard hydrogenases (Fig. 1, Fig. S1) and is too large for these two species to exhibit magnetically coupled EPR spectra (17). Therefore, the complex signals are attributed to an interaction pattern of the two reduced proximal and distal clusters (S=1/2) mediated by the high spin [3Fe4S]0 cluster (S = 2). These signals are well resolved in contrast to the broad and featureless spectrum of the reduced iron-sulfur clusters in standard hydrogenases (14, 15, 18, 19) (Fig. S6).
Identification of the HP Species.
Fig. 5 shows spectra of Hase I poised at different positive potentials between +44 and +346 mV at pH 6.4. At mildly reducing conditions (+44 mV) the proximal [4Fe4S] is reduced and is magnetically coupled to the [NiFe] site leading to a splitting of the Ni-B EPR signal. At +176 mV the signal of the proximal iron-sulfur cluster disappears due to its oxidation to the 2+ state (S = 0) and, concomitantly, the Ni-B signal attains its “unsplit” form. The [3Fe4S]1+ cluster is too far away from the [NiFe] site (∼18 Å) to induce interaction fingerprints on the Ni-B signals.
Fig. 5.

cw X-Band EPR spectra at pH 6.4 poised at +44, +176, and +346 mV ambient redox potentials. The low-field regions between the gray brackets have been amplified. Mod. amplitude 1 mT, mw power 2 mW at 9.436 GHz and T = 15 K.
Unexpectedly, an additional redox transition occurs at higher redox potentials, assigned to a high-potential (HP) species denoted as 1 (Fig. 3E); at +346 mV the Ni-B components are broadened and shifted and exhibit an astonishing relaxation enhancement. In addition, the [3Fe4S]1+ signal is now converted into a more complex pattern (Fig. 5). These spectral features show that the paramagnetic HP species must be positioned sufficiently close to both the [NiFe] and the [3Fe4S] cluster to allow magnetic interactions. The HP center is thus identified as the proximal iron-sulfur cluster lying in between these centers (8, 15, 17), implying that it has become oxidized to the 3+ state.
The proximal cluster in Hase I thus undergoes two distinct one-electron transitions (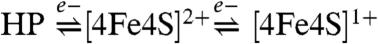 ). Both, the superoxidized and reduced forms of the proximal cluster are spin coupled with the [NiFe] site (Fig. 5), however, the magnitude of interaction is markedly different. At very oxidizing potentials the isotropic exchange coupling is in the intermediate range. Measurements at W-Band frequencies (∼94 GHz) set the upper limit for the interaction of the HP species with the [NiFe] site to be 0.021(2) cm-1 consistent with the significant relaxation enhancement observed for the Ni-B signals (Fig. S7, Fig. S8, Fig. S9, Fig. S10). In contrast, the exchange coupling between the [4Fe4S]1+ species and the Ni-B state of the [NiFe] site is about an order of magnitude weaker (0.0036(3) cm-1) (Fig. S9, Fig. S10).
). Both, the superoxidized and reduced forms of the proximal cluster are spin coupled with the [NiFe] site (Fig. 5), however, the magnitude of interaction is markedly different. At very oxidizing potentials the isotropic exchange coupling is in the intermediate range. Measurements at W-Band frequencies (∼94 GHz) set the upper limit for the interaction of the HP species with the [NiFe] site to be 0.021(2) cm-1 consistent with the significant relaxation enhancement observed for the Ni-B signals (Fig. S7, Fig. S8, Fig. S9, Fig. S10). In contrast, the exchange coupling between the [4Fe4S]1+ species and the Ni-B state of the [NiFe] site is about an order of magnitude weaker (0.0036(3) cm-1) (Fig. S9, Fig. S10).
Electronic Properties and Characterization of the HP Species.
The proximal cubane cluster is able to participate in two successive redox transitions implying that this center can be oxidized to a (3+) state (21). W-Band EPR shows that this HP species has a very small g-anisotropy, in contrast to a typical high-potential iron-sulfur (HiPIP) center (22) (Fig. S8, Fig. S9). The small g-anisotropy, however, could represent a different valence delocalization on this cluster, affecting the ground state properties and yielding different g-factors. The relaxation properties of the HP center resemble an iron-sulfur cluster rather than a radical signal (Fig. S2), excluding the possibility that the spin is localized on a ligand (e.g., covalently bound to a [4Fe4S]2+ moiety) (23).
Therefore, 57Fe Mössbauer experiments have been performed on the as-isolated and the fully oxidized Hase I to ascertain the oxidation state of the proximal cluster in the HP state. The analysis first corroborated the presence of a three-iron and two four-iron clusters. Fig. 6 shows the Mössbauer spectrum of the as-isolated enzyme recorded at zero magnetic field. This spectrum differs from the one reported for standard hydrogenases (10, 19) by two main features: (i) a substantially higher amount of Fe3+ that can be best ascribed to oxidation of the proximal cluster from the (2+) state with average valence Fe2.5+ to the (3+) state and (ii) a subspectrum with an isomer shift of 0.46 mms-1 and an unusually large quadrupole splitting ΔEQ = 2.41 mms-1 (Table S3). The latter indicates a special Fe subsite with an additional coordinating ligand in a distorted geometry (24–26). The average isomer shift for this cluster in this state is δav = 0.34 mms-1 at 160 K, being in the range of those reported for HiPIP-like cubane clusters (22).
Fig. 6.

57Fe Mössbauer spectrum of the as-isolated Hase I at pH 7.4 recorded at T = 160 K without applied magnetic field. The spectrum can be well fitted accounting for a [3Fe4S]1+, a [4Fe4S]2+, a low-spin Fe2+ of the [NiFe] site, and a [4Fe4S]3+ with a special site. The doublet 1 indicates the ferric contribution in the spectrum and the doublet 2 the Fe site with the large quadrupole splitting. The structure of the modeled proximal cubane cluster with the additional cysteine residues is shown. The spectra are deconvoluted to the respective components, whose contributions are indicated by the color of the doublets.
Discussion
The Redox Potentials of the Three [FeS] Clusters.
The components of the electron transfer chain in Hase I and their redox potentials have been determined and compared to those occurring in other homologous hydrogenases (Table 1). The distal [4Fe4S]2+/1+ cluster has a reduction potential of -78 mV that is pH-dependent (20). The estimated pKa(ox) is 7.1 and may be attributed to the protonation of the histidine ligand of this cluster (8, 18). The midpoint potential of the distal [FeS] cluster is approximately 300 mV more positive than the one in standard hydrogenases, which can be rationalized in terms of the higher potential redox partner of Hase I, a diheme cytb (Em = -20, -110 mV) (27).
The medial [3Fe4S]1+/0 cluster is found to have a reduction potential of +68 mV. There is a large variation in the values reported for the medial clusters in standard and O2-tolerant hydrogenases (Table 1) (14, 18, 19, 28). In general, in O2-tolerant hydrogenases there is a trend towards more positive potentials that may be related to the surrounding of this cluster. In the pH range between 6.4 and 8.3, the midpoint potential for the [3Fe4S] is independent of the proton concentration, in agreement with previous results on O2-sensitive enzymes (20).
The proximal cluster is able to access three redox states: [4Fe4S]3+ (HP state), [4Fe4S]2+, and [4Fe4S]1+. The [4Fe4S]3+/2+ couple has a reduction potential of +232 mV that is essentially pH-independent in the range between 6.4 and 7.4. Such a high-potential species is so far found only in O2-tolerant hydrogenases (Table 1), suggesting its possible role in protection against O2-inactivation. The second [4Fe4S]2+/1+ redox couple of the proximal cluster has an apparent midpoint potential of +87 mV, which is about +300 to +440 mV more positive than the respective ones for the hydrogenases from Desulfovibrio and Allochromatium species (10, 18–20). Such a very positive potential is not entirely atypical for [4Fe4S]2+/1+ centers. In the MBH hydrogenases from R. eutropha H16 and R. metallidurans CH34, the reduction potential for this cluster is also very positive (28, 29), while in the nitrate reductase from E. coli, both [3Fe4S] and [4Fe4S] clusters have comparable reduction potentials with those in Hase I (29). For pH values greater than 7.4 the Em of the [4Fe4S]2+/1+ couple approaches a dependence of 57 mV/pH and the proton association constant inferred is between 6.9 and 7.1 (Fig. S5), indicating a proton-coupled electron transfer involving a coordinating residue of this cluster.
The Dual Redox Character of the Proximal Cluster.
The proximal cluster in Hase I allows two redox transitions (i.e., 3 + /2+, 2 + /1+), which usually are mutually exclusive and do not occur in biological systems. This high redox compliance is proposed to be related to an unusual ligation by five or six cysteines, as may be inferred from homology modeling. Such additional thiol ligand(s) can easily alter the cluster geometry as well as the redox properties (24, 26, 30).
Also, oxidation of cysteines to disulfides has been associated with a protective mechanism against reactive oxygen species (31). Such a chemistry could be similar to that of the Ferredoxin:Thioredoxin Reductase (FTR) (32) and the heterodisulfide reductase (HDR) (33), in which a [4Fe4S] cluster mediates the cleavage of a disulfide. If in a similar manner formation of a disulfide bridge would occur upon the one-electron oxidation of the proximal cluster to the HP state, the cluster should be reduced, because bond formation is a two-electron process. However, this scheme disagrees with the present Mössbauer experiments that show iron oxidation. Moreover, formation of a disulfide bridge would presumably rupture the Fe-S bond at that site and affect the cluster stability.
A more likely possibility is that one or more of these additional cysteines are capable of coordinating the proximal [4Fe4S] cluster. A similar situation has been observed for the N-ethylmaleimide (NEM)-modified FTR that represents the only biological example of a [4Fe4S]3+, in which one Fe is coordinated by two cysteine residues (32, 34, 35). The large quadrupole splitting assigned to one of the Fe ions of this cluster in Hase I is indicative of a unique subsite with two-coordinating protein ligands in a distorted geometry that deviates from the conventional quasitetrahedral symmetry. Such high coordination leads to a large quadrupole splitting for the distorted Fe subsite and shifts the 3 + /2+ reduction potential of the HiPIP-like core to lower values, as has been shown for synthetic five-coordinated [FeS] cubanes (24). The latter originates from the increased negative charge of the cluster, which in biological systems, however, may be partially counterbalanced by additional hydrogen bonds leading overall to less covalent Fe-S bonds (30, 36). In addition, synthetic five-coordinated clusters can also carry out two redox steps in a close range of potentials. However, in contrast to the case of Hase I, the oxidation products in synthetic clusters are unstable and the transitions are often irreversible (24, 26).
The proximal [FeS] cubane shuttles reversibly through three different oxidation states in a very small potential range (∼150 mV). Up to date, attempts to overoxidize a [4Fe4S]1+ cluster resulted in loss of an iron, whereas reversible superreduction of [4Fe4S]3+ clusters is possible but the potential range between the [4Fe4S]3+ and the [4Fe4S]1+ spans more than 1,000 mV (37). The ability of the proximal cluster in Hase I to carry out two single-electron transitions is rather unusual. From the Mössbauer experiments this cluster is shown to have a unique Fe subsite in a distorted coordination environment indicating that this is coordinated by two cysteine residues (24–26). Such coordination should lead to an enhancement in the electron donation from the ligands to the iron-sulfur cluster core, particularly to molecular orbitals that have metal character and are slightly antibonding (34). The enhanced ligand-to-metal donation should result in an elongation of Fe-Fe distances and to a more distorted structure of the proximal [FeS] cluster (Fig. S11). Because stability of five or six coordinated clusters relates to the presence of terminal thiol ligands (24, 26), there may also be an active role for the second additional cysteine, as demonstrated in recent mutation experiments (13). The presence of the second additional cysteine is thus likely to be important for the stability of the induced HiPIP-like core.
Implications for Oxygen Tolerance.
The first redox transition (2 + /1+) of the proximal cluster serves in the canonical electron transfer reaction of the enzyme and its midpoint potential is far more positive as compared to the ones reported for standard hydrogenases. This upshift in the reduction potential is expected to result from an adaptation of the whole electron transfer chain to the more positive final electron acceptor (quinone as compared to cytc3). As a consequence, the [FeS] cofactors in Hase I are poor electron donors for oxidizing species (i.e., O2) and thus render the enzyme partially O2-tolerant. The second transition of the proximal cluster (3 + /2+) appears to be associated with a redox switch against oxidative stress rather than participating in the catalytic function of the enzyme. An increase in O2-sensitivity was reported in mutagenesis experiments of the additional cysteines surrounding the proximal cluster in R. eutropha MBH (13). It is intriguing to understand the presence of easily oxidizable cysteines in enhancing the oxygen tolerance of a hydrogenase, because these could readily react with O2. Formation of disulfide bonds could protect against oxidative stress but on the other hand also increase the generation of reactive oxygen species. This scenario is not supported by the present Mössbauer experiments.
It is not clear how oxygen diffuses in and interacts with Hase I, and whether there is a specific sequence of oxidation events following its interaction with the metal centers in their different redox states. Nevertheless, O2-tolerance of enzymes such as Hase I is understood to comprise a concerted effect of redox (thermodynamic) and kinetic factors (7, 38). Considering the redox potentials of the “activated” [NiFe] site and the reduced [FeS] clusters (7), the [NiFe] site is likely to be the primary target of O2. It is assumed that reduction of the attacking O2 molecules then proceeds via electrons supplied from the [NiFe] site that are also delivered from oxidation of the [FeS] clusters. Models for these reactions have been proposed (38). In this sense, the proximal cluster may represent a cofactor that is situated sufficiently close so that it can provide an additional reducing equivalent by forming the superoxidized state, for efficient neutralization of reactive oxygen species at the active site (38). The occurrence of two redox transitions for the proximal cluster, of which at least one is coupled to a proton transfer, contributes but is not solely responsible for the observed O2-tolerance (13) of membrane bound hydrogenases harboring these two extra cysteines. In conclusion and following the scheme proposed earlier for O2 inactivation (38), it is suggested that in enzymes such as Hase I from A. aeolicus the superoxidized state of the proximal cluster promoted by its cysteine surplus coordination is a key intermediate in a protection reaction against attack by molecular oxygen.
Materials and Methods
Detailed descriptions are available in SI Text.
The hyperthermophilic bacterium A. aeolicus was grown at 85 °C in two-liter bottles under a CO2/H2/O2 atmosphere. Hydrogenase I was purified at room temperature under semianaerobic conditions in a 50 mM Tris-HCl buffer pH 7.0, in the presence of 5–10% glycerol and 0.01% n-dodecyl-β-D-maltoside (DDM) as previously described (6). Redox potentials were determined by cw EPR potentiometric titrations. The titration was carried out anaerobically by adjusting the potential with substoichiometric amounts of solutions of sodium dithionite (Na2S2O4) and potassium ferricyanide (K3[Fe(CN)6]).
Supplementary Material
Acknowledgments.
The authors are grateful to Dr. Thomas Weyhermüller (MPI Mülheim) and Dr. Oliver Lenz (HU Berlin) for helpful discussions. This work was supported by EU/Energy Network project SOLAR-H2 (FP7 Contract 212508), Bundesministerium für Bildung and Forschung (BMBF) (03SF0318B, 03SF0355C), Max Planck Society, CNRS, French National Research Agency, the city of Marseilles, Région Provence-Alpes-Côte d’Azur, “Pôle de compétitivité CapEnergies.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1100610108/-/DCSupplemental.
References
- 1.Cammack R, Frey M, Robson R. Hydrogen as a fuel: learning from Nature. London: Taylor and Francis; 2001. [Google Scholar]
- 2.Mertens R, Liese A. Biotechnological applications of hydrogenases. Curr Opin Biotech. 2004;15:343–348. doi: 10.1016/j.copbio.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 3.Vincent KA, et al. Electrocatalytic hydrogen oxidation by an enzyme at high carbon monoxide or oxygen levels. Proc Natl Acad Sci USA. 2005;102:16951–16954. doi: 10.1073/pnas.0504499102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo XJ, Brugna M, Tron-Infossi P, Giudici-Orticoni MT, Lojou E. Immobilization of the hyperthermophilic hydrogenase from Aquifex aeolicus bacterium onto gold and carbon nanotube electrodes for efficient H2 oxidation. J Biol Inorg Chem. 2009;14:1275–1288. doi: 10.1007/s00775-009-0572-y. [DOI] [PubMed] [Google Scholar]
- 5.Deckert G, et al. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- 6.Brugna-Guiral M, et al. [NiFe] hydrogenases from the hyperthermophilic bacterium Aquifex aeolicus: properties, function, and phylogenetics. Extremophiles. 2003;7:145–157. doi: 10.1007/s00792-002-0306-3. [DOI] [PubMed] [Google Scholar]
- 7.Pandelia ME, et al. Membrane-bound hydrogenase I from the hyperthermophilic bacterium Aquifex aeolicus: enzyme activation, redox intermediates and oxygen tolerance. J Am Chem Soc. 2010;132:6991–7004. doi: 10.1021/ja910838d. [DOI] [PubMed] [Google Scholar]
- 8.Volbeda A, et al. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature. 1995;373:580–587. doi: 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]
- 9.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases. Chem Rev. 2007;107:4273–4303. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- 10.Surerus KK, et al. Further characterization of the spin coupling observed in oxidized Hydrogenase from Chromatium vinosum. A Mössbauer and multifrequency EPR Study. Biochemistry. 1994;33:4980–4993. doi: 10.1021/bi00182a029. [DOI] [PubMed] [Google Scholar]
- 11.Saggu M, et al. Spectroscopic insights into the oxygen-tolerant membrane-associated [NiFe] hydrogenase of Ralstonia eutropha H16. J Biol Chem. 2009;284:16264–16276. doi: 10.1074/jbc.M805690200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lukey MJ, et al. How Escherichia coli is equipped to oxidize Hydrogen under different redox conditions. J Biol Chem. 2010;285:3928–3938. doi: 10.1074/jbc.M109.067751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lenz O, et al. H2 conversion in the presence of O2 as performed by the membrane-bound [NiFe]-hydrogenase of Ralstonia eutropha. ChemPhysChem. 2010;11:1107–1119. doi: 10.1002/cphc.200901002. [DOI] [PubMed] [Google Scholar]
- 14.Asso M, Guigliarelli B, Yagi T, Bertrand P. EPR and redox properties of Desulfovibrio vulgaris Miyazaki hydrogenase-comparison with the NiFe enzyme from Desulfovibrio gigas. Biochim Biophys Acta. 1992;1122:50–56. doi: 10.1016/0167-4838(92)90126-x. [DOI] [PubMed] [Google Scholar]
- 15.Guigliarelli B, et al. Structural organization of the Ni and (4Fe-4S) centers in the active form of Desulfovibrio gigas hydrogenase. Analysis of the magnetic-interactions by Electron Paramagnetic Resonance spectroscopy. Biochemistry. 1995;34:4781–4790. doi: 10.1021/bi00014a036. [DOI] [PubMed] [Google Scholar]
- 16.Bencini A, Gatteschi D. Electron Paramagnetic Resonance of Exchange Coupled Systems. New York: Springer-Verlag New York, Inc; 1990. [Google Scholar]
- 17.Coffman RE, Buettner GR. General magnetic dipolar interaction of spin-spin coupled molecular dimers—application to an EPR spectrum of xanthine oxidase. J Phys Chem. 1979;83:2392–2400. [Google Scholar]
- 18.Rousset M, et al. [3Fe-4S] to [4Fe-4S] cluster conversion in Desulfovibrio fructosovorans [NiFe] hydrogenase by site-directed mutagenesis. Proc Natl Acad Sci USA. 1998;95:11625–11630. doi: 10.1073/pnas.95.20.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teixeira M, et al. Redox intermediates of Desulfovibrio gigas [NiFe] hydrogenase generated under hydrogen—Mössbauer and EPR characterization of the metal centers. J Biol Chem. 1989;264:16435–16450. [PubMed] [Google Scholar]
- 20.Roberts LM, Lindahl PA. Stoichiometric reductive titrations of Desulfovibrio gigas hydrogenase. J Am Chem Soc. 1995;117:2565–2572. [Google Scholar]
- 21.Beinert H, Holm RH, Münck E. Iron-sulfur clusters: nature’s modular, multipurpose structures. Science. 1997;277:653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 22.Bertini I, Campos AP, Luchinat C, Teixeira M. A Mössbauer investigation of oxidized Fe4S4 HiPIP II from Ectothiorhodospira halophila. J Inorg Biochem. 1993;52:227–234. [Google Scholar]
- 23.Hu ZG, Jollie D, Burgess BK, Stephens PJ, Münck E. Mössbauer and EPR Studies of Azotobacter vinelandii Ferredoxin-I. Biochemistry. 1994;33:14475–14485. doi: 10.1021/bi00252a014. [DOI] [PubMed] [Google Scholar]
- 24.Ciurli S, et al. Subsite-differentiated analogs of native [4Fe-4S]2+ clusters—preparation of clusters with 5-coordinate and 6-coordinate subsites and modulation of redox potentials and charge distributions. J Am Chem Soc. 1990;112:2654–2664. [Google Scholar]
- 25.Johnson RE, Papaefthymiou GC, Frankel RB, Holm RH. Effects of secondary bonding interactions on the [Fe4S4]2+ core of ferredoxin site analogs—Fe4S4(SC6H4 - o - OH)4]2-, a distorted cubane-type cluster with one five-coordinate iron atom. J Am Chem Soc. 1983;105:7280–7287. [Google Scholar]
-
26.Kanatzidis MG, Coucouvanis D, Simopoulos A, Kostikas A, Papaefthymiou V. Synthesis, structural characterization, and electronic properties of the tetraphenylphosphonium salts of the mixed terminal ligand cubanes
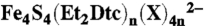 (X = Cl-, PhS-) (n = 1, 2). Two different modes of ligation on the [Fe4S4]2+ core. J Am Chem Soc. 1985;107:4925–4935. [Google Scholar]
(X = Cl-, PhS-) (n = 1, 2). Two different modes of ligation on the [Fe4S4]2+ core. J Am Chem Soc. 1985;107:4925–4935. [Google Scholar] - 27.Infossi P, et al. Aquifex aeolicus membrane hydrogenase for hydrogen biooxidation: role of lipids and physiological partners in enzyme stability and activity. Int J Hydrogen Energy. 2010;35:10778–10789. [Google Scholar]
- 28.Knüttel K, et al. Redox properties of the metal centers in the membrane-bound hydrogenase from Alcaligenes eutrophus CH34. Bull Pol Acad Sci Chem. 1994;42:495–511. [Google Scholar]
- 29.Guigliarelli B, et al. EPR and redox characterization of iron-sulfur centers in nitrate reductases-A and Z- from Escherichia-coli. Evidence for a high-potential and a low-potential class and their relevance in the electron-transfer mechanism. Eur J Biochem. 1992;207:61–68. doi: 10.1111/j.1432-1033.1992.tb17020.x. [DOI] [PubMed] [Google Scholar]
- 30.Dey A, et al. Solvent tuning of electrochemical potentials in the active sites of HiPIP versus Ferredoxin. Science. 2007;318:1464–1468. doi: 10.1126/science.1147753. [DOI] [PubMed] [Google Scholar]
- 31.Vita N, Hatchikian EC, Nouailler M, Dolla A, Pieulle L. Disulfide bond-dependent mechanism of protection against oxidative stress in pyruvate-ferredoxin oxidoreductase of anaerobic Desulfovibrio bacteria. Biochemistry. 2008;47:957–964. doi: 10.1021/bi7014713. [DOI] [PubMed] [Google Scholar]
- 32.Dai SD, Schwendtmayer C, Schürmann P, Ramaswamy S, Eklund H. Redox signaling in chloroplasts: cleavage of disulfides by an iron-sulfur cluster. Science. 2000;287:655–658. doi: 10.1126/science.287.5453.655. [DOI] [PubMed] [Google Scholar]
- 33.Duin EC, Madadi-Kahkesh S, Hedderich R, Clay MD, Johnson MK. Heterodisulfide reductase from Methanothermobacter marburgensis contains an active-site [4Fe-4S] cluster that is directly involved in mediating heterodisulfide reduction. FEBS Lett. 2002;512:263–268. doi: 10.1016/s0014-5793(02)02281-0. [DOI] [PubMed] [Google Scholar]
- 34.Walters EM, et al. Spectroscopic characterization of site-specific [Fe4S4] cluster chemistry in Ferredoxin:Thioredoxin Reductase: implications for the catalytic mechanism. J Am Chem Soc. 2005;127:9612–9624. doi: 10.1021/ja051909q. [DOI] [PubMed] [Google Scholar]
- 35.Dai S, et al. Structural snapshots along the reaction pathway of Ferredoxin-Thioredoxin Reductase. Nature. 2007;448:92–96. doi: 10.1038/nature05937. [DOI] [PubMed] [Google Scholar]
- 36.Niu S, Ichiye T. Probing ligand effects on the redox energies of [4Fe4S] clusters using broken-symmetry density functional theory. J Phys Chem A. 2009;113:5671–5676. doi: 10.1021/jp809446q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heering HA, Bulsink YBM, Hagen WR, Meyer TE. Reversible super-reduction of the cubane [4Fe4S](3+,2+,1+) in the high-potential iron-sulfur protein under non-denaturing conditions—EPR spectroscopic and electrochemical studies. Eur J Biochem. 1995;232:811–817. [PubMed] [Google Scholar]
- 38.Cracknell JA, Wait AF, Lenz O, Friedrich B, Armstrong FA. A kinetic and thermodynamic understanding of O2 tolerance in [NiFe]-hydrogenases. Proc Natl Acad Sci USA. 2009;106:20681–20686. doi: 10.1073/pnas.0905959106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.