

Abstract
We describe a phage display methodology to engineer synthetic antigen binders (sABs) that recognize either the apo- or the ligand-bound conformation of maltose binding protein (MBP). sABs that preferentially recognize the maltose-bound form of MBP act as positive allosteric effectors by significantly increasing the affinity for maltose. A crystal structure of a sAB bound to the closed form of MBP reveals the basis for the exhibited allosteric effect. We show that sABs which recognize the bound form of MBP can rescue the function of a binding-deficient mutant by restoring its natural affinity for maltose. Further, the sABs can enhance maltose binding in vivo by providing a growth advantage to bacteria under low maltose conditions. The results demonstrate that structure-specific sABs can be engineered to dynamically control ligand-binding affinities by modulating the transition between different conformations.
Numerous biological processes are regulated through the interactions of proteins with small molecules or other proteins. In many cases, these interactions induce conformational changes that directly modulate activities or provide new binding sites that facilitate building higher order organizations. Thus, it is recognized that the ability to engineer systems that can precisely control these processes will have extensive applications in broad areas in biomedical research and biotechnology. Both rational and combinatorial approaches to modulating protein-ligand interactions have relied mainly on introducing mutations within the protein of interest at residues that directly involve the ligand binding site1–3. An alternative strategy to modulating protein-ligand interactions is the use of allosteric effectors. While mutations permanently change the affinity for the ligand, an attribute of allostery is the ability to “dial in” a range of affinities based on the concentration of the allosteric effector in a completely controlled fashion. Many naturally occurring allosteric effectors share a common feature, namely the ability to bind preferentially to one form or conformation of the protein4,5. Effectors that favor the ligand-bound form of a protein enhance binding and act as positive allosteric effectors, whereas those that favor the apo form of the protein act as negative allosteric effectors.
Previous studies have shown that monoclonal antibodies (mAbs) can sometimes function as enhancers or inhibitors of ligand binding6,7. However, to produce a panel of mAbs to perform a specified function is a non-trivial undertaking, because there is little control over the conformational states in an animal’s bloodstream. Consequently, finding a mAb that can induce the desired function or binds to an intended epitope is not a matter of design, but an expensive exercise in screening a multitude of mAbs for the desired function.
To explore the potential of allosteric control of molecular processes, we sought to develop a general strategy to rationally engineer reagents that can dynamically modulate the binding affinity of proteins for their ligands. As a model system, we utilized maltose-binding protein (MBP), a member of the bacterial periplasmic binding protein superfamily8. MBP is the soluble component of the maltodextrin transport system and resides in the periplasm of Gram-negative bacteria where it shuttles its ligands; maltose, maltoriose and maltotetraose; to the membrane bound transporter complex9,10. The ligand binding site of MBP is positioned between two domains separated by a hinge region11. In the absence of ligand, MBP is present almost exclusively in an open (ligand-free) conformation. Upon ligand binding, a large conformational change takes place via a hinge-bending motion of roughly 35 degrees as MBP adopts a closed (ligand-bound) conformation12,13.
Here, we engineered allosteric effectors based on a novel class of antibody-like affinity reagents referred to as synthetic antigen binders or sABs14–16. The sABs were generated using a phage display methodology where a library of a humanized antibody Fab fragment was displayed on the surface of the M13 bacteriophage17. In contrast to mAbs, sABs are generated in vitro allowing exquisite control over the conformational state of the target protein. The phage display strategy takes advantage of the structural differences between the apo and ligand-bound forms of MBP12 to generate sABs that preferentially bind either form. These selections were carried out in the presence or absence of maltose to generate sABs that bind to the open or closed form of MBP, respectively. Further, the ability of the sABs to influence maltose binding was investigated in vitro and in vivo. The crystal structure of a maltose-bound MBP in complex with one sAB was determined, revealing the mechanism of the allosteric effect and confirming the selective conformational trapping capability of the sAB. The results demonstrate the ability to use sAB-based affinity modulators to dynamically control (enhance or diminish) ligand binding without introducing mutations within the binding protein itself. The data also suggest that simple in vitro selection conditions for sAB generation can translate to in vivo function.
Results
Phage display and selection strategy
To generate synthetic antigen binders (sABs) for MBP, we utilized a synthetic library built on a single antibody fragment (Fab) fused to the minor coat protein pIII of the M13 bacteriophage. The framework maximizes the number of randomized positions by introducing limited diversity into the three heavy chain complementarity-determining regions (CDR-H1, -H2, and -H3), and one light chain CDR (CDR-L3)17. The library over-represents Tyr, Ser, Gly, Trp and Phe side chains (Tyr-30%; Ser-15%; Gly-10%; Trp-5%; Phe-5%) at solvent accessible positions responsible for antigen recognition (Koide et al. in preparation). The library also includes all other amino acids at a lower percentage (2.5%) in CDR-H3, excluding cysteines to prevent formation of random disulfide bonds. The combined naïve library contained >1010 unique clones.
To generate sABs that preferentially bind to the closed form of MBP (closed-specific sABs), the selection was carried out in the presence of 10 μM maltose (Figure 1). After four rounds of selection, the isolated colonies were subjected to competitive phage ELISA to provide an estimate of their affinity for MBP and specificity for the closed form (Figure S1). The 48 sABs with the highest specificity for the closed form of MBP were sequenced resulting in 17 unique clones. Of the 17 MBP closed-specific sABs, 8 sABs designated MCS1 – MCS8 with the highest ELISA signal were expressed and purified for binding characterization. In addition, a control sAB (MOS1), which was generated from a selection carried out in the absence of maltose, was used as an example of an open-specific sAB.
Figure 1. Phage display selection strategy.
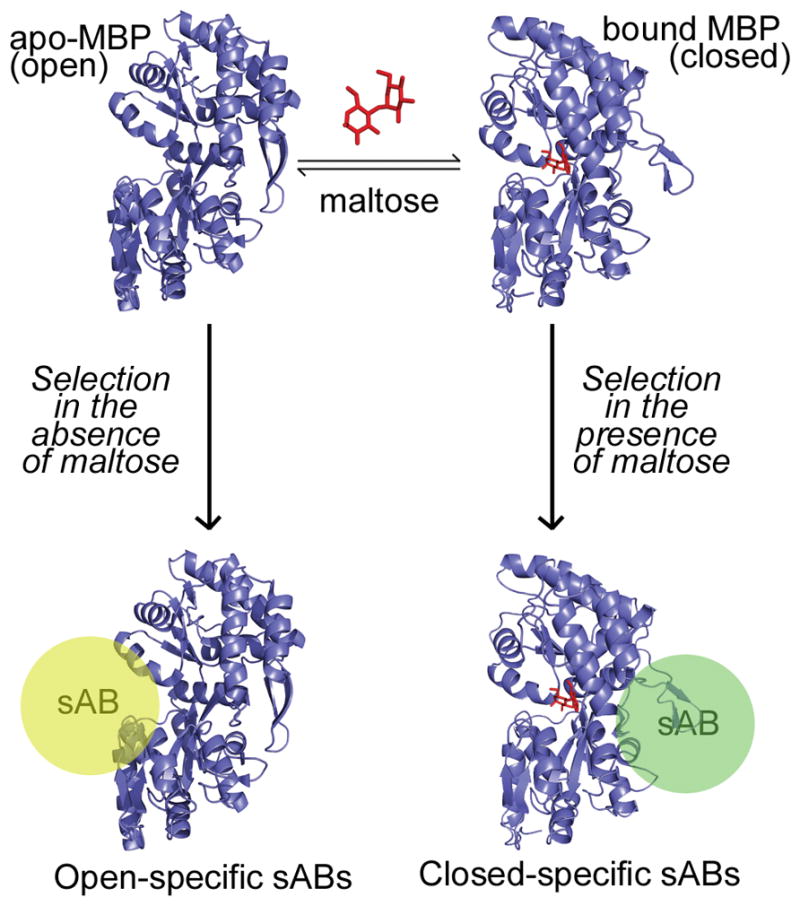
Maltose binding protein (MBP) undergoes a conformational change through a hinge-bending motion upon binding to maltose (red). Carrying out the phage display selection in the absence of maltose generates sABs (yellow sphere) that bind preferentially to the open form of MBP, whereas selection in the presence of maltose results in closed-specific sABs (green sphere). Note: placement of spheres indicate postulated binding modes of sABs to the different forms of MBP.
The affinity of each of the sABs for MBP in the presence and absence of maltose was determined by surface plasmon resonance (Table 1). Representative sensograms for sAB binding to either form of MBP are shown in Figure S2. Analysis of the binding data indicates that all sABs generated in the presence of maltose preferentially bind to the closed form of MBP with as much as a 2.3 kcal/mol difference in binding energy between the two forms. In contrast, MOS1, which was generated in the absence of maltose, exhibits a high affinity (3 nM) for MBP in the absence of maltose (Table 1). In the presence of 1 μM maltose, the affinity was significantly decreased (30 nM), and no binding was detected in the presence of 1 mM maltose.
Table 1.
Sequence of CDRs and affinity of characterized sABs for the apo and bound forms of MBP.
| sAB | Loop Sequence | Kd, apo (nM) | Kd, bound (nM) | ΔΔG closed - open (Kcal/mol)a | |||
|---|---|---|---|---|---|---|---|
| L3 | H1 | H2 | H3 | ||||
| MCS1 | WSTYKYLV | ISSSSI | SIYPYSGSTS | RYWWPKAM | 175 | 4b | −2.2 |
| MCS2 | SSYIPV | FSSSYI | YISSYSGYTS | TPWWYWSGL | 67 | 5b | −1.5 |
| MCS3 | SSSSLI | FSSSSI | SISSSSGSTS | YHWGWWWAL | 200 | 12b | −1.7 |
| MCS4 | SSYWPI | VYSSSI | YIYPSSGSTS | WHWGWSWAM | 140 | 3b | −2.3 |
| MCS5 | YWETLI | VYSSSI | SIYPYSGSTY | WRWEAL | 34 | 9b | −0.79 |
| MCS6 | YPFGLI | IYSSSI | SIYPYSGSTS | YRWEAM | 70 | 2b | −2.1 |
| MCS7 | NSGYPL | FYSSYI | SISSYSGYTS | AGWGWWGF | 90 | 4b | −1.8 |
| MCS8 | SYSSPL | FSSSYI | SISPSSGYTS | YVWYWWWAL | 46 | 4b | −1.4 |
| MOS1 | HYTTPP | ISYYSI | SIYSYYSSTS | VYVPYISQYYSWSYRGI | 3 | 30c | +1.4 |
ΔG of sAB binding with maltose -ΔG of sAB binding without maltose
Kd in the presence of 1 mM Maltose
Kd in the presence of 1 μM maltose
The effect of sABs on the affinity of MBP for maltose
We reasoned that sABs that selectively bind to the closed form of MBP can increase the affinity for maltose by shifting the equilibrium towards the bound form and vice versa. A convenient method for measuring the affinity of MBP for maltose is to monitor the ligand-dependant change in the emission of a single fluorophore attached at an introduced cysteine mutation18,19 (Figure 2a). We utilized the MBP-233C mutant conjugated to Alexa 488 to measure the effect of the sABs on the affinity of MBP for maltose. Using the fluorescence emission assay, the affinity of MBP-233C for maltose in the absence and in the presence of each of four closed-specific sABs (MCS1, MCS2, MCS3 and MCS4) was determined. As predicted, the presence of excess concentration of each of the sABs resulted in an increase in the affinity of MBP for maltose (Figure 2b). MCS1 and MCS4, which exhibit the highest preference for the closed form of MBP, induced the largest increases in the affinity of MBP for maltose (Table 2). A similar increase in affinity for maltotriose was observed (data not shown). We also determined the effect of the open-specific sAB MOS1 on maltose affinity. We utilized the change in intrinsic tryptophan fluorescence of MBP as a function of maltose binding to determine the affinity for maltose in the presence or absence of MOS1 (Figure 2c). In contrast to the closed-specific sABs, MOS1 induced a large decrease in the affinity of MBP (Figure 2d) for maltose with a ΔΔG resembling that of its preference for the open form of MBP (Table 2).
Figure 2. Influence of sABs on maltose binding.

(A) The change in fluorescence of the MBP-233C Alexa 488 conjugate (solid line) upon addition of 1 mM maltose (dashed line). (B) Fluorescence maltose binding curves of MBP-233C Alexa 488 in the absence (●) or presence of 200 nM sAB MCS1 (◇) or 200 nM sAB MCS4 (○). (C) Intrinsic tryptophan fluorescence of MBP in the absence (solid line) or presence of 1 mM maltose (dashed line). (D) Fluorescence maltose binding curves of MBP the absence (●) or presence of 200 nM sAB MOS1 (○).
Table 2.
The effect of sABs on the affinity of MBP for maltose.
| Kd maltose (μM) | ΔΔG maltose (Kcal/mol)a | ΔΔG closed-open (Kcal/mol) b | |
|---|---|---|---|
| MBP-233C | 67c | ||
| MBP-233C + MCS1 | 1.9c | −2.1 | −2.2 |
| MBP-233C + MCS2 | 9.6c | −1.1 | −1.5 |
| MBP-233C + MCS3 | 3.6c | −1.7 | −1.7 |
| MBP-233C + MCS4 | 2.1c | −2.0 | −2.3 |
| wt-MBP | 0.63d | ||
| wt-MBP + MOS1 | 10.5d | +1.6 | +1.4 |
ΔG of maltose binding with sAB -ΔG of maltose binding without sAB
ΔΔG values from Table 1
Affinity measured using fluorescence of Alexa 488-labeled MBP-233C mutant
Affinity measured using intrinsic tryptophan fluorescence of wt MBP
Both closed- and open-specific sABs influenced the shape of the maltose binding curves. In the absence of sABs, the binding of maltose to MBP fits a single-ligand binding function. However, in the presence of sABs, best fits were obtained by adding the Hill coefficient to the equation. The closed-specific sABs MCS1 and MCS4 exhibit Hill coefficients of 1.7 and 1.3, respectively, indicating positive co-operativity, whereas the Hill coefficient for MOS1 is 0.45, indicating negative co-operativity. The maltose binding data in the presence and absence of sABs were used to generate Scatchard plots (Figure 3), which can be used to determine the allosteric contributions to ligand binding based on the shape of the resulting curve20. In the absence of sABs, maltose binding shows a linear plot indicating no allostery (Figure 3a). In contrast, both MCS1 and MCS4 result in convex curves (Figure 3c, d), a characteristic of positive allosteric effectors. In the case of MOS1, which binds preferentially to the open form, the curve is concave (Figure 3b), indicating that MOS1 acts as a negative allosteric effector21.
Figure 3. Scatchard analysis of maltose binding.

Data points from figure 2 were re-plotted as r vs. r/[Maltose], where r is the fractional saturation of MBP with maltose. (A) Binding of maltose to MBP in the absence of sABs showing no co-operativity. (B) Maltose binding in the presence of 200 nM sAB MOS1 showing negative co-operativity. (C) Maltose binding in the presence of 200 nM sAB MCS1 or (D) 200 nM sAB MCS4 showing positive co-operativity.
Structure of the sAB-MBP complex
To investigate the mechanism of the allosteric effect of the sABs, we crystallized the complex of MBP with sAB MCS2 in the presence of maltose. The maltose bound MBP complexed with sAB MCS2 crystal contained one complex per asymmetric unit. Density was continuous for the heavy chain between residues 2–214, for the light chain between residues 2–213 and for MBP between residues 1–165 and 175–367. As expected, the sAB primarily interacts with MBP through the CDR loops at the opposite side of the ligand binding pocket and makes no contacts to the bound maltose (Figure 4). The scaffold of the sAB resembles that of previously-reported Fab fragments15,16 and the MBP structure closely overlaps with that of the maltose bound structures of the protein11,12,22. The only significant change is the shift of loop 166–185 in MBP, which in previously reported structures forms an extended β-strand loop that wraps around to the back face. In this structure, part of the loop (166–174) appears disordered, and the other portion forms a more compact structure, which, in part, fills a void left by the C-terminal truncation of our MBP construct.
Figure 4. Crystal structure of MBP-MCS2 complex.

MBP (blue) is in the closed form, bound to 1 molecule of maltose (red). The sAB (Heavy chain: green, Light chain: yellow) interacts with MBP at the opposite side of the binding pocket forming a wedge that favors the closed form.
The majority of the interactions of the sAB with MBP are formed by residues from the heavy chain. The buried surface area is 864 Å2 of the heavy chain compared to only 322 Å2 of the light chain. A set of tryptophans and tyrosines in CDR-H1, -H2, and -H3 (Tyr33, Tyr53, Tyr56, Trp97, Trp98, Tyr99 and Trp100) form a wedge that makes extensive hydrophobic and aromatic interactions with MBP (Figure 5a, b). The residues from CDR-H3 (97–100) occupy a cleft between residues 90–98 and 314–327 of MBP that is present in the closed but not the open conformation. CDR-H2 (52–56) occupies a surface along the helix between residues 90–98 of MBP and the following turn that in the open conformation is masked by the tip of the β-strand loop 166–185. CDR-L3 (91–94) and the unvaried CDR-L1 (30–31) make contacts with Pro 91, Lys305 and Glu309 of MBP along a surface of MBP that does not change between the open and closed conformations. There are several ordered waters located all along the interface of the sAB and MBP, as well as between the varied loops of the sAB itself. An overlay of the open structure of MBP12 with our structure shows a clash between the CDR loops and the apo MBP molecule (Figure 5c) demonstrating the basis of the selectivity of the sAB for the maltose bound form of MBP. The allosteric binding of the sAB to the closed conformation of MBP sheds light on the cooperative behavior of maltose binding exhibited in the presence of closed-specific sABs. In contrast to the MBP-MCS2 complex reported here, a previous structure of a sAB based on a monobody scaffold generated in the absence of maltose shows that the monobody interacts with an open form of MBP at the maltose binding site23.
Figure 5. The “wedge” formed by the CDR loops of MCS2.

(A) A group of bulky side-chain residues within CDRH-3 of the sAB (green sticks) form the wedge structure, which interacts with a region within MBP (blue) that is only exposed in the closed, maltose-bound conformation. (B) Sequence of the CDR loops of MCS2. Bold letters indicate residues that interact with the MBP molecule. (C) An overlay of the MCS2-MBP complex with the open form of MBP (purple, PDB code: 1OMP 12) indicates that the apo form of MBP clashes with the sAB CDR loops.
Functional rescue of an MBP mutant using closed-specific sABs
The ability to modulate protein-ligand interactions using an alloesteric effector can serve as a powerful tool for restoring the affinity of mutant proteins where the ligand binding activity has been affected by the mutation. Using the crystal structure of the maltose-bound form of MBP, we generated a binding pocket mutant where the tryptophan at position 62 was replaced with phenylalanine. While the mutation did not completely abolish maltose binding, it resulted in a ~ 2 kcal/mol decrease in binding energy (Figure 6). By adding a 5-fold excess of MCS1 to the W62F mutant, the affinity for maltose was restored to near wild type value, while addition of a 5-fold-exccess of MCS4 resulted in a slightly higher affinity than the wild type protein. The sABs do not affect the overall change in fluorescence between the apo and bound forms (Figure 6), but shift the equilibrium to favor maltose binding and restore binding affinity.
Figure 6. Rescuing binding function of an MBP mutant.

The effect of the binding pocket mutation W62F on affinity of MBP for maltose was assessed using the emission change of Alexa 488 attached to a cysteine at position 233. In the absence of sAB (●), the mutant exhibits low binding affinity. The affinity is restored by addition of either 200 nM sAB MCS4 (○) or 200 nM sAB MCS1 (◇). The binding curve of MBP-233C with no binding pocket mutations is shown (dashed line) as a reference.
Effect of sABs on maltose uptake in vivo
MBP is involved in the uptake of maltose and other sugars in the periplasm of gram-negative bacteria. Recently, a crystal structure of the maltose transporter complex revealed key elements in the mechanism of maltose transport and the role of MBP in this process9. Upon binding to maltose, the closed form of MBP is recognized by the MalF/MalG dimer, and an ATP hydrolysis step at the MalK protein allows the release of the sugar to the transporter by forcing the transition of MBP from the closed to the open form24. We reasoned that our closed specific sABs can provide a growth advantage for bacteria at low concentrations of maltose by increasing the maltose affinity within the periplasm. We utilized E. coli 55244 cells to periplasmically express either sAB MCS1, MCS4 or a control sAB (sAB-27), which was previously generated to bind actin filaments14. The cells were grown in minimal media containing various concentrations of maltose as the sole carbon source, incubated with shaking at 30°C, and growth curves were obtained by monitoring the OD595 values over 48 hrs. Cells expressing MCS1, MCS4 and sAB-27 showed similar growth rates under high concentrations of maltose. However, at low concentrations of maltose, the growth rates of cells expressing sAB-27 were significantly decreased, while the growth rates for cells expressing MCS1 or MCS4 were unaffected (Figure 7). These results indicate that cells expressing closed-specific sABs can thrive under low concentrations of maltose presumably due to the ability of the sABs to shift the equilibrium of the open-to-closed transition of the intrinsic MBP within the periplasm. This demonstrates the ability to dynamically change the affinity of a protein in vivo without introducing mutations in the protein itself, but rather by introducing an external effector to modulate its activity.
Figure 7. Alloesteric activity of sABs in vivo.

E. coli cells expressing sABs in the periplasm were grown in minimal media containing maltose as the sole carbon source. Cells expressing sAB MCS1 (○) or sAB MCS4 (◇) show no change in the growth rate at low maltose concentrations. Control cells expressing sAB-27 (■), which is an actin binding sAB (from reference14), show a lower growth rate at low maltose concentrations.
Discussion
Here, we describe a rational approach to engineer a set of allosteric effectors that can modulate the affinity of a protein by generating conformational-selective sABs that favor either the ligand-free or ligand-bound form of a protein. The sABs exhibit nanomolar binding affinities for MBP and can discriminate between the two forms of the protein by as much as 2.3 kcal/mol. As a result, the sABs influence the affinity of MBP for its maltose ligand by shifting the equilibrium to either the open or the closed form of the protein. Since many proteins undergo conformational changes upon binding to a cognate ligand, the strategy presented here provides a general approach to engineering sABs that can manipulate the activity of a protein by dynamically influencing its conformation. Such conformational-selective sABs can serve as molecular switches for the regulation of protein function, and represent a powerful tool in studying protein-ligand interactions.
Our results indicate that the ability of the sABs to enhance the affinity of MBP for maltose is not related to the absolute affinity of the sAB for either conformational form of MBP, but rather the difference in affinity of the sABs for the two forms of the protein. In all four sABs tested (MCS1, MCS2, MCS3 and MCS4), the degree of enhancement in maltose affinity (ΔΔGmaltose) corresponds approximately to the difference in binding energy of each sAB (ΔΔGclosed - open) between the open and the closed forms of MBP (Table 2). Additionally, maltose binding in the presence of the sABs shows cooperative behavior. Closed-specific sABs induce positive cooperativity, acting as classic heterotropic positive allosteric effectors21, as both the sAB and the maltose preferentially bind to the same form of MBP. On the other hand, the open-specific sAB MOS1 acts as a negative allosteric effector of maltose binding. Scatchard analysis of the binding behavior indicates that MOS1 does not simply act as a competitive antagonist (i. e. competing with maltose for the same binding site), instead MOS1 and maltose compete for two different conformations of the protein.
An important application for the rational design of alloesteric effectors is the ability to rescue binding function of an impaired mutant, which is a common feature of disease. We demonstrate here the ability of the closed-specific sABs to restore the affinity of the binding pocket mutant MBP-W62F. Interestingly, MCS4 has a greater effect on the affinity of the mutant for maltose than MCS1. The opposite effect was observed with wild type MBP, where MCS1 has a greater effect on affinity. This phenomenon is probably due to small changes in the overall structure that may result from the mutation within the binding pocket. As a result of the large surface of interaction between the sABs and MBP, such small changes can influence the binding of each sAB differently. This observation suggests that the interactions of sABs with a target protein can be sensitive to subtle changes in structure or sequence. This high specificity of sAB-protein interactions can serve as a tool for probing minor fluctuations in the target protein conformation. Importantly, the ability of the sABs to restore the affinity of a mutant protein may prove to be a powerful approach to rescue the function of proteins where mutations have decreased binding affinity, enzymatic activity or overall protein stability. Conceivably, sABs can be engineered as therapeutic reagents that stabilize the active form of a mutant protein, providing an alternative approach to gene therapy.
In addition to influencing the affinity of MBP for maltose in vitro, we sought to determine whether this set of sABs when expressed in cells could influence the effectiveness of maltose uptake in vivo. We showed that sABs, which recognize the closed form of MBP, produce a growth advantage for E. coli cells under low levels of maltose. The closed-specific sABs MCS1 and MCS4 are expressed in the periplasm, where each presumably interacts with the intrinsic MBP and shifts the equilibrium towards the closed form, making it a potent scavenger in a low maltose environment. According to the current model for maltose transport, the closed form of MBP is recognized by the MalF/MalG transporter complex, and the energy from ATP hydrolysis by the MalK dimer is used to convert MBP into the open form allowing maltose to pass through the transporter. Interestingly, it appears that little thermodynamic penalty is paid in the closed to open transition facilitating maltose transfer from the binding protein to the transporter due to the presence of the sAB suggesting that this is not the rate determining step in the process.
The structure of the MCS2-MBP complex described here shows that sAB binding to MBP does not interfere with the interactions between MBP and the transporter. While MCS2 does not enhance bacterial growth, limited epitope mapping studies (data not shown) suggest that sABs, which enhance growth rates (MCS1 and MCS4), bind to a similar region on MBP. This suggests that the ability to engineer sABs that influence nutrient uptake in bacteria provides a powerful tool for studying bacterial transporters in vivo, and may result in a greater understanding of the underlying mechanisms by which bacteria respond to their environment. Importantly, the results demonstrate that relatively simple selection strategies in vitro can translate into functional affinity modulators in vivo.
The data presented here demonstrate the power of library selection through phage display in generating sABs with highly specific binding interactions. In contrast to traditional methods for generating mAbs, this approach allows direct control over the conformational state of the target protein during the selection process. Indeed, mAbs have been found to influence the interactions of proteins with their ligands. However, since it is extremely difficult to control the conformation of a target protein in the bloodstream of an immunized animal, such mAbs were either serendipitously discovered or were the result of extensive screening efforts6,7. The approach described here allows production of highly specific binding reagents, as clearly demonstrated by the observation that all closed-specific sABs chosen for characterization showed a preference to the closed form of MBP (see Table 1). A panel of sABs with a desired function can be generated by several rounds of selection, as a result, little to no screening is required to identify sABs with the desired function. Therefore, as a technology, conformational-selective phage display is not only cheaper, less time and labor intensive than mAb generation; it is a superior approach that allows rational engineering of allosteric effectors by applying a selection instead of “fishing” for a mAb with the desired function. The resulting sABs act as modulators of protein affinity by shifting the equilibrium towards a specific structure of a protein in vitro and in vivo. This strategy is not limited to modulation of protein-ligand interactions. Similar selection strategies have lead to the design of novel enhancers or inhibitors of enzymatic activity25. Furthermore, the ability to rescue the function of a mutant protein demonstrated here shows the importance of engineering conformational specificity. This approach may provide a general method for the rational design of protein-based therapeutics for many diseases where mutations inhibit binding of a receptor to its natural ligand or the activity of a critical enzyme.
Methods
Phage display
Maltose binding protein was expressed from the pHFT2_MBP plasmid (kindly provided by A. Koide from the University of Chicago), which contains a 10-histidine tag, followed by a flag tag at the N-terminal of MBP lacking its periplasmic signal sequence and four amino acids at the c-terminus. All mutants were constructed by Kunkel mutagenesis and verified by DNA sequencing. MBP variants were expressed in BL-21(DE3) cells grown in 1 L of LB medium supplemented with kanamycin and induced with 1 mM IPTG at OD600 of 0.6 for 3 hrs at 37°C. Cells were lysed by sonication, and the proteins were purified on a Ni-NTA column. MBP was biotinylated using sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate (EZ-Link Sulfo-NHS-SS-Biotin, Thermo Scientific). The reaction contained 5 μM MBP in 50 mM MOPS buffer pH 7.0 and a 15-fold molar excess of biotinylation reagent for 45 min at room temperature, followed by extensive dialysis into TBS (150 mM NaCl, 50 mM Tris–HCl pH 7.5). Biotinylation was verified by mass spectrometry and a pull down assay using using strepatavidin Magneshpere magnetic beads (Promega).
Biotinylated MBP (0.1 nmol) saturated with 10 μM maltose was adsorbed to a minimal quantity of Streptavidin MagneSphere Paramagnetic Particles and free streptavidin binding sites was blocked with biotin to prevent nonspecific binding. After manual washing of the magnetic beads, 1 ml of phage library solution (1012–1013 colony-forming units (cfu)) was added and incubated at room temperature for 15 min, then the phage solution was discarded and the beads were washed twice with TBST-BSA (TBS, 0.5% Tween 20, 0.5% BSA, 10 μM maltose). The captured phages on the magnetic beads were used directly to infect E. coli XL1-Blue cells (Stratagene) for phage amplification. The phage particles captured in round 1 were cycled through additional rounds of binding and selection for immobilized MBP using an automated magnetic bead manipulator (KingFisher, Thermo Scientific). In the second round, phage solution was incubated with 100 μL of 50 nM biotinylated MBP for 15 min and then captured using streptavidin magnetic beads. After a total of five washing steps with 100 μL TBST-BSA with 10 μM maltose, the captured phages were eluted from the beads with 100 mM DTT in 20 mM Tris pH 8.0 for 10 min and amplified in E. coli XL1-Blue. In the third round, the concentration of the biotinylated MBP was reduced to 10 nM. In the fourth round, the amplified phages were tested in two separate sorting experiments, one with 10 nM biotinylated MBP and the other with no MBP as a negative control. Recovered phages were tittered, and the enrichment ratio was determined as the number of phage from the MBP-containing selection over the number of phage from the control selection. We obtained a significant enrichment ratio after a total of three rounds in all experiments with an average above 100.
Phages from the third round were produced from individual clones grown in a 96-well format. Culture supernatants were used directly in a single-point competitive phage ELISA in the absence or presence of maltose to estimate the affinity and the conformational specificity of each clone. E.coli XL1-blue colonies (Stratagene) harboring phagemids were inoculated directly into 400 μL of 2YT broth supplemented with ampicilin 100 μg/mL and M13-KO7 helper phage (New England Biolabs); the cultures were grown overnight at 37°C in a 96-well format. Culture supernatants containing sAB-phage were diluted five-fold in phosphate-buffered saline, 0.5% (w/v) BSA, 0.1% (v/v) Tween 20 (PBT buffer) either with or without MBP and maltose (20 nM MBP, 1 μM maltose). After 1 h incubation at room temperature, the mixtures were transferred to neutravidin coated plates preloaded with 50 μL of 20 nM biotinylated MBP and incubated for 15 min. The plates were washed with phosphate-buffered saline, 0.05% (v/v) Tween 20 and incubated for 30 min with horse radish peroxidase/anti-M13 antibody conjugate (1:5000 dilution in PBT buffer). The plates were washed, developed with 3,3′,5,5′-Tetramethyl-benzidine/H2O2 peroxidase substrate (Thermo Scientific), quenched with 1 M H3PO4, and the absorbance at 450 nm (A450) was determined. For each clone, the competition percentage was determined by dividing the A450 signal in the presence of MBP by the A450 in the absence of MBP. Clones that bound to MBP in the presence of 1 μM maltose but not in the absence of maltose were subjected to DNA sequencing. We obtained 17 unique clones from 48 colonies picked.
sAB expression and purification
The phagemid DNA from the selected sAB clones was purified and used as the template for Kunkel mutagenesis to introduce a stop codon between the sAB and the phage coat protein pIII, then verified by DNA sequencing. sAB clones were used for large scale protein expression in E. coli 55244 cells. The transformants were grown in 1L of the CRAP media (27 mM (NH4)2SO4, 14 mM KCl, 2.4 mM sodium citrate 5.4 g/L yeast extract, 5.4 g/L HyCase SF Casein, 0.11 M MOPS buffer pH 7.3, 0.55% (w/v) glucose and 7 mM MgSO4) supplemented with 100 μg/mL ampicillin. The cells were lysed using a microfluidizer in PBS supplemented with hen egg lysozyme (0.1 mg/mL; Sigma) and deoxyribonuclease I (0.01 mg/mL; Sigma). sABs were purified on rProteinA Sepharose 4 Fast Flow (GE Healthcare) using 20 mM phosphate buffer supplemented with 0.5 M NaCl as a wash solution. After elution with 0.1 M acetic acid, the sABs were dialyzed into running buffer (10 mM HEPES, 150 mM NaCl, 50 μM EDTA. 0.005% Tween 20, pH 7.4).
Surface plasmon resonance
Binding kinetics were determined using a BIAcore-2000 with sABs immobilized on a Ni-NTA chip (GE Healthcare). The purified protein samples were diluted at least 20-fold in the running buffer with and without 1 mM maltose. Serial dilutions of MBP with and without maltose were injected, and binding responses were corrected by subtraction of responses on a blank flow cell. The association and dissociation kinetics of MBP dilutions (60 μL each) were injected at 25 μL/min. For kinetic analysis, a 1:1 Langmuir model of global fittings of kon and koff was used. The Kd values were determined from the ratios of kon and koff values.
Maltose binding assays
Maltose binding was determined using either intrinsic tryptophan fluorescence or Alexa 488 fluorescence, both performed on a FluoroMax-3 fluoremeter. MBP233C was labeled with a 10-fold molar excess of Alexa 488 maleimide (Invitrogen) in 20 mM MOPS, 100 mM NaCl, pH 6.9 (labeling buffer) at room temperature for 1 hour or overnight at 4°C, followed by gel filtration chromatography using an Econopac 10-DG column (Bio-rad) to remove excess fluorophore. For intrinsic tryptophan fluorescence measurements, the protein was excited at 270 nm, and for Alexa 488 fluorescence measurements, the protein was excited at 488 nm. Maltose was directly titrated into 3 mL of labeling buffer containing 1–10 nM MBP or MBP233C-Alexa 488 in the absence or presence of 5-fold excess of sAB. Changes at the emission maximum were monitored as a function of maltose concentration. Binding curves were fitted using Kaleidagraph.
X-ray crystallography
His-tagged MBP was dialyzed into TBS with 1mM maltose and 1mM DTT. The MBP-MCS2 complex was prepared by combining a 1.5 molar excess of MBP with MCS2 and purified using a Superdex 200 column (GE Healthcare) equilibrated with 20 mM HEPES pH7.5, 100mM NaCl 1mM maltose 1mM DTT. The complex was crystallized by hanging drop method over 1 mL well solution containing 19% PEG 3400 and 8% Tacimate pH 6.0 (Hampton Research). Crystals formed after 1 month at 19°C. Crystals were cryoprotected with a solution of 20 % PEG 3400, 10% Tacimate pH 6.0, 23% glycerol and flash frozen. Data were collected at 21-ID-D LS-CAT at APS, and processed with HKL200026. Initial phases were determined using Phaser27 in CCP4 by molecular replacement28 with a closed MBP structure22 and the Fab scaffold16. Manual addition of CDR loops was done in Coot29, followed by atomic position refinement using alternating rounds of manual rebuilding in Coot and restrained refinement in Refmac530. TLS domains were determined using the TLSMD server and TLS parameters were also refined in Refmac531–33. Waters were added using Arp/Warp34. Figures were created in PyMol35.
In vivo assay
E. coli 55244 cells were transformed with plasmids coding for either MCS1, MCS4, or sAB-27. Overnight cultures in LB (1 mL) were used to inoculate cultures in a 96-deep-well plate containing 1.2 mL of CRAP media supplemented with ampicillin and grown overnight. The cultures were diluted into another deep-well plate containing 1.2 mL minimal media (2 g of (NH4) 2SO4, 0.5 mg of FeSO4·7H2O, 75 mg of KCl, 125 mg of thiamine, 50 mg of MgCl2 and 7.5 g of triethanolamine per liter) in each well with 0.02, 0.04, 0.2, 0.4 % maltose or 0.4% glucose. The growth of the cultures was monitored by measuring the OD595 (TECAN plate reader) of 100 μL aliquots removed from each well at various time points over 48 hours.
Supplementary Material
Table 3.
X-ray data collection, structure determination and refinement statistics
| Wavelength (Å) | 1.07817 |
| Data reduction resolution (Å) | 50.00-1.83 |
| Space group | C2221 |
| Total no. of observations | 340096 |
| No. of unique observations | 74833 |
| Completeness (2.17–2.11, 1.86–1.83) | 85.2% (84.1%, 39.7%) |
| I/σI (2.17–2.11, 1.86–1.83) | 19 (6.0, 1.4) |
| Rsyma (2.17–2.11, 1.86–1.83) | 0.060 (0.219, 0.516) |
| Refinement resolution (Å) | 38.0-2.1 |
| No. reflections used in refinement | 53794 |
| Completeness (2.15-2.10 Å) | 97.0% (82.8%) |
| No. of non-hydrogen atoms | 6592 |
| No. of waters | 400 |
| Rworkingb(%) (2.15-2.10 Å) | 17.7 (21.4) |
| Rfree (%) (2.15-2.10 Å) | 22.7 (27.3) |
| r.m.s deviation bonds (Å) | 0.025 |
| r.m.s deviation bonds (°) | 2.0 |
Rsyn is the unweighted R value on I of symmetry–related reflections (Σhkl|Ihkl−<Ihkl>|/(ΣhklIhkl).
Rworking = Σhkl|Fobshkl− Fcalchkl|/Σhkl| Fobshkl|, Rfree is calculated from 5% of reflections not used in model refinement.
Acknowledgments
This work was supported by National Institutes of Health Grants GM 072688 and F32DK080619-02 (to S.S.R.). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Abbreviations
- CDR
complementarity-determining region
- mAb
monoclonal antibody
- MBP
maltose binding protein
- sAB
synthetic antigen binder
Footnotes
Atomic coordinates have been deposited in the Protein Data Bank, PDB code: 3PGF
References
- 1.Murphy PM, Bolduc JM, Gallaher JL, Stoddard BL, Baker D. Alteration of enzyme specificity by computational loop remodeling and design. Proc Natl Acad Sci U S A. 2009;106:9215–20. doi: 10.1073/pnas.0811070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bowerman NA, et al. Engineering the binding properties of the T cell receptor:peptide:MHC ternary complex that governs T cell activity. Mol Immunol. 2009;46:3000–8. doi: 10.1016/j.molimm.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lowman HB, Wells JA. Affinity maturation of human growth hormone by monovalent phage display. J Mol Biol. 1993;234:564–78. doi: 10.1006/jmbi.1993.1612. [DOI] [PubMed] [Google Scholar]
- 4.Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: A Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 5.Koshland DE, Jr, Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–85. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 6.Du X, et al. Long range propagation of conformational changes in integrin alpha IIb beta 3. J Biol Chem. 1993;268:23087–92. [PubMed] [Google Scholar]
- 7.Kimura T, Imai Y, Irimura T. Calcium-dependent conformation of a mouse macrophage calcium-type lectin. Carbohydrate binding activity is stabilized by an antibody specific for a calcium-dependent epitope. J Biol Chem. 1995;270:16056–62. doi: 10.1074/jbc.270.27.16056. [DOI] [PubMed] [Google Scholar]
- 8.Medintz IL, Deschamps JR. Maltose-binding protein: a versatile platform for prototyping biosensing. Curr Opin Biotechnol. 2006;17:17–27. doi: 10.1016/j.copbio.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–21. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- 10.Oldham ML, Davidson AL, Chen J. Structural insights into ABC transporter mechanism. Curr Opin Struct Biol. 2008;18:726–33. doi: 10.1016/j.sbi.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quiocho FA, Spurlino JC, Rodseth LE. Extensive features of tight oligosaccharide binding revealed in high-resolution structures of the maltodextrin transport/chemosensory receptor. Structure. 1997;5:997–1015. doi: 10.1016/s0969-2126(97)00253-0. [DOI] [PubMed] [Google Scholar]
- 12.Sharff AJ, Rodseth LE, Spurlino JC, Quiocho FA. Crystallographic evidence of a large ligand-induced hinge-twist motion between the two domains of the maltodextrin binding protein involved in active transport and chemotaxis. Biochemistry. 1992;31:10657–63. doi: 10.1021/bi00159a003. [DOI] [PubMed] [Google Scholar]
- 13.Evenas J, et al. Ligand-induced structural changes to maltodextrin-binding protein as studied by solution NMR spectroscopy. J Mol Biol. 2001;309:961–74. doi: 10.1006/jmbi.2001.4695. [DOI] [PubMed] [Google Scholar]
- 14.Rizk SS, et al. An engineered substance P variant for receptor-mediated delivery of synthetic antibodies into tumor cells. Proc Natl Acad Sci U S A. 2009;106:11011–5. doi: 10.1073/pnas.0904907106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uysal S, et al. Crystal structure of full-length KcsA in its closed conformation. Proc Natl Acad Sci U S A. 2009;106:6644–9. doi: 10.1073/pnas.0810663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye JD, et al. Synthetic antibodies for specific recognition and crystallization of structured RNA. Proc Natl Acad Sci U S A. 2008;105:82–7. doi: 10.1073/pnas.0709082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fellouse FA, et al. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol. 2007;373:924–40. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Marvin JS, et al. The rational design of allosteric interactions in a monomeric protein and its applications to the construction of biosensors. Proc Natl Acad Sci U S A. 1997;94:4366–71. doi: 10.1073/pnas.94.9.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Lorimier RM, et al. Construction of a fluorescent biosensor family. Protein Sci. 2002;11:2655–75. doi: 10.1110/ps.021860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scatchard G. THE ATTRACTIONS OF PROTEINS FOR SMALL MOLECULES AND IONS. Annals of the New York Academy of Sciences. 1949;51:660–672. [Google Scholar]
- 21.Hammes GG. Thermodynamics and kinetics for the biological sciences. John Wiley and Sons, Inc; New York: 2000. [Google Scholar]
- 22.Sharff AJ, Rodseth LE, Quiocho FA. Refined 1.8-A structure reveals the mode of binding of beta-cyclodextrin to the maltodextrin binding protein. Biochemistry. 1993;32:10553–9. doi: 10.1021/bi00091a004. [DOI] [PubMed] [Google Scholar]
- 23.Koide A, Gilbreth RN, Esaki K, Tereshko V, Koide S. High-affinity single-domain binding proteins with a binary-code interface. Proc Natl Acad Sci U S A. 2007;104:6632–7. doi: 10.1073/pnas.0700149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gould AD, Shilton BH. Studies of the maltose transport system reveal a mechanism for coupling ATP hydrolysis to substrate translocation without direct recognition of substrate. J Biol Chem. 2010;285:11290–6. doi: 10.1074/jbc.M109.089078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao J, Sidhu SS, Wells JA. Two-state selection of conformation-specific antibodies. Proc Natl Acad Sci U S A. 2009;106:3071–6. doi: 10.1073/pnas.0812952106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 27.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collaborative Computational Project, n. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 29.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.Painter J, Merritt EA. A molecular viewer for the analysis of TLS rigid-body motion in macromolecules. Acta Crystallogr D Biol Crystallogr. 2005;61:465–71. doi: 10.1107/S0907444905001897. [DOI] [PubMed] [Google Scholar]
- 32.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–50. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 33.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–33. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 34.Lamzin V, Perrakis A, Wilson K. The ARP/WARP suite for automated construction and refinement of protein models. In: Rossmann M, Arnold E, editors. Crystallography of biological macromolecules. F. Dordrecht, Kluwer Academic Publishers; The Netherlands: 2001. pp. 720–722. [Google Scholar]
- 35.DeLano W. The PyMOL Molecular Graphics System. 0.99. DeLano Scientific LLC; 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.