

Abstract
During each catalytic cycle, DNA polymerases select deoxyribonucleoside triphosphate (dNTP) substrates complementary to a templating base with high fidelity from a pool that includes noncomplementary dNTPs and both complementary and noncomplementary ribonucleoside triphosphates (rNTPs). The Klenow fragment of Escherichia coli DNA polymerase I (KF) achieves this through a series of conformational transitions that precede the chemical step of phosphodiester bond formation. Kinetic evidence from fluorescence and FRET experiments indicates that discrimination of the base and sugar moieties of the incoming nucleotide occurs in distinct, sequential steps during the selection pathway. Here we show that KF-DNA complexes formed with complementary rNTPs or with noncomplementary nucleotides can be distinguished on the basis of their properties when captured in an electric field atop the α-hemolysin nanopore. The average nanopore dwell time of KF-DNA complexes increased as a function of complementary rNTP concentration. The increase was less than that promoted by complementary dNTP, indicating that the rNTP complexes are more stable than KF-DNA binary complexes but less stable than KF-DNA-dNTP ternary complexes. KF-DNA-rNTP complexes could also be distinguished from KF-DNA-dNTP complexes on the basis of ionic current amplitude. In contrast to complementary rNTPs, noncomplementary dNTPs and rNTPs diminished the average nanopore dwell time of KF-DNA complexes in a concentration-dependent manner, suggesting that binding of a noncomplementary nucleotide keeps the KF-DNA complex in a less stable state. These results imply that nucleotide selection proceeds through a series of complexes of increasing stability in which substrates with the correct moiety promote the forward transitions.
Keywords: DNA Enzymes, DNA Polymerase, DNA-Protein Interaction, DNA Replication, Single Molecule Biophysics
Introduction
To ensure the fidelity of replication, DNA polymerases preferentially incorporate nucleotide substrates complementary to a templating residue and select deoxyribonucleoside triphosphates (dNTPs)3 rather than ribonucleoside triphosphates (rNTPs) in each catalytic cycle. This selection is achieved through a series of conformational transitions that precede the covalent step of phosphodiester bond formation (Fig. 1) (1–5). One of these transitions is well characterized through the comparison of the crystal structures of polymerase-DNA complexes formed in the absence or presence of dNTP substrate complementary to the template residue at N = 0 in the polymerase active site. These crystal structures reveal a major conformational difference between the two functional states. The polymerase domain has a conserved architecture that resembles a partially closed right hand (6, 7) comprising three subdomains. The palm subdomain contains residues required for the chemistry of catalysis, including the ligands for the two magnesium ions (metals A and B) that are essential for the reaction. The thumb subdomain positions the primer/template duplex in the active site, and the fingers subdomain contains residues essential for binding incoming nucleotide substrates. In complexes containing complementary dNTP, elements of the fingers subdomain rotate in toward the active site cleft to achieve a tight steric fit with the nascent base pair. Fluorescence resonance energy transfer (FRET) studies have shown for A family DNA polymerases that the transition between the open and closed states occurs rapidly in response to nucleotide binding (Fig. 1, Step 2.2) and is not rate-limiting for the catalytic cycle (2, 5). For the Klenow fragment of Escherichia coli DNA polymerase I (KF), stabilization of the fingers-closed state requires the metal ligand, Asp-882, presumably to form an essential contact with the Mg2+ ion that is escorted into the closed complex with the incoming nucleotide substrate, but this step does not require the other metal ligand, Asp-705 (8).
FIGURE 1.

Proposed pathway for the steps in nucleotide selection by KF preceding phosphodiester bond formation. EO indicates the KF-DNA complex in which the fingers-open conformation dominates, and EC is the complex in which the fingers-closed conformation dominates. DNA* refers to the DNA rearrangement reported by 2-aminopurine (2-AP) at position N = +1 of the template strand (4). The scheme was adapted from Refs. 1–4.
Pre-steady-state ensemble fluorescence and FRET experiments have revealed additional conformational changes that occur in response to nucleotide binding for KF (2, 4) (Fig. 1). After an initial rapid step that is reported by a change in the environment of the templating base at N = 0 (Fig. 1, Step 2), a subsequent step that precedes fingers closing is reported as a change in the environment of the N = +1 template base (Fig. 1, Step 2.1). This step is promoted by dNTPs and rNTPs that are complementary to the templating base but not by noncomplementary dNTPs. In contrast to the fingers-closing step, progression through this step requires neither Asp-705 nor Asp-882. These findings imply that base complementarity is detected in an open or partially open complex in which the ribose substrate is not rejected. Such a potential “preview” complex was observed in single molecule FRET (smFRET) experiments that examined the fingers-closing transition using donor and acceptor fluorophores attached to the fingers and thumb subdomains, respectively (9). This complex, detected in the presence of noncomplementary dNTPs, had a FRET intensity intermediate in value between the open and closed complexes (9).
In contrast to the step reported by rearrangement of the +1 template residue, the fingers-closing step (Fig. 1, Step 2.2) was not supported by complementary rNTP in ensemble FRET experiments (2). Therefore, complementary rNTP may advance KF-DNA complexes along the selection pathway past an initial preview complex in which substrates with noncomplementary bases are rejected but not support full fingers closing, consistent with steric exclusion of the ribose 2′-OH in the closed state (10). The smFRET experiments also revealed a complex elicited by complementary rNTPs that differed in FRET intensity from the closed complex formed with complementary dNTP (9). The complexes promoted by noncomplementary dNTP and complementary rNTP had similar FRET efficiencies. Thus, while ensemble fluorescence experiments revealed kinetically distinct steps at which the incorrect base and incorrect sugar were recognized, the donor-acceptor pair used in the smFRET study did not readily distinguish between these complexes.
The α-hemolysin (α-HL) nanopore can be used to measure the properties of polymerase-DNA complexes at the single molecule level (11–17). Fig. 2A illustrates a single nanopore inserted in a lipid bilayer that separates two chambers (termed cis and trans) containing buffer solution. Capture of a KF-DNA complex results in an ionic current blockade with a characteristic structure. An initial current decrease from the open channel current (Io) corresponds to capture of DNA in the enzyme-bound state (EBS). In this state, KF, which is too large to enter the pore vestibule, holds the duplex portion of the DNA substrate atop the pore (11) (Fig. 2B, i, EBS). The DNA template strand is suspended in the pore lumen, which is wide enough to accommodate single-stranded but not duplex DNA. The current trace shown in gray results from capture of a KF-DNA complex formed with a primer/template substrate composed of standard DNA residues. The black current trace results from capture of a KF-DNA complex formed with a substrate bearing a block of consecutive abasic (1′,2′-H) residues (shown in Fig. 2B as red circles) positioned in the DNA template strand such that they reside in the narrow lumen of the nanopore during the EBS and augment the amplitude of this state (13). Complexes reside in the EBS until the force of the electric field pulling on the template strand causes dissociation of KF from the DNA. Upon this voltage-promoted polymerase dissociation, the duplex DNA segment is drawn into the pore vestibule, which is just wide enough to accommodate it, causing a further current decrease (Fig. 2B, ii, terminal step). In this state, the abasic residues are outside the most sensitive region of the pore and thus have negligible influence on the amplitude of this terminal step. Following electrophoresis of the DNA through the pore, the open channel current is restored (Fig. 2B, iii). Amplitude and duration of the EBS (Fig. 2C, black dots) and the terminal steps (Fig. 2C, blue dots) of the captured KF-DNA complex events can be quantified for thousands of events in each experiment and represented in a dwell time versus amplitude plot. Captured DNA molecules that are not bound to KF have the same amplitude and duration of the terminal current step (Fig. 2C, red dots), and these events are also quantified.
FIGURE 2.

Detection of KF-DNA complexes with the α-HL nanopore. A, the nanopore instrument. A single α-HL nanopore is inserted in a ∼25-μm-diameter lipid bilayer, which separates two chambers (cis and trans) containing buffer solution. Current through the nanopore is carried by K+ and Cl− ions. A patch clamp amplifier applies voltage and measures ionic current. B, ionic current blockade structure for the capture of a KF-DNA complex. The gray current trace corresponds to capture of a KF-DNA complex formed with a substrate composed solely of standard DNA residues. The black trace is for capture of a KF-DNA complex formed with a substrate bearing an insert of abasic residues in the template strand. The molecular events corresponding to each current level are illustrated in schematics i–iii (11, 13). The abasic residues are indicated as red circles. The initial longer blockade (i) is the EBS observed upon capture of a KF-DNA complex with the duplex DNA held atop the pore vestibule by the polymerase (11). The amplitude of this initial segment is increased when abasic residues are positioned to reside in the nanopore lumen during the EBS (13). The shorter, lower amplitude segment (terminal step; ii) occurs when the force of the electric field pulling on the template strand causes dissociation of KF from the DNA and the duplex DNA is drawn into the nanopore vestibule (11). Electrophoresis of the unbound DNA through the nanopore (iii) restores the open channel current (60 ± 2 pA at 180 mV in buffer containing 0.3 m KCl). C, representative dwell time versus amplitude plot for an experiment in which KF-DNA complexes were captured at 180-mV applied potential. Ionic current blockades were quantified with software developed in our laboratory that identifies the dwell time and amplitude of the EBS segments of events (black points) and their corresponding terminal step segments (blue points). The red points indicate events for which a terminal step in the ionic current blockade was not detected. These unbound DNA events have the same amplitude and duration as the terminal current steps.
KF-DNA binary complexes and KF-DNA ternary complexes formed in the presence of complementary dNTP can be distinguished on the basis of their dwell time atop the nanopore (11, 14). While the event structure shown in Fig. 2B is observed for KF-DNA complexes captured in the absence or presence of complementary dNTP, the average duration of the EBS segment for KF-DNA complexes formed with DNA substrates bearing a 3′-H-terminated primer increases without saturating as a function of complementary dNTP concentration across 4 orders of magnitude. We have shown that this kinetic behavior is due to iterative cycles of complementary dNTP binding and unbinding to KF-DNA complexes while they reside atop the pore (14). The Kd value determined for complementary dNTP binding to the captured complexes was 7.52 μm, which is on the same order as the Kd determined for the complexes in the bulk phase (4.2 μm) in the same study (14). This value is in good agreement with the range of Kd values determined for dNTP binding to DNA-KF complexes (5–20 μm) in pre-steady-state kinetic studies (1, 10, 18), suggesting that the active site of captured KF-DNA complexes remains largely undistorted under the conditions of the nanopore experiments. This reversible, iterative binding of complementary dNTP also implies that KF can transition between the open and closed states while atop the pore.
Here we show that complementary rNTPs also extended the nanopore dwell time of KF-DNA complexes. Dwell time increases promoted by rNTP were concentration-dependent but of much shorter duration than increases supported by complementary dNTP, indicating that the rNTP complex is more stable than the binary complex but less stable than the dNTP complex. KF-DNA-rNTP complexes could also be distinguished from KF-DNA-dNTP complexes on the basis of ionic current amplitude. In contrast to complementary dNTPs and rNTPs, noncomplementary dNTPs and rNTPs diminished the nanopore dwell time of KF-DNA complexes in a concentration-dependent manner. Thus, complexes essential to fidelity that occur early in the KF reaction pathway in which the base or sugar moieties of incoming substrates are recognized in sequential steps prior to the formation of the closed complex can be distinguished based on their properties when captured in an electric field atop the α-HL nanopore.
EXPERIMENTAL PROCEDURES
Enzymes and DNA Oligonucleotides
The D355A,E357A exonuclease-deficient variant of KF (100,000 units ml−1; specific activity, 20,000 units mg−1) was from New England Biolabs. DNA oligonucleotides were synthesized at the Stanford University Protein and Nucleic Acid Facility and purified by denaturing PAGE.
Nanopore Methods
The nanopore device and insertion of a single α-HL nanopore into a lipid bilayer have been described (11, 19, 20). An Axopatch 200B patch clamp amplifier (Molecular Devices) was used to apply the transmembrane voltage and measure the resulting ionic current through the nanopore. The patch clamp amplifier was used in voltage clamp mode, whole-cell configuration, with the built-in Bessel filter set to 5 kHz. The applied transmembrane voltage and measured ionic current were sampled at 100 kHz using a Digidata 1440A (Molecular Devices) analog-to-digital converter.
Experiments were conducted at 23 °C in buffer containing 15 mm K-Hepes, pH 8.0, 1 mm EDTA, 0.3 m KCl, and 11 mm MgCl2. Experiments shown in Fig. 3C and supplemental Fig. S2B were conducted using 1 μm primer/template DNA substrate, 2 μm KF, and complementary dNTPs or rNTPs at the indicated concentrations. Experiments in Fig. 5 were conducted with 1 μm primer/template DNA substrate, 1 μm KF, and an equimolar mixture of three noncomplementary dNTPs or rNTPs at the indicated concentrations. Primer/template substrates were annealed by heating at 95 °C for 3 min followed by slow cooling to room temperature.
FIGURE 3.

KF-DNA complexes formed with complementary nucleotides. A, the DNA primer/template substrate used to compare the effect of rGTP and dGTP on nanopore dwell time of KF-DNA complexes. This substrate features a 23-base pair primer/template duplex with a 3′-H primer terminus. The residue at position N = 0 of the template strand is dCMP (shown in blue). Six consecutive abasic (1′,2′-H) residues (shown as red Xs) span positions +12 to +17 of the template strand. Thirty-two residues at the 5′-end of the 79-mer template strand are not shown. Complete sequences for all of the DNA substrates used in this study are shown in supplemental Fig. S1. B, structure of an abasic (1′,2′-H) residue used in the template strands of DNA substrates in this study to optimize detection and quantification of the EBS (13); the 1′-H that replaces the nucleobase of a normal DNA residue is highlighted in red. C, complementary nucleotide concentration dependence of EBS mean nanopore dwell times for KF-DNA complexes. i, mean dwell times on the nanopore for complexes captured at 180-mV applied potential in the presence of 1 μm primer/template DNA, 2 μm KF, and the indicated concentrations of dGTP (red circles) or rGTP (blue circles). Error bars indicate the 95% confidence interval. The solid and dotted gray lines indicate the mean dwell time for the binary complex of KF and DNA and its 95% confidence interval, respectively. ii, 1/(mean dwell time) versus 1/[dGTP] and the linear fitting. iii, 1/(mean dwell time) versus 1/[rGTP] and the linear fitting. Each linear fitting determines the dissociation rate of KF from the ternary complex and the binding affinity of dGTP or rGTP (the estimated values with standard errors are described in the text). Mathematical methods for the determination of parameter values and statistical quantities in this study are detailed under “Experimental Procedures” and in the supplemental data.
FIGURE 5.

KF-DNA complexes formed in the presence of noncomplementary dNTPs or rNTPs. An equimolar mixture of all three noncomplementary dNTPs or rNTPs (each present at the indicated concentrations) was added to the nanopore cis chamber with KF and the primer/template substrate shown in Fig. 3A each present at 1 μm. The templating residue at position N = 0 is dCMP. A, the effect of an equimolar mixture of dATP, dCTP, and dTTP on KF-DNA complexes captured at 180-mV applied potential on mean dwell time (i) and the fraction of captured DNA molecules that were KF-bound (ii). The total number of capture events for each of the concentrations of noncomplementary dNTPs from which the KF-bound fractions were determined was as follows: 0 μm, 2003; 50 μm, 829; 200 μm, 907; 600 μm, 1364; 800 μm, 1667; 1000 μm, 2285; and 1200 μm, 1998. iii, 1/(enzyme bound fraction) versus (incorrect dNTP concentration) and the linear fitting, which yields the binding affinity of incorrect dNTPs (the estimated value with standard error is described in the text). B, the effect of applied voltage on the mean EBS dwell time of KF-DNA complexes in the presence of a 0 (red circles) or 800 μm (blue circles) concentration of each of dATP, dCTP, and dTTP. C and D, noncomplementary nucleotide concentration dependence of EBS dwell times for KF-DNA complexes. C, mean dwell times on the nanopore at 40-mV applied potential for KF-DNA complexes in the presence of an equimolar mixture of dATP, dCTP, and dTTP (blue circles) or rATP, rCTP, and rUTP (red circles) at the indicated concentrations. D, mean dwell times on the nanopore at 40-mV applied potential for KF-DNA complexes in the presence of an equimolar mixture of dATP, dCTP, and dGTP (blue circles) or rATP, rCTP, and rGTP (red circles) at the indicated concentrations. In this panel, the residue at position N = 0 of the template strand is dAMP; this DNA substrate is otherwise identical to the substrate used in A–C. In C and D, the solid and dotted gray lines indicate the mean dwell time for the binary complex of KF and DNA and its 95% confidence interval, respectively. In all panels, error bars indicate the 95% confidence interval. Measurements at voltages lower than 180 mV (B–D) were made using the method illustrated in supplemental Fig. S4 and described in the legend to supplemental Fig. S4.
Active Voltage Control Experiments
The experiments in Fig. 5, B–D, were conducted as described in the legend to supplemental Fig. S4 and in Refs. 15 and 16. Finite state machine logic was programmed with LabVIEW software (Version 8, National Instruments) on a field programmable gate array (PCI-7831R, National Instruments). The tethering oligonucleotide (4 μm), which is complementary to 20 residues at the 5′-end of the template strand (highlighted in green in supplemental Fig. S1, A–D) of the DNA substrates, was present in the trans chamber.
Data Analysis for Constant Voltage Experiments
Nanopore capture events, each resulting in a blockade of the measured ionic current, were quantified off line with software developed in our laboratory (13, 16). For capture experiments at 180 mV, an event was identified as a portion of the current signal where consecutive data samples (spanning a minimum of 200 μs) had a current amplitude more than ∼7 pA below the open channel current. An event resulting from capture of an unbound DNA molecule typically results in a blockade to a single amplitude. Capture of KF-bound DNA results in a blockade with two segments where the transition from the first, higher amplitude segment (EBS) to the second, lower amplitude segment (terminal step) corresponds to dissociation of KF from the captured complex (Fig. 2B). Thus, subsequent to initial event detection, each event was searched for the presence of a best fit step change in the ionic current that met minimum step size and duration requirements. Once a KF-bound event was divided into an EBS and terminal step, the dwell time and mean amplitude for the EBS and the terminal step were calculated separately. Events lacking an amplitude step change were counted as unbound DNA, and a single dwell time and mean amplitude were calculated.
Data Analysis for Active Voltage Control Experiments
In experiments at voltages lower than 180 mV, we used active voltage control (for details, see supplemental Fig. S4) to alternately expose the primer/template junction of a tethered DNA molecule to the bulk phase in the cis chamber and then to draw it back onto the top of the nanopore with submillisecond precision (15, 16). Due to capacitance in the nanopore system, the voltage changes required for active voltage control cause a transient spike in the measured ionic current at the beginning of each probing event. For less than 1 ms, the capacitive transient obscures the true nanopore current, and thus, in voltage control experiments, the start of an event was identified as the time of the voltage switch that draws the primer/template junction back to the pore instead of being identified from the measured current amplitude.
Before searching for the amplitude step change that characterizes an event as KF-bound, we used two off-line techniques to address the capacitive transient. First, the initial, most severe portion of the transient (100–800 μs) was omitted when searching for a step change because information about the captured complex is obscured during this period. Second, the effect of the remaining portion of the transient was minimized by subtracting an exponential fit to the transient decay. Using a representative event, the three-parameter exponential function
was fit to the transient decay in MATLAB (2008a, The MathWorks, Inc., Natick, MA) where t is the time from the voltage switch. Non-linear least squares was used to determine a0, a1, and a2. The empirical transient decay was then subtracted from the beginning of the measured current amplitude after each voltage switch. A search for the step change in ionic current corresponding to dissociation of KF from the captured complex (supplemental Fig. S4B) was then performed identically to the case with 180-mV capture, and the dwell time and mean amplitude for the EBS and the terminal step were calculated separately. Events lacking an amplitude step change were counted as unbound DNA.
Removing Outliers from Dwell Time Samples by Truncation
In analyzing the dwell time samples measured for various KF-DNA complexes captured atop the nanopore, we noticed that each set of dwell time samples generally follows an exponential distribution except that sometimes it has some extra samples of relatively long dwell times. That is, each set of dwell time samples may be polluted by some outliers of long dwell times. Because both good samples and outliers are randomly distributed, it is not possible to pinpoint which one is a good sample and which one is an outlier. However, we know that outliers tend to have large values. To minimize the damage caused by the outliers on the estimated parameters, we use the strategy of truncating the samples at the high end. Truncating samples has two conflicting effects on reducing the error in the estimated parameters. (i) Truncating tends to weed out more outliers than good samples so the samples left after truncating are less polluted by outliers. (ii) But if the sample set is unpolluted, fitting truncated samples to a truncated model will increase the statistical uncertainty in the estimated parameters. It is clear that when the sample set is polluted no truncation will lead to large biases caused by outliers, whereas too much truncation will lead to large statistical errors associated with fitting to a truncated model. In between, there is an optimal truncation. To determine the optimal truncation, we introduce a measure of the overall error in the estimated parameters. Let q be the fraction of samples truncated. We consider the following equation.
 |
Here the first part of Error(q) measures whether or not the truncated sample set is consistent with the truncated model. We solve for the optimal truncation fraction q by minimizing Error(q). Then we use maximum likelihood estimation (21) to estimate the parameter values and their statistical errors. More mathematical details are presented in the supplemental data. Key statistical quantities, including the number of data points (before and after outlier removal) for the data plotted in Figs. 3C and 5, Ai and B–D, and supplemental Fig. S2B, are given in supplemental Table S1.
RESULTS
KF-DNA Complexes with Complementary rNTP
Throughout this study, we have used the D355A,E357A variant of KF, which is deficient in 3′–5′ exonuclease activity (22) and which we refer to for simplicity as KF. To test the effect of complementary rNTP on KF-DNA complexes, we used a primer/template substrate with a 23-base pair duplex bearing a dCMP residue at N = 0 of the template strand (N = 0 = C) and a 3′-H primer terminus (Fig. 3A and supplemental Fig. S1A). KF complexes formed with DNA substrates with 3′-H-terminated primers have been shown to undergo the transitions that occur during nucleotide selection up to and including fingers closing (Fig. 1, Steps 1–2.2) but cannot undergo phosphodiester bond formation (2, 4, 9). The DNA substrate we used also featured an insert of six consecutive abasic (1′,2′-H; Fig. 3B) residues spanning positions +12 to +17 in the template strand to optimize detection and quantification of the EBS. KF-DNA complexes formed with this substrate and captured at 180-mV applied potential showed the concentration-dependent, unsaturable increase in nanopore dwell time in response to dGTP (Fig. 3C, i, red circles) that was observed previously with a 3′-H-terminated DNA hairpin substrate (14).
When rGTP was titrated into the nanopore chamber, the dwell time of KF-DNA complexes captured at 180 mV increased in a concentration-dependent manner (Fig. 3C, i, blue circles). These rGTP ternary complexes differed from dGTP ternary complexes. Much higher rGTP concentrations were required to increase the dwell time of complexes, and the magnitude of the increases was significantly smaller than obtained with dGTP.
From the titration data in Fig. 3C, i, we estimated the dissociation rate of KF from complexes held atop the pore at 180 mV for the KF-DNA binary complex and the KF-DNA-dGTP or KF-DNA-rGTP ternary complex. We also estimated the binding affinity of dGTP or rGTP for KF-DNA complexes when they reside atop the pore.
We first introduce the notation. Let k1 = the dissociation rate of KF from the binary state (state 1), k2 = the dissociation rate of KF from the ternary state (state 2) with dGTP (or rGTP) bound, Kd = the binding affinity of dGTP (or rGTP) when the KF-DNA complex is held atop the pore, and <T> = the mean dwell time of the KF-DNA complex held atop the pore. In the supplemental data, we show the following.
 |
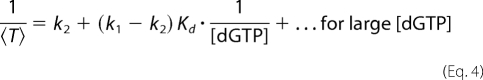 |
k1 is estimated from the mean dwell time at [dGTP] = 0. Then, k2 and Kd are estimated by fitting a linear function to data points of (1/[dGTP], 1/<T>) at high dGTP concentrations. Fig. 3C, ii, shows the fitting to the dGTP titration data, and Fig. 3C, iii, shows the fitting to the rGTP titration data. The following parameter values are reported in the format of estimated value ± S.E.: k1 = (1.001 ± 0.025) × 10−1 ms−1, k2(KF-DNA-dGTP) = (1.996 ± 0.213) × 10−4 ms−1, k2(KF-DNA-rGTP) = (1.138 ± 0.078) × 10−2 ms−1, Kd(dGTP) = 7.094 ± 0.466 μm, and Kd(rGTP) = 203.5 ± 24.2 μm.
Thus, for KF-DNA complexes held atop the nanopore at 180-mV applied potential, the Kd of rGTP (complementary rNTP) is ∼30-fold larger than that of dGTP (complementary dNTP), and the dissociation rate of KF from DNA is ∼50-fold higher for the KF-DNA-rGTP complexes than for the KF-DNA-dGTP complexes.
The marked difference in the stability of complexes promoted by complementary rNTP and dNTP was not unique to the primer/template with N = 0 = C and incoming guanine nucleotides. When dTTP or rUTP was assayed using KF-DNA complexes formed with a primer/template substrate bearing a dAMP residue at the N = 0 template position (N = 0 = A; supplemental Fig.S1B) that was otherwise identical to the N = 0 = C substrate, similar behavior was observed (supplemental Fig. S2). Complementary dNTP promoted dwell time increases in a manner that appeared unsaturated over at least 3 orders of magnitude in dTTP concentration, whereas rUTP yielded measurable but much more modest increases. The nanopore dwell time of the binary complexes formed with this N = 0 = A DNA substrate and the dwell times supported by either dTTP or rUTP were notably shorter than the corresponding dwell times for the N = 0 = C primer/template substrate. For both DNA substrates, the extension of the nanopore dwell time by either rNTP or dNTP required base complementarity and was not supported by noncomplementary nucleotide substrates (see below).
When dGTP was added to the nanopore chamber in the presence of rGTP (with the N = 0 = C substrate), the dwell time was further extended. For example, while the mean dwell time (with the 95% confidence interval of the mean in parentheses) for complexes captured in the presence of 2400 μm rGTP was 52.9 ms (48.8, 57 ms), it increased to 523.6 ms (462.3, 584.9 ms) following the subsequent addition of 400 μm dGTP. This compares well with the dwell time value of 509.2 ms (455.6, 562.8 ms) obtained in the presence of 400 μm dGTP alone and indicates that the influence of complementary dNTP dominates when both complementary rNTP and dNTP are present.
Structural Difference between KF-DNA Complexes with Complementary rNTP or dNTP
We noted that in the data files recorded after dGTP was added to experiments subsequent to rGTP the amplitude of the EBS population was consistently higher (by ∼1 pA) than the amplitude recorded for the rGTP complexes in the immediately preceding data files. This suggested that the KF-DNA-rNTP complexes could be distinguished structurally from KF-DNA-dNTP complexes on the nanopore.
To study this further, we exploited two DNA substrates bearing dCMP or dAMP residues at template position N = 0 (Fig. 4A, i and ii, and supplemental Fig. S1, A and B). In experiments in which we examined each of these two DNA substrates individually in the presence of KF, we established that the EBS amplitude of their respective binary complexes did not differ (supplemental Fig. S3). Fig. 4, B–D, i, shows dwell time versus amplitude plots of the EBS populations for three experiments in which KF and both of the DNA substrates are present. In the experiment in Fig. 4B, 4000 μm rGTP (complementary for the N = 0 = C substrate) was added to the cis chamber (Fig. 4B, ii). An increase in dwell time occurred with no change in amplitude. When 1000 μm dTTP (complementary for the N = 0 = A substrate) was next added, a population of complexes emerged with longer dwell time than when only rGTP was present (Fig. 4B, iii). This population was ∼1 pA higher in amplitude than the population in Fig. 4B, ii, and than the remainder of the complexes in Fig. 4B, iii. The dwell time distribution within the population that remained at the lower amplitude changed; the addition of dTTP appeared to selectively recruit complexes from the shorter dwell time end of the distribution in Fig. 4B, ii, which we therefore presume corresponds primarily to the KF-DNA binary complexes with the N = 0 = A substrate. In contrast, the longer dwell time end of the distribution within this lower amplitude population, which likely corresponds to the KF-DNA-rGTP complex with the N = 0 = C substrate, appeared unchanged upon the addition of dTTP. The dwell time of the higher amplitude population was selectively increased when the dTTP concentration was raised to 2000 μm (Fig. 4B, iv), confirming its assignment to the KF-DNA-dNTP complex with the N = 0 = A substrate.
FIGURE 4.

Current amplitudes for KF-DNA complexes formed with complementary rNTP or dNTP captured on the nanopore. A, primer/template DNA substrates used to compare the amplitudes of KF-DNA complexes. Key features of the template strands that permit complexes to be distinguished in the experiments are the identity of the residue at N = 0 (highlighted in blue) and the position of a block of six abasic (1′,2′-H) residues. The abasic residues are shown as red Xs, and their position in the template strands relative to the N = 0 residue is indicated above the sequences. Forty-seven residues starting from the 3′-end of each of the template strands are shown. The binding site for the 23-mer 3′-H-terminated primer is underlined. Complete sequences for all of the DNA substrates used in this study are shown in supplemental Fig. S1. B, C, and D show dwell time versus amplitude plots of the EBS populations from individual experiments with the plots for data files collected after each of a series of sequential substrate additions labeled above each subpanel. The three experiments in B, C, and D started with the same components in the nanopore chamber; that is, subpanels i in B, C, and D each had 2 μm KF and a 1 μm concentration of each of the primer/template substrates in A (i and ii). The subsequent additions in the experiment in B were 4000 μm rGTP (ii), 1000 μm dTTP (iii), and an additional 1000 μm dTTP to a final concentration of 2000 μm dTTP (iv). In the experiment in C, the additions were 5000 μm rUTP (ii) and 100 μm dGTP (iii). In the experiment in D, the additions were 30 μm dGTP (ii) followed by 2000 μm dTTP (iii). The red lines across the panels are placed through the lower amplitude population clusters (KF-DNA and KF-DNA-complementary rNTP complexes) as a visual guide. The amplitude range in B–D is restricted to 30–40 pA to highlight the EBS events. Dwell time versus amplitude plots for the experiments in B–D showing the amplitude range between 0 and 50 pA, which includes the terminal steps from the EBS events and the unbound DNA events, are shown in supplemental Fig. S3. Supplemental Fig. S3 also includes the initial steps from the experiments in B–D, showing the binary complexes formed with the two DNA substrates individually and confirming that these binary complexes have the same EBS amplitude. E, dwell time versus amplitude plot for KF-DNA complexes captured in the presence of 3 μm KF, 300 μm dGTP, and a 1 μm concentration of each of the primer/template substrates in A (i, iii, and iv). The KF-DNA-dNTP ternary complex populations corresponding to each of the three DNA substrates are labeled; assignment of the populations was determined in experiments in which the DNA substrates were added to the nanopore chamber sequentially (not shown).
To examine whether the difference in the amplitude of the KF-DNA-rGTP complexes and KF-DNA-dTTP complexes was due to the identity of the sugar moiety of the nucleotide, we performed the experiment in Fig. 4C. When 5000 μm rUTP was added to the chamber in the presence of binary complexes formed with both the N = 0 = C and N = 0 = A substrates, the dwell time was increased without a change in amplitude (Fig. 4C, ii). The subsequent addition of 100 μm dGTP caused a longer lived, ∼1-pA higher amplitude population to emerge, whereas the amplitude of the remaining population corresponding to the KF-DNA-rUTP complex with the N = 0 = A substrate was unchanged (Fig. 4C, iii). Thus, in two different experiments, the addition of rNTP complementary to N = 0 of one of the DNA substrates increased the dwell time of at least a portion of the complexes without an effect on amplitude (Fig. 4, B, ii and C, ii), and the addition of dNTP complementary to N = 0 of the other of the DNA substrates elicited an additional population with both increased dwell time and amplitude (Fig. 4, B, iii and C, iii). These data indicate that the difference in amplitude between the complexes formed in response to complementary rNTP or complementary dNTP corresponds to the difference in the sugar moiety of the incoming nucleotide. That is, the structural difference in the captured ternary complexes that results in an increase in amplitude relative to the binary complex is not promoted by the complementary ribose substrates.
Finally, in the experiment in Fig. 4D, we added 30 μm dGTP (which causes an increase in dwell time for the N = 0 = C substrate of magnitude similar to that caused by 4000 μm rGTP) to the nanopore chamber in the presence of binary complexes formed with both the N = 0 = C and N = 0 = A substrates. The dwell time of a portion of the complexes increased (Fig. 4D, ii). However, in contrast to when rGTP or rUTP was added at this point in the experiment (Fig. 4, B, ii and C, ii), the amplitude of the events with longer dwell time increased by ∼1 pA (Fig. 4D, ii). This indicates that the amplitude increase is specific to KF-DNA-dNTP complexes rather than due to the duration of events. Subsequent addition of 2000 μm dTTP caused both the dwell time and amplitude of the shorter lived, lower amplitude complexes (presumably binary complexes with the N = 0 = A substrate) that were present in Fig. 4D, ii, to increase (Fig. 4D, iii). Thus, in contrast to the cases where KF-DNA-rNTP and KF-DNA-dNTP complexes were both present and differed in amplitude by ∼1 pA (Fig. 4, B, iii and iv and C, iii), KF-DNA-dNTP complexes formed with the two DNA substrates had the same amplitude. Taken together, the experiments in Figs. 3C and 4, B–D, indicate that KF-DNA-rNTP complexes can be distinguished from KF-DNA binary complexes by dwell time and from KF-DNA-dNTP complexes by dwell time and amplitude. In addition, KF-DNA and KF-DNA-dNTP complexes can be distinguished from one another on the basis of nanopore dwell time and amplitude.
Mechanism That Yields Amplitude Difference between KF-DNA Complexes with Complementary rNTP or dNTP
The difference in EBS amplitude between KF-DNA-dNTP complexes and both the KF-DNA and KF-DNA-rNTP complexes (Fig. 4, B–D) could be caused by distinct physical mechanisms. First, the conformation of the KF-DNA-dNTP complex atop the pore could differ from the binary and KF-DNA-rNTP complexes such that it affects the extent to which ion flow into the channel is occluded. For example, fingers closing upon correct dNTP binding could render the complex more compact, allowing more current to flow into the pore and yielding the higher amplitude observed for complexes in this state. Alternatively, the amplitude difference between complexes could be due to movement of the template strand in the nanopore lumen. We have shown that the register of captured polymerase-DNA complexes on a primer/template substrate can be reported with precision by a block of three to six abasic residues in the template strand (13, 15, 17). Single nucleotide displacements of the template strand yield measurable differences in EBS amplitude when polymerase-DNA-dNTP complexes are formed with substrates in which the position of the abasic block in the template is varied (13) or when DNA synthesis is catalyzed by polymerase-DNA complexes atop the pore and the template strand is consequently drawn through the nanopore lumen in single nucleotide increments (15, 17). The abasic block thus reports template displacements in the lumen that occur as a result of concerted movement of the duplex and single-stranded template DNA with respect to the pore and the polymerase, such as those caused by the translocation step during DNA synthesis. Finally, the abasic block might also report differences in the arrangement of the template strand in and near the polymerase active site for complexes in different functional states that result in displacement of the abasic block in the lumen but do not necessarily involve changes in the register of the enzyme along the duplex.
We performed experiments aimed at distinguishing the contributions of these mechanisms to the ∼1-pA higher amplitude of KF-DNA-dNTP complexes relative to both the KF-DNA and KF-DNA-rNTP complexes (Fig. 4, B–D). Detection of a conformational change in the polymerase complex that affects ion flow into the nanopore should not be dependent upon the presence of an abasic insert in the template strand. In experiments that were conducted identically to the experiment in Fig. 4B but in which we used two primer/template substrates bearing dCMP or dAMP at N = 0 containing only normal DNA residues in the template strand, we were unable to detect any differences in EBS amplitude among KF-DNA binary complexes or complexes formed with complementary rNTP or dNTP (not shown). This requirement for the abasic block reporter implicates a template movement in the nanopore lumen as the cause of the amplitude difference between the complexes.
We therefore tested whether the magnitude of the amplitude difference observed between the complexes in Fig. 4, B–D, was consistent with displacement of the DNA template in the nanopore lumen by the distance of a single nucleotide. We compared the amplitudes of KF-DNA ternary complexes formed with dGTP and three different N = 0 = C primer/template substrates. These DNA substrates had abasic blocks spanning template positions +12 to +17 (Fig. 4A, i), positions +11 to +16 (Fig. 4A, iii), or positions +13 to +18 (Fig. 4A, iv). The KF-DNA-dGTP ternary complexes formed with these three substrates are in equivalent functional states, but in the complexes formed with the latter two DNA substrates, the position of the abasic block in the nanopore lumen is displaced by a single nucleotide in either direction relative to the +12 to +17 substrate. A dwell time versus amplitude plot of capture events for these three DNA substrates in the presence of KF and dGTP is shown in Fig. 4E. Average amplitudes of the populations corresponding to KF-DNA-dGTP ternary complexes bearing the +13 to +18, +12 to +16, and +11 to +17 abasic blocks were 30.6, 35.1, and 38.9 pA, respectively.
Using the amplitude of KF-DNA-dGTP ternary complexes formed with the substrate in which the abasic block spans positions +12 to +17 as a reference point, the lower amplitude of the KF-DNA binary and KF-DNA-rNTP complexes formed with the +12 to +17 substrates in Fig. 4, B–D, indicates that the direction of displacement of the template in those complexes is the same as it is in the KF-DNA-dGTP complexes with the +13 to +18 substrate in Fig. 4E. That is, in comparison with the binary complex or the complex with complementary rNTP, in the complexes with complementary dNTP, the template is moved with respect to the nanopore lumen toward the enzyme. However, the magnitude of the amplitude difference (∼1 pA) is considerably smaller than the 4.5 pA between the KF-DNA-dGTP complexes formed with the +12 to +17 and the +13 to +18 substrates (Fig. 4E). Thus, the ∼1-pA difference between the binary or rNTP complexes and the dNTP complexes in the experiments in Fig. 4, B–D, is consistent with a structural difference among the KF complexes that alters the position of the template strand in the nanopore lumen by a distance of less than a full nucleotide.
KF-DNA Complexes with Noncomplementary dNTPs and rNTPs
Noncomplementary dNTPs have been shown to destabilize KF-DNA complexes in several different assays (2, 23–25). We previously found that the dwell time of KF-DNA complexes formed with a DNA hairpin substrate bearing a dCMP residue at N = 0 was unaffected by the addition of dTTP when they were captured at 180 mV (14). We tested whether an equimolar mixture of noncomplementary dATP, dCTP, and dTTP affected the nanopore dwell time of KF-DNA complexes formed with the N = 0 = C primer/template substrate shown in Fig. 2A. When complexes were captured at 180 mV, the mixture of incorrect dNTPs had a negligible effect on dwell time even when present at 1200 μm each (Fig. 5A, i). However, as the mixture of incorrect dNTPs was titrated into the nanopore chamber, we observed a concentration-dependent decrease in the fraction of DNA molecules captured in the KF-bound state (Fig. 5A, ii). From the data in Fig. 5A, ii, we estimated the bulk phase binding affinity for the composite mixture of noncomplementary dNTPs. Let Cincorrect = the concentration of each of three incorrect dNTPs, p1(Cincorrect) = the observed enzyme-bound fraction under 180-mV voltage, and Kd(B) = the effective binding affinity of incorrect dNTPs (when we view the three incorrect dNTPs as one composite species). Fig. 5A, ii, shows experimental results of Cincorrect versus p1(Cincorrect).
In the supplemental data, we show the following.
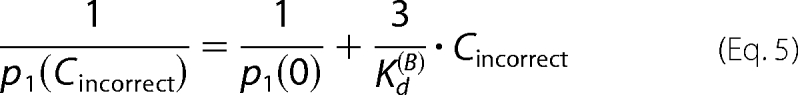 |
The binding affinity Kd(B) is estimated by fitting a linear function to data points of (Cincorrect, 1/p1). After the least square fitting, we obtain Kd(B)(dNTPincorrect) = 515.0 ± 30.1 μm. Note that Kd(B)(dNTPincorrect) is the effective bulk phase binding affinity of the composite species dNTPincorrect when all three noncomplementary dNTPs are present at equimolar concentrations, and the binding affinity of each individual noncomplementary dNTP may be higher or lower.
The data in Fig. 5A, i–iii, imply that when a KF-DNA-dNTPincorrect ternary complex is captured atop the nanopore at 180 mV KF dissociates quickly, and as a result, most KF-DNA-dNTPincorrect ternary complexes are not recognized as enzyme-bound events. Thus, at 180-mV applied potential and in the presence of only incorrect dNTPs, only the KF-DNA binary complexes are recognized as enzyme-bound events.
We examined whether an effect of noncomplementary dNTPs on the nanopore dwell time of KF-DNA complexes could be detected at lower voltages. To circumvent the problem of infrequent capture of DNA and complexes at lower voltages, we used active voltage control of the DNA primer/template substrate non-covalently tethered in the nanopore (15, 16) (for details, see supplemental Fig. S4). The dwell time of KF-DNA binary complexes increased as voltage was lowered (Fig. 5B, red circles) as expected when the electric field force pulling on the template strand is attenuated. The presence of an 800 μm concentration of each noncomplementary dNTP had a negligible effect on nanopore dwell time when measured at 120 or 100 mV. When the voltage was lowered to 80 mV, the dwell time of complexes in the presence of the mixture of incorrect dNTPs (Fig. 5B, blue circles) was slightly shorter than that of binary complexes (Fig. 5B, red circles). The destabilization effect of noncomplementary dNTPs was increasingly pronounced when complexes were compared at 60 mV and then at 40 mV. Thus, the ability of noncomplementary dNTPs to diminish the dwell time of complexes was revealed at voltages lower than 100 mV, consistent with formation of a complex more labile than the binary complex that could not survive the force of capture at higher voltages.
The nanopore dwell time of KF-DNA complexes at 40 mV was diminished as a function of noncomplementary dNTP concentration (Fig. 5C, blue circles). The concentration-dependent decrease in dwell time at 40 mV caused by noncomplementary dNTPs mirrored the concentration-dependent decrease in the fraction of molecules that could be captured in the KF-bound state at 180 mV (Fig. 5A, ii), providing further support for the proposed destabilized complex.
The mean dwell time at 40 mV of KF-DNA complexes also decreased as a function of the concentration of an equimolar mixture of noncomplementary rNTPs (Fig. 5C, red circles). While complementary rNTP was significantly less potent than complementary dNTP at increasing the nanopore dwell time of complexes (Fig. 3C, i), destabilization of complexes by noncomplementary rNTPs or by noncomplementary dNTPs followed a similar concentration dependence (Fig. 5C). This indicates that the destabilization of KF-DNA complexes occurs in a complex in which the nucleobase is the primary recognition determinant and is consistent with a mechanism in which the base is recognized prior to sugar discrimination.
The nanopore dwell time at 40 mV for complexes formed with the primer/template substrate bearing dAMP at template position N = 0 was also diminished as a function of the concentration of a mixture of all three noncomplementary dNTPs (Fig. 5D, blue circles) or rNTPs (Fig. 5D, red circles). The extent to which noncomplementary dNTPs or rNTPs decreased dwell time was similar. The mean dwell time on the nanopore at 40 mV for binary complexes formed with the N = 0 = A substrate (Fig. 5D, gray line) was about half as long as the dwell time for binary complexes formed with the N = 0 = C primer/template substrate, consistent with observations at 180 mV (Fig. 3 and supplemental Fig. S2).
When dGTP was added to the nanopore chamber in the presence of incorrect dNTPs (with the N = 0 = C substrate), the nanopore dwell time at 40 mV was increased to a level similar to that elicited by dGTP alone. For example, the mean dwell time (with the 95% confidence interval of the mean in parentheses) for complexes captured in the presence of a 200 μm concentration of each incorrect dNTP was 80.1 ms (76.9, 83.3 ms). The dwell time increased to 327.6 ms (262, 393.2 ms) upon the subsequent addition of 50 μm dGTP. This is similar to the value of 326.9 ms (277.9, 375.9 ms) obtained in the presence of 50 μm dGTP alone and indicates that the influence of dGTP, which supports fingers closing, dominates in the presence of incorrect nucleotides.
DISCUSSION
We have used the α-HL nanopore to distinguish between KF-DNA complexes in different states during which the base complementarity (Fig. 6A) or correct sugar structure (Fig. 6B) of an incoming NTP substrate is detected by the polymerase. Compelling kinetic evidence from ensemble fluorescence and FRET experiments indicates that these substrate features are selected in distinct steps prior to phosphodiester bond formation (2, 4), and smFRET experiments revealed candidate complexes for these steps (9). In the smFRET study, the FRET efficiencies of complexes elicited by complementary rNTPs and noncomplementary dNTPs were both consistent with conformations in which the distance between the donor probe in the fingers subdomain and the acceptor probe in the thumb subdomain was intermediate between the fingers-open state that dominates in the KF-DNA binary complex and the fingers-closed state that dominates in complexes formed with complementary dNTPs (9). The results of this nanopore study indicate that KF-DNA complexes with complementary rNTPs or noncomplementary dNTPs differ markedly in their resistance to voltage-promoted dissociation when captured on the α-HL nanopore. While complexes formed with noncomplementary dNTPs were more labile than KF-DNA binary complexes, complexes formed with complementary rNTPs were more stable than KF-DNA binary complexes but less stable than KF-DNA ternary complexes formed with complementary dNTPs.
FIGURE 6.

Distinct complexes for base and sugar discrimination during nucleotide substrate selection by KF. Shown are steps in the pathway for nucleotide selection by KF discerned in pre-steady-state kinetic and fluorescence studies (2, 4) with the complexes detected in this study for base discrimination (A) or sugar discrimination (B) highlighted against a gray background. A, binding of incorrect (noncomplementary) dNTPs or rNTPs yields a complex that is less stable than the KF-DNA binary complex and that does not undergo the transition reported by a change in the environment of the N = +1 template base (Step 2.1) (2). B, binding of correct (complementary) dNTPs or rNTPs yields complexes that are more stable than KF-DNA binary complexes, but the complexes formed with correct rNTPs are less stable than those formed with correct dNTPs as they do not undergo the fingers-closing transition (Step 2.2). DNA* refers to the DNA rearrangement reported by 2-aminopurine (2-AP) at position N = +1 of the template strand (4).
Sugar Discrimination
The concentration-dependent increase in dwell time for KF-DNA complexes in the presence of complementary rNTP (Fig. 3 and supplemental Fig. S2) indicates that a specific complex can be formed with KF-DNA and this substrate (Fig. 6B). This complex is more stable atop the nanopore at 180 mV than KF-DNA binary complexes but much less stable than complexes formed in the presence of equivalent concentrations of complementary dNTP. In contrast to KF-DNA complexes with complementary dNTP for which voltage-promoted dissociation of KF occurs predominantly when dNTP dissociates and the complexes pass through the binary state (14), the ∼50-fold faster dissociation rate of KF from KF-DNA-rNTP complexes suggests that voltage-promoted dissociation of KF directly from the KF-DNA-rNTP complex can occur. Nonetheless, the dissociation rate of KF from the complex with complementary rNTP is ∼9 times slower than from the KF-DNA binary complex.
KF-DNA complexes formed with complementary rNTP are structurally distinguishable from KF-DNA-dNTP complexes on the basis of the amplitude of captured complexes. Using primer/template substrates with a block of six abasic residues in the template strand, the EBS of complexes formed with complementary dNTP was ∼1 pA higher in amplitude than the EBS of KF-DNA binary complexes or complexes formed with complementary rNTP (Fig. 4, B–D). This difference in EBS amplitude is smaller than the amplitude difference of KF-DNA-dNTP ternary complexes that corresponds to shifting the abasic block by a full nucleotide in the nanopore lumen (Fig. 4E). This suggests a difference in the conformation of KF complexes that either displaces the template strand in the lumen by a distance less than a full nucleotide or alters the extent of ion flow into the nanopore. We were unable to detect an EBS amplitude difference between the rNTP and dNTP complexes when they were formed with primer/template substrates composed of only normal DNA in the absence of an abasic reporter in the template strand. Thus, the magnitude of the amplitude difference between the complexes with complementary rNTP or dNTP is consistent with a difference in the conformation of KF complexes as they reside atop the pore that changes the position of the template strand with respect to the nanopore lumen by a distance smaller than a full nucleotide.
If this movement of the template strand in the nanopore lumen occurs due to a rearrangement in the enzyme complex, then this rearrangement presumably occurs after the step reported as a change in environment of 2-aminopurine at the N = +1 position of the template; that step is promoted by complementary rNTP (2), but the amplitude shift we detect requires both the correct base and sugar in the incoming substrate. The rNTP complexes may progress beyond a state in which base complementarity is sampled to achieve partial fingers closure but be inhibited from complete closure by the steric gate residue Glu-710 that is encountered as the complex approaches the closed state (10). This is consistent with the FRET state that is intermediate between the open and closed conformations promoted by complementary rNTP in smFRET experiments (9). Thus a movement of the template strand detected as an amplitude difference between the rNTP and dNTP complexes might occur in concert with fingers closing. Perhaps this movement occurs when Tyr-766, which occupies the templating site in crystal structures of A family DNA polymerase binary complexes, is displaced by the N = 0 base that occupies this position in crystal structures of polymerase-DNA-dNTP ternary complexes (26–28). Finally, it is possible the movement we detect as an amplitude shift corresponds to a recently reported change in circular dichroism spectra that occurs in response to complementary dNTP binding to KF-DNA complexes and that is detected using probes at template positions N = 0 and N = +1 (29).
Base Discrimination
The finding that complementary rNTP, but not noncomplementary dNTP, promotes the template rearrangement that is reported by a change in the environment of the N = +1 base (Fig. 1, Step 2.1) indicates that the base moiety of the incoming nucleotide is detected in a distinct step prior to the detection of the sugar moiety during nucleotide selection by KF (2). Base complementarity may be tested in a preview complex possibly similar to the complex observed by x-ray crystallography for bacteriophage T7 RNA polymerase, another A family polymerase (30). It is possible that in this putative preview state KF-DNA complexes are inherently less stable than in the binary state. Furthermore, the smFRET study demonstrated that KF-DNA binary complexes are in equilibrium between the open and closed states, favoring the fingers-open conformation by a factor of ∼2:1 (9). When complementary dNTP is not present, noncomplementary dNTPs, which lack the ability to promote subsequent steps in the pathway toward fingers closing, could increase the fraction of time spent in the preview state and thus decrease the frequency with which the complex samples the closed binary state, which may be more stable than the open binary state.
Incorrect dNTPs diminished, in a concentration-dependent manner, the proportion of complexes that were captured and recognized as KF-bound atop the nanopore at 180 mV (Fig. 5A, ii), which suggests that noncomplementary substrates promote the formation of a species that is less stable than the KF-DNA binary complex and that cannot survive long enough in the field force pulling on the template strand at higher voltages to be detected. Consistent with this proposal, when the voltage is lower, the more labile complexes do survive nanopore capture long enough for their effect on dwell time to be detected (Fig. 5, B–D). The decrease in mean dwell time of captured complexes observed in the presence of noncomplementary dNTP at low voltages implies that dissociation of KF directly from the KF-DNA-noncomplementary dNTP complex can occur and that it occurs at a rate faster than the rate of dissociation of KF from the KF-DNA binary complex (Fig. 6A) because if the dissociation of KF from DNA had to be preceded by the dissociation of noncomplementary dNTP the presence of noncomplementary dNTP would lead to an increase in the mean dwell time of captured complexes.
The destabilizing effect of noncomplementary nucleotides on KF-DNA complexes that can be detected upon capture under force atop the nanopore may be negligible in the bulk phase under optimal conditions (2, 9) and may require perturbation to be revealed (Fig. 6A, dashed blue arrow). Consistent with this, although an increase in the rate of dissociation of KF from DNA in the presence of noncomplementary dNTPs has been observed in several studies, it is often under conditions in which binary complexes may already be destabilized, such as in the presence of modifications to the DNA substrate (2, 25), during gel electrophoresis (23, 24), or in the presence of a competitor DNA trap (2). In cells, rather than promoting dissociation of KF from the DNA, it is more probable that noncomplementary dNTP will dissociate first, allowing the complex to resolve to the stability of the binary state. Critically, binding of complementary dNTP will drive the complex forward to the fingers-closing step. In accord with this, we find that the increased dwell time promoted by complementary dNTP dominates in the presence of noncomplementary dNTPs.
If base complementarity is detected in a step that precedes sugar discrimination and if the decrease in KF-DNA dwell time observed at 40 mV in the presence of noncomplementary dNTPs is due to formation of a complex in which base discrimination occurs, then it would be predicted that noncomplementary rNTPs would also destabilize KF-DNA complexes. Consistent with this, we found that the mean dwell times of complexes at 40 mV (Fig. 5, C and D) decreased as a function of noncomplementary rNTP concentration. The fact that we used an equimolar mixture of all three noncomplementary substrates may account for the variation in details of the response to noncomplementary dNTPs or rNTPs between the N = 0 = C and N = 0 = A primer/template substrates (Fig. 5, C and D). Individual mismatches may differ in their efficacy in destabilizing the KF-DNA complex because complexes with different mismatched base pairs in the polymerase active site have non-ideal, but nonequivalent geometry (31). A detailed understanding of the concentration dependence and influence of sequence on the relative extent to which incorrect dNTPs elicit a destabilized complex or even occasionally promote fingers closing will require future analysis using individual noncomplementary dNTPs.
Conclusions
The findings in this study support the proposal that the nucleobase and sugar moieties of incoming substrates are recognized in different steps during the KF nucleotide selection pathway (2). Ribose substrates with complementary bases or deoxyribose substrates with noncomplementary bases elicit distinct complexes that are readily distinguishable from one another based upon their properties when captured in an electric field atop the α-HL nanopore. Furthermore, both of these complexes are distinct from the more stable complexes promoted by the binding of complementary dNTP. Although the assays in this study do not address the rates at which complexes form and evolve, the results imply that nucleotide selection proceeds through a series of complexes of increasing stability in which substrates with the correct moiety permit the forward transitions (Fig. 6). This suggests a scheme featuring a progression through at least three distinct complexes. The first is a complex in which base complementarity is sampled (Fig. 6A, highlighted in gray). Recognition of base complementarity in this complex permits progression through Step 2.1 to the second complex in which the 2′ constituent of the sugar moiety is recognized (Fig. 6B, the complex highlighted in gray). The deoxyribose, but not ribose, sugar moiety selectively permits progression to the third complex, in which full fingers closing dominates, that can progress through the rate-limiting active site rearrangement that precedes catalysis.
Acknowledgments
We are grateful to Ai Mai and Felix Olasagasti for oligonucleotide purification; Felix Olasagasti, Gerald M. Cherf, and Robin Abu-Shumays for helpful discussions; and Peter Walker (Stanford University Protein and Nucleic Acid Facility) for expert oligonucleotide synthesis.
This work was supported, in whole or in part, by National Institutes of Health Grants 1RC2HG005553 from the NHGRI (to M. A.) and 1R01GM087484-01A2 from the NIGMS (to K. R. L. and M. A.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1, Figs. S1–S4, and additional mathematical methods.
- dNTP
- deoxyribonucleoside triphosphate
- rNTP
- ribonucleoside triphosphate
- KF
- Klenow fragment of E. coli DNA polymerase I
- EBS
- enzyme-bound state
- smFRET
- single molecule FRET
- α-HL
- α-hemolysin.
REFERENCES
- 1. Dahlberg M. E., Benkovic S. J. (1991) Biochemistry 30, 4835–4843 [DOI] [PubMed] [Google Scholar]
- 2. Joyce C. M., Potapova O., Delucia A. M., Huang X., Basu V. P., Grindley N. D. (2008) Biochemistry 47, 6103–6116 [DOI] [PubMed] [Google Scholar]
- 3. Kuchta R. D., Mizrahi V., Benkovic P. A., Johnson K. A., Benkovic S. J. (1987) Biochemistry 26, 8410–8417 [DOI] [PubMed] [Google Scholar]
- 4. Purohit V., Grindley N. D., Joyce C. M. (2003) Biochemistry 42, 10200–10211 [DOI] [PubMed] [Google Scholar]
- 5. Rothwell P. J., Mitaksov V., Waksman G. (2005) Mol. Cell 19, 345–355 [DOI] [PubMed] [Google Scholar]
- 6. Brautigam C. A., Steitz T. A. (1998) Curr. Opin. Struct. Biol. 8, 54–63 [DOI] [PubMed] [Google Scholar]
- 7. Rothwell P. J., Waksman G. (2005) Adv. Protein Chem. 71, 401–440 [DOI] [PubMed] [Google Scholar]
- 8. Bermek O., Grindley N. D., Joyce C. M. (2011) J. Biol. Chem. 286, 3755–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Santoso Y., Joyce C. M., Potapova O., Le Reste L., Hohlbein J., Torella J. P., Grindley N. D., Kapanidis A. N. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Astatke M., Ng K., Grindley N. D., Joyce C. M. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 3402–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Benner S., Chen R. J., Wilson N. A., Abu-Shumays R., Hurt N., Lieberman K. R., Deamer D. W., Dunbar W. B., Akeson M. (2007) Nat. Nanotechnol. 2, 718–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cockroft S. L., Chu J., Amorin M., Ghadiri M. R. (2008) J. Am. Chem. Soc. 130, 818–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gyarfas B., Olasagasti F., Benner S., Garalde D., Lieberman K. R., Akeson M. (2009) ACS Nano. 3, 1457–1466 [DOI] [PubMed] [Google Scholar]
- 14. Hurt N., Wang H., Akeson M., Lieberman K. R. (2009) J. Am. Chem. Soc. 131, 3772–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Olasagasti F., Lieberman K. R., Benner S., Cherf G. M., Dahl J. M., Deamer D. W., Akeson M. (2010) Nat. Nanotechnol. 5, 798–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilson N. A., Abu-Shumays R., Gyarfas B., Wang H., Lieberman K. R., Akeson M., Dunbar W. B. (2009) ACS Nano. 3, 995–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lieberman K. R., Cherf G. M., Doody M. J., Olasagasti F., Kolodji Y., Akeson M. (2010) J. Am. Chem. Soc. 132, 17961–17972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DeLucia A. M., Chaudhuri S., Potapova O., Grindley N. D., Joyce C. M. (2006) J. Biol. Chem. 281, 27286–27291 [DOI] [PubMed] [Google Scholar]
- 19. Akeson M., Branton D., Kasianowicz J. J., Brandin E., Deamer D. W. (1999) Biophys. J. 77, 3227–3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vercoutere W., Winters-Hilt S., Olsen H., Deamer D., Haussler D., Akeson M. (2001) Nat. Biotechnol. 19, 248–252 [DOI] [PubMed] [Google Scholar]
- 21. Savage L. J. (1976) Ann. Stat. 4, 441–500 [Google Scholar]
- 22. Derbyshire V., Freemont P. S., Sanderson M. R., Beese L., Friedman J. M., Joyce C. M., Steitz T. A. (1988) Science 240, 199–201 [DOI] [PubMed] [Google Scholar]
- 23. Dzantiev L., Romano L. J. (1999) J. Biol. Chem. 274, 3279–3284 [DOI] [PubMed] [Google Scholar]
- 24. Dzantiev L., Alekseyev Y. O., Morales J. C., Kool E. T., Romano L. J. (2001) Biochemistry 40, 3215–3221 [DOI] [PubMed] [Google Scholar]
- 25. Stengel G., Gill J. P., Sandin P., Wilhelmsson L. M., Albinsson B., Nordén B., Millar D. (2007) Biochemistry 46, 12289–12297 [DOI] [PubMed] [Google Scholar]
- 26. Doublié S., Tabor S., Long A. M., Richardson C. C., Ellenberger T. (1998) Nature 391, 251–258 [DOI] [PubMed] [Google Scholar]
- 27. Johnson S. J., Taylor J. S., Beese L. S. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 3895–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Y., Korolev S., Waksman G. (1998) EMBO J. 17, 7514–7525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Datta K., Johnson N. P., von Hippel P. H. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 17980–17985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Temiakov D., Patlan V., Anikin M., McAllister W. T., Yokoyama S., Vassylyev D. G. (2004) Cell 116, 381–391 [DOI] [PubMed] [Google Scholar]
- 31. Minnick D. T., Liu L., Grindley N. D., Kunkel T. A., Joyce C. M. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 1194–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]