

Abstract
Objective
To identify plasma membrane ion channels mediating calcium influx at the blood-brain barrier in response to disrupting stimuli.
Methods
We examined the expression and function of candidate transient receptor potential channels using RT-PCR, Fura-2 calcium imaging and permeability assays.
Results
Immortalized mouse brain microvessel endothelial cells expressed multiple transient receptor potential isoforms: transient receptor potential C1, C2, C4, and C7, M2, M3, M4 and M7, and V2 and V4. Similar profiles were observed in freshly isolated cerebral microvessels and primary cultured rat brain endothelial cells. Thrombin-stimulated calcium influx in brain endothelial cells was blocked by transient receptor potential C inhibitors. Transient receptor potential V activating stimuli also increased intracellular calcium. This increase was inhibited by a transient receptor potential V blocker or by removal of extracellular calcium. Barrier integrity was compromised by thrombin, hypo-osmolar stress, and PMA treatment. The increase in barrier permeability induced by transient receptor potential V activators was blocked by transient receptor potential V inhibition, while thrombin effects were inhibited by transient receptor potential C inhibitors.
Conclusions
These results demonstrate that transient receptor potential C and transient receptor potential V channels mediate calcium influx at the blood-brain barrier, and as a consequence, may modulate barrier integrity.
Keywords: brain endothelial cell, thrombin, osmolar stress, calcium flux, phorbol esters
INTRODUCTION
The blood-brain barrier (BBB) is a critical organ in the regulation of central nervous system homeostasis (37). Permeability of the barrier is dependent on tight cell-cell contacts mediated by tight junction complexes, low levels of paracellular diffusion, and low transcellular endocytosis. The uptake of ions, peptides, and drugs into the central nervous system is largely mediated by specific transport processes and carrier proteins differentially expressed on the blood (luminal) and brain (abluminal) sides of brain capillary endothelial cells. Regulation of barrier function relies on numerous processes, but recent evidence has implied an important role for intracellular calcium in modulating BBB paracellular permeability (11, 54).
Although calcium has been implicated in mediating endothelial paracellular permeability both at the BBB and in other endothelial tissues (36, 45), the mechanism by which calcium enters BBB endothelial cells is as yet unknown. BBB endothelial cells express many different types of calcium channels, including calcium transporters like the sodium-calcium exchanger (15), the plasma membrane calcium ATPase (26), P2X receptors (5), as well as calcium influx triggered by peptide receptor activation (45, 52). Recent work has identified a new superfamily of cation-permeable channels called the transient receptor potential (TRP) family. There are currently seven identified families of TRP channels (33), and they mediate calcium influx in response to a diverse range of stimuli, including receptor activation, store-operated calcium release, temperature, shear or mechanical stress, and pH (38). Of particular interest to our studies are the TRPC (canonical), and TRPV (vanilloid) and TRPM (melastatin) subfamilies. TRPC (canonical) channels are the best characterized, and have been proposed to mediate multiple receptor-operated calcium influxes (33, 47); there are seven TRPC isoforms. TRPC channels are also considered potential candidates for mediating store-operated calcium influx (31, 58, 61). The TRPV subfamily is named after the founding member, the capsaicin or vanilloid receptor (TRPV1). There are six TRPV isoforms, which respond to a wide variety of environmental stimuli (9, 39), including stimuli that are important to consider at the BBB, such as shear stress (60). The TRPM channels, named after the founding member, the melastatin receptor (TRPM1), are involved in mediating responses to cold (23) and cytosolic alkalinization (27). The TRPMs also respond to intracellular signals such as increased intracellular calcium (38) and ADP-ribose (25). Members of each of these families have been identified in vascular endothelial cells (28, 38).
While calcium is acknowledged to be important in modulating BBB integrity (11), there is no consensus on the mechanism by which changes in intracellular calcium occur. We hypothesize that TRPC and TRPV channels mediate calcium influx in BBB endothelial cells in response to numerous stimuli that also disrupt BBB function. In this study we examined the expression of three TRP channel families in BBB endothelial cells, and characterized the calcium response of BBB endothelial cells to known TRPC and TRPV channel activators or disrupting stimuli.
MATERIALS AND METHODS
Cell Culture
bEnd3 cells, an immortalized mouse cell line generated from brain capillary endothelial cells (34), were grown according to the supplier’s instructions (American Type Culture Collection, Manassas, VA) in DMEM with 4.5 g/L glucose, 3.7 g/L sodium bicarbonate, 4 mM glutamine, 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were maintained in a humidified cell culture incubator at 37°C and 10% CO2/90% room air as instructed by the manufacturer. The higher CO2 concentration maintains appropriate pH and accounts for the difference in sodium bicarbonate concentrations between the commercial media used and the recommended media. For all experiments, cells were trypsinized and seeded at a density of 0.5–1.0 × 104 cells/cm2 (13, 42) onto glass coverslips or Transwell™ permeable supports. Cells seeded onto Transwells™ were allowed to reach confluence (6–7 days). The media in the lower chamber was then removed, filters were washed with PBS, and media in the lower chamber was replaced with serum-free DMEM. Cells were further cultured for at least 4 days before experiments were performed (13).
RNA isolation and RT-PCR
Confluent bEnd3 monolayers grown on glass coverslips or permeable supports were lysed with TRIzol® Reagent (Invitrogen, Carlsbad, CA). The expression of RNA for TRPC, TRPV and TRPM proteins was examined using SuperScript one-step RT-PCR kit (Invitrogen) and specific primers (Table 1). RT-PCR was performed on a Gene Amp PCR System 2400 (Perkin Elmer, Wellesley, MA). Products were separated by electrophoresis on 1% agarose/TBE gels and stained with ethidium bromide. Gels were photographed using the ChemiGenius2 bioimaging system (Syngene, Frederick, MD).
Table 1.
RT-PCR Primers for TRP channels in BBB endothelial cells.
| Forward primer | Reverse primer | Amplicon size (bp) | |
|---|---|---|---|
| TRPC1 | gta ccc gag cac gga cct | aac agc att tct ccc aag ca | 250 |
| TRPC2 | ttg ctg gga gag tct ctg gt | tca atc atc ttg cca atg ga | 452 |
| TRPC3 | att ctt cga agc ccc ttc at | ctc ctt gca ctc aga cca ca | 234 |
| TRPC4 | gct gga gga gaa gac cat gg | gac ctg tcg atg tgc tga ga | 211 |
| TRPC5 | taa cct gca tgc cca ttg ga | ccc ttg gac gag aac cat ta | 405 |
| TRPC6 | caa tcg cgg tgg ttt taa gt | cgc atc atc ctc aat ttc ct | 416 |
| TRPC7 | cac gac atc aca ccc atc at | gag gac agg gtc ttc act gg | 234 |
| TRPV1 | tga cta ccg gtg gtg ttt ca | tga tcc ctg cat agt gtc ca | 250 |
| TRPV2 | tga tga agg ctg tgc tga ac | cag agc atg cag gac tgt gt | 452 |
| TRPV3 | ctg gcc aag gaa gaa cag ag | gcc tct tcc gtg tac tca gc | 234 |
| TRPV4 | atc aac tcg ccc ttc aga ga | ggt gtt ctc tcg ggt gtt gt | 211 |
| TRPV5 | act gca tgt agc tgc cct ct | aca ggc aaa ggt ttt gtt gg | 405 |
| TRPV6 | gca ctg ttc agc acc ttt ga | gac cat act ctc gcc cac at | 416 |
| TRPM1 | gct tcg aag aca gct gga aa | cgc agg gac tcg ttt atg ac | 246 |
| TRPM2 | cag agc aaa cga agg aaa gg | tca gta acg cct gca gaa tg | 387 |
| TRPM3 | tgg gcc gaa aaa tct atg ag | gat cat tcc cac cga gaa ga | 299 |
| TRPM4 | gag agg atc atg acc cga aa | gtc att cag cag agc atc ca | 255 |
| TRPM5 | atg tcc tgc tgg tgg act tc | agc cat acg ctc agg aag aa | 400 |
| TRPM6 | tga cca gtt gaa tgc aga gc | gag gct ctt gag ggc ttt tt | 302 |
| TRPM7 | gct cca tgg gga gtg ata ga | atc aaa gcc acc aca gga ac | 254 |
| TRPM8 | ttc tgg tca acc tcc tgg tc | taa acc gat gcc tca tct cc | 339 |
All primers are listed in 5′-3′ orientation. The size of the expected amplified fragment is included. For details on the RT-PCR procedure, see Materials and Methods.
Calcium imaging
Changes in intracellular calcium were monitored in cultured cells using the fura-2 fluorescent indicator technique in ratiometric mode (18) as previously described (17). Cells on coverslips were loaded with 2–5 μM fura-2/AM in growth media for 60 min at 37°C. In some experiments, 100 μM probenicid and 0.67% pluronic were added to the media to enhance Fura-2/AM loading. Coverslips were attached to the bottom of a custom designed perfusion chamber, and placed on the microscope stage of an InCa workstation (Intracellular Imaging, Inc.) at 37°C. Intracellular calcium levels were estimated from fura-2 fluorescence images by ratioing the emission images at 510 nm upon excitation at 340 nm and 380 nm. In all studies, cells were initially bathed in aerated modified balanced salt solution (ISO, containing in mM: 140 NaCl, 5.4 KCl, 0.5 MgCl2, 0.4 MgSO4, 3.3 NaHCO3, 2.0 CaCl2, 10 HEPES, 5.5 glucose, pH 7.4, adjusted to 310 mOsm/L with mannitol).
To assess TRP channel activity and calcium influx in response to environmental and pharmacological stimuli, coverslips were prepared as described above. Drugs were added to the chamber in the indicated final concentrations. For hypo-osmotic studies, the MBSS in the chamber was removed and replaced with hypo-osmotic MBSS (ISO minus 40 mM NaCl, HYPO, 220 mOsm/L). The change in fura-2 fluorescence in response to these treatments was followed over time. In experiments where TRPC (SK&F 96365 or LOE 908) or TRPV (ruthenium red, RR) inhibitors were used, these drugs were added shortly before the addition of the stimulating agent. GdCl3, a general TRP channel blocker, was also added prior to the addition of the stimulating agent. Histamine, bradykinin, thrombin, phorbol 12-myristate 13-acetate (PMA), and 4α-Phorbol 12,13-didecanoate (4α-PDD) were added as indicated.
Monolayer permeability
Paracellular solute permeability studies were performed using [14C]sucrose to determine paracellular diffusion at 37°C across confluent bEnd3 monolayers grown on permeable supports. Apical-to-basolateral diffusion was determined by dividing the pmoles of radioactive marker appearing in the receiver chamber by the time in minutes from which the apparent permeability coefficient was calculated:
where volume is the volume of media in the receiving chamber, SA is the surface area of the cell monolayer, CD is the initial concentration of radioactive marker in the donor chamber, and CR is the concentration of the radioactive marker in the receiving chamber at a specific time. The concentration of marker in the receiving chamber at the end of the experiments presented here was 8.6% of the initial donor concentration.
Effects of various TRP channel activators on paracellular permeability were examined over time. For thrombin studies, thrombin was added at the same time as the radiolabeled sucrose. Diffusion of radiolabeled sucrose was followed over time. For studies examining the effects of osmolar stress, monolayers were washed and incubated in ISO, ISO + 1 μM RR, HYPO or HYPO + 1 μM RR for 120 min. Radiolabeled sucrose was added at the beginning of the time course, and permeability was followed over time. For studies examining the effects of phorbol esters, radiolabeled sucrose was added and the cells were returned to the incubator for 1 hr to allow for determination of basal permeability. PMA or 4α-PDD (100 nM, from a 1 mM stock in DMSO) was then added and permeability changes were followed over time. In studies were inhibitors were used, the inhibitors were added at the same time as the radiolabeled sucrose.
Statistics
All data are presented as mean +/− SEM of raw data (340/380 ratios) or percent of time=0 (permeability data). Results were analyzed using Sigma Stat 3.5 (SPSS Inc, Chicago, IL) with a significance level set at p<0.05. Data were analyzed using one-way or two-way analysis of variance (ANOVA) with post-hoc tests as appropriate, or by t-test as indicated.
RESULTS
BBB endothelial cells express RNA for TRPC, TRPV and TRPM isoforms
We investigated the expression of TRPC, TRPV and TRPM isoforms at the BBB using RT-PCR and specific primers. RT-PCR was performed on an immortalized mouse brain endothelial cell line (bEnd3) that has been recently characterized as a BBB model system (13) and freshly isolated mouse brain microvessels (24), Figure 1). The mouse brain microvessel RNA was tested for the expression of GFAP (astrocyte marker) and desmin (vascular smooth muscle marker); neither marker was detected (data not shown). bEnd3 endothelial cells and brain microvessels express TRPC1, C2, C4, and C7. Mouse brain microvessels also express TRPC3, C5 and C6; the expression of these other isoforms may be due to contamination from other cell types. Both source tissues express TRPV2 and TRPV4; mouse brain microvessels also express TRPV6. bEnd3 and mouse brain microvessels cells express TRPM2, M3, M4, and M7 (Figure 1). TRPC, TRPV and TRPM RNA expression was also assessed in primary cultures of rat brain microvessel endothelial cells. Rat primary brain endothelial cells expressed TRPC1, C3, C4, and C7, TRPV2, V3 and V4, and TRPM3, M4 and M7 (data not shown). These results indicate presence or absence of a transcript, and do not reflect relative expression levels of each transcript within the tissue.
FIGURE 1.

Expression of TRP channels in bEnd3 cells and mouse brain microvessels. Total RNA was reverse transcribed with primers specific to the TRPC, TRPV and TRPM isoforms. Both bEnd3 cells and freshly isolated mouse brain microvessels express message for TRPC1, C2, C4 and C7, while mouse brain microvessels also express message for TRPC3, C5 and C6. TRPV2 and TRPV4 are expressed in both tissues, with message for TRPV6 also seen in the mouse brain microvessels. Finally, both source tissues express TRPM2, M3, M4 and M7 only. These results do not reflect relative expression levels of each transcript. Gels shown are representative blots for 3–4 separate experiments.
BBB endothelial cell calcium responses to TRPC and TRPV-activating stimuli
We examined bEnd3 cell intracellular calcium responses to a number of stimuli known to activate TRPC and TRPV receptors and to cause disruption of endothelial barrier systems. Histamine activates TRPC4 through the histamine H1 receptor (40), and disrupts the BBB (1). In our hands, histamine increased intracellular calcium in a very small number of bEnd3 cells (13–20%, Table 2, Figure 2A). There was no significant difference in the amplitude of intracellular calcium response between 100 and 300 μM doses, and no significant difference in the percentage of cells activated. Bradykinin, another TRPC activator (41) and disruptor of BBB function (16), also increased intracellular calcium in a small percentage of bEnd3 cells (Figure 2B, Table 2), with no significant variation in response for doses ranging from 1–10 μM. Thrombin activates TRPC channels (3), and increased intracellular calcium levels in bEnd3 cells (Figure 2C). Thrombin is known to disrupt endothelial barriers (10), but in our hands only about half of the cells showed a response to thrombin application (Table 2), although the response tended to trend upward at higher doses (65% at 10 U/ml). Significantly more cells responded to the 10 U/ml dose then to 1 U/ml [one-way ANOVA, F3,32=3.390, p=0.031].
Table 2.
Response of bEnd3 cells to TRPC and TRPV stimuli.
| Stimulus | Percent of cells responding with increased intracellular calcium SEM, (n) |
|---|---|
| Histamine | |
| 100 mM | 12.9 ± 2.6 (5) |
| 300 mM | 20.4 ± 5.9 (5) |
| Bradykinin | |
| 1 mM | 27.1 ± 4.7 (7) |
| 3 mM | 22.8 ± 7.9 (7) |
| 5 mM | 32.9 ± 7.2 (4) |
| 10 mM | 42.4 ± 17.9 (4) |
| Thrombin | |
| 1 U/ml | 32.3 ± 4.8 (4) |
| 3 U/ml | 53.9 ± 2.3 (19) |
| 5 U/ml | 58.8 ± 16.6 (4) |
| 10 U/ml | 64.9 ± 8.8 (6)* |
| Hypo-osmolarity | 61.4 ± 5.8 (21) |
| PMA | 61.4 ± 8.4 (7) |
| 4α-PDD | 67.9 ± 4.8 (20) |
bEnd3 cells were grown on glass coverslips and loaded with 2–5 μM Fura-2/AM. Increases in intracellular calcium after various treatments was assessed as detailed in the Materials and Methods. The percent of responding cells represents an average from n coverslips.
p<0.05 versus 1 U/ml treatment.
n numbers reflect the number of coverslips investigated in each treatment group.
FIGURE 2.

bEnd3 cell calcium responses to TRPC stimuli. bEnd3 cells were grown on glass coverslips and loaded with 2–5 μM Fura-2/AM as described. Peptides were added in the indicated doses and changes in intracellular calcium were assessed. A) Histamine increases [Ca]i in bEnd3 cells at doses of 100 and 300 μM, n=5 imaging experiments for each dose. B) Bradykinin also increase bEnd3 [Ca]i, but there is no dose-response noted with increasing concentrations of peptide from 1–10 μM, n=4–11. C) Thrombin causes an increase in [Ca]i, and this response is dose-dependent, n=4–10. D) The increase in [Ca]i to 3 U/ml of thrombin is inhibited by LOE 908 (LOE), SK&F 96365 (SKF) and GdCl3 (Gd), all TRPC or non-selective cation channel inhibitors (t test, * p<0.05 vs. control, *** p<0.001 vs. control), n=3–10.
We further examined the effects of thrombin on bEnd3 cell calcium levels using 3 U/ml of thrombin as a stimulus in conjunction with TRPC and TRPV channel inhibitors (Figure 2D). The TRPV inhibitor ruthenium red (RR, (7) had no effect on thrombin-stimulated calcium influx, even at a high concentration. A low dose of the TRPC inhibitor SKF 96365 (29) had no effect, but at a 10 μM, calcium influx was significantly reduced. Another general TRPC inhibitor, LOE 908 (29) significantly decreased calcium influx after thrombin stimulation. The non-specific cation channel blocker GdCl3 also significantly decreased the thrombin-induced calcium influx (Figure 2D).
Since bEnd3 cells also express TRPV channels, we examined bEnd3 calcium responses to stimuli associated with TRPV activation: increasing temperature (9), hypoosmolar stress (17), and phorbol esters (17). For temperature experiments, bEnd3 cells were loaded with Fura-2 and placed in the imaging chamber; the heating system was then turned on, and levels of intracellular calcium were measured over time as the temperature inside the imaging chamber rose. Intracellular calcium levels rose with increasing temperature over time, with a marked increase occurring at around 28°C (Figure 3). This is within the threshold range of activation temperatures reported for the TRPV channels (9), particularly TRPV3 and TRPV4, and likely reflects activation of TRPV channels in bEnd3 cells.
FIGURE 3.
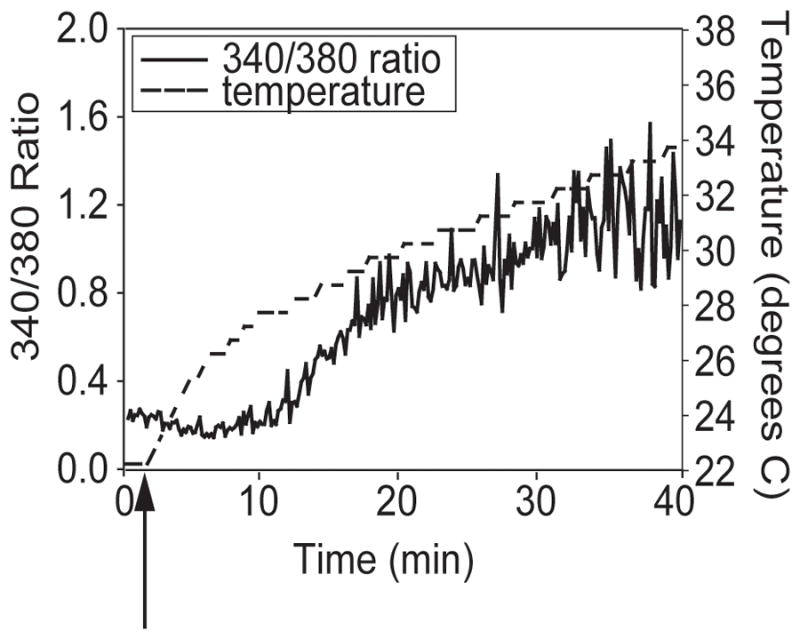
bEnd3 cell calcium responses to increasing temperature. bEnd3 cells were plated on glass coverslips and loaded with Fura-2/AM as described. Coverslips were placed in a room-temperature imaging system and fluorescent imaging was started. After 2–3 min, the heat in the imaging chamber was turned on (arrow), and [Ca]i was assessed as the temperature increased. bEnd3 cell calcium levels remained steady until the imaging chamber reached 26–30°C (a representative experiment is shown, n=5). This is the temperature range that has been found to activate TRPV channels in heterologous expression systems, indicating the presence of functional endogenous TRPV channels.
bEnd3 cells also showed an increase in intracellular calcium in response to other TRPV-selective stimuli. Exposure to hypo-osmolar solution significantly increased intracellular calcium (Figure 4A). This increase was significantly inhibited by application of RR. bEnd3 cell intracellular calcium levels were increased by treatment with phorbol 12-myristate 13-acetate (PMA, Figure 4B), a PKC-activating phorbol ester which has been shown to activate TRPV4 (17) and TRPV1 (14). 4α-Phorbol 12,13-didecanoate (4α-PDD), a non-PKC activating phorbol ester that is a specific activator of TRPV4 (59), also increased intracellular calcium in bEnd3 cells (Figure 4C), demonstrating functional TRPV4 channels. The effects of both PMA and 4α-PDD were significantly inhibited by RR. Furthermore, the increase in intracellular calcium after phorbol ester treatment was dependent on the presence of extracellular calcium; when cells were exposed to either PMA or 4α-PDD in Ca++-free ISO solution, there was no increase in intracellular calcium as detected by changes in Fura-2 fluorescence (data not shown).
FIGURE 4.

TRPV stimulation in bEnd3 cells. We assessed the bEnd3 cell [Ca]i responses to TRPV selective stimuli in the presence or absence of 1 μM ruthenium red (RR), a TRPV channel blocker. Cells were exposed to A) HYPO (220 mOsm/L), B) PMA (100 nM) or C) 4α-PDD (4α-P, 100 nM) and changed in [Ca]i were measured as a change in the final 340/380 nm ratio. All three stimuli triggered an increase in [Ca]I that could be significantly inhibited by co-treatment with 1 μM RR (t test, *** p<0.001), n=10–16 separate imaging experiments per group.
BBB endothelial cell monolayer disruption after TRPC and TRPV-activating stimuli
Levels of intracellular calcium have been implicated in mediating BBB integrity (11). Since a number of stimuli increased intracellular calcium levels in bEnd3 cells, we investigated whether these same stimuli can alter monolayer permeability by loosening tight junctions between adjacent endothelial cells and increasing paracellular permeability of sucrose. The mean basal monolayer permeability for all permeability experiments was 5.12 ± 0.7 × 10−4 cm/min.
We exposed confluent bEnd3 cell monolayers to increasing doses of thrombin and followed monolayer permeability over time. Thrombin treatment significantly increased monolayer permeability as compared to control [two-way ANOVA, F3,98=5.949, p=0.001, Figure 5A]; this effect was due to the increase in permeability seen with 10 U/ml thrombin, which was significantly different from control (p=0.001), 1 U/ml (p=0.011) and 3 U/ml (p=0.027). This increase in permeability appeared 60 min after the beginning of thrombin exposure. There was also a significant effect of time [two-way ANOVA, F4,98=9.627, p<0.001], due to the increase in permeability over time with the 10 U/ml treatment. Control monolayers had no significant change in permeability over the time course of the experiment [one-way ANOVA, F4,17=1.105, p=0.395].. We also investigated the ability of the TRPC inhibitors SK&F 96365 (10 μM) and LOE 908 (1 μM) to block thrombin-induced barrier disruption. Both inhibitors were effective (Figure 5B) at doses similar to those needed to block intracellular calcium influx in response to thrombin (Figure 2D). There was a significant effect of treatment [two-way ANOVA, F4,152=6.732, p<0.001]; treatment with 10 U/ml thrombin significantly increased monolayer permeability. This increase was significantly inhibited by co-treatment with either SK&F 96365 and LOE 908. Interestingly, LOE 908 treatment appears to increase permeability at later time points, but this increase was not statistically significant at 120 min [one-way ANOVA, F3,30=2.519, p=0.079].
FIGURE 5.

TRPC activation alters bEnd3 monolayer permeability. bEnd3 cells were grown on permeable Transwell™ filters and barrier function was assessed by following the diffusion of [14C]sucrose across the monolayers. A) 10 U/ml thrombin caused a significant increase in barrier permeability 1 hr after the addition of the peptide, indicating that TRPC activation can lead to barrier disruption. n for each time point = 3–6. B) The increase in barrier permeability induced by 10 U/ml thrombin could be inhibited by simultaneous treatment with either 10 μM SK&F 96365 or 1 μM LOE 908, both inhibitors of the TRPC family. n for each time point = 4–9. The mean basal monolayer permeability for all experiments was 5.12 ± 0.7 × 10−4 cm/min.
We also investigated the effects of TRPV activators on bEnd3 cell monolayer permeability. To examine the effects of hypo-osmolar stress, we exposed confluent bEnd3 monolayers to HYPO solution (225 mOsm/L) and followed diffusion of radiolabeled [14C]sucrose for 2 hrs. There was a significant effect of HYPO treatment [two-way ANOVA, F3,327=5.701, p<0.001]. HYPO treatment rapidly increased paracellular permeability after only 5 min of exposure (Figure 6). This increase was partially blocked by co-treatment with 1 μM RR. This increase in permeability resolved quickly, and the permeability of HYPO treated monolayers was not significantly different from control monolayers after 30 min. There was some increase in permeability in all monolayers, including the control. This likely reflects disruption of the barrier due to mechanical stress involved in changing the solution in the Transwells prior to the start of the experiment.
FIGURE 6.
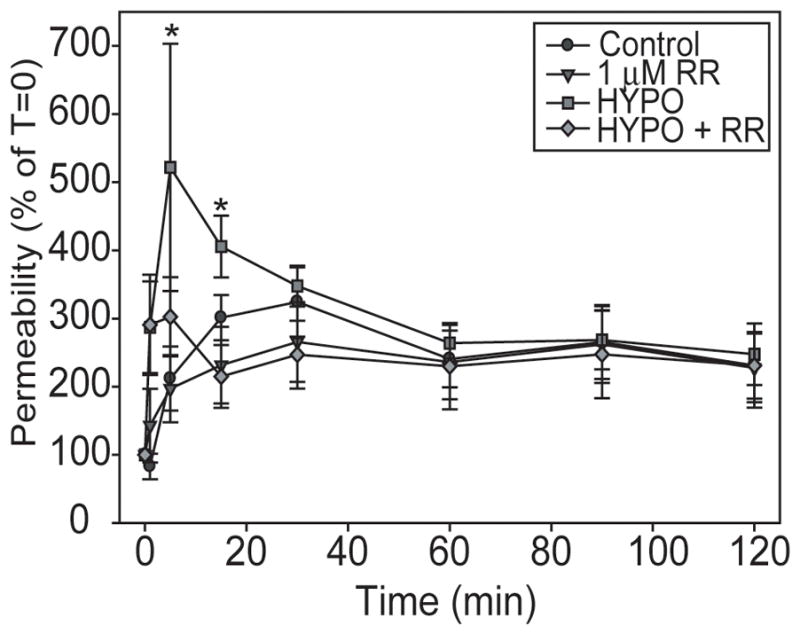
Hypo-osmolar stress rapidly and transiently disrupts BBB monolayers. Confluent bEnd3 monolayers were exposed to continuous ISO or continuous HYPO, with or without 1 μM RR for 2 hours and paracellular permeability was measured. HYPO caused a rapid increase in [14C]sucrose permeability that was partially inhibited by co-treatment with RR. This increase from 5–15 min was gone after 30 min. Graphs represent data from 4 separate permeability experiments, n = 6–16, * p<0.05 versus time 0.
We examined the effects of TRPV-activating phorbol esters on bEnd3 monolayer permeability. In monolayers exposed to 100 nM PMA with and without 1 μM RR, there was a significant effect of treatment [two-way ANOVA, F3,463=46.346, p<0.001] and of time [F10,463=4.439, p<0.001], with no interaction between the two factors. 100 nM PMA caused an increase in monolayer permeability (p<0.001, Figure 7) that was partially inhibited by treatment with 1 μM RR. This increase was initially significant after 45 min of exposure to PMA, and continued to increase throughout the 120 min monitoring period. Monolayers exposed to PMA and RR still exhibited a significant increase in paracellular permeability, although this increase was not as dramatic as in the monolayers treated with PMA alone (p<0.001). RR alone had no effect on permeability over time. In contrast, monolayers exposed to 100 nM 4α-PDD did not exhibit increased permeability in response to the phorbol ester (Figure 7).
FIGURE 7.

TRPV activation alters bEnd3 monolayer permeability. We also assessed barrier function after exposure of confluent monolayers to 100 nM PMA or 4α-PDD. Monolayers were first incubated in media with [14C]sucrose for one hour to determine basal permeability before the addition of phorbol esters. 100 nM PMA also caused an increase in barrier permeability at 120 min (*** p<0.001 vs control, * p<0.05 vs control, ^^ p<0.01 vs HYPO + RR). This increase was prolonged, and partially inhibited by 1 μM RR. Interestingly, under the conditions used in these experiments, 100 nM 4α-PDD did not alter barrier permeability. n = 5–9.
DISCUSSION
Calcium is a critical and ubiquitous signaling molecule in cells. In addition to directly regulating the function of many proteins and enzymes, calcium serves as a trigger for a multitude of signaling cascades that modulate diverse processes ranging from cell growth and development to apoptosis and cell death. Calcium ions involved in signaling can be derived from two main sources in response to a stimulus: release from intracellular stores such as the endoplasmic (sarcoplasmic) reticulum or mitochondria, or influx from the extracellular milieu through plasma membrane cation channels (48).
In many tissues, calcium-permeable channels have been extensively studied and characterized. However, in the endothelial cells of the blood-brain barrier (BBB), the molecular identity of calcium influx channels has not been specifically described. BBB endothelial cells are typically characterized as “epithelial-like”, and exhibit a polarity of plasma membrane protein expression. Potential candidates for calcium transport across the plasma membrane include P2X receptors, cation exchange proteins and the transient receptor potential (TRP) superfamily of cation channels. While P2X receptors are found in other types of endothelium (49, 56), there are few reports of their presence in the BBB (4). There are a number of calcium transporter proteins expressed by BBB endothelial cells, including the sodium-calcium exchanger (NCX) and the plasma membrane Ca++-ATPase (PMCA). The NCX normally transports calcium out of the cell, but under certain conditions, such as hypoxic stress, the NCX reverses (30, 46), causing an increase in intracellular calcium. PMCA is expressed in a calcium-sensitive manner in brain barrier systems (26), and is important for clearance of high intracellular calcium levels after signaling events (53). Under normal conditions however, these transporters are not good candidates for mediating Ca++ influx at the BBB.
In this study we investigated the potential for the transient receptor potential (TRP) superfamily of cation permeable channels to mediate calcium influx in BBB endothelial cells. BBB endothelial cells express RNA for a number of channels in the TRPC, TRPV and TRPM families. We tested BBB endothelial calcium responses to a number of stimuli known to disrupt the BBB and activate TRPC channels, including histamine (52), bradykinin (8, 52) and thrombin (8). While we did see increases in intracellular calcium in response to these three compounds, the percentage of responding cells was not high, ranging from 13–65% (see Table 1). bEnd3 cell responses to histamine and bradykinin were lower then was expected, but not out of the range previously described for brain endothelial cells in culture (45). bEnd3 calcium responses to thrombin showed dose-dependence, increasing from 32% of cells responding at a dose of 1 U/ml up to 65% of cells responding at a dose of 10 U/ml. While there was no significant dose response (in terms of the magnitude of the increase in intracellular calcium) for histamine, bradykinin or thrombin, there was a trend towards a dose response with thrombin at the highest doses. This trend, coupled with a significant increase in the percent of responding cells with increasing thrombin levels, may explain why the only change in monolayer permeability seen was with the highest dose of thrombin, even though there were increases in intracellular calcium at the lower doses for a subpopulation of cells.
A role for TRP channels in mediating receptor-mediated stimulation of calcium influx is likely. A number of inflammatory-related peptides, including histamine, bradykinin and thrombin, have been shown to activate TRPC and TRPV cation channels in a range of tissues (2, 40, 41), leading to calcium influx. bEnd3 cell responses to an intermediate concentration of thrombin (3 U/ml) were inhibited by the general TRPC channel inhibitors, SK&F 96365 and LOE 908, and by the non-selective cation channel blocker, GdCl3, but not by RR, a TRPV family inhibitor. These results suggest that bEnd3 cell responses to thrombin are mediated through TRPC channels. This is in agreement with studies implicating TRPC1 (3) and TRPC4 (55) in calcium influx into lung endothelial cells after exposure to thrombin. Since RR had no effect on the calcium influx after thrombin treatment, TRPV channels do not mediate this response. As expected, thrombin transiently increased BBB endothelial monolayer permeability. This is similar to other studies where thrombin increased endothelial permeability both in vitro (10) and in vivo (19). This increase was significantly inhibited by SK&F 96365 and LOE 908 at early time points, further supporting a role for TRPC channels in mediating BBB permeability in response to thrombin.
TRPV channels are also strategically placed to mediate BBB endothelial cell response to a number of physiological and pathological stimuli. TRPV2 and V4 have both been shown to have mechanosensitive properties (17, 35), which may be activated by changes in shear stress or cell volume that occur within the brain capillaries after changes in blood flow. TRPV4, in addition to being mechanosensitive, is activated by heat, phorbol esters and osmolarity (9, 17), and has been shown to mediate barrier permeability in other epithelial (51) and endothelial (6, 22) cell systems. In the BBB, TRPV2 and TRPV4 may be very important in mediating changes in cell calcium after changes in flow, as in a stroke; studies are currently underway to address this question. In our hands, BBB endothelial cells are sensitive to temperature, with spontaneous activation of calcium influx above 27°C, the gating temperature for TRPV4. Calcium influx through TRPV channels is also triggered by exposure to a hypo-osmolar solution, a mechanical stimulus for both TRPV2 and V4, by a PKC-activating phorbol ester, PMA, and by a non-PKC activating phorbol ester, 4α-PDD, a specific activator for TRPV4. These responses are dependent on the presence of extracellular calcium, indicating that the effects of these stimuli are not due to release of calcium from intracellular stores.
In order to determine the role of TRPV channels in mediating BBB endothelial cell permeability, we investigated changes in BBB monolayer permeability over time after exposure to TRPV2 and TRPV4 activating stimuli. Confluent bEnd3 monolayers were exposed to hypo-osmolar solution and permeability was assessed at multiple time points over a two hour period. Exposure to hypo-osmolar solution, and presumably activation of TRPV2 and TRPV4 channels, rapidly and transiently increased monolayer permeability. The involvement of TRPV channels is further supported by the finding that this transient increase in permeability was blocked by co-treatment with ruthenium red (RR), a TRPV family inhibitor. While many previous studies have examined the effect of hyper-osmolar stress on BBB permeability both in vitro (45) and in vivo (12), and hyper-osmolar disruption of the BBB is used clinically to treat brain tumors (20), there are fewer studies looking at the effect of hypo-osmolar stress on BBB permeability (44). This study is the first demonstration of hypo-osmolar disruption of the BBB in vitro, and may represent a good model for more fully investigating the effects of hypo-osmolar solutions on barrier structure and function.
Ruthenium red, while used in these studies as an inhibitor of TRPV channels, has many other targets. Originally used as a contrast agent for electron microscopy (50), RR will block mitochondrial calcium uptake (21), stimulate calcium loading by the sarcoplasmic reticulum (57), and inhibit activity of the ryanodine receptor (43). The doses used in the above models range from 2.5 – 10 μM RR, and the IC50 for inihibition of transmembrane calcium fluxes is 7 μM. Since we are using a lower dose of ruthenium red in our studies (1 μM), the results we see are not likely due to these other off-target actions of RR. Furthermore, the inhibition of calcium influxes in response to TRPV activating stimuli by RR seen in BBB endothelial cells is identical to the inhibition of calcium responses mediated by TRPV4 channels in a heterologous expression system using either RR (17)or siRNA directed against TRPV4 (60).
Barrier permeability was increased by treatment with PMA, a PKC activator that has been shown to regulate TRPV4 (17). The increase in monolayer permeability was inhibited by treatment with RR, although this treatment did not completely protect the monolayer. This suggests that activation of PKC modulates BBB permeability through multiple mechanisms, including the activation of TRPV channels. Surprisingly, treatment with 4α-PDD, a selective activator of TRPV4 (59), did not alter monolayer permeability at any of the time points investigated. This suggests several possible explanations: firstly, the possibility that TRPV4 channels are not important in mediating BBB monolayer permeability under the conditions of our experiments, potentially implicating TRPV2 in this process, since this is the only other TRPV channel expressed in this system. Alternatively, the 4α-PDD response of TRPV4 after the relatively low dose of 4α-PDD employed may be too transient for a change in endothelial permeability to be observed, similar to the results seen with lower doses of thrombin. Furthermore, in contrast to TRPV4 heterologous expression systems, endogenously expressed functional TRPV4 channels may be made up of heterotetrameric complexes that, while containing TRPV4 proteins, do not respond to 4α-PDD in the same fashion as homomeric TRPV4 channels investigated in expression systems. We have preliminary evidence that TRPV4 and TRPV2 are associated in kidney cortical collecting duct cells (data not shown), suggesting that the endogenous channels in BBB endothelial cells could consist of both isoforms. This may account for the differences in our data versus those studies done in expression systems, and explain why TRPV4 activation alone is not sufficient to disrupt the BBB.
Endothelial cells of the BBB also express a number of the TRPM isoforms. Although we did not specifically address TRPM activation in our studies, these channels may also be critical in regulating BBB permeability under both normal and pathophysiological conditions. TRPM channels are activated by a number of stimuli. Some isoforms of TRPM3 are activated by hypo-tonicity (23), but we do not know if these isoforms are expressed in bEnd3 cells. However, hypo-osmolar activation of TRPM3 can be inhibited by lanthanides or gadolinium, but is not inhibited by SK&F 96365 (23). Further studies will determine if SK&F 96365 can inhibit hypo-osmolar disruption of BBB monolayers in our system. TRPM4 is a calcium-activated non-selective cation channel (23), which could certainly be activated at the BBB under conditions that increase intracellular calcium, such as those demonstrated in this study. TRPM 2 and TRPM7 are very interesting in that they are channels with kinase activity (32). TRPM2 has been suggested as a sensor for oxidative stress, and is activated by ADP-ribose and hydrogen peroxide (23). TRPM7 is thought to be involved in cellular Mg++ homeostasis, and is activated by Mg-ATP, breakdown of PIP2 or increases in cellular cAMP levels (23). Clearly these channels are also important to consider in the regulation of BBB permeability and function.
In summary, we examined the potential role for TRP channels in mediating calcium influx in BBB endothelial cells after treatment with multiple stimuli shown to disrupt the BBB in cell culture and in vivo models. BBB endothelial cells express numerous isoforms of the TRPC, TRPV and TRPM channels, which may play an important role in mediating calcium flux and barrier function at the BBB in many pathophysiological conditions, such as stroke. We investigated the importance of the TRPV channels in BBB endothelial cells and found RR-sensitive calcium influxes triggered by TRPV activating stimuli. These calcium influxes in response to hypo-osmotic stress or treatment with PMA resulted in increases in barrier permeability that could be inhibited by treatment with RR, implicating TRPV channels in modulation of barrier function. These data implicate TRPV channels at the BBB as potentially important players in the regulation of barrier function in response to changes in osmolarity, shear stress and pH that may occur following stroke. The exact stoichiometry of the endogenous channels remains to be determined, as does the specific triggers of channel function in vivo. However, TRP channels of multiple families potentially play an important role in mediating BBB endothelial cell calcium influx, and in modulating barrier function. It will also be necessary to further examine the role of TRPV channels in mediating BBB function through in vivo studies to confirm our results. Although a number of studies have investigated the in vivo effects of hyper-osmolar stimulation of the BBB (12), there is little known about the in vivo effects of hypo-osmolar stimulation. Furthermore, in vivo phorbol ester treatment is problematic due to the many sites of action for these compounds in vivo. With a better understanding of the distribution of TRPV channels at the BBB, and the development of selective agonists and antagonists, we will be able to further elucidate the role of these channels in mediating BBB disruption in physiological and pathophysiological conditions.
Acknowledgments
This work was supported by RO1 DK70950 to RGO, and AHA 0635066N to RCB.
References
- 1.Abbott JN. Inflammatory mediators and modulation of the blood-brain barrier permeability. Cell Mol Neurobiol. 2000;20:131–147. doi: 10.1023/a:1007074420772. [DOI] [PubMed] [Google Scholar]
- 2.Ahmmed G, Mehta D, Vogel S, Holinstat M, Paria B, Tiruppathi C, Malik AB. Protein kinase Calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem. 2004;279:20941–20949. doi: 10.1074/jbc.M313975200. [DOI] [PubMed] [Google Scholar]
- 3.Ahmmed GU, Mehta D, Vogel S, Holinstat M, Paria BC, Tiruppathi C, Malik AB. Protein kinase Calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem. 2004;279:20941–20949. doi: 10.1074/jbc.M313975200. [DOI] [PubMed] [Google Scholar]
- 4.Albert J, Boyle J, Roberts J, Challis R, Gubby S, Boarder M. Regulation of brain capillary endothelial cells by P2Y receptors coupled to Ca2+, phospholipase C and mitogen-activated protein kinase. Br J Pharmacol. 1997;122:935–941. doi: 10.1038/sj.bjp.0701453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Albert JL, Boyle JP, Roberts JA, Challiss RA, Gubby SE, Boarder MR. Regulation of brain capillary endothelial cells by P2Y receptors coupled to Ca2+, phospholipase C and mitogen-activated protein kinase. Br J Pharmacol. 1997;122:935–941. doi: 10.1038/sj.bjp.0701453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvarez DF, King JA, Weber D, Addison E, Liedtke W, Townsley MI. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res. 2006;99:988–995. doi: 10.1161/01.RES.0000247065.11756.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amann R, Maggi C. Ruthenium red as a capsaicin antagonist. Life Sci. 1991;49:849–856. doi: 10.1016/0024-3205(91)90169-c. [DOI] [PubMed] [Google Scholar]
- 8.Aschner JL, Lum H, Fletcher PW, Malik AB. Bradykinin- and thrombin-induced increases in endothelial permeability occur independently of phospholipase C but require protein kinase C activation. J Cell Physiol. 1997;173:387–396. doi: 10.1002/(SICI)1097-4652(199712)173:3<387::AID-JCP11>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 9.Benham CD, Gunthorpe MJ, Davis JB. TRPV channels as temperature sensors. Cell Calcium. 2003;33:479–487. doi: 10.1016/s0143-4160(03)00063-0. [DOI] [PubMed] [Google Scholar]
- 10.Bogatcheva N, Garcia JG, Verin AD. Molecular mechanisms of thrombin-induced endothelial cell permeability. Biochemistry (Mosc) 2002;67:75–84. doi: 10.1023/a:1013904231324. [DOI] [PubMed] [Google Scholar]
- 11.Brown RC, Davis TP. Calcium modulation of adherens and tight junction function: A potential mechanism for blood-brain barrier disruption after stroke. Stroke. 2002;33:1706–1711. doi: 10.1161/01.str.0000016405.06729.83. [DOI] [PubMed] [Google Scholar]
- 12.Brown RC, Egleton RD, Davis TP. Mannitol opening of the blood-brain barrier: regional variation in the permeability of sucrose, but not 86Rb+ or albumin. Brain Res. 2004;1014:221–227. doi: 10.1016/j.brainres.2004.04.034. [DOI] [PubMed] [Google Scholar]
- 13.Brown RC, Morris AP, O’Neil RG. Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res. 2007;1130:17–30. doi: 10.1016/j.brainres.2006.10.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Correll CC, Phelps PT, Anthes JC, Umland S, Greenfeder S. Cloning and pharmacological characterization of mouse TRPV1. Neurosci Lett. 2004;370:55–60. doi: 10.1016/j.neulet.2004.07.058. [DOI] [PubMed] [Google Scholar]
- 15.Domotor E, Abbott NJ, Adam-Vizi V. Na+-Ca2+ exchange and its implications for calcium homeostasis in primary cultured rat brain microvascular endothelial cells. J Physiol. 1999;515:147–155. doi: 10.1111/j.1469-7793.1999.147ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Easton A, Abbott N. Bradykinin increases permeability by calcium and 5-lipoxygenase in the ECV304/C6 cell culture model of the blood-brain barrier. Brain Res. 2002;953:157–169. doi: 10.1016/s0006-8993(02)03281-x. [DOI] [PubMed] [Google Scholar]
- 17.Gao X, Wu L, O’Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem. 2003;278:27129–27137. doi: 10.1074/jbc.M302517200. [DOI] [PubMed] [Google Scholar]
- 18.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 19.Guan J, Sun S, Cao X, Chen Z, Tong E. Effect of thrombin on blood brain barrier permeability and its mechanism. Chin Med J (Engl) 2004;117:1677–1681. [PubMed] [Google Scholar]
- 20.Gumerlock MK, Belshe BD, Madsen R, Watts C. Osmotic blood-brain barrier disruption and chemotherapy in the treatment of high grade malignant glioma: patient series and literature review. J Neurooncol. 1992;12:33–46. doi: 10.1007/BF00172455. [DOI] [PubMed] [Google Scholar]
- 21.Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium. 2006;40:553–560. doi: 10.1016/j.ceca.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamanaka K, Jian M, Weber D, Alvarez DF, Townsley MI, Al-Mehdi A, King JA, Liedtke W, Parker J. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol. 2007 doi: 10.1152/ajplung.00221.2007. [DOI] [PubMed] [Google Scholar]
- 23.Harteneck C. Function and pharmacology of TRPM cation channels. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:307–314. doi: 10.1007/s00210-005-1034-x. [DOI] [PubMed] [Google Scholar]
- 24.Hawkins BT, Abbruscato TJ, Egleton RD, Brown RC, Huber JD, Campos CR, Davis TP. Nicotine increases in vivo blood-brain barrier permeability and alters cerebral microvascular tight junction protein distribution. Brain Res. 2004;1027:48–58. doi: 10.1016/j.brainres.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 25.Heiner I, Radukina N, Eisfeld J, Kuhn F, Luckhoff A. Regulation of TRPM2 channels in neutrophil granulocytes by ADP-ribose: a promising pharmacological target. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:325–333. doi: 10.1007/s00210-005-1033-y. [DOI] [PubMed] [Google Scholar]
- 26.Keep RF, Ulanski LJ, 2nd, Xiang J, Ennis SR, Lorris Betz A. Blood-brain barrier mechanisms involved in brain calcium and potassium homeostasis. Brain Res. 1999;815:200–205. doi: 10.1016/s0006-8993(98)01155-x. [DOI] [PubMed] [Google Scholar]
- 27.Kozak JA, Matsushita M, Nairn AC, Cahalan MD. Charge screening by internal pH and polyvalent cations as a mechanism for activation, inhibition, and rundown of TRPM7/MIC channels. J Gen Physiol. 2005;126:499–514. doi: 10.1085/jgp.200509324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwan HY, Huang Y, Yao X. TRP channels in endothelial function and dysfunction. Biochim Biophys Acta. 2007 doi: 10.1016/j.bbadis.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 29.Leung Y, Kwan C. Current perspectives in the pharmacological studies of store-operated Ca2+ entry blockers. Jpn J Pharmacol. 1999;81:253–258. doi: 10.1254/jjp.81.253. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Jiang Q, Stys PK. Important role of reverse Na(+)-Ca(2+) exchange in spinal cord white matter injury at physiological temperature. J Neurophysiol. 2000;84:1116–1119. doi: 10.1152/jn.2000.84.2.1116. [DOI] [PubMed] [Google Scholar]
- 31.Lievremont J, Bird G, Putney J. Canonical transient receptor potential TRPC7 can function as both a receptor- and store-operated channel in HEK-293 cells. Am J Physiol Cell Physiol. 2004;287:C1709–C1716. doi: 10.1152/ajpcell.00350.2004. [DOI] [PubMed] [Google Scholar]
- 32.Minke B, Cook B. TRP channel proteins and signal transduction. Physiol Rev. 2002;82:429–472. doi: 10.1152/physrev.00001.2002. [DOI] [PubMed] [Google Scholar]
- 33.Montell C. The TRP superfamily of cation channels. Sci STKE. 2005:re3. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- 34.Montesano R, Pepper M, Mohle-Steinlein U, Risau W, Wagner E, Orci L. Increased proteolytic activity is responsible for the aberrant morphogenetic behavior of endothelial cells expressing the middle T oncogene. Cell. 1990;62:435–445. doi: 10.1016/0092-8674(90)90009-4. [DOI] [PubMed] [Google Scholar]
- 35.Muraki K, Iwata Y, Katanosaka Y, Ito T, Ohya S, Shigekawa M, Imaizumi Y. TRPV2 is a component of osmotically sensitive cation channels in murine aortic myocytes. Circ Res. 2003;93:829–838. doi: 10.1161/01.RES.0000097263.10220.0C. [DOI] [PubMed] [Google Scholar]
- 36.Nagashima T, Ikeda K, Wu S, Kondo T, Yamaguchi M, Tamaki N. The mechanism of reversible osmotic opening of the blood-brain barrier: role of intracellular calcium ion in capillary endothelial cells. Acta Neurochir Suppl. 1997;70:231–233. doi: 10.1007/978-3-7091-6837-0_71. [DOI] [PubMed] [Google Scholar]
- 37.Neuwelt EA. Mechanisms of disease: the blood-brain barrier. Neurosurg. 2004;54:131–142. doi: 10.1227/01.neu.0000097715.11966.8e. [DOI] [PubMed] [Google Scholar]
- 38.Nilius B, Droogmans G, Wondergem R. Transient receptor potential channels in endothelium: solving the calcium entry puzzle? Endothelium. 2003;10:5–15. doi: 10.1080/10623320303356. [DOI] [PubMed] [Google Scholar]
- 39.O’Neil RG, Brown RC. The Vanilloid Receptor Family of Calcium-Permeable Channels (TRPV): Molecular Integrators of Microenvironmental Stimuli. News Physiol Sci. 2003;18:226–231. doi: 10.1152/nips.01468.2003. [DOI] [PubMed] [Google Scholar]
- 40.Obukhov AG, Nowycky MC. TRPC4 can be activated by G-protein-coupled receptors and provides sufficient Ca(2+) to trigger exocytosis in neuroendocrine cells. J Biol Chem. 2002;277:16172–16178. doi: 10.1074/jbc.M111664200. [DOI] [PubMed] [Google Scholar]
- 41.Ohta T, Morishita M, Mori Y, Ito S. Ca2+ store-independent augmentation of [Ca2+]i responses to G-protein coupled receptor activation in recombinantly TRPC5-expressed rat pheochromocytoma (PC12) cells. Neurosci Lett. 2004;358:161–164. doi: 10.1016/j.neulet.2004.01.028. [DOI] [PubMed] [Google Scholar]
- 42.Omidi Y, Campbell L, Barar J, Connell D, Akhtar S, Gumbleton M. Evaluation of the immortalised mouse brain capillary endothelial cell line, b. End3, as an in vitro blood-brain barrier model for drug uptake transport studies. Brain Res. 2003;990:95–112. doi: 10.1016/s0006-8993(03)03443-7. [DOI] [PubMed] [Google Scholar]
- 43.Ozawa T. Ryanodine-sensitive Ca2+ release mechanism in non-excitable cells. Int J Mol Med. 2001;7:21–25. [PubMed] [Google Scholar]
- 44.Oztas B, Kaya M, Kucuk M, Tugran N. Influence of hypoosmolality on the blood-brain barrier permeability during epileptic seizures. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:701–704. doi: 10.1016/S0278-5846(03)00084-8. [DOI] [PubMed] [Google Scholar]
- 45.Paemeleire K, de Hemptinne A, Leybaert L. Chemically, mechanically, and hyperosmolarity-induced calcium responses of rat cortical capillary endothelial cells in culture. Exp Brain Res. 1999;126:473–481. doi: 10.1007/s002210050755. [DOI] [PubMed] [Google Scholar]
- 46.Park SI, Park EJ, Kim NH, Baek WK, Lee YT, Lee CJ, Suh CK. Hypoxia delays the intracellular Ca2+ clearance by Na+-Ca2+ exchanger in human adult cardiac myocytes. Yonsei Med J. 2001;42:333–337. doi: 10.3349/ymj.2001.42.3.333. [DOI] [PubMed] [Google Scholar]
- 47.Plant T, Schaefer M. TRPC4 and TRPC5: receptor-operated Ca2+-permeable nonselective cation channels. Cell Calcium. 2003;33:441–450. doi: 10.1016/s0143-4160(03)00055-1. [DOI] [PubMed] [Google Scholar]
- 48.Putney J, McKay R. Capacitative calcium entry channels. BioEssays. 1999;21:38–46. doi: 10.1002/(SICI)1521-1878(199901)21:1<38::AID-BIES5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 49.Ramirez AN, Kunze DL. P2X purinergic receptor channel expression and function in bovine aortic endothelium. Am J Physiol Heart Circ Physiol. 2002;282:H2106–2116. doi: 10.1152/ajpheart.00892.2001. [DOI] [PubMed] [Google Scholar]
- 50.Reimann B. On the value of ruthenium red as a contrast medium for electron microscopy. Mikroskopie. 1961;16:224–226. [PubMed] [Google Scholar]
- 51.Reiter B, Kraft R, Gunzel D, Zeisseg S, Schulzke JD, Fromm M, Harteneck C. TRPV4-mediated regulation of epithelial permeability. Faseb J. 2006;20:1802–1812. doi: 10.1096/fj.06-5772com. [DOI] [PubMed] [Google Scholar]
- 52.Revest PA, Abbott NJ, Gillespie JI. Receptor-mediated changes in intracellular [Ca2+] in cultured rat brain capillary endothelial cells. Brain Res. 1991;549:159–161. doi: 10.1016/0006-8993(91)90614-2. [DOI] [PubMed] [Google Scholar]
- 53.Sedova M, Blatter LA. Dynamic regulation of [Ca2+]i by plasma membrane Ca(2+)-ATPase and Na+/Ca2+ exchange during capacitative Ca2+ entry in bovine vascular endothelial cells. Cell Calcium. 1999;25:333–343. doi: 10.1054/ceca.1999.0036. [DOI] [PubMed] [Google Scholar]
- 54.Tiruppathi C, Freichel M, Vogel SM, Paria B, Mehta D, Flockerzi V, Malik AB. Impairment of store-operated Ca2+ entry in TRPC4−/− mice interferes with increase in lung microvascular permeability. Circ Res. 2002;91:70–76. doi: 10.1161/01.res.0000023391.40106.a8. [DOI] [PubMed] [Google Scholar]
- 55.Tiruppathi C, RDM, Paria B, Vogel S, Malik AB. Role of Ca2+ signaling in the regulation of endothelial permeability. Vasc Pharm. 2003;39:173–185. doi: 10.1016/s1537-1891(03)00007-7. [DOI] [PubMed] [Google Scholar]
- 56.Townsley MI, King JA, Alvarez DF. Ca2+ channels and pulmonary endothelial permeability: insights from study of intact lung and chronic pulmonary hypertension. Microcirculation. 2006;13:725–739. doi: 10.1080/10739680600930362. [DOI] [PubMed] [Google Scholar]
- 57.Volpe P, Salciati G, Chu A. Calcium-gated calcium channels in sarcoplasmic reticulum of rabbit skinned skeletal muscle fibers. J Gen Physiol. 1986;87:289–303. doi: 10.1085/jgp.87.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X, Pluznick J, Wei P, Padanilam B, Sansom S. TRPC4 forms store-operated Ca2+ channels in mouse mesangial cells. Am J Physiol Cell Physiol. 2004;287:C357–C364. doi: 10.1152/ajpcell.00068.2004. [DOI] [PubMed] [Google Scholar]
- 59.Watanabe H, Davis J, Smart D, Jerman J, Smith G, Hayes P, Vriens J, Cairns W, Wissenbach U, Prenen J, Flockerzi V, Droogmans G, Benham CD, Nilius B. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002;277:13569–13577. doi: 10.1074/jbc.M200062200. [DOI] [PubMed] [Google Scholar]
- 60.Wu L, Gao X, Brown RC, Heller S, O’Neil RG. Dual role of TRPV4 channel as a sensor of flow and osmlolality in renal epithelial cells. Am J Physiol Renal Physiol. 2007 doi: 10.1152/ajprenal.00462.2006. in press. [DOI] [PubMed] [Google Scholar]
- 61.Wu X, Zagranichnaya T, Gurda G, Eves E, Villereal M. A TRPC1/TRPC3-mediated increase in store-operated calcium entry is required for differentiation of H19–7 hippocampal neuronal cells. J Biol Chem. 2004;279:43392–43402. doi: 10.1074/jbc.M408959200. [DOI] [PubMed] [Google Scholar]