

Abstract
Prion diseases are believed to propagate by the mechanism involving self-perpetuating conformational conversion of the normal form of the prion protein, PrPC, to the misfolded, pathogenic state, PrPSc. One of the most intriguing aspects of these disorders is the phenomenon of prion strains. It is believed that strain properties are fully encoded in distinct conformations of PrPSc. Strains are of practical relevance to human prion diseases, as their diversity may explain the unusual heterogeneity of these disorders. The first insight into the molecular mechanisms underlying heterogeneity of human prion diseases was provided by the observation that two distinct disease phenotypes, and their associated PrPSc conformers, co-distribute with distinct PrP genotypes as determined by the methionine/valine polymorphism at codon 129 of the PrP gene. Subsequent studies identified six possible combinations of the three genotypes (determined by the polymorphic codon 129) and two common PrPSc conformers (named types 1 and 2) as the major determinants of the phenotype in sporadic human prion diseases. This scenario implies that each 129 genotype-PrPSc type combination would be associated with a distinct diseases phenotype and prion strain. However, notable exceptions have been found. For example, two genotype-PrPSc type combinations are linked to the same phenotype and, conversely, the same combination was found to be associated with two distinct phenotypes. Furthermore, in some cases, PrPSc conformers naturally associated with distinct phenotypes appear, upon transmission, to lose their phenotype-determining strain characteristics. Currently it seems safe to assume that typical sporadic prion diseases are associated with at least six distinct prion strains. However the intrinsic characteristics that distinguish at least four of these strains remain to be identified.
Keywords: Creutzfeldt-Jakob disease, sporadic fatal insomnia, variably protease-sensitive prionopathy, 129 polymorphism, PrPSc type, PrP sequencing
Introduction
The existence of prion strains is one of the most controversial and poorly understood facets of prion diseases. The concept of strain, borrowed from virology, was introduced to of the field of prion diseases at the time when it was believed that these disorders, known as transmissible spongiform encephalopathies, were caused by viruses or other infectious agents yet to be identified. The idea that prion diseases can be associated with different strains of the infectious agent emerged from the observation that animals inoculated with brain homogenates from different scrapie-infected donors consistently developed a disease with distinct incubation times and histopathological lesions, and these differences could be stably propagated in subsequent passages into syngenic hosts. More recently, the apparent analogy between strain phenomena in viruses and prion diseases has been pushed even further with the demonstration that prion strains can undergo apparent mutations and selective amplification, which may lead to drug resistance [43].
While such phenomena are easily explained for viruses carrying nucleic acid that confers upon them the propensity to mutate and undergo selection, the existence of different prion strains has for many years presented a major challenge to the protein-only hypothesis of prion diseases. The latter model asserts that the infectious pathogen is not a virus but a misfolded form of the prion protein, PrPSc, which self-perpetuates by a mechanism that entails binding to the normal prion protein, PrPC, and inducing its conversion to the PrPSc state. A series of recent studies has provided conclusive evidence in support of this protein-only hypothesis [15,32,41,45,73]. Within the context of this model, prion strains are believed to be encrypted in distinct conformations of PrPSc. This conformational diversity may arise from a number of factors, including the amino acid sequence of substrate PrPC, the cell and tissue environment where the conversion takes place, and the process leading to the selection of the successful strain from the PrPSc population initially engendered [13].
Human prion diseases: Heterogeneity
The issue of strains is of particular significance in the case of human prion diseases, for the ultimate cause of the extraordinary diversity of these disorders may lie in strain variability (Table 1 and 2). Human prion diseases are unique in that they may be acquired by infection, be inherited, or arise spontaneously. The critical initial event in the pathogenesis of the form acquired by infection is the interaction between the exogenous PrPSc and the endogenous PrPC of the recipient. This interaction results in the formation of endogenous PrPSc which propagates further by autocatalytic conversion of additional PrPC molecules, while the original exogenous PrPSc is cleared. The physicochemical characteristics of the exogenous PrPSc, the site of the initial conversion, and the tissues and organs involved on the pathway towards brain invasion are among many factors that may contribute to strain variability of the acquired cases. In the inherited prion diseases, the presence of a mutation is thought to alter the properties of PrPC, promoting its conversion to the PrPSc state [37]. The combination of specific mutation with the methionine/valine (M/V) polymorphism at codon 129 is the major determinant of strain variation in familial cases [20]. The nature of the etiological events which lead to the idiopathic or sporadic form of prion diseases is less clear. Several scenarios, not mutually exclusive, have been proposed as explanations of the initial PrPC misfolding and PrPSc formation. One scenario posits an error in PrP post-translational processing, or the occurrence of a somatic mutation coupled with a weakening in the stringency of the quality control system which is known to occur in aging [21,33,60]. Another scenario presupposes the presence of PrPSc-like oligomers (denoted PrP* or silent prions) under normal conditions [75]. Following accidental stabilization likely facilitated by aging, PrP* would acquire the capability to convert PrPC to PrPSc thereby triggering the disease [16]. Regardless of the etiology, all three forms of prion disease present a remarkable degree of phenotypic heterogeneity, which is likely to be closely related to the diversity of prion strains.
Table 1. Human Prion Diseases: General classification.
| Form or Etiology | Phenotype |
|---|---|
| Acquired by infection | Kuru |
| vCJD | |
| Iatrogenic CJD | |
| Familial | Creutzfeldt-Jakob disease |
| Fatal familial insomnia | |
| Gerstmann-Sträussler-Scheinker d. | |
| Non specific or mixed phenotype | |
| Sporadic | Creutzfeldt-Jakob disease (with five phenotypes) |
| Fatal insomnia (FI) | |
| Variably protease-sensitive prionopathy (VPSPr)? (with three phenotypes)? |
Table 2. Classification of sCJD and sFI according to the 129 PrP genotype and PrP type with the corresponding previous nomenclature.
| Subtype | Phenotypic features | Onset (yrs)/Duration (mo) | Distribution1 (%) | Previous Nomenclature |
|---|---|---|---|---|
| sCJD | ||||
| M/M1 M/V1 | Typical CJD clinically and pathologically - Typical EEG (83%) - “Synaptic” immunostaining pattern | 63.2/3.8 | 58 | Myoclonic Heidenhain |
| V/V1 | Early onset - No typical EEG - Cerebellum spared - Weak “synaptic” immunostaining | 46.0/15.3 | 4 | Not described |
| M/M2 cortical | No typical EEG - Large and confluent vacuoles -Coarse PrP immunostaining - Cerebellum spared | 60.3/15.7 | 9 | Not described |
| M/V2 | Ataxia at onset - Rarely typical EEG - No cerebellar atrophy but kuru plaques - Plaque-like pattern of immunostaining | 60.3/17.0 | 14 | Cerebellar or ataxic |
| V/V2 | As M/V2 but no kuru plaques and cerebellar atrophy | 60.3/6.6 | 15 | Cerebellar or ataxic |
| sFI | ||||
| M/M2 | Clinically and pathologically indistinguishable from FFI | 60.3/14 | 1 | Thalamic |
Based on 609 cases examined by the National Prion Disease Pathology Surveillance Center, Cleveland, OH, USA
History of human prion strains
The very first evidence of diversity in human prion strains was reported in 1992 in a study describing the linkage of the D178N PrP gene mutation (leading to the substitution of aspartic acid with asparagine at PrP position 178) to a phenotype named fatal familial insomnia (FFI) [44,46,47]. It was observed that the PrPSc associated with FFI is characterized by an electrophoretic mobility different from that of PrPSc in sporadic CJD (sCJD) [47]. Subsequent studies described the linkage of the D178N mutation not only to FFI but also to yet another CJD-like phenotype, which was identified as familial (f) CJDD178N [20]. These two phenotypes were found to co-segregate with the genotype of codon 129 present in the mutated allele: FFI was associated with the methionine codon (129M), whereas fCJDD178N segregated with the valine codon (129V) (Figure 1) [20]. Therefore, while the D178N mutation may trigger the disease, codon 129 on the mutated allele appears to determine the basic characteristics of the disease phenotype. These observations provided the first evidence that the 129M/V polymorphism plays an important role in determining the prion disease phenotype. Furthermore, immunoblot examination demonstrated that the protease-resistant PrPSc in the two conditions had distinct electrophoretic mobilities: in FFI the unglycosylated form of the protease-resistant PrPSc had an electrophoretic mobility of 19 kDa, whereas in fCJDD178N the corresponding mobility was approximately 21 kDa. These two PrPSc conformers were subsequently named types 1 (the 21 kDa) and type 2 (the 19 kDa) (Figure 1) [48,55].
Figure 1. Representation of the original identification of two PrPSc conformers as distinct strains subsequently named types 1 and 2.
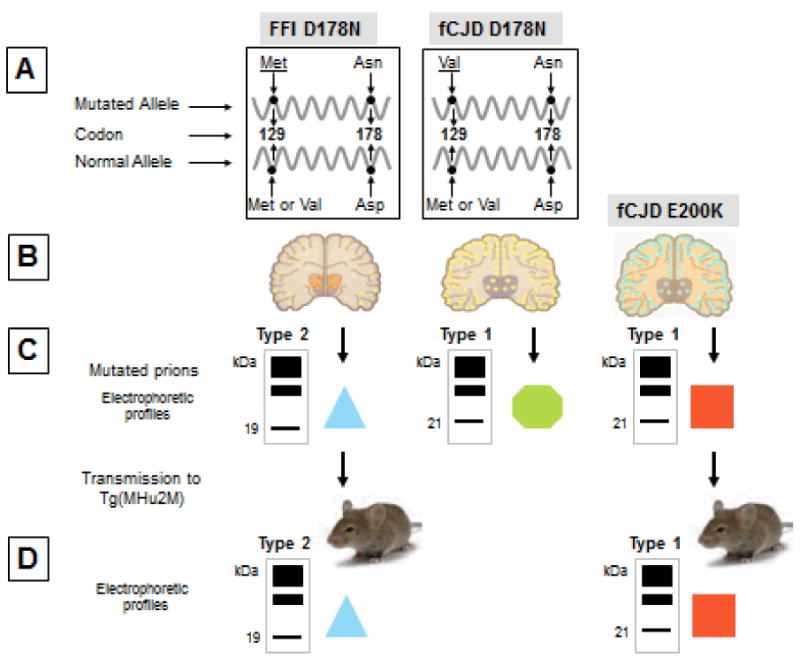
The two PrPSc conformers, referred to as types 2 and 1, were respectively associated with fatal familial insomnia (FFI) and Creutzfeldt-Jakob disease (fCJDD178N) (A), two familial prion diseases that share the same D178N mutation in the PrP gene but have distinct phenotypes as demonstrated by the opposite topography of the lesions in thalamus and cerebral cortex (B). PrPSc types 2 and 1 also show distinct electrophoretic profiles (C), which can be appreciated by the distinct mobilities to kDa 19 and kDa 21 of the lowest electrophoretic bands corresponding to the unglycosylated fragments of PrP resistant to protease digestion. The different electrophoretic mobilities of the two PrPSc conformers support the notion that they have different conformations represented as different geometric forms (C). PrPSc types 2 and 1 associated with FFI and fCJDE200K another form of familial CJD was replicated upon inoculation to receptive mice (D), even in the absence of the original PrP gene mutation. Familial CJDD178N could not be transmitted to the same transgenic mice probably because 129M/129V mismatch.  : The light blue triangle represents the conformation of the PrPSc conformer from FFID178N;
: The light blue triangle represents the conformation of the PrPSc conformer from FFID178N;  : The green hexagon represents the conformation of PrPSc from fCJDD178N;
: The green hexagon represents the conformation of PrPSc from fCJDD178N;  : The red square represents the conformation of PrPSc from fCJDE200K which although being type one as fCJDD178N- associated PrPSc is presumably different based on the fCJDE200K distinct phenotype.
: The red square represents the conformation of PrPSc from fCJDE200K which although being type one as fCJDD178N- associated PrPSc is presumably different based on the fCJDE200K distinct phenotype.
FFI, along with sCJDMM and fCJDE200K, were then transmitted to transgenic (Tg) mice expressing chimera human/mouse PrP with 129M (fCJDD178N could not be transmitted likely because of the mismatch in the 129 residue with the Tg mouse PrP) (Figure 1). The PrPSc in these infected mice faithfully reproduced electrophoretic mobilities of the inoculum (i.e., 19 kDa in FFI and 21 kDa in fCJDE200K and sCJDMM1), and these characteristics were maintained upon second passage [38,71]. Furthermore, the pattern of PrPSc deposition in the brain was also different in the affected mice inoculated with brain homogenates from FFI, sCJDMM1 or fCJDE200K. These findings clearly indicate that the PrPSc isoforms associated with FFI and fCJDE200K are distinct, representing stable prion strains which can be reproduced even in the absence of the original PrP gene mutations in the host animals.
The realization that the polymorphic codon 129 of the PrP gene could modify the phenotype of familial prion diseases raised the question as to whether the 129 genotype could also affect the phenotype in sporadic prion diseases and thereby explain, at least in part, the phenotypic heterogeneity of these diseases.
Cases with proven sCJD were then subdivided into six groups according to the six possible combination of their PrPSc type (either 1 or 2) with each of the three possible 129 genotypes (MM, MV or VV). The phenotypes of the six possible combinations were examined for homogeneity within the group and diversity between groups. This exercise led to a new classification of sporadic prion diseases based on molecular rather than clinical or histopathological features, making possible the identification of six distinct phenotypes of sporadic prion disease (Table 2) [18,55,56]. The first and most common phenotype is associated with both the 129MM and 129MV genotypes and with PrPSc type 1 [sCJDMM(MV)1]; it is often referred to as “classic” CJD. The second and third phenotypes associated with sCJDMM2 and sCJDVV1 had not been identified as separate subtypes in previous clinical or pathological classifications. The sCJDMM2 phenotype is characterized by the very large and often confluent vacuoles of the spongiform degeneration (SD), whereas the frequently early age at onset defines sCJDVV1. The two phenotypes labeled sCJDMV2 and sCJDVV2 match the phenotype previously labeled as cerebellar or ataxic. Despite their similar clinical features, these two phenotypes can easily be distinguished by the presence in the cerebellum of kuru-like plaques in sCJDMV2 (but not in sCJDVV2), and their different patterns of cerebellar PrP immunostaining. The sixth phenotype is associated with sFI. This phenotype is virtually indistinguishable from that of FFI and matches the sCJD form previously identified as thalamic. This classification goes a long way towards providing a molecular basis for the phenotypic heterogeneity and in facilitating identification of the PrPSc strains in sCJD (Table 2).
An alternative classification of PrPSc types and their pairing with CJD phenotypes has been proposed by Collinge and collaborators [12,26,42]. This classification differs from that of Parchi and colleagues discussed above in two major aspects: 1) It recognizes three (not two) PrPSc types based on electrophoretic mobility, and 2) it proposes the same nomenclature, i.e. with progressive PrPSc type numbers, also for PrPSc isoforms with different ratios of the three PrP glycoforms. In the classification by Parchi and coworkers, PrPSc types 1 and 2 that show different glycoform ratios are differentiated with letters (Figure 2). Including variant CJD, six human PrPSc types have been identified according to Collinge and colleagues. As for the associations with the sCJD phenotypes, PrPSc type 1 would be present in a subtype characterized by a slightly shorter disease duration, which includes sCJDMM and sCJDMV; type 2 and 3 would be found in all three 129 phenotypes in cases generally of long duration. However, the PrPSc electrophoretic profile in sCJDMM3 and sCJDVV3 apparently differed from that of sCJDMV3.
Figure 2. Western blots of PrPSc types 1 and 2, and of PrPDis associated with VPSPr-129MM.
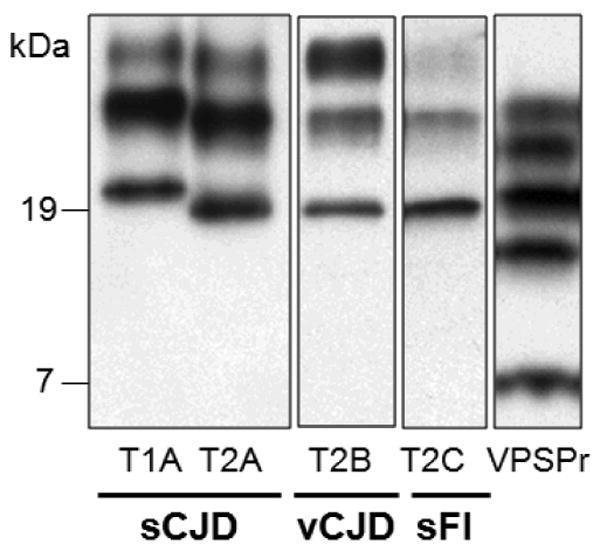
The different electrophoretic mobilities of the PrPSc types 1 (T1A) and 2 (T2A-C) are evident. The capital letters identify the glycoform ratios that may occur in PrPSc types 1 and 2. The overrepresentation of the middle band containing the monoglycosylated isoform is indicated with A; B indicates the overrepresentation of the upper band (diglycosylated isoform) and C that of the lowest band (unglycosylated isoform). The ladder-like profile formed by the PrPDis indicated as VPSPr is from VPSPr-129MM but this profile is shared by the three VPSPr 129 genotypes. All preparations have been treated with proteinase K. The sporadic Creutzfeldt-Jakob disease (sCJD) and variant (v) CJD preparations have been probed with 3F4, while the sporadic fatal insomnia (sFI) and the variably protease-sensitive prionopathy (VPSPr) preparations have been probed with IE4, both monoclonal antibodies to the prion protein.
The studies on classification of sporadic prion diseases and PrPSc typing also provided insight into the relationship between the 129 genotype and the PrPSc type. In the original study of Parchi et al over 95% of cases homozygous for methionine at codon 129 (129MM) were found to be associated with PrPSc type 1, while 86% of sCJD cases associated with PrPSc type 2 were found to be homozygous for valine or methionine/valine heterozygous (129VV and 129MV) [18,56]. Therefore, the 129MM genotype appears to favor the formation of PrPSc type 1, whereas type 2 formation is favored by the presence of one or two 129V alleles. The study of the recently discovered cases of sCJD that actually carries both types 1 and 2 further support this notion (Table 3 and see below) [8,59].
Table 3. Percent distribution of PrPSc types in sCJD.
| sCJD subtype | Type 1 or 2 (%) | Type 1-2 (%) |
|---|---|---|
| MM1 | 52 | 43 |
| MM2 | 5 | |
| MV1 | 6 | 23 |
| MV2 | 71 | |
| VV1 | 5 | 15 |
| VV2 | 80 |
From an unselected series of 200 sCJD cases, modified from [63].
Furthermore, amino acid sequencing has revealed that while the major N-termini of PrPSc types 1 and 2 are at residue 82 and 97, respectively, secondary N-termini are present in both PrPSc types 1 and 2 (Figure 3) [58]. Interestingly, the secondary N-termini appear to be non-random but rather to correlate with the PrPSc types and the 129 genotype. They show three main characteristics. First, they are more prevalent in PrPSc type 2 than in type 1. Second, within each PrPSc type, secondary cleavages are more prominent in the 129MV heterozygous than homozygous 129 genotypes. Third, they appear to be distributed toward opposite directions in the PrP sequence: in PrPSc type 1 they appear to involve to a greater extent the PrP sequence that is C-terminal to the main cleavage site, whereas in PrPSc type 2 they extend more toward the region that is N-terminal to the main cleavage site (Figure 3). Therefore, the 129 genotype may affect not only the main cleavages of the protease resistant fragments of PrPSc types 1 and 2 but also the number of the secondary cleavages and, therefore, the degree of conformational heterogeneity within each PrPSc type.
Figure 3. Diagram of the cleavage pattern of PrPSc types 1 and 2 associated with the three 129 genotypes.
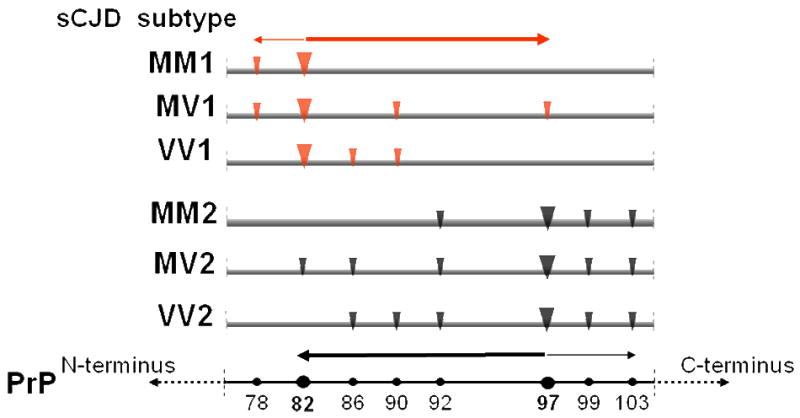
The large arrowheads indicate the major sites of cleavage by proteinase K, which are further identified with the residue numbers of the prion protein (PrP) (lowest bar). The smaller arrowheads indicate the minor cleavage sites. Minor cleavage sites extend more towards the C-terminus in PrPSc type 1, while they extend more toward the N-terminus in at least 2 of the PrPSc type 2 preparations (MV2 and VV2). This phenomenon is more prominent when PrPSc types 1 and 2 are associated with the 129MV genotype indicating that in this association, PrPSc 1 and 2 N-termini are more ragged.
Altogether, available data strongly suggest that PrPSc type formation and finer variations within each type are influenced by the genotype at codon 129.
Residue 129 polymorphism and strain determination: A biophysical perspective
The fundamentally important role of M/V polymorphism at codon 129 in human PrPC prompted studies aimed at understanding the effect of this polymorphism on the structure and biophysical properties of the recombinant prion protein. However, extensive NMR and X-ray crystallographic studies have failed to detect any significant differences in the global and local protein fold between the 129M-and 129V-containing structure [27,40]. Furthermore, residue 129M/V polymorphism was found to have no measurable effect on the global thermodynamic stability, folding or dynamics of the PrP monomer [27]. These findings suggest that the effect of residue 129 on prion diseases is mediated by events taking place at the time of PrPSc formation, events which probably influence the conformation of the PrPSc oligomer [27].
Consistent with this view, structural differences which were dependent upon polymorphism were detected by FTIR spectroscopy for amyloid fibrils formed by recombinant PrP 90-231 [2] and recombinant PrP23-144 [30]. An interesting insight into the potential nature of these structural effects was provided by recent crystallographic data for six- residue segments of PrP centered on residue 129. These studies revealed that the crystal structures for M- and V-containing peptides differ in packing within the so-called ‘steric zipper’ motif [3]. It was suggested that these packing effects could account for differences in disease susceptibility between individuals who are homozygous or heterozygous for either 129M or 129V [3]. However, it is not known whether the structures of these short segments adequately represent structural features within the corresponding region of PrPSc.
The effect of 129 polymorphism on the structure and biophysical properties of PrP has also been explored in the context of the pathogenic mutation D178N. From early NMR measurements, it was inferred that, in wild-type PrP, Asp178 forms a salt bridge with Arg164 and has the potential to form a hydrogen bond with Tyr128, a residue sequentially adjacent to position 129 [65]. It was also proposed that the Asp→Asn replacement at position 178 would affect this network of hydrogen bonds differentially depending on residue 129 Met/Val polymorphism, providing a rationale for polymorphism-dependent phenotypic variability of inherited prion diseases with D178N mutation. A more detailed insight into structural effects of D178N mutation was provided by recent crystallographic data, which revealed mutation-dependent differences in the loop region (residues R164-S170), as well as polymorphism-dependent differences in the local environment surrounding the altered D178N residue [40]. It was also shown that the polymorphism at residue 129 strongly affects conversion of the D178N variant of recombinant PrP to amyloid fibrils, with the 129M polymorph undergoing substantially faster conversion than the 129V polymorph [2]. Furthermore, fibrils formed by the D178N/129V variant appear to be structurally distinct from those formed by the D178N/129M protein [2]. Altogether, this biophysical insight strongly suggests that by modulating both the conversion process and the conformation of PrPSc, residue 129V/M polymorphism governs susceptibility to prion disease and prion strain properties.
Prion strains in human prion diseases
Sporadic prion diseases: Creutzfeldt-Jakob disease and fatal insomnia
From the perspective of the disease, prion strains may be viewed as PrPSc conformers which often (but not necessarily always) are associated with distinct disease phenotypes [4,6,12,61,68,71]. Within the context of this definition, PrPSc types 1 and 2 definitely fulfill the requirements of distinct prion strains for several reasons. First, they have different physicochemical characteristics such as proteolytic cleavage sites and conformational stability profiles, the features implying different conformations [9, 11,36,51,58]. Second, upon inoculation they cause diseases characterized by different incubation times, histopathologies and PrPSc distributions [5,38,49,69,70]. Finally, they can be faithfully replicated by protein misfolding cyclic amplification (PMCA) [10,31,67]. However, the identification of six phenotypes, five in sCJD and one in sFI (according to the classification of Parchi and colleagues [56]), raises the question of the number of distinct strains of sCJD [18]. Distinctive clinical, histopathological and PrP immunostaining features of the six phenotypes point to an association with at least six distinct PrPSc strains. Should this be the case, the strains of sporadic prion diseases would far exceed types 1 and 2. This implies that PrPSc types 1 and 2, rather than just representing two strains, actually define two categories of strain subtypes, each associated with a distinct phenotype.
Several studies of sCJD subtypes, including transmission to transgenic mice expressing human PrPC or chimera human/mouse PrP have been reported in the last 15 years [12,25,35,70,71,72]. These studies provide strong evidence that some sCJD subtypes are indeed associated with distinct PrPSc strains. However, only one recent study has systematically attempted to determine the number of PrPSc strains associated with the major subtypes of sCJD [5].
Bishop and collaborators tested the strain characteristics of PrPSc isoforms associated with sCJD (but not sFI) by inoculating sCJD brain homogenates representing the six possible combinations of the 129 genotype and PrPSc type (i.e., MM, MV and VV each associated with PrPSc 1 or 2) into knock-in Tg mice expressing human PrPC with each of the three 129 genotypes [5]. Using incubation times, lesion profiles, and the pattern of PrPSc intracerebral deposition, this study identified four strains and found them to be associated with the five phenotypes identified in sCJD by Parchi et al [5,56]. Specifically, by bioassay, MM1PrPSc could not be distinguished from MV1PrPSc nor could MV2PrPSc be distinguished from VV2PrPSc. VV1PrPSc and MM2PrPSc could be identified as distinct strains, although both strains appeared to be unusual and difficult to replicate even in their homonymous 129 backgrounds.
The study by Bishop et al [5] has several implications. First, the inability to separate MM1PrPSc and MV1PrPSc is consistent with the strong similarity of the sCJD phenotypes associated with these two PrPSc isoforms. It also supports the finding that these two sCJD PrPSc isoforms (but not other PrPSc isoforms including VV1PrPSc) share the same PK-resistance profile and sensitivity to pH as well as the presence of the same truncated anchorless fragment of 18.5 kDa [50,52]. The lack of distinguishing features suggests two possibilities which are not mutually exclusive. Thus, it is possible that in sCJDMV1 the PrPSc strain originates exclusively or overwhelmingly from the conversion of 129MPrPC. Alternatively, the difference in strain between MM1PrPSc and MV1PrPSc could be too subtle to generate distinct phenotypes. Indistinguishable physicochemical features have been recently reported for some prion strains, leading to the concept of substrain [13,43]. Indeed, as mentioned above, N-terminal sequencing of PrPSc isoforms has shown that while MM1PrPSc and MV1PrPSc share PK cleavage sites at residues 82 and 78, only MV1PrPSc is cleaved at the more C-terminal residues 90 and 97, the latter being the major cleavage site of PrPSc type 2 [58]. Although additional sCJDMV1 cases need to be examined, these extra cleavage sites argue for the presence of a small amount of PrPSc type 2 in MV1PrPSc.
A second consideration suggested by the data of Bishop and colleagues is that although by bioassay MV2PrPSc and VV2PrPSc were found to have identical strain features, the clinical and pathological differences between sCJDMV2 and sCJDVV2 — the two phenotypes to which they are associated — suggests otherwise. Major neuropathological differences occur in the cerebellum, where in the case of sCJDMV2 PrPSc kuru plaque-like deposits with the tinctorial characteristics of the amyloid are found along with good preservation of neurons [17,18,55,56]. In contrast, in sCJDVV2 the PrP deposits are loose and amyloid-free, and there is a significant loss of neurons. These contrasting features argue that the mode by which the PrPSc aggregates form, and possibly the toxicity associated with these aggregates, differ in these two sCJD subtypes. Consistent with the presumed higher toxicity of the PrPSc aggregates in sCJDVV2, the clinical duration of the disease is also significantly shorter in this sCJD subtype (6.5 months with 3-18 month range in compared with 17 months with a 5-72 month range in sCJDMV2) [13,56]. According to a recently proposed model of strain pathogenicity, the diverse features that distinguish sCJDMV2 from sCJDVV2 could result from different kinetics of PrPSc propagation [13]. Furthermore, sequencing has shown that the N-terminus of PK-resistant core of MV2PrPSc is somewhat more ragged than that of VV2PrPSc. The presence in MV2PrPSc of a fragment starting at residue 82, the major cleavage site of PrPSc type 1, is consistent with the presence of small amounts of PrPSc type 1 [58]. In agreement with this finding, the electrophoretic profiles of MV2PrPSc and VV2PrPSc from most brain regions are easily distinguished by the presence of two highly PK-resistant bands (corresponding to the unglycosylated isoform) in the MV2PrPSc blot profile and only one such band in the profile of VV2PrPSc [50,51,59]. Furthermore, the electrophoretic profile of VV2PrPSc shows an additional fragment of approximately 18 kDa [59]. These findings suggest that, although not distinguishable after transmission to receptive animals, the MV2PrPSc and VV2PrPSc isoforms behave differently in the human brain. Distinct physicochemical features may be revealed upon more extensive examination.
The poor transmissibility of both MM2PrPSc and VV1PrPSc to Tg mice of all three 129 genetic backgrounds sets these two strains apart, suggesting that they form differently from the other sCJD strains. According to a prevailing hypothesis, the primary structure of the host's PrPC determines the spectrum of PrPSc conformations that can be generated in a given species [13,29,43]. However, it is likely that several distinct isoforms of PrPSc are initially generated when PrPC converts to PrPSc. The successful strain is viewed as the result of a selection dictated by the extent to which the rate of conversion and propagation exceeds the rate of clearance for that particular PrP isoform [13]. According to this hypothesis, VV1PrPSc and MM2PrPSc might represent isoforms which under some exceptional conditions ‘escape’ the selection process. This would explain the extreme rarity of these strains (sCJDMM2 and sCJDVV1 combined account for only ∼3% of all cases of sCJD [25,56]) as well as the difficulty encountered in replicating them in humanized Tg mice regardless of the 129 background [5].
Finally, it is noteworthy that neither the study of Bishop et al nor any other study on sCJD transmission to humanized Tg mice has found the PrPSc type 1 described by Collinge et al (that displays a slightly slower gel mobility than the PrPSc type 1 described by Parchi et al) [5,12,56].
Sporadic CJD with co-occurrence of PrPres type 1 and type 2
It has now been clearly established that, although sCJD subtypes exclusively associated with PrPSc type 1 or type 2 account for the majority of the cases, the concurrent presence of both PrPres types is significant, varying between 15% and 43% according to the sCJD 129 genotype (Table 3) [8,22,42, 59,64,69,72]. The co-occurrence of PrPres types 1 and 2 (hereafter identified as type 1-2) seems to follow certain general patterns. The two PrPres types can be found together in the same brain region or separately in different regions. Regardless whether combined or separate, the topographic distribution of PrPres 1-2 is non-random: it favors specific brain regions such as the cerebral neocortex and the thalamus, but is less common in the cerebellum. Therefore, every brain region can contain both PrPSc types, though some regions, such as the thalamus, appear to be more apt to propagate both PrPSc types [8,59]. Again, the representation of types 1 and 2 appears to correlate with the 129 genotype; in sCJDMM1-2 type 1 dominates, whereas in sCJDMV and sCJDVV the opposite is observed, indicating that the strong association of PrPSc type 1 with the 129MM homozygosity and of PrPSc type 2 with the presence of at least one 129V allele is also maintained in sCJD1-2 [56,59]. In general, the concomitant presence of PrPSc types 1 and 2 has the tendency to modify the clinical and histopathological phenotype which exhibits features that are intermediate between the phenotypes associated with the corresponding sCJD1 and sCJD2 variants [8,59]. This is particularly noticeable in the sCJDMM group, in which case the shift of the phenotype from sCJDMM1 to sCJDMM2 is directly related to the amount of PrPSc type 2 present in the brain [8]. The PrPSc type-related shift of the phenotype is much less noticeable for sCJDMV1-2 and sCJDVV1-2, perhaps because the presence of one or two 129V alleles makes the phenotypic features of sCJDMV2 and sCJDVV2 dominant over those of sCJDMV1 and sCJDVV1 [7]. Additional studies are needed to more clearly define the phenotype and PrPSc characteristics of sCJDMV1-2 and sCJDVV1-2 [7,59]. With regard to PrPSc strain characteristics in sCJDMM1-2, it is intriguing that the co-existence of PrPSc types 1 and 2 in the same brain locale seems to alter the characteristics of each of the two PrPSc types compared to those displayed when they occur separately. For example, the resistance to denaturation of PrPSc type 1, as determined by the conformation stability immunoassay (CSI), becomes similar to that of PrPSc type 2, although the cleavage sites of the shifted PrPSc do not change (Figure 4). A similar effect is also seen after PrPSc types 1 and 2 are mixed in vitro [8]. Therefore, type 1 and 2 co-occurrence in vivo, as well as the interaction of the two types in vitro, may change some conformational characteristics of PrPSc type 1 [8]. These findings suggest yet another potential mechanism of strain mutation [1,13,43]. On the whole, the co-occurrence of PrPSc types 1 and 2 is likely to shed light on the effect that co-occurring strains may exert upon one another as well as on the selection process leading to the presence of two major strains [1,8].
Figure 4. Conformation stability immunoassay (CSI) of the unglycosylated isoform of PrPSc from sCJDMM1, sCJDMM2 and sCJDMM1-2.
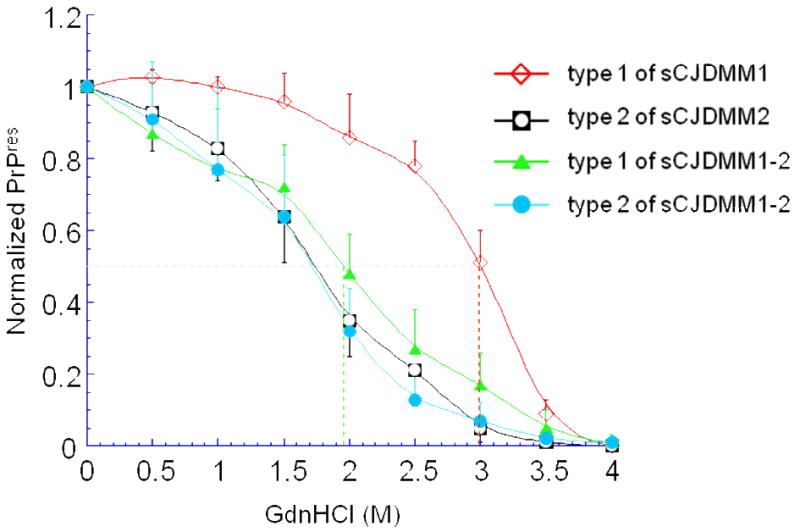
To measure the stability of PrPSc, increasing concentrations of GdnHCl are used to unfold PrPSc, which in turn becomes progressively more sensitive to digestion with PK. The concentration of guanidine applied to achieve the digestion of 50% of the PrPSc with proteinase K (PK) is indicated by [GdnHCl]1/2 (shown by the vertical color coded broken lines). The CSI curve shows that the stability of the PrPSc type 1 when it is present alone as in sCJDMM1 (red hollow diamond) is significantly different from the stability that it shows when it co-occurs with PrPSc type 2 as in sCJDMM1-2 (green solid triangle) ([GdnHCl]1/2 values 2.96M vs. 1.96M, respectively; p<10-5). Furthermore, the CSI curve of the PrPSc type 1 from sCJDMM1-2 becomes indistinguishable from that of PrPSc type 2 from sCJDMM2 (black square with hollow circle) and sCJDMM1-2 (blue solid circle), which are similar indicating no change in CSI characteristics between PrPSc 2 when alone or co-occurring with type 1. CSI profiles are smooth curves best matching data (mean ± SD) obtained at different concentrations of guanidine.
Sporadic fatal insomnia (also occasionally referred to as sCJDMM2 thalamic)
Presents a special challenge in the assessment of prion strains associated with sporadic prion diseases. Although the clinical and histopathological phenotypes of sFI and sCJDMM2 are quite distinct, both conditions are associated with the 129MM genotype and PrPSc type 2 [89,56-58]. A most striking variation between these two diseases lies in their contrastive lesion topography. In sFI the thalamus is very severely affected and the cerebral cortex is often almost spared, while the opposite is true for SCJDMM2. This suggests that the histopathology starts, and the pathogenic PrPSc initially acts, in different brain regions. The lack of correlation of the phenotype with the 129 genotype and the PrPSc type in these two diseases raises questions regarding the role and nature of additional factors (beyond the 129 genotype and PrPSc type) as potential determinants of prion strains.
A possible feature distinguishing PrPSc conformers associated with sFI and sCJDMM2 is the pattern of glycosylation as revealed by two dimensional WB analysis [54]. Whether differences in glycosylation can explain variations in the phenotype remains, however, to be determined [9,63]. Furthermore, variations in the PK-resistant PrP fragments associated with sFI and sCJDMM2 have been reported [52].
Atypical prion diseases: Variably protease-sensitive prionopathy (VPSPr)
A disease affecting the prion protein but exhibiting characteristics different from those of the classic prion diseases was first reported in 2008 [19]. The study of eleven original cases revealed several major features which distinguished this disorder from common sporadic prion diseases. These include: the relatively long duration of nearly two years; clinical presentation more consistent with that of an atypical dementia than sporadic prion disease; the distinctive ladder-like electrophoretic profile and relatively low protease-resistance of the insoluble, disease-associated PrP (PrPDis) (Figure 2). Due to these characteristics as well as the uncertainty regarding transmissibility, this condition was originally named “protease-sensitive prionopathy”. Another peculiar feature of this condition is that six out of ten cases that could be investigated had a positive family history of cognitive impairment. However, none of these cases was associated with a mutation in the open reading frame of the PrP gene.
All eleven cases of the original report were 129VV. However, a subsequent study revealed that the same disease also affects subjects with the 129MV and 129MM genotypes and the disease was renamed “variably protease-sensitive prionopathy” (VPSPr) (see below) [77]. Currently, 29 cases of VPSPr have been reported: 20 are 129VV, 6 are 129MV, and 3 are 129MM [19, 23, 28, 39, 77]. Thus, after the original report of CJD in 1921, VPSPr appears to be the second presumably sporadic disease involving the prion protein that affects all three 129 genotypes [34].
The finding that VPSPr affects the three 129 genotypes raises a number of intriguing questions. One of them is whether each 129 genotype is paired with a distinct phenotype and distinct PrPDis characteristics as is the case with subtypes of sCJD [18,56]. Clinically the 129VV and 129MV VPSPr cases (the availability of only two 129MM symptomatic cases precludes the comparative clinical study of this subset) differ significantly in duration and the prevalence of myoclonus. Though histopathologically these cases differ only slightly with respect to PrP immunostaining patterns, the major difference lies in the PK resistance of PrPDis [77]. As originally reported, PrPDis from 129VV cases is very sensitive to PK digestion, whereas the PK-resistance of PrPDis from 129MM cases is comparable to that of PrPSc in classic prion diseases, and the proteolytic resistance of 129MV cases lies somewhere between those of 129 MM and 129 VV cases. This variability in PK resistance that co-distributes with the 129 genotype prompted the addition of “variably” to the original label of “protease-sensitive prionopathy” [77]. Furthermore, although the ladder-like electrophoretic profile is shared by all three VPSPr 129 genotypes, the ratios of the individual bands are different, suggesting that though the same PrPDis sites are cleaved by PK, their accessibility is dissimilar. Finally, the PrPDis conformers associated with the three VPSPr genotypes also differ in the degree of immunoreactivity with monoclonal antibodies 3F4 (epitope within residues 106-110) and 1E4 (epitope within residues 97-105) [76,78]. Although these variations may not be as distinctive as they are between the subtypes of sCJD, the three 129 genotypes of VPSPr are associated with distinguishable phenotypes and PrPDis characteristics.
Of note, there appears to be a drastic difference between VPSPr and classic prion diseases with respect to the prevalence of the three 129 genotypes. In VPSPr the 129MM, 129MV and 129VV genotypes account for 10%, 23% and 67% of all cases, respectively. In contrast 129MM, 129MV and 129VV patients account for 72%, 11% and 17%, respectively, of all sCJD and sFI cases combined [18,56]. Furthermore, in VPSPr, as well as in sCJD, the representations of the three 129 genotypes markedly differ from the prevalence of these genotypes in the general Caucasian population [53]. In sCJD, the overrepresentations of the 129MM and (to a lesser extent) the 129VV genotypes, combined with the underrepresentation of the 129MV genotype, have been attributed to more efficient conformational conversion of PrPC to PrPSc in individuals homozygous at codon 129 compared to those heterozygous at this codon [3,53]. This mechanism does not seem to play a role in VPSPr, in which the 129VV genotype appears to pose by far the highest risk, whereas the 129MM genotype appears to be protective.
Altogether, available data argue that PrPDis associated with VPSPr is distinct from the PrPSc strains associated with the various subtypes of sCJD, and that the 129 genotype may play a different role in PrPDis formation [19]. Furthermore, the distinctive phenotypic and PrPDis features observed among the three 129 genotypes suggest that PrPDis types associated with these genotypes represent distinct strains (or, at least, sub-strains according to the recently proposed terminology [14,74]). More detailed characterization of these strains awaits the results of ongoing studies on amplification and transmissibility.
Concluding remarks
The assessment of the number of strains associated with human sporadic prion diseases must take into account the observation that both the 129 genotype and the PrPSc type are the major determinants of the disease phenotype. However, the role of these two determinants is complex and interrelated. First, the genotype at codon 129 appears to be a major determinant of PrPSc types 1 and 2 which, in turn, are also associated with different phenotypes. However, the three 129 genotypes also may act as determinants of the phenotype in the sCJD associated with PrPSc types 1 as well as type 2. Since there are three 129 genotypes and two PrPSc types, one would expect that, at least theoretically, up to six distinct strains could be associated with sCJD and sFI combined, each of them corresponding to one of the six possible pairings of three codon129 genotypes with the two PrPSc types. However, as discussed in this article, there might be a number of exceptions to this general scenario.
First exception is exemplified by sCJDMM1 and sCJDMV1. Despite the different 129 genotype, these two subtypes of sCJD share the same phenotype and appear to be associated with same strain, suggesting that the 129V allele plays no role in determining the phenotype or the PrPSc characteristics in sCJDMV1. The opposite exception is sCJDMM2 and sFI, which, despite being associated with two very different phenotypes, share the same combination of 129MM genotype and PrPSc type 2. Although the mechanism of phenotypic determination in these two diseases is currently unknown, it is reasonable to expect that specific features which discriminate between the two PrPSc strains associated with sCJDMM2 and sFI do exist and will eventually be found.
Another layer of complexity arises from the inability of propagating to receptive mice the distinct strain properties apparently exhibited by the PrPSc conformers associated with sCJDMV2 and sCJDVV2 [5]. Again, although the phenotype-determining characteristics of some strains are apparently evident in the human disease, they might be difficult to propagate experimentally, perhaps because of their instability.
Yet another interesting situation is presented by sCJDVV1, in which PrPSc appears to have all the features of a distinct strain. However, sCJDVV1, like sCJDMM2 and sFI, has a very low prevalence and, like sCJDMM2 is difficult to transmit to humanized Tg mice [5]. Therefore, the strains associated with these three phenotypes are unusual. They might represent variant strains which have escaped the selection process believed to occur during strain formation [13].
Altogether, the picture emerging from these considerations suggests that despite the exceptions previously noted, there are at least six different strains associated with sCJD and sFI, three of which are common while the other three are rare or variant strains (Figure 5).
Figure 5. Proposed relationships between the 129 genotypes, PrPSc types and subtypes, phenotypes and strains in sporadic Creutzfeldt-Jakob (sCJD) and sporadic fatal insomnia (sFI).

The three 129 genotypes (and possibly other polymorphisms) are believed to influence the formation of PrPSc type 1 or 2 as the 129MM genotype is preferentially associated with type 1 while 129MV and 129VV are associated with type 2. The six possible combinations of the three 129 genotypes with PrPSc types 1 and 2 are associated with six distinct disease phenotypes, five belonging to sCJD and one to sFI. The pairing of 129 genotype-PrPSc type combinations with the phenotypes is not stringent. Notable exceptions are the association of the distinct MM1 and MV1 combinations with one phenotype (MM1/MV1) and the association of the single MM2 combination with two phenotypes (MM2 sCJD and MM2 sFI). It is proposed that the six phenotypes are paired with six distinct prion strains, three of which are relatively prevalent, while three are rare and might result from variations in the strain selection process. The distinct strain proposed to be associated with sCJDMV2 is questionable because of the inability to maintain its distinctive characteristics after transmission [5]. Coexistence of types 1 and 2 associated with the three 129 genotypes and the strains possibly associated with the novel disease variably protease-sensitive prionopathy (VPSPr), are not shown for simplicity, and because the presence of true strains in VPSPr is currently unclear.
Despite recent progress in strain-dependent characterization of sporadic human prion diseases, there are still a number of unresolved questions: Can the presumed strains associated with sCJDMM2 and sFI be distinguished based on their physicochemical properties? What mechanism governs the reciprocal influence of PrPSc types 1 and 2 when they co-occur in sCJDMM1-2? Are some strains inherently unstable and, thus, unable to propagate some of their phenotype-determining characteristics upon transmission? Does PrPDis associated with VPSPr qualify as a true strain which encompasses three distinguishable sub-strains? Clearly, additional studies are needed to dissect the intricate details of human prion strains. Such studies have potentially broader implications, for recent data suggest that the strain phenomenon may also be relevant to other neurodegenerative diseases associated with protein misfolding and aggregation [62,66].
Acknowledgments
We are grateful to Dr. Jiri Safar for the critical reading of the manuscript. This research was supported by the National Institutes of Health (NIH) grants AG14359 (to P.G., Q.K. and W.K.S.), AG08702 (to P.G.), NS062787 (to W.Q.Z.), NS44158 (to W.K.S.), NS38604 (to W.K.S.); the Centers for Disease Control and Prevention grant CCU 515004 (to P.G.); the Britton Fund; CJD Foundation, Alliance BioSecure and the University Center on Aging and Health with the support of the McGregor Foundation and the President's Discretionary Fund (Case Western Reserve University).
References
- 1.Angers RC, Kang HE, Napier D, et al. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science. 2010;328:1154–1158. doi: 10.1126/science.1187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Apetri AC, Vanik DL, Surewicz WK. Polymorphism at residue 129 modulates the conformational conversion of the D178N variant of human prion protein 90-231. Biochemistry. 2005;44:15880–15888. doi: 10.1021/bi051455+. [DOI] [PubMed] [Google Scholar]
- 3.Apostol MI, Sawaya MR, Cascio D, Eisenberg D. Crystallographic studies of PrP segments suggest how structural changes encoded by polymorphism at residue 129 modulate susceptibility to human prion disease. J Biol Chem. 2010 Aug 4; doi: 10.1074/jbc.C110.158303. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bessen RA, Marsh RF. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol. 1992;73:329–334. doi: 10.1099/0022-1317-73-2-329. [DOI] [PubMed] [Google Scholar]
- 5.Bishop MT, Will RG, Manson JC. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A. 2010;107:12005–12010. doi: 10.1073/pnas.1004688107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breusing N, Grune T. Regulation of proteasome-medicated protein degradation during oxidative stress and aging. Biol Chem. 2008;386:203–209. doi: 10.1515/BC.2008.029. [DOI] [PubMed] [Google Scholar]
- 7.Budka H. In: The Encyclopedia of Movement Disorders (MOVE) Jennifer G, editor. Elsevier Ltd.; London, UK: 2009. In press. [Google Scholar]
- 8.Cali I, Castellani R, Alshekhlee A, et al. Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: its effect on the phenotype and prion-type characteristics. Brain. 2009;132:2643–2658. doi: 10.1093/brain/awp196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancellotti E, Wiseman F, Tuzi NL, et al. Altered glycosylated PrP proteins can have different neuronal trafficking in brain but do not acquire scrapie-like properties. J Biol Chem. 2005;280:42909–42918. doi: 10.1074/jbc.M509557200. [DOI] [PubMed] [Google Scholar]
- 10.Castilla J, Morales R, Saá P, et al. Cell-free propagation of prion strains. EMBO J. 2008;27:2557–2566. doi: 10.1038/emboj.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen SG, Zou W, Parchi P, Gambetti P. PrP(Sc) typing by Nterminal sequencing and mass spectrometry. Arch Virol Suppl. 2000;16:209–216. [PubMed] [Google Scholar]
- 12.Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 13.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 14.Collinge J. Prion strain mutation and selection. Science. 2010;328:1111–1112. doi: 10.1126/science.1190815. [DOI] [PubMed] [Google Scholar]
- 15.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edgeworth JA, Gros N, Alden J, et al. Spontaneous generation of mammalian prions. Proc Natl Acad Sci U S A. 2010;107:14402–14406. doi: 10.1073/pnas.1004036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faucheux BA, Privat N, Brandel JP, et al. Loss of cerebellar granule neurons is associated with punctate but not with large focal deposits of prion protein in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol. 2009;68:892–901. doi: 10.1097/NEN.0b013e3181af7f23. [DOI] [PubMed] [Google Scholar]
- 18.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–239. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- 19.Gambetti P, Dong Z, Yuan J, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008;63:697–708. doi: 10.1002/ana.21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldfarb LG, Petersen RB, Tabaton M, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science. 1992;258:806–808. doi: 10.1126/science.1439789. [DOI] [PubMed] [Google Scholar]
- 21.Gorbunova V, Seluanov A. Making ends meet in old age: DSB repair and aging. Mech Ageing Dev. 2005;126:621–628. doi: 10.1016/j.mad.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 22.Head MW, Bunn TJ, Bishop MT, et al. Prion protein heterogeneity in sporadic but not variant Creutzfeldt-Jakob disease: UK cases 1991-2002. Ann Neurol. 2004;55:851–859. doi: 10.1002/ana.20127. [DOI] [PubMed] [Google Scholar]
- 23.Head MW, Knight R, Zeidler M, et al. A case of protease sensitive prionopathy in a patient in the UK. Neuropathol Appl Neurobiol. 2009;35:628–632. doi: 10.1111/j.1365-2990.2009.01040.x. [DOI] [PubMed] [Google Scholar]
- 24.Heinemann U, Krasnianski A, Meissner B, et al. Creutzfeldt-Jakob disease in Germany: a prospective 12-year surveillance. Brain. 2007;130:1350–1359. doi: 10.1093/brain/awm063. [DOI] [PubMed] [Google Scholar]
- 25.Hill AF, Desbruslais M, Joiner S, et al. The same prion strain causes vCJD and BSE. Nature. 1997;389:448–450. 526. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 26.Hill AF, Joiner S, Wadsworth JD, et al. Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain. 2003;126:1333–1346. doi: 10.1093/brain/awg125. [DOI] [PubMed] [Google Scholar]
- 27.Hosszu LL, Jackson GS, Trevitt CR, et al. The residue 129 polymorphism in human prion protein does not confer susceptibility to Creutzfeldt-Jakob disease by altering the structure or global stability of PrPC. J Biol Chem. 2004;279:28515–28521. doi: 10.1074/jbc.M313762200. [DOI] [PubMed] [Google Scholar]
- 28.Jansen C, Head MW, van Gool WA, et al. The first case of protease-sensitive prionopathy (PSPr) in The Netherlands: a patient with an unusual GSS-like clinical phenotype. J Neurol Neurosurg Psychiatry. 2010 Jun 14; doi: 10.1136/jnnp.2009.175646. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 29.Jones EM, Surewicz WK. Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell. 2005;121:63–72. doi: 10.1016/j.cell.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 30.Jones EM, Surewicz K, Surewicz WK. Role of N-terminal familial mutations in prion protein fibrillization and prion amyloid propagation in vitro. J Biol Chem. 2006;281:8190–8196. doi: 10.1074/jbc.M513417200. [DOI] [PubMed] [Google Scholar]
- 31.Jones M, Peden AH, Wight D, et al. Effects of human PrPSc type and PRNP genotype in an in-vitro conversion assay. Neuroreport. 2008;19:1783–1786. doi: 10.1097/WNR.0b013e328318edfa. [DOI] [PubMed] [Google Scholar]
- 32.Kim JI, Cali I, Surewicz K, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem. 2010;285:14083–14087. doi: 10.1074/jbc.C110.113464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirkwood TB, Austad SN. Why do we age? Nature. 2000;408:233–238. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- 34.Kirschbaum WR. Jakob-Creuzfeldt Disease. American Elzevier Publishing Company, Inc.; New York: 1968. p. 229. [Google Scholar]
- 35.Kitamoto T, Mohri S, Ironside JW, et al. Follicular dendritic cell of the knock-in mouse provides a new bioassay for human prions. Biochem Biophys Res Commun. 2002;294:280–286. doi: 10.1016/S0006-291X(02)00476-X. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi A, Satoh S, Ironside JW, Mohri S, Kitamoto T. Type 1 and type 2 human PrPSc have different aggregation sizes in methionine homozygotes with sporadic, iatrogenic and variant Creutzfeldt-Jakob disease. J Gen Virol. 2005;86:237–240. doi: 10.1099/vir.0.80389-0. [DOI] [PubMed] [Google Scholar]
- 37.Kong Q, Surewicz WK, Petersen RB, et al. Inherited prion diseases. In: Prusiner SB, editor. Prion biology and diseases. 2nd. Cold Spring Harbor Laboratory Press; New York: 2004. pp. 673–776. [Google Scholar]
- 38.Korth C, Kaneko K, Groth D, et al. Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc Natl Acad Sci U S A. 2003;100:4784–4789. doi: 10.1073/pnas.2627989100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krebs B, Bader B, Klehmet J, et al. A novel subtype of Creutzfeldt-Jakob disease characterized by a small 6 kDa PrP gragment. Acta Neruopathol. 2007;114:195–199. doi: 10.1007/s00401-007-0242-5. [DOI] [PubMed] [Google Scholar]
- 40.Lee S, Antony L, Hartmann R, et al. Conformational diversity in prion protein variants influences intermolecular beta-sheet formation. EMBO J. 2010;29:251–262. doi: 10.1038/emboj.2009.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Legname G, Baskakov IV, Nguyen HO, et al. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 42.Lewis V, Hill AF, Klug GM, et al. Australian sporadic CJD analysis supports endogenous determinants of molecular-clinical profiles. Neurology. 2005;65:113–118. doi: 10.1212/01.wnl.0000167188.65787.a0. [DOI] [PubMed] [Google Scholar]
- 43.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. Darwinian evolution of prions in cell culture. Science. 2010;327:869–872. doi: 10.1126/science.1183218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lugaresi E, Medori R, Montagna P, et al. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med. 1986;315:997–1003. doi: 10.1056/NEJM198610163151605. [DOI] [PubMed] [Google Scholar]
- 45.Makarava N, Kovacs GG, Bocharova O, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010;119:177–187. doi: 10.1007/s00401-009-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Medori R, Montagna P, Tritschler HJ, et al. Fatal familial insomnia: a second kindred with mutation of prion protein gene at codon 178. Neurology. 1992;42:669–670. doi: 10.1212/wnl.42.3.669. [DOI] [PubMed] [Google Scholar]
- 47.Medori R, Tritschler HJ, LeBlanc A, et al. Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N Engl J Med. 1992;326:444–449. doi: 10.1056/NEJM199202133260704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Monari L, Chen SG, Brown P, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad USA. 1994;91:2839–2842. doi: 10.1073/pnas.91.7.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nonno R, Di Bari MA, Cardone F, et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog. 2006;2:e12. doi: 10.1371/journal.ppat.0020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Notari S, Capellari S, Giese A, et al. Effects of different experimental conditions on the PrPSc core generated by protease digestion: implications for strain typing and molecular classification of CJD. J Biol Chem. 2004;16;279(16):16797–804. doi: 10.1074/jbc.M313220200. Epub 2004 Jan 29. [DOI] [PubMed] [Google Scholar]
- 51.Notari S, Capellari S, Langeveld J, et al. A refined method for molecular typing reveals that co-occurrence of PrP(Sc) types in Creutzfeldt-Jakob disease is not the rule. Lab Invest. 2007;87:1103–12. doi: 10.1038/labinvest.3700676. [DOI] [PubMed] [Google Scholar]
- 52.Notari S, Strammiello R, Capellari S, et al. Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. J Biol Chem. 2008;283:30557–30565. doi: 10.1074/jbc.M801877200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palmer MS, Dryden AJ, Hughes JT, Collinge J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature. 1991;352:340–342. doi: 10.1038/352340a0. [DOI] [PubMed] [Google Scholar]
- 54.Pan T, Colucci M, Wong BS, et al. Novel differences between two human prion strains revealed by two-dimensional gel electrophoresis. J Biol Chem. 2001;276:37284–37288. doi: 10.1074/jbc.M107358200. [DOI] [PubMed] [Google Scholar]
- 55.Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 56.Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- 57.Parchi P, Capellari S, Chin S, et al. A subtype of sporadic prion disease mimicking fatal familial insomnia. Neurology. 1999;52:1757–1763. doi: 10.1212/wnl.52.9.1757. [DOI] [PubMed] [Google Scholar]
- 58.Parchi P, Zou W, Wang W, et al. Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci U S A. 2000;97:10168–10172. doi: 10.1073/pnas.97.18.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parchi P, Strammiello R, Notari S, et al. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol. 2009;118:659–71. doi: 10.1007/s00401-009-0585-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Partridge L, Gems D. Mechanisms of ageing: public or private? Nat Rev Genet. 2002;3:165–175. doi: 10.1038/nrg753. [DOI] [PubMed] [Google Scholar]
- 61.Peretz D, Scott MR, Groth D, et al. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 2001;10:854–863. doi: 10.1110/ps.39201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Petkova AT, Leapman RD, Guo Z, et al. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 63.Piro JR, Harris BT, Nishina K, et al. Prion protein glycosylation is not required for strain-specific neurotropism. J Virol. 2009;83:5321–5328. doi: 10.1128/JVI.02502-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Puoti G, Giaccone G, Rossi G, et al. Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrP(Sc) in the same brain. Neurology. 1999;53:2173–2176. doi: 10.1212/wnl.53.9.2173. [DOI] [PubMed] [Google Scholar]
- 65.Riek R, Wider G, Billeter M, et al. Prion protein NMR structure and familial human spongiform encephalopathies. Proc Natl Acad Sci U S A. 1998;95:11667–11672. doi: 10.1073/pnas.95.20.11667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Russo C, Schettini G, Saido TC, et al. Presenilin-1 mutations in Alzheimer's disease. Nature. 2000;405:531–532. doi: 10.1038/35014735. [DOI] [PubMed] [Google Scholar]
- 67.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 68.Safar J, Wille H, Itri V, et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 69.Schoch G, Seeger H, Bogousslavsky J, et al. Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med. 2006;3:e14. doi: 10.1371/journal.pmed.0030014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taguchi Y, Mohri S, Ironside JW, Muramoto T, Kitamoto T. Humanized knock-in mice expressing chimeric prion protein showed varied susceptibility to different human prions. Am J Pathol. 2003;163:2585–2593. doi: 10.1016/S0002-9440(10)63613-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Telling GC, Parchi P, DeArmond SJ, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 72.Uro-Coste E, Cassard H, Simon S, et al. Beyond PrPres type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog. 2008;4:e1000029. doi: 10.1371/journal.ppat.1000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–1135. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weissmann C. Thoughts on mammalian prion strains. Folia Neuropathol. 2009;47:104–113. [PubMed] [Google Scholar]
- 75.Yuan J, Xiao X, McGeehan J, et al. Insoluble aggregates and protease-resistant conformers of prion protein in uninfected human brains. J Biol Chem. 2006;281:34848–34858. doi: 10.1074/jbc.M602238200. [DOI] [PubMed] [Google Scholar]
- 76.Yuan J, Dong Z, Guo JP, et al. Accessibility of a critical prion protein region involved in strain recognition and its implications for the early detection of prions. Cell Mol Life Sci. 2008;65:631–643. doi: 10.1007/s00018-007-7478-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zou W, Puoti G, Xiao X, et al. Protease-sensitive prionopathy: A new sporadic disease of the prion protein. Annals Neurol. 2010;68:162–172. doi: 10.1002/ana.22094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zou WQ, Langeveld J, Xiao X, et al. PrP conformational transitions alter species preference of a PrP-specific antibody. J Biol Chem. 2010;285:13874–13884. doi: 10.1074/jbc.M109.088831. [DOI] [PMC free article] [PubMed] [Google Scholar]