

Abstract
Objectives
Targeting the COX-2/prostanoid pathway is considered an intriguing approach for therapy and prevention of several cancers. However, the molecular mechanisms that underlie the pro-tumorigenic properties of COX-2 in pancreatic cancer (PaCa) are still poorly understood. The purpose of the present study was to characterize the phenotype of COX-2 expressing syngeneic PaCa cells.
Methods
COX-2 negative MIA PaCa-2 cells were stably transduced with COX-2 or control viruses (MP2+COX-2 and MP2−COX-2). Prostaglandin E2 (PGE2) production was measured by liquid chromatography and tandem mass spectrometry. Anchorage-dependent and -independent cell growth was analyzed by cell count and 3D collagen cell culture system, respectively. Changes in apoptotic gene expression were measured by a PCR array. The growth of tumors in vivo was evaluated in a xenograft animal model.
Results
Stable expression of COX-2 increased anchorage-dependent and -independent cell growth, which was accompanied by elevated PGE2 production. Several significant differences in apoptotic gene expression were detected between MP2+COX-2 and MP2−COX-2 cells. Furthermore, MP2+COX-2 cells grew faster than MP2−COX-2 cells in a xenograft animal model.
Conclusions
Our results will provide the basis for more mechanistic studies on the role of COX-2 in PaCa and may help to develop novel therapeutic strategies aiming at the COX-2/prostanoid pathway.
Keywords: Cyclooxygenase-2, prostaglandin E2, pancreatic cancer, proliferation, arachidonic acid
Introduction
Of all gastrointestinal carcinomas, pancreatic cancer (PaCa) has the most unfavorable prognosis. Despite some progress made in other epithelial tumors, pancreatic cancer still remains an almost universally fatal disease, with mortality rates approaching the number of newly diagnosed cases. An estimated 21,050 men and 21,420 women will be diagnosed with pancreatic cancer in the year 2009 in the United States, and the majority of these patients will die within 6 months (1). For the past decade, gemcitabine has been considered the standard chemotherapeutic agent in the treatment of pancreatic cancer. However, studies using gemcitabine either as monotherapy or as combination therapy with cytotoxic or molecular targeted agents revealed only a marginal overall survival benefit of 1 to 2 months at best (2).
Cyclooxygenase-2 (COX-2), the rate-limiting enzyme for prostanoid production, is over-expressed in many types of cancer, including PaCa (3–8). There is substantial evidence that COX-2 contributes to the phenotype of human malignancies. The pro-tumorigenic properties of COX-2 are thereby generally thought to be mediated by COX-2 generated prostanoids, of which prostaglandin E2 (PGE2) is the most abundant prostanoid species in many tumors (9, 10). PGE2 has been shown to promote tumor growth and spread by multiple mechanisms, including stimulation of cellular proliferation, invasion, and angiogenesis, inhibition of apoptotic cell death, and modulation of immune response (11–14). In pancreatic cancers, COX-2 and COX-2 derived PGE2 have been found to stimulate growth, invasion, and angiogenesis (15–17). We have previously shown that enzymes involved in PGE2 biosynthesis, including cytoplasmic phospholipase A2 (cPLA2), COX-2, microsomal and cytoplasmic prostaglandin E synthases (mPGES-1 and -2, cPGES), are variably over-expressed in the majority of human pancreatic cancers (18). In addition, we have previously demonstrated that selective COX-2 inhibitors attenuate the growth PaCa and delay the progression of PaCa precursor lesions in mouse models (19, 20), indicating that the COX-2/prostanoid pathway is an intriguing target for PaCa therapy and prevention. However, the precise molecular signals that underlie the pro-tumorigenic properties of COX-2 in PaCa are still poorly described. The aim of the present study was to characterize the phenotype of COX-2 expressing syngeneic human PaCa cells to clearly delineate the phenotypic changes brought upon pancreatic cancer cells by expressing COX-2. Our data provide strong evidence that COX-2 confers profound growth advantage to human pancreatic cancer cells in vitro and in vivo.
Materials and Methods
Reagents
The following antibodies were used: mPGES-1, mPGES-2, cPGES, COX-2 (Cayman Chemical Company, Ann Arbor, MI); cPLA2, GAPDH (Cell Signaling Technology, Inc., Danvers, MS); β-actin (Sigma Chemical, St Lois, MO). COX-2 electrophoresis standard was purchased from Cayman Chemical Company. Arachidonic acid was obtained from Sigma Chemical). Other reagents were purchased from common commercial sources.
Cell culture
The undifferentiated human pancreatic cancer cell line MIA PaCa-2 (COX-1 positive, COX-2 negative) was obtained from the American Type Culture Collection (ATCC, Rockville, MD). Cells were cultured and maintained in Dulbecco modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin G (100 U/ml), and streptomycin (100 μg/ml) at 37 °C in a 10% CO2.
Creation of COX-2 expressing stable cell lines
The full length human COX-2 cDNA was cloned into a third generation, self-inactivating pRRL-sin-cPPT-hCMV-MCS-IRES-GFP lentiviral vector. COX-2 negative MIA PaCa-2 cells were transduced with COX-2/GFP (MP2+COX-2) or GFP alone (MP2−COX-2) encoding lentiviruses. Transduced MIA PaCa-2 cells were subsequently sorted by FACS to yield >95% pure MP2+COX-2 and MP2−COX-2 cell clones.
Western blot analysis
Total cell lysates from cultures were prepared using ice-cold modified radioimmunoprecipitation assay (RIPA) buffer. Equal amounts of protein were fractionated on 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Proteins were detected using specific primary antibodies and horseradish peroxidase-conjugated secondary antibodies (Pierce, Rockford, IL). Protein-antibody complexes were visualized with the SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Immunofluorescent staining
Cells were seeded into a Lab-Tek®II Chamber Slide™ System (Nalge Nunc International, Naperville, IL). At subconfluence, cells were fixed in 4% paraformaldehyde at room temperature for 15 minutes. Cells were then washed three times and incubated with 5% BSA for 30 minutes to block nonspecific binding. Slides were incubated with monoclonal anti-COX-2 antibodies for 1 hour. After three washes with PBS, slides were incubated with secondary antibody conjugates in the dark for 1 hour. After three washes with PBS, slides were mounted with VECTASHIELD containing DAPI (Vector Laboratories Inc., Burlingame, CA). Fixed cells were observed under a fluorescence microscope.
Liquid chromatography and tandem mass spectrometry
Prostaglandin levels in the culture medium were determined by liquid chromatography and tandem mass spectrometry (LC-MS/MS). Prostaglandins were extracted using a solid-phase method. A solution of 10% butylated hydroxytoluene and deuterated prostaglandins as internal standards were added to the culture medium. The solution was then applied to a Sep-Pak C18 cartridge (Waters Corp, Milford, MA) that had been preconditioned with methanol and water. Prostaglandins were eluted with methanol, and the eluate was evaporated under a stream of nitrogen. The residue was dissolved in methanol/10mM ammonium acetate buffer (70:30, v/v, pH 8.5). The extracted prostaglandins were quantified by LC-MS/MS using a Quattro Ultima MS/MS (Micromass, Beverly, MA) equipped with an Agilent HP 1100 binary pump high-performance liquid chromatography inlet. Prostaglandins were separated using a Luna 3K Phenyl-Hexyl 2- × 150-mm liquid chromatography column (Phenomenex, Torrance, CA). Prostaglandins were detected using electrospray negative ionization and multiple-reaction monitoring of the transition ions for the metabolites and their internal standards as described previously (21).
Cell growth assay
Anchorage-dependent cell growth of MP2+COX-2 and MP2−COX-2 cells treated with arachidonic acid (AA) (10 μM) was analyzed by cell count. Briefly, cells were seeded on 24-well plates and incubated overnight. The medium was replaced with serum free medium, and the COX-2 substrate AA was added to the cultures once daily. Cell number was determined by counting the cells under a light microscope.
Colony formation assay
Anchorage-independent cell growth was assessed by a 3D Collagen Cell Culture System (Chemicon, Temecula, CA). 20,000 cells were seeded on matrigel-coated 24-well plates and were treated with AA (10 μM). AA was added to the Matrigel and the culture medium containing 0.5% FBS and 0.1% BSA. The medium was changed every other day. After one week, colonies were counted and photographed under a light microscope.
RNA isolation and apoptosis gene array
Total RNA was extracted using the Aurum total RNA mini kit (Bio-Rad Laboratories, Hercules, CA). RNA quality and quantity were measured using the Experion Automated Electrophoresis System (Bio-Rad). RNA concentrations were additionally confirmed spectrophotometrically. RNA was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad). The expression of 84 genes involved in apoptosis was analyzed by the human apoptosis RT2 Profiler™ PCR array according to the manufacturers (SABiosciences, Frederick, MD) on an iQ5 Real-Time PCR Detection System (Bio-Rad).
Animal model
Animal studies were approved by the Chancellor’s Animal Research Committee of the University of California, Los Angeles, in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals. MP2+COX-2 and MP2−COX-2 pancreatic cancer cells (2×106) were injected subcutaneously into the flank of nude mice (n = 5 each). After 2 weeks, animals were sacrificed and the tumor volume was assessed using the formula for a hemi-ellipsoid (2/3*π*r1*r2*r3) as described previously (20).
COX-2 immunohistochemistry
Formalin-fixed, paraffin-embedded tissues were sectioned (4 μm), deparaffinized in xylene, and rehydrated in a graded ethanol series. Sections were subjected to heat-induced epitope retrieval using boiling citrate buffer (pH 6.0). Endogenous peroxidase activity was quenched by 0.3% H2O2 and 5% bovine serum albumin was applied for 10 minutes to block nonspecific binding. Sections were then incubated with anti-COX-2 antibody. Immunoreactivity was detected by the UltraVision Detection System using 3-amino-9-ethylcarbozol as the chromogen according to the manufacturer’s instructions (Lab Vision, Fremont, CA).
Western blot analysis of tumor homogenates
Tumor extracts were prepared by homogenizing and sonicating non-necrotic areas of pancreatic cancers in anti-protease lysis buffer (Roche Applied Science, Indianapolis, IN). Tumor homogenates were cleared by centrifugation. COX-2 protein expression was determined by sodium dodecyl sulfate-gel electrophoresis and immunoblotting as described above.
Statistical analysis
Data are presented as means ± SD, and statistical comparisons were made using the Student’s t test for paired observations. Comparisons of more than two groups were made by a one-way ANOVA with post hoc Holm-Sidak analysis for pairwise comparisons and comparisons versus control. An alpha value of 0.05 was used to determine significant differences. All statistics were done in SigmaStat 3.1 (Systat Software, Inc.).
Results
Generation of MP2+COX2 and MP2−COX-2 cells
To clearly delineate the role of COX-2 on the malignant phenotype of human pancreatic cancer cells, MIA PaCa-2 cells were stably transduced with lentiviruses encoding for the full length human COX-2. Previous studies have demonstrated that MIA PaCa-2 cells express the COX-1 isoform but not COX-2 (17, 22). The lentiviruses also contained the sequence for the green fluorescent protein (GFP). After transduction, COX-2 expressing MIA PaCa-2 cells (MP2+COX2) and control cells (MP2−COX2) were sorted to yield >95% pure cell populations. Expression of COX-2 in MP2+COX2 and absence of COX-2 in MP2−COX2 was confirmed by immunofluorescence (Fig. 1A) and Western blotting (Fig. 1B). To confirm stable expression of COX-2 over several passages, MP2+COX2 cells were cultured over 6 weeks. As shown in Fig. 1C, COX-2 expression was stable in MP2+COX2 cells over 6 weeks.
Figure 1. Generation of MP2+COX2 and MP2−COX-2 cells.

A, COX-2 protein expression was detected by immunofluorescence staining (top panel). Nuclei were stained with DAPI (bottom panel).
B, The expression of COX-2 in MP2+COX2 and the absence of COX-2 in MP2−COX2 was confirmed by western blotting. Equal protein loading was confirmed by β-actin.
C, Successful stable expression of COX-2 over 6 weeks in MP2+COX2 cells was confirmed by western blot.
Characterization of PGE2 production in MP2+COX2 and MP2−COX2 cells
There is strong evidence that the pro-tumorigenic effects of COX-2 are mediated largely by the generation of prostaglandins (9, 10). Among the various prostaglandin species, PGE2 is commonly found to be the most abundant in human cancers (9, 10). We have previously demonstrated that PGE2 is generated by COX-2 in COX-2 positive human pancreatic cancer cells and stimulates growth and angiogenesis in pancreatic cancer cells (17, 22). Having generated MP2+COX2 cells we first confirmed production of PGE2 in these cells. PGE2 levels in the culture medium of MP2+COX2 andMP2−COX2 cells under serum free conditions were measured by LC-MS/MS. In the absence of AA, the substrate for COX-2, PGE2 production was not detected in either cell line. However, exposure of MP2+COX2 cells to AA (10 μM) dramatically increased PGE2 production. AA-induced PGE2 production was elevated within minutes in MP2+COX2, reached its maximum at ~30 min, and was still enhanced after 6 hours. In contrast, only a slight increase in PGE2 production was found in MP2−COX2 cells after stimulation with AA (Fig. 2). The maximal AA-stimulated PGE2 production in MP2−COX2 cells was >2-fold lower than those in MP2+COX2 cells. These results are completely consistent with our previous data that AA robustly stimulated PGE2 production in COX-2 positive cell lines (BxPC-3, Capan-2, and HPAF-II), but only slightly in COX-2 negative cell lines (MIA PaCa-2 and Panc-1) (17).
Figure 2. PGE2 production in MP2+COX2 and MP2−COX-2 cells.
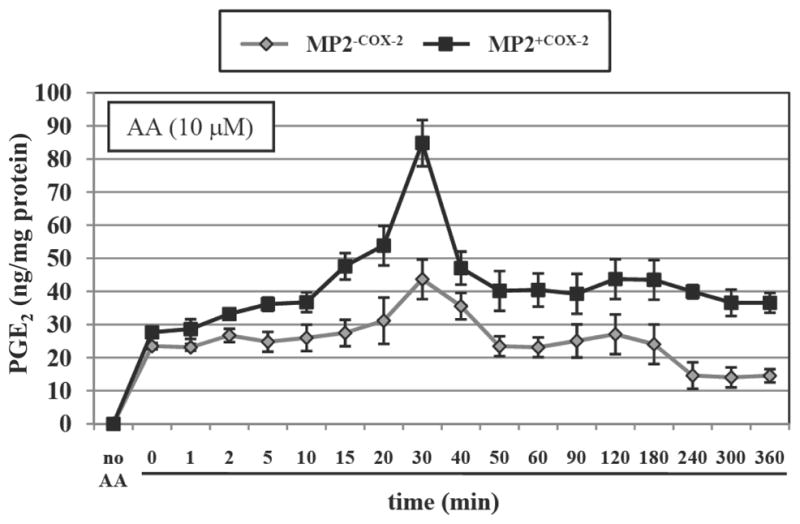
The level of PGE2 in the cell culture supernatant was measured by LC-MS/MS and expressed as ng/mg protein. Treatment of MP2+COX2 cells with arachidonic acid (10 μM) greatly increased PGE2 production in a time-dependent manner, while only a slight increase was seen in the MP2−COX2 cells. Results are representative of three independent experiments and are presented as means ± SD (bars).
The biosynthesis of PGE2 is initiated by the release of the polyunsaturated fatty acid AA from membrane phospholipids by cytoplasmic phospholipase A2 (cPLA2), and proceeds through cyclooxygenases and prostaglandin E synthases (23, 24). There are three different prostaglandin E synthases, namely cytoplasmic prostaglandin E synthase (cPGES), microsomal prostaglandin E synthase-1, and -2 (mPGES1, mPGES2). We have previously demonstrated that the aforementioned enzymes are variably expressed in human pancreatic cancers (18). To evaluate whether forced expression of COX-2 in MP2+COX2 cells alters expression of these enzymes, which could affect PGE2 synthesis, we measured protein expression of cPLA2, COX-2, cPGES, mPGES1, and mPGES2 by Western blotting. We found no detectable changes in protein expression of any enzyme in MP2+COX2 and MP2−COX2 cells with and without AA (Fig. 3). Notably, there was no expression of cPLA2 in these cells, underscoring the necessity of adding AA to the cell cultures to stimulate PGE2 production.
Figure 3. Expression of proteins involved in PGE2 biosynthesis.
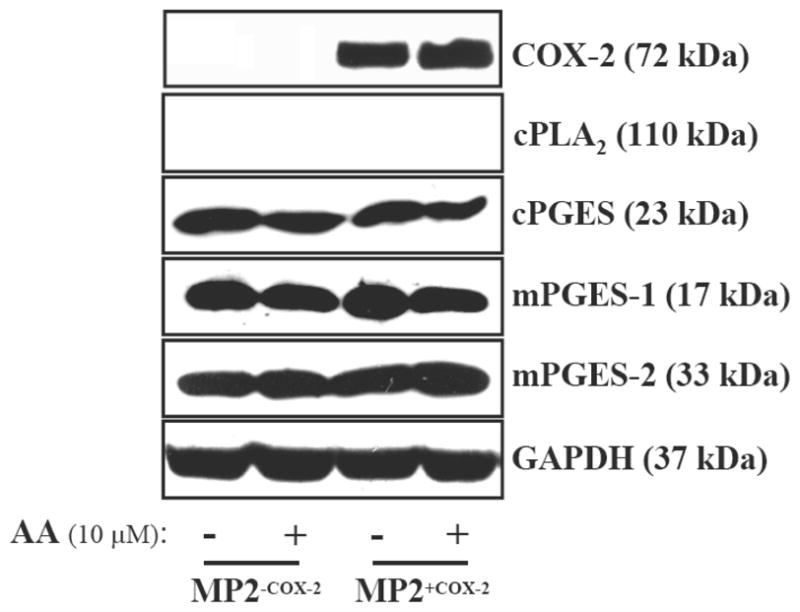
Western blot analysis of COX-2, cPLA2, mPGES-1, mPGES-2, and cPGES were performed in MP2+COX2 and MP2−COX2 cells. GAPDH expression was used as a loading control. Results are representative of three independent experiments.
Overexpression of COX-2 enhances growth of MIA PaCa-2 cells in vitro
We have previously showed that exogenous AA stimulated growth of COX-2 positive PaCa cell lines, but not of COX-2 negative PaCa cell lines (22). To clearly delineate whether COX-2 is responsible for the more robust growth-stimulating effect of AA in human pancreatic cancer cells, MP2+COX2 and MP2−COX2 cells were exposed to AA and anchorage-dependent and -independent cell growth was measured. In the absence of exogenous AA there was no difference in the anchorage-dependent cell growth between MP2+COX2 and MP2−COX2 cells (data not shown). However, exposure of MP2+COX2 cells to AA (10 μM) resulted in a significant increase in anchorage-dependent cell growth compared to MP2−COX2 cells treated with AA (Fig. 4A). The differences in the number of MP2+COX2 and MP2−COX2 cells were significant from day 4. In addition, using an anchorage-independent 3D Collagen Cell Culture System, AA increased the number of MP2+COX2 colonies after 7 days by 170% while it increased the number of colonies of MP2−COX2 cells only by 35% (Fig. 4B and C).
Figure 4. Anchorage-dependent and –independent growth of MP2+COX2 and MP2+COX2 cells.

A, MP2+COX2 and MP2−COX2 cells were treated with 10 μM arachidonic acid for 6 days, and anchorage-dependent cell proliferation were measured by cell count. Results are representative of three independent experiments and are presented as mean±SD (bars). *p<0.01 vs. MP2−COX2.
B, Anchorage-independent cell growth of MP2+COX2 and MP2−COX2 cells was measured by in 3D collagen cell culture system. Results are representative of three independent experiments and are presented as means ± SD (bars). *p<0.01 vs. MP2−COX2.
C, Representative images of colony formation. Top panel, MP2−COX2 cells; bottom panel, MP2+COX2 cells. There were no differences between MP2+COX2 and MP2−COX2 cells without AA (left panel).
Expression of COX-2 alters apoptotic gene expression in MIAPaCa-2 cells
It has been reported that pharmacological inhibition or genetic deletion of COX-2 overcomes the resistance to apoptotic cell death in several cancer cell lines, including PaCa (25–29), suggesting that COX-2 and COX-2 derived prostaglandins protect cancer cells from apoptosis. To gain first insight whether the increase in cell growth in MP2+COX2 cells was mediated by inhibition of apoptotic cell death, we evaluated changes in apoptotic gene expression in MP2+COX2 and MP2−COX2 cells. MP2+COX2 and MP2−COX2 cells were treated with AA (10 μM), after which total RNA was extracted and changes in apoptotic gene expression was measured using the human apoptosis RT2 Profiler™ PCR array kit. As shown in Figure 5, several apoptosis-related genes were significantly differentially expressed in MP2+COX2 and MP2−COX2 cells. Those differences (MP2+COX2 relative to MP2−COX2 cells) included up-regulation of genes of the TNF ligand family (TNFSF10), TNF receptor family (TNFRSF21), Bcl-2 family (BNIP1 and HRK), and CIDE domain family (CIDEA), as well as down-regulation of other genes in the TNF ligand family (FASLG), TNF receptor family (TNFRSF9, TNFRSF11B), and caspase family (CASP1 and CASP5).
Figure 5. Apoptosis PCR array.

Total RNA was isolated from MP2+COX2 and MP2−COX2 cells, and the relative expression of 84 cell death-related genes was measured by Human Apoptosis PCR Array. Several significant differences (>2 fold increase (red) or decrease (green) in gene expression) were detected between MP2+COX2 and MP2−COX2 cells. Results are representative of three independent experiments.
Expression of COX-2 confers growth advantage of MIA PaCa-2 cells in vivo
Having demonstrated that COX-2 expression stimulated growth of MP2+COX2 cells in vitro, we sought to determine whether a similar observation could be made in a xenograft animal model, in which MP2+COX2 and MP2−COX2 cells were injected subcutaneously into nude mice. After 2 weeks, MP2+COX2 tumors were significantly larger than MP2−COX2 tumors (294 ± 135 mm3 vs. 122 ± 48 mm3, p<0.05) (Fig. 6A). Immunohistochemistry and Western blot confirmed COX-2 protein expression in MP2+COX2 tumors but not in MP2−COX2 tumors (Fig. 6B and C). Further histo-pathological examination revealed that MP2−COX2 tumors were comprised of variably-sized solid nests of tumor cells that were circumscribed by thin fibrous or fibrovascular septa with accompanying mild acute and chronic inflammatory cells. The solid tumor nests also had a delicate fibrovascular framework and were associated with scattered neutrophilic inflammation. In addition, there was prominent central necrosis (up to 30–40% of the tumor) and additional scattered smaller necrotic abscesses with associated neutrophilic inflammation. In striking contrast, MP2+COX2 tumors were comprised of densely packed sheets of tumor cells, lacking the nested arrangement and discrete fibrous septa of the MP2−COX2 tumors. In addition, the extent of central necrosis was significantly less prominent in MP2+COX2 tumors (Fig. 6D).
Figure 6. COX-2 confers growth advantage in an animal model.


A, Tumor volume of subcutaneous MP2+COX2 and MP2−COX2 tumors in nude mice. Data are presented as the means ± SD of 5 MP2+COX2 tumors and of 4 MP2−COX2 tumors.
B, Immunohistchemistry of COX-2 in representative MP2+COX2 and MP2−COX2 tumors.
C, Analysis of COX-2 protein expression in MP2+COX2 andMP2−COX2 tumors by Western blot. For positive control (PC), cell lysates of BxPC-3 cells were used. GAPDH expression was used as a loading control.
D, Representative H.E. staining. Left panel, MP2−COX2 tumors; right panel, MP2+COX2 tumors. In MP2−COX2 tumors, there is prominent central necrosis and additional scattered smaller necrotic abscesses with associated neutrophilic inflammation.
Discussion
There is substantial evidence that COX-2, the rate-limiting enzyme in the biosynthesis of prostanoids, is critical for tumor development and growth (3–8, 30). Tissue-specific expression of COX-2 in genetically-engineered mouse models increased the risk of developing certain types of cancer (31), suggesting a role in early tumor development. In this regard, we have previously shown that oral administration of a selective COX-2 inhibitor delayed the progression of pancreatic cancer precursor lesions in a conditional Kras mouse model of pancreatic cancer (19). In addition, genetic ablation of COX-2 or pharmacologic inhibition of its activity commonly results in decreased tumor growth in a variety of animal models (32–35), demonstrating the potential of COX-2 as a therapeutic target. The pro-tumorigenic properties of COX-2 are thereby believed to be mediated by the generation of prostanoids, of which PGE2 is commonly the most abundant species in human cancers (9, 10). The majority of human pancreatic cancers express COX-2 but a substantial proportion is considered COX-2 negative. To clearly delineate the role of COX-2 to the malignant phenotype of human pancreatic cancer cells, we generated syngeneic cell lines with and without COX-2 expression. This model allows a better understanding of the role of COX-2 in pancreatic cancer biology than using different cell lines with varying COX-2 expression. Precise knowledge about the pro-tumorigenic mechanisms of COX-2 is necessary to understand and predict the efficacy of targeted therapies aimed at inhibiting the COX-2 enzyme.
Our studies demonstrated that in the absence of the COX-2 substrate, arachidonic acid, MP2+COX2 cells grew in a similar fashion than MP2−COX2 cells. This could be explained by the lack of detectable expression of cPLA2, the enzyme that releases AA from membrane phospholipids. In accordance to that finding was the observation that PGE2 production was very low and virtually identical between MP2+COX2 and MP2−COX2 cells in the absence of AA. However, addition of AA to the cultures robustly enhanced the growth of MP2+COX2 cells, which was significantly more pronounced than the effect of AA in MP2−COX2 cells. This was accompanied by a significantly higher production of PGE2 in MP2+COX2 cells compared to MP2−COX2 cells. There was a slight increase in AA-induced PGE2 production in MP2−COX2 cells, which may have been caused by constitutively expressed COX-1 in MIA PaCa-2 cells. Since there were no differences among all other enzymes involved in PGE2 biosynthesis between MP2+COX2 and MP2−COX2 cells, the increased production of PGE2 in MP2+COX2 cells was clearly caused by COX-2. The lack of cPLA2 expression in MIA PaCa-2 cells thereby provides a unique opportunity to functionally “turn on” or “turn off” COX-2 activity.
An increase in cellular growth can be generally accomplished by stimulation of cell division (proliferative effect) or inhibition of cell death (survival signal). In order to gain first insight into the mechanisms of the growth-promoting effects of COX-2 expression in MIA PaCa-2 cells, we employed an apoptosis array that covers 84 apoptosis-related genes. We found several significantly up- and down-regulated genes in MP2+COX2 compared to MP2−COX2 cells, indicating that COX-2 expression alters the apoptotic/survival pathways in pancreatic cancer cells. The importance of COX-2 and COX-2 derived prostanoids in cell death and survival have been demonstrated in many reports. Tjiu and colleagues revealed that COX-2 induces anti-apoptotic activity in basal cell carcinoma by up-regulation of Bcl-2 and Mcl-1. (36) Singh and coworkers also reported that COX-2 transfected MCF7 cells produced a significantly higher expression level of Bcl-2 than the parental cells, and that COX-2 expression correlated with the increased resistance to doxorubicin. (37) Having demonstrated here that COX-2 changes the expression of several apoptosis-related genes, further studies are clearly necessary to delineate the precise signaling network that is affected by COX-2.
Similar to our in vitro studies, the growth-promoting effect of COX-2 could also be observed in a xenograft mouse model. MP2+COX2 tumors contained significantly higher levels of PGE2 than MP2−COX2 tumors (data not shown), suggesting a pivotal role of COX-2 derived PGE2 in tumor growth; a notion which has been corroborated by many reports (11, 15, 32, 38–40). In this context, our previous data demonstrated that inhibition of PGE2 production by a selective COX-2 inhibitor decreased tumor angiogenesis (17). As a source of exogeneous AA in our animal study, the animal diet contains significant amounts of linoleic acid, which can be converted in vivo to AA. In our current study MP2+COX2 tumors lacked the prominent central necrosis that was present in MP2−COX2 tumors, suggesting sufficient blood supply (and hence tumor angiogenesis) in MP2+COX2 cancers. In addition, MP2+COX2 tumors lacked the strong presence of inflammatory infiltrates and fibrous septae that were seen in MP2−COX2. The inflammatory infiltration and fibrous septae may be reactions to the central necrosis. However, the lack of both in MP2+COX2 tumors could also indicate a direct immune-suppressive effect of COX-2, as has been described in other reports (41, 42). In summary, we present strong evidence that expression of COX-2 in pancreatic cancer cells confers a distinct growth advantage in vitro and in vivo. This effect may be mediated by modulating the apoptotic/survival machinery, by increasing tumor angiogenesis, and by immunosuppression. The syngeneic cell lines generated in our studies will provide an ideal basis for detailed mechanistic studies deciphering the precise role of COX-2 in pancreatic cancer pathobiology.
Acknowledgments
Financial support: This work was supported by the National Institutes of Health (CA104027, CA122042, and AT003960).
The stable cell lines were generated at the UCLA Vector Core (Kasahara N.). Prostaglandins were measured by Peiying Yang (M.D. Anderson Cancer Center, Houston, TX).
Abbreviations
- COX-2
Cyclooxygenase-2
- PGE2
prostaglandin E2
- PaCa
pancreatic cancer
- LC-MS/MS
liquid chromatography and tandem mass spectrometry
- AA
arachidonic acid
- cPLA2
cytoplasmic phospholipase A2
- mPGES
microsomal prostaglandin E synthases
- cPGES
cytoplasmic prostaglandin E synthases
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Hochster HS, Haller DG, de Gramont A, et al. Consensus report of the international society of gastrointestinal oncology on therapeutic progressin advanced pancreatic cancer. Cancer. 2006;107:676–685. doi: 10.1002/cncr.22036. [DOI] [PubMed] [Google Scholar]
- 3.Petkova DK, Clelland C, Ronan J, et al. Overexpression of cyclooxygenase-2 in non-small cell lung cancer. Respir Med. 2004;98:164–172. doi: 10.1016/j.rmed.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Wang D, Dubois RN. Cyclooxygenase-2: a potential target in breast cancer. Semin Oncol. 2004;31:64–73. doi: 10.1053/j.seminoncol.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 5.Eberhart CE, Coffey RJ, Radhika A, et al. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 6.Gupta S, Srivastava M, Ahmad N, et al. Over-expression of cyclooxygenase-2 in human prostate adenocarcinoma. Prostate. 2000;42:73–78. doi: 10.1002/(sici)1097-0045(20000101)42:1<73::aid-pros9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 7.Okami J, Yamamoto H, Fujiwara Y, et al. Overexpression of cyclooxygenase-2 in carcinoma of the pancreas. Clin Cancer Res. 1999;5:2018–2024. [PubMed] [Google Scholar]
- 8.Yip-Schneider MT, Barnard DS, Billings SD, et al. Cyclooxygenase-2 expression in human pancreatic adenocarcinomas. Carcinogenesis. 2000;21:139–46. doi: 10.1093/carcin/21.2.139. [DOI] [PubMed] [Google Scholar]
- 9.Rigas B, Goldman IS, Levine L. Altered eicosanoid levels in human colon cancer. J Lab Clin Med. 1993;122:518–523. [PubMed] [Google Scholar]
- 10.Uefuji K, Ichikura T, Mochizuki H. Cyclooxygenase-2 expression is related to prostaglandin biosynthesis and angiogenesis in human gastric cancer. Clin Cancer Res. 2000;6:135–138. [PubMed] [Google Scholar]
- 11.Jain S, Chakraborty G, Raja R, et al. Prostaglandin E2 regulates tumor angiogenesis in prostate cancer. Cancer Res. 2008;68:7750–7759. doi: 10.1158/0008-5472.CAN-07-6689. [DOI] [PubMed] [Google Scholar]
- 12.Krysan K, Reckamp KL, Dalwadi H, et al. Prostaglandin E2 activates mitogen-activated protein kinase/Erk pathway signaling and cell proliferation in non-small cell lung cancer cells in an epidermal growth factor receptor-independent manner. Cancer Res. 2005;65:6275–6281. doi: 10.1158/0008-5472.CAN-05-0216. [DOI] [PubMed] [Google Scholar]
- 13.Singh B, Berry JA, Shoher A, et al. COX-2 overexpression increases motility and invasion of breast cancer cells. Int J Oncol. 2005;26:1393–1399. [PubMed] [Google Scholar]
- 14.Sheng H, Shao J, Morrow JD, et al. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res. 1998;58:362–366. [PubMed] [Google Scholar]
- 15.Chu J, Lloyd FL, Trifan OC, et al. Potential involvement of the cyclooxygenase-2 pathway in the regulation of tumor-associated angiogenesis and growth in pancreatic cancer. Mol Cancer Ther. 2003;2:1–7. [PubMed] [Google Scholar]
- 16.Ito H, Duxbury M, Benoit E, et al. Prostaglandin E2 Enhances Pancreatic Cancer Invasiveness through an Ets-1–Dependent Induction of Matrix Metalloproteinase-2. Cancer Res. 2004;64:7439–7446. doi: 10.1158/0008-5472.CAN-04-1177. [DOI] [PubMed] [Google Scholar]
- 17.Eibl G, Bruemmer D, Okada Y, et al. PGE2 is generated by specific COX-2 activity and increases VEGF production in COX-2-expressing human pancreatic cancer cells. Biochem Biophys Res Commun. 2003;306:887–897. doi: 10.1016/s0006-291x(03)01079-9. [DOI] [PubMed] [Google Scholar]
- 18.Hasan S, Satake M, Dawson DW, et al. Expression analysis of the prostaglandin E2 production pathway in human pancreatic cancers. Pancreas. 2008;37:121–127. doi: 10.1097/MPA.0b013e31816618ba. [DOI] [PubMed] [Google Scholar]
- 19.Funahashi H, Satake M, Dawson DW, et al. Delayed progression of pancreatic intraepithelial neoplasia in a conditional Kras(G12D) mouse model by a selective cyclooxygenase-2 inhibitor. Cancer Res. 2007;67:7068–7071. doi: 10.1158/0008-5472.CAN-07-0970. [DOI] [PubMed] [Google Scholar]
- 20.Eibl G, Takata Y, Boros LG, et al. Growth stimulation of COX-2-negative pancreatic cancer by a selective COX-2 inhibitor. Cancer Res. 2005;65:982–990. [PubMed] [Google Scholar]
- 21.Yang P, Felix E, Madden T, et al. Quantitative high-performance liquid chromatography/electrospray ionization tandem mass spectrometric analysis of 2- and 3-series prostaglandins in cultured tumor cells. Anal Biochem. 2002;308:168–177. doi: 10.1016/s0003-2697(02)00218-x. [DOI] [PubMed] [Google Scholar]
- 22.Funahashi H, Satake M, Hasan S, et al. Opposing effects of n-6 and n-3 polyunsaturated fatty acids on pancreatic cancer growth. Pancreas. 2008;36:353–362. doi: 10.1097/MPA.0b013e31815ccc44. [DOI] [PubMed] [Google Scholar]
- 23.Smith WL, Marnett LJ, DeWitt DL. Prostaglandin and thromboxane biosynthesis. Pharmacol Ther. 1991;49:153–179. doi: 10.1016/0163-7258(91)90054-p. [DOI] [PubMed] [Google Scholar]
- 24.Chell S, Kaidi A, Williams AC, et al. Mediators of PGE2 synthesis and signalling downstream of COX-2 represent potential targets for the prevention/treatment of colorectal cancer. Biochim Biophys Acta. 2006;1766:104–119. doi: 10.1016/j.bbcan.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 25.Li M, Wu X, Xu XC. Induction of apoptosis in colon cancer cells by cyclooxygenase-2 inhibitor NS398 through a cytochrome c-dependent pathway. Clin Cancer Res. 2001;7:1010–1016. [PubMed] [Google Scholar]
- 26.Dandekar DS, Lokeshwar BL. Inhibition of cyclooxygenase (COX)-2 expression by Tet-inducible COX-2 antisense cDNA in hormone-refractory prostate cancer significantly slows tumor growth and improves efficacy of chemotherapeutic drugs. Clin Cancer Res. 2004;10:8037–8047. doi: 10.1158/1078-0432.CCR-04-1208. [DOI] [PubMed] [Google Scholar]
- 27.Hida T, Kozaki K, Muramatsu H, et al. Cyclooxygenase-2 inhibitor induces apoptosis and enhances cytotoxicity of various anticancer agents in non-small cell lung cancer cell lines. Clin Cancer Res. 2000;6:2006–2011. [PubMed] [Google Scholar]
- 28.Ding XZ, Tong WG, Adrian TE. Blockade of cyclooxygenase-2 inhibits proliferation and induces apoptosis in human pancreatic cancer cells. Anticancer Res. 2000;20:2625–2631. [PubMed] [Google Scholar]
- 29.Levitt RJ, Pollak M. Insulin-like growth factor-I antagonizes the antiproliferative effects of cyclooxygenase-2 inhibitors on BxPC-3 pancreatic cancer cells. Cancer Res. 2002;62:7372–7376. [PubMed] [Google Scholar]
- 30.Sinicrope FA. Targeting cyclooxygenase-2 for prevention and therapy of colorectal cancer. Mol Carcinog. 2006;45:447–454. doi: 10.1002/mc.20232. [DOI] [PubMed] [Google Scholar]
- 31.Müller-Decker K, Fürstenberger G, Annan N, et al. Preinvasive duct-derived neoplasms in pancreas of keratin 5-promoter cyclooxygenase-2 transgenic mice. Gastroenterology. 2006;130:2165–2178. doi: 10.1053/j.gastro.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Chen W, Xie X, et al. Celecoxib inhibits tumor growth and angiogenesis in an orthotopic implantation tumor model of human colon cancer. Exp Oncol. 2008;30:42–51. [PubMed] [Google Scholar]
- 33.Qin J, Yuan J, Li L, et al. In vitro and in vivo inhibitory effect evaluation of cyclooxygenase-2 inhibitors, antisense cyclooxygenase-2 cDNA, and their combination on the growth of human bladder cancer cells. Biomed Pharmather. 2009;63:241–248. doi: 10.1016/j.biopha.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 34.Stasinopoulos I, O’Brien DR, Wildes F, et al. Silencing of cyclooxygenase-2 inhibits metastasis and delays tumor onset of poorly differentiated metastatic breast cancer cells. Mol Cancer Res. 2007;5:435–442. doi: 10.1158/1541-7786.MCR-07-0010. [DOI] [PubMed] [Google Scholar]
- 35.Gregor JI, Kilian M, Heukamp I, et al. Effects of selective COX-2 and 5-LOX inhibition on prostaglandin and leukotriene synthesis in ductal pancreatic cancer in Syrian hamster. Prostaglandins Leukot Essent Fatty Acids. 2005;73:89–97. doi: 10.1016/j.plefa.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 36.Tjiu JW, Liao YH, Lin SJ, et al. Cyclooxygenase-2 overexpression in human basal cell carcinoma cell line increases antiapoptosis, angiogenesis, and tumorigenesis. J Invest Dermatol. 2006;126:1143–1151. doi: 10.1038/sj.jid.5700191. [DOI] [PubMed] [Google Scholar]
- 37.Singh B, Cook KR, Vincent L, et al. Cyclooxygenase-2 induces genomic instability, BCL2 expression, doxorubicin resistance, and altered cancer-initiating cell phenotype in MCF7 breast cancer cells. J Surg Res. 2008;147:240–246. doi: 10.1016/j.jss.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 38.Toomey DP, Murphy JF, Conlon KC. COX-2, VEGF and tumour angiogenesis. Surgeon. 2009;7:174–180. doi: 10.1016/s1479-666x(09)80042-5. [DOI] [PubMed] [Google Scholar]
- 39.Cianchi F, Cortesini C, Bechi P, et al. Up-regulation of cyclooxygenase 2 gene expression correlates with tumor angiogenesis in human colorectal cancer. Gastroenterology. 2001;121:1339–1347. doi: 10.1053/gast.2001.29691. [DOI] [PubMed] [Google Scholar]
- 40.Kim HS, Youm HR, Lee JS, et al. Correlation between cyclooxygenase-2 and tumor angiogenesis in non-small cell lung cancer. Lung Cancer. 2003;42:163–170. doi: 10.1016/s0169-5002(03)00290-3. [DOI] [PubMed] [Google Scholar]
- 41.Stolina M, Sharma S, Lin Y, et al. Specific inhibition of cyclooxygenase 2 restores antitumor reactivity by altering the balance of IL-10 and IL-12 synthesis. J Immunol. 2000;164:361–370. doi: 10.4049/jimmunol.164.1.361. [DOI] [PubMed] [Google Scholar]
- 42.Huang M, Stolina M, Sharma S, et al. Non-small cell lung cancer cyclooxygenase-2-dependent regulation of cytokine balance in lymphocytes and macrophages: up-regulation of interleukin 10 and down-regulation of interleukin 12 production. Cancer Res. 1998;58:1208–1216. [PubMed] [Google Scholar]