

Abstract
Protein tyrosine phosphatase 1B (PTP1B) is a key regulator of metabolism and cell growth by its ability to dephosphorylate tyrosine kinase receptors and modulate the intensity of their signaling cascades. Because liver regeneration involves tyrosine phosphorylation–mediated signaling, we investigated the role of PTP1B in this process by performing partial hepatectomy in wild-type (PTP1B+/+) and PTP1B-deficient (PTP1B−/−) mice. The expression of PCNA and cyclins D1 and E (cell proliferation markers) was enhanced in PTP1B−/− regenerating livers, in parallel with 5′-bromo-2′-deoxyuridine incorporation. Phosphorylation of JNK1/2 and STAT3, early triggers of hepatic regeneration in response to TNF-α and IL-6, was accelerated in PTP1B−/− mice compared with PTP1B+/+ mice. These phosphorylations were increased in PTP1B−/− hepatocytes or by silencing PTP1B in wild-type cells and decreased further after the addition of recombinant PTP1B. Enhanced EGF– and HGF receptor–mediated signaling was observed in regenerating livers lacking PTP1B and in EGF- or HGF–stimulated PTP1B−/− hepatocytes. Moreover, PTP1B−/− mice displayed a more rapid increase in intrahepatic lipid accumulation than PTP1B+/+ control mice. Late responses to partial hepatectomy revealed additional divergences because stress-mediated signaling was attenuated at 24 to 96 hours in PTP1B−/− mice compared with PTP1B+/+ mice. Finally, PTP1B deficiency also improves hepatic regeneration in mice fed a high-fat diet. These results suggest that pharmacological inhibition of PTP1B would improve liver regeneration in patients with acute or chronic liver injury.
The protein tyrosine phosphatase 1B (PTP1B) is a widely expressed nonreceptor PTP that is associated with the endoplasmic reticulum and other intracellular membranes via a hydrophobic interaction of its C-terminal 35–amino acid targeting sequence.1 Protein tyrosine phosphatase 1B is a key regulator of metabolism and cell growth by its ability to dephosphorylate receptors of the tyrosine kinase superfamily and modulate the duration and intensity of the signals emerging from these activated receptors. As demonstrated by genetic and biochemical approaches, PTP1B dephosphorylates and inactivates the epidermal growth factor receptor (EGFR),2 the platelet-derived growth factor receptor,3 the hepatocyte growth factor receptor (HGFR),4 the insulin receptor (IR),5 and the insulin-like growth factor-1 receptor.6 All of them are implicated in the control of proliferation, survival, and/or metabolic functions of hepatocytes7,8 and many other cellular models.
Protein tyrosine phosphorylation plays key roles in many physiological processes and is deregulated in pathological conditions. Hepatocyte proliferation is induced by different mitogens, including HGF and ligands of the EGFR.9,10 Thus, rapid activation of EGFR and HGFR promotes cellular replication during liver regeneration after partial hepatectomy (PH). This is a unique process that requires the re-entry of quiescent hepatocytes into the cell cycle to proliferate and, therefore, restore the original liver mass after surgery.11,12 More important, cell cycle progression induced by growth factors after PH is preceded by a priming step in which the pro-inflammatory cytokines tumor necrosis factor-α (TNF-α) and IL-6 activate nuclear factor κB, activator protein-1, and signal transducer and activator of transcription 3 (STAT3) transcription factors, resulting in the G0 to G1 transition triggering the complex regeneration of liver mass.13–15 Binding of IL-6 to its receptor is followed by homodimerization of the 130-kDa transmembrane signal transducing component (gp130) and stimulates the tyrosine kinase activity of the Janus-associated kinase. Activated Janus-associated kinase then phosphorylates the associated gp130 and STAT3 on the Tyr705 residue.16 Thus, like EGFR- and HGFR-mediated signaling, tyrosine phosphorylation is essential for IL-6–mediated mitogenic effect in hepatocytes. More recently, dephosphorylation of the Tyr705 residue of STAT3 was elicited by PTP1B.17 Finally, on completion of the repair process and restoration of liver mass, hepatocyte proliferation is inhibited, most likely through transforming growth factor (TGF) β and activin signaling.18
Extensive analysis of the PTP1B-deficient (PTP1B−/−) mice has confirmed that the IR is a key physiological target of PTP1B.19,20 In fact, PTP1B inhibition is actually being assayed as a potential therapy for type 2 diabetes mellitus. Despite the negative modulation of activation of receptors of the tyrosine kinase superfamily by PTP1B, PTP1B−/− mice lack any obvious signs of increased activity of these receptors, such as susceptibility to tumor development through life. However, the role of PTP1B in the regulation of the mitogenic capacity of the regenerating liver in response to physiological activators of tyrosine phosphorylation–mediated signaling cascades remains unexplored. Accordingly, in the present article, we performed PH in wild-type (PTP1B+/+) and PTP1B−/− mice and analyzed the hepatic proliferation rate and the early and late signaling pathways that are activated through regeneration. We demonstrated by in vivo and in vitro experiments that PTP1B plays an essential role in the modulation of multiple signaling pathways that trigger hepatocyte proliferation in response to PH and in the control of the termination phase of hepatic regeneration.
Materials and Methods
PH in Mice
Animals used in the current study were male PTP1B+/+ and PTP1B−/− mice, aged 10 to 14 weeks, on a mixed genetic background (129 SV × C57/BL6), as described previously.21 Animals included in the study were controlled following the recommendations of the Federation of European Laboratory Animal Science Associations on health monitoring; use of animals in experimental procedures was approved by the Consejo Superior de Investigaciones Científicas Animal Care and Use Committee. Mice were fed a standard chow diet or a high-fat diet (HFD) (TD-88137; Harland-Teklad, Boxmeer, The Netherlands). Three groups of seven mice of both genotypes were fed with an HFD for 12 weeks from weaning. Mice were subjected to a standard 70% PH during isoflurane anesthesia, as described by Higgins and Anderson22 and adapted to mice.23 The remaining livers were collected between 0.5 hours and 7 days after PH. The livers were removed, weighed, and normalized to body weight. Samples of tissue were rinsed in ice-cold PBS and frozen immediately in liquid nitrogen or fixed in 4% paraformaldehyde for immunohistochemistry analysis.
Metabolic Measurements
For fed and fasting (20- to 24-hour fast) glucose determinations, blood was withdrawn from the tail and glucose levels were measured with a glucometer (Accu-Check Aviva; Hoffmann-La Roche, Basilea, Switzerland). The i.p. glucose tolerance tests were performed on mice that fasted for 20 to 24 hours and were injected with 2 g D-glucose/kg body weight; the blood glucose level was measured at 30, 60, 90, and 120 minutes after injection. The i.p. insulin tolerance tests were performed on mice that fasted for 4 hours and were injected with 0.75 U human regular insulin/kg body weight; the blood glucose level was measured at 30, 60, and 90 minutes after injection.
Analysis of Serum TNF-α, IL-6, and Alanine Aminotransferase
Serum cytokines (IL-6 and TNF-α) were measured using a commercially available kit (Multiplex Kit; Millipore, Billerica, MA) following manufacturer's instructions. Serum transaminase (alanine aminotransferase) activity was determined with another kit (catalog No. 11832; Biosystems, Barcelona, Spain) according to manufacturer's instructions.
Measurement of Free Glycerol in Serum
Blood was obtained by retro-orbital bleeding, and free glycerol contained in serum was determined by colorimetric methods with Free Glycerol Reagent (F6428; Sigma-Aldrich, Madrid, Spain).
Magnetic Resonance Images
Animals were subjected to nuclear magnetic resonance (NMR) scan analysis in fed conditions to avoid weight loss induced by overnight fasting. Animals were weighed, anesthetized with isoflurane, and scanned in toto using gadolinium diethylenetriaminopentaacetic acid (Magnevist). The images were acquired with a spectrometer (Bruker Pharmascan, Bruker Biospin, Billerica, MA) (horizontal magnet, 7.0 T/16-cm diameter) interfaced with a workstation operating under the Linux environment (Hewlett-Packard, Palo Alto, CA). T2-weighted images were obtained with a rapid acquisition with relaxation enhancement sequence in axial or coronal orientation (Repetition Time, 3000 milliseconds; Inter-Echo Time, 59 milliseconds; average, 3; field of view, 3.80 cm; acquisition matrix, 256 × 256; slice thickness, 1.50 mm). Images meet the digital imaging and communications in medicine format. Fat and lean body mass values were determined.
BrdU Labeling
For in vivo labeling, mice were injected i.p. with 150-mg/kg 5′-bromo-2′-deoxyuridine (BrdU; Sigma-Aldrich) and sacrificed after 3 hours. The preparation of liver samples for immunohistochemistry assays was performed as recommended by the manufacturer (G.E. Healthcare product booklet: Amersham Cell Proliferation Kit. Piscataway, NJ). Livers were fixed in 4% para formaldehyde, dehydrated, embedded in paraffin, and sectioned (10 μm). Then, samples were processed following manufacturer's instructions. All microscopy images were taken in an Eclipse 90i microscope (Nikon Corp., Tokyo, Japan). Capture, management, and quantification of images were performed with software (NIS-Elements Software; Nikon). Nuclei were identified automatically by roundness shape threshold. The BrdU-stained nuclei were assessed by applying color-intensity threshold.
Oil Red O Staining
Portions of regenerating liver were fixed, included in OCT (Tissue Tek; Sakura Finetek Europe, Alphen aan den Rijn, The Netherlands), and flash frozen in liquid nitrogen. Tissue slices (5 μm) were stained with oil red O and counterstained with hematoxylin.24
Homogenization and Preparation of Tissue Extracts
Frozen livers were homogenized in 16 volumes (w/v) of ice-cold lysis buffer [50 mmol/L Tris-HCl, 1% Triton X-100, 2 mmol/L EGTA, 10 mmol/L EDTA, 100 mmol/L NaF (sodium fluoride), 1 mmol/L Na4P2O7 (sodium pyrophosphate), 2 mmol/L Na3VO4 (sodium orthovanadate), 100 μg/mL phenylmethylsulphonyl fluoride, 1 μg/mL aprotinin, 1 μg/mL pepstatin A, and 1 μg/mL leupeptin], using a disperser (IKA T10 basic ULTRA-TURRAX). Extracts were kept ice cold at all times. Liver extracts were cleared by microcentrifugation at 40,000 × g for 40 minutes at 4°C. The supernatant was fractionated and stored at −80°C.
Protein Determination
Protein determination was performed by the Bradford dye method using a reagent (Bio-Rad, Berkeley, CA) and bovine serum albumin as the standard.
Western Blot Analysis
After SDS–polyacrylamide gel electrophoresis, gels were transferred to polyvinylidene fluoride membranes and blocked using 5% nonfat dried milk or 3% bovine serum albumin in 10 mmol/L Tris-HCl and 150 mmol/L NaCl (pH 7.5); they were incubated overnight with several antibodies, as indicated, in 0.05% Tween 20, 10 mmol/L Tris-HCl, and 150 mmol/L NaCl (pH 7.5). Immunoreactive bands were visualized using enhanced chemiluminescent HRP Western blotting (Millipore). The antibodies used in this study were as follows: anti-human PTP1B (sc-14021), PCNA (sc-56), cyclin D1 (sc-718), cyclin E (sc-25303), phospho-Akt1/2/3 (sc-16646-R), Met(B-2) (sc-8057), SMAD2/3 (sc-6032), pSMAD2/3 (sc-11769-R), TGF-β–RI (sc-398), TGF-β–RII (sc-220), SnoN (sc-9141), plasminogen activated inhibitor (PAI)-1 (sc-8979), and secondary antibodies [goat anti-mouse (sc-2005) and goat anti-rabbit (sc-2004)]. These antibodies were purchased from Santa Cruz Biotechnology, Palo Alto, CA. The Akt (9272), phospho-c-Jun (9164), JNK (9252), phospho-JNK (4668), phospho-STAT3 (9131), STAT3 (9132), p44/42 extracellular signal-regulated kinases (ERK) (9102), phospho-p44/42 ERK (9101), phospho-Met (3129), phospho-AMP-activated protein kinase (AMPK) (2531), AMPK (2532), phospho-EGFR (2220), EGFR (2232), and gp130 (3732) antibodies were purchased from Cell Signaling Technology (Danvers, MA). The anti-mouse PTP1B (07-088) and the anti-p85α (06-195) antibodies were purchased from Upstate (Millipore). The anti–β-actin antibody (A3854) was purchased from Sigma. The anti–glyceraldehyde-3-phosphate dehydrogenase antibody (AM4300) was purchased from Ambion (Austin, TX).
Primary Culture of Adult Hepatocytes
Hepatocytes were isolated from nonfasting male PTP1B+/+ and PTP1B−/− mice (aged 8 to 12 weeks) by perfusion with collagenase, as described previously.25 Cells were plated on 60 mm primaria dishes (Falcon; BD Biosciences, San Jose, CA) and cultured in William's E medium supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal serum for 48 hours. Then, cells were used for experiments.
Immortalization of PTP1B+/+ and PTP1B−/− Neonatal Hepatocytes
The generation of immortalized PTP1B+/+ and PTP1B−/− hepatocyte cell lines is described in detail by Gonzalez-Rodriguez et al.21 Cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% FS and then used for experiments.
Quantification of Apoptotic Cells by Flow Cytometry
After induction of apoptosis, adherent and nonadherent cells were collected by centrifugation, washed with PBS, and fixed with cold ethanol (70%, vol/vol). The cells were then washed, resuspended in PBS, and incubated with RNAse (25 µg/106 cells) for 30 min at 37°C. After addition of 0.05% propidium iodide, cells were analyzed by flow cytometry using a FacsCanto (Becton Dickinson, Franklin Lakes, NJ).
Transient Transfection with Small-Interfering RNA
The small-interfering RNA (siRNA) oligonucleotide was synthesized by Dharmacon RNAi Technologies (Thermo Fisher Scientific, Waltham, MA) for gene silencing of mouse PTP1B. Wild-type immortalized hepatocytes or the HuH-7 human liver cancer cell line (JCRB Cell Bank, JCRB0403, JTC-39) was seeded in 6-cm dishes and incubated at 37°C with 5% CO2 overnight. When 40% to 50% confluence was reached, cells were transfected with 75-nmol/L PTP1B siRNA or a scrambled control siRNA, following the manufacturer's protocol (Dharmacon RNAi Technologies protocol circular; DharmaFECT General Transfection Protocol, Lafayette, CO). After 48 hours, cells were used for experiments.
RNA Isolation and Analysis of IR A and B Isoforms by PCR
Total RNA was isolated from approximately 100 mg of mouse liver using a reagent (TRIzol; Invitrogen, Carlsbad, CA). To analyze mouse and IR A isoform (IRA) and IR B isoform (IRB) in regenerating livers by PCR, 5 μg of total RNA was primed with oligo(dT) in the presence of murine mammary tumor virus reverse transcriptase (Invitrogen) to synthesize cDNA. The samples were diluted fivefold, and 5% of the total volume was used for subsequent PCR. The primers used were as follows: mouse IR exon 11, 5′-ATCAGAGTGAGTATGACGACTCGG-3′ (forward) and 5′-TCCTGACTTGTGGGCACAATGGTA-3′ (reverse). The PCR reactions were performed as described.26 For amplification of mouse β-actin, the primers used were as follows: 5′-GGTATGGAATCCTGTGGCATCCATGAAA-3′ (forward) and 5′-GTGTAAAACGCAGCTCAGTAACAGTCC-3′ (reverse). Reaction products were resolved on 2% agarose gels.
Real-Time PCR
The PTP1B+/+ and PTP1B−/− immortalized hepatocytes were grown to 80% confluence and then stimulated with TGF-β (5 ng/mL) for several periods. Total RNA (1 μg), extracted with reagent (TRIzol), was reverse transcribed using a first-strand synthesis system (SuperScriptTM III) following the indications of the manufacturer (Invitrogen). Real-time PCR was conducted with SYBR Green (Applied Biosystems, Foster City, CA) on a real-time PCR system (MyiQ; Bio-Rad). The primers used were as follows: PAI-1, 5′-CGGCAGATCCAAGATGCTATG-3′ (forward) and 5′-GACCAGCTCTAGGTCCCGCT-3′ (reverse); and 36B4, 5′-AGATGCAGCAGATCCGCAT-3′ (forward) and 5′-GTTCTTGCCCATCAGCACC-3′ (reverse). The PCR thermocycler parameters were 94°C for 3 minutes; 40 cycles of 94°C for 20 seconds, 60°C for 20 seconds, and 72°C for 20 seconds; and a final cycle of 72°C for 5 minutes. Each sample was run in duplicate and was normalized with the expression of 36B4. The fold induction was determined using ΔΔCt-based fold change calculations. The relative quantity is as follow: 2 - ΔΔCt.
Analysis of PTP1B-Mediated Dephosphorylation
The PTP1B−/− hepatocytes were stimulated with TNF-α (10 ng/mL) and IL-6 (10 ng/mL) for 10 minutes and EGF (20 ng/mL) or HGF (25 ng/mL) for 30 minutes and immediately homogenized in buffer containing 20 mmol/L imidazole-HCl, 2 mmol/L EGTA, and 2 mmol/L EDTA (pH 7.0), supplemented with protease inhibitors (10 μg/mL leupeptin, 10 μg/mL aprotinin, and 1 mmol/L phenylmethylsulfonyl fluoride). Samples were sonicated three times for 15 seconds at 1.5 mA, and lysates were clarified by centrifugation at 12,000 × g for 10 minutes. After protein content determination, 60 μL of cell lysates (1 μg/mL) was incubated with 10 U recombinant PTP1B (Upstate and Millipore) for 30 minutes at 37°C. Then, the reaction was discontinued by adding LaemmLi buffer and samples were analyzed by Western blotting.
Data Analysis
Data are expressed as mean ± SEM. Comparisons between groups were made using one-way analysis of variance. Differences were considered statistically significant at P < 0.05. The data were analyzed by a computer program (SPSS for Windows statistical package, version 9.0.1).
Results
Increased Liver/Body Weight Ratio and Hepatocyte Proliferation after PH in PTP1B−/− Mice
To investigate the role of PTP1B in liver regeneration in vivo, we performed 70% PH on 10- to 14-week-old male PTP1B+/+ and PTP1B−/− mice. Before PH, there was no significant difference in the liver/body weight ratio between PTP1B+/+ and PTP1B−/− mice (Figure 1A). However, the liver/body weight ratio was persistently higher in mice lacking PTP1B from 36 hours to 7 days after PH (Figure 1B). After surgery, liver mass is restored predominantly by the proliferation of existing hepatocytes. By using BrdU incorporation studies, we found increased liver cell proliferation in PTP1B−/− mice compared with PTP1B+/+ mice (Figure 1, C and D). In mice of both genotypes, proliferating hepatocytes were almost undetectable before PH. Proliferation peaked 48 hours after PH in both genotypes of mice, but the percentage of BrdU-labeled cells was higher in PTP1B−/− mice. At 96 hours after surgery, the number of proliferating cells remained elevated in PTP1B−/− mice. To confirm further our findings, additional markers for cell cycle progression were analyzed. PCNA is an auxiliary protein of DNA polymerase delta. Its expression is cell cycle dependent, being detected in late G1 and remaining increased throughout the S phase and into the early postreplicative G2 period.27 The PCNA levels increased at 48 hours after PH in both genotypes of mice (Figure 1E). However, the magnitude of induction was significantly higher in the absence of PTP1B. The PCNA levels declined at 96 hours after PH in both genotypes of mice. Moreover, the induction of cyclins D1 and E after PH occurred at 36 hours in regenerating liver lacking PTP1B compared with the PTP1B+/+ mice that elicited this response at 48 hours.
Figure 1.
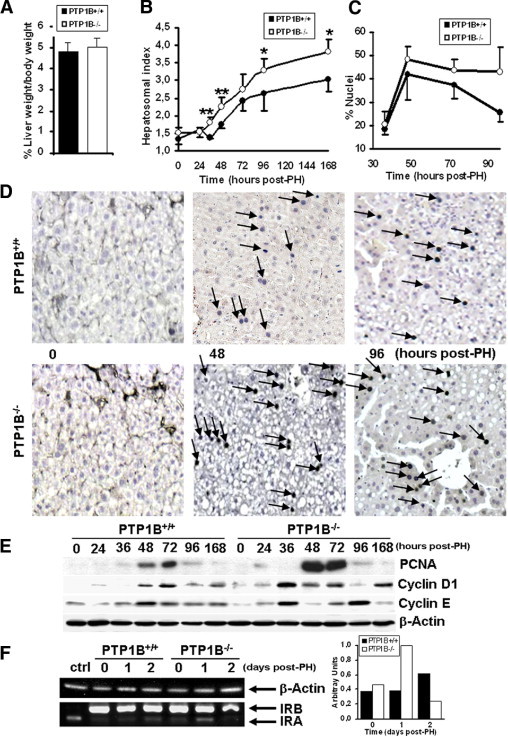
Increased liver/body weight ratio and hepatocyte proliferation after PH in PTP1B−/− mice. A: Comparison of liver/body weight ratio in PTP1B+/+ and PTP1B−/− mice. Results are given as the mean ± SEM (n = 9–11). B: The PH was performed in PTP1B+/+ and PTP1B−/− mice, and regenerating livers were removed at the indicated periods. The liver regeneration index (liver/body weight ratio) at the indicated periods after PH was calculated. Results are given as the mean ± SEM (n = 6–7). *P < 0.05; **P < 0.01 for PTP1B−/− versus PTP1B+/+ mice. C: Quantification of BrdU-positive nuclei in regenerating livers from PTP1B+/+ and PTP1B−/− mice at 24 to 96 hours after PH. Values are given as the mean ± SEM (n = 4–6). D: Representative microscopic images of BrdU incorporation at 48 and 96 hours after PH. The BrdU-positive nuclei are indicated by arrows. Original magnification, ×10. E: Total protein (50 μg) was analyzed by Western blot, with the antibodies against PCNA, cyclins D1 and E, and β-actin as a loading control. Representative autoradiograms from three independent experiments with similar results are shown. F: Total RNA was isolated from regenerating livers from each genotype, and RT-PCR was performed as described in the corresponding section of Materials and Methods. The IR-deficient immortalized neonatal hepatocytes reconstituted with IRA were used as the control for the IRA isoform. RT-PCR with mouse β-actin primers was performed as a loading control. A representative experiment is shown. Similar results were obtained in two independent series of each genotype of mice.
Our recent data have demonstrated increased expression of the IRA and, therefore, an increased IRA/IRB isoform ratio in the livers of PTP1B−/− mice in the suckling period, a stage related to high hepatocyte proliferation.28 Because IRA represents the predominant isoform in proliferative cells,29 we analyzed the pattern of IR isoforms after PH. Before surgery, adult liver expressed mostly the IRB isoform28 (Figure 1F). However, regenerating livers lacking PTP1B re-expressed IRA 24 hours after PH; in PTP1B+/+ mice, a slight increase in IRA was detected at 48 hours. Altogether, these findings demonstrated that livers of PTP1B−/− mice can regenerate more efficiently than those of PTP1B+/+ controls.
Effect of PTP1B Deficiency on the TNF-α– and IL-6–Mediated Early Responses after PH
The early events that occur in response to PH are mediated, in part, by a rapid increase in serum TNF-α and IL-6, triggering the activation of cytokine-mediated signaling pathways in regenerating liver.12,30 These pro-inflammatory cytokines are key players of the priming step, resulting in G0 to G1 transition and survival of liver parenchymal cells. Serum levels of TNF-α peaked at 2 hours after PH in PTP1B−/− mice, whereas maximum levels of this cytokine were reached at 4 hours in PTP1B+/+ mice (Figure 2A). Likewise, serum levels of IL-6 were higher in PTP1B−/− mice, reaching a significant difference at 6 hours after PH. In regenerating liver, TNF-α–mediated JNK and c-Jun phosphorylations were maximal 2 hours after PH in both genotypes of mice. However, these responses were higher in the absence of PTP1B (Figure 2B). Similarly, IL-6–induced phosphorylation of STAT3 was enhanced in PTP1B−/− regenerating liver compared with the PTP1B+/+ liver.
Figure 2.
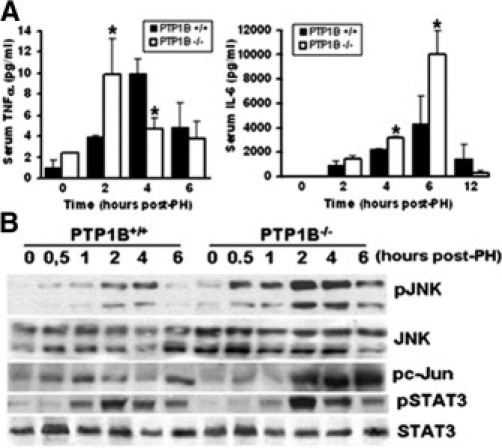
Effect of PTP1B deficiency on the TNF-α– and IL-6–mediated early responses after PH. The PH was performed in PTP1B+/+ and PTP1B−/− mice, and liver and serum samples were collected at the indicated periods. A: Serum levels of TNF-α (left) and IL-6 (right) after PH. Results are given as the mean ± SEM (n = 4–6). *P < 0.05 for PTP1B−/− versus PTP1B+/+ mice. B: Western blot analysis of regenerating livers was performed in PTP1B+/+ and PTP1B−/− mice at the indicated periods. Total protein (50 μg) was analyzed by Western blot with the antibodies against phospho-JNK (Thr183/Tyr185), JNK, phospho-c-Jun (Ser73), phospho-STAT3 (Tyr705), and STAT3. Representative autoradiograms of three independent series of PH in each genotype are shown.
PTP1B−/− Hepatocytes Are Hypersensitive to TNF-α– and IL-6–Mediated Signaling
To determine whether the increased responses to pro-inflammatory signaling in regenerating liver are a direct consequence of PTP1B inhibition, we treated primary hepatocytes from both genotypes of mice with TNF-α or IL-6 for several periods; then, the phosphorylation of JNK and STAT3 was analyzed. Tumor necrosis factor-α–induced JNK phosphorylation was enhanced and more sustained in PTP1B−/− primary hepatocytes compared with PTP1B+/+ controls (Figure 3A). To obtain further support for ERK mediation of the TNF-α action, we examined its activation during the same experimental conditions. Treatment with TNF-α led to increased and prolonged ERK phosphorylation in PTP1B−/− hepatocytes. To reinforce these data, immortalized wild-type hepatocytes were transfected with PTP1B or scrambled siRNA oligos and stimulated with TNF-α. Silencing of PTP1B in wild-type cells resulted in increased JNK and ERK phosphorylations in response to TNF-α. Similarly, IL-6 stimulation led to a transient increase in STAT3 and ERK phosphorylation in primary hepatocytes from PTP1B+/+ mice that was enhanced in PTP1B−/− hepatocytes or immortalized PTP1B+/+ cells transfected with the PTP1B siRNA oligo (Figure 3B).
Figure 3.
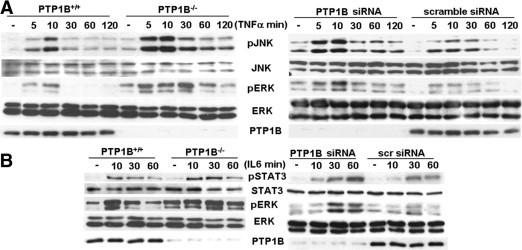
The PTP1B−/− hepatocytes are hypersensitive to TNF-α– and IL-6–mediated signaling. A: Left: Primary hepatocytes (PTP1B+/+ and PTP1B−/−) were stimulated with 10-ng/mL TNF-α for the indicated periods. Control cells were maintained in the absence of cytokines. At the end of the culture time, cells were lysed and 50 μg of total protein was analyzed by Western blot with antibodies against phospho-JNK (pJNK) (Thr183/Tyr185), JNK, phospho-c-Jun (Ser73), phospho-ERK (pERK) (Thr202/Tyr204), and ERK. Right: Immortalized PTP1B+/+ hepatocytes were transfected with PTP1B or scramble siRNA oligos. After 48 hours, cells were stimulated with 10-ng/mL TNF-α or left untreated. Total protein was analyzed by Western blot with the indicated antibodies. Results shown are representative of two independent experiments. B: Left: Primary hepatocytes were cultured as described in A and stimulated with 10-ng/mL IL-6 for the indicated periods. Control cells were maintained in the absence of cytokines. At the end of the culture time, cells were lysed and 50 μg of total protein was analyzed by Western blot with antibodies against phospho-STAT3 (pSTAT3) (Tyr705), STAT3, pERK (Thr202/Tyr204), and ERK. Right: Immortalized PTP1B+/+ hepatocytes were transfected with PTP1B or scramble siRNA oligos. After 48 hours, cells were stimulated with 10-ng/mL IL-6 or left untreated. Total protein was analyzed by Western blot with the indicated antibodies. Representative autoradiograms are shown. Similar results were obtained in two series of independent experiments.
PTP1B Decreased TNF-α–Induced JNK Phosphorylation in Human and Mouse Hepatocytes
Next, in human liver cells (HuH-7), we investigated the effect of PTP1B silencing on JNK and STAT3 phosphorylation after stimulation with TNF-α and IL-6, respectively. Figure 4A shows that, in HuH-7 cells, both JNK and STAT3 phosphorylations, in response to TNF-α and IL-6 stimulation, respectively, were increased when the expression of PTP1B was reduced by siRNA. Because JNK contains the dual phosphorylation motif Thr-Pro-Tyr,31 and our results demonstrated that PTP1B deficiency increased TNF-α–induced JNK phosphorylation in mouse and human hepatocytes, we analyzed whether PTP1B directly dephosphorylates JNK in liver cells. For this goal, PTP1B−/− hepatocytes were stimulated with TNF-α (10 ng/mL) for 10 minutes, when phosphorylation on Thr183/Tyr185 residues was maximally induced (Figure 3A). Then, cell lysates were incubated with purified PTP1B, and JNK phosphorylation was analyzed with the phospho-specific antibody recognizing Tyr185. The TNF-α–induced phosphorylation of JNK was decreased dramatically in the presence of active PTP1B (Figure 4B). Similarly, PTP1B reduced IL-6–induced phosphorylation of Tyr705 residue of STAT3, a well-known substrate of this phosphatase.17
Figure 4.
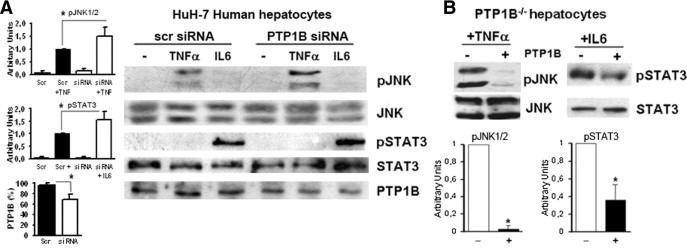
The PTP1B decreased TNF-induced JNK phosphorylation in human and mouse hepatocytes. A: The HuH-7 human liver cells were transfected with PTP1B or scramble (scr) siRNA oligos for 48 hours before TNF-α or IL-6 stimulation. The phosphorylation of JNK (pJNK) and STAT3 (pSTAT3) was analyzed with the indicated antibodies. A representative Western blot is shown. Autoradiograms corresponding to three independent experiments were quantified by scanning densitometry. Results show arbitrary units of JNK and STAT3 phosphorylation, and average PTP1B levels in scr or PTP1B siRNA-transfected cells are expressed as the mean ± SEM. *P < 0.05 for HuH-7 cells transfected with the PTP1B siRNA versus cells transfected with scr siRNA. B: Immortalized PTP1B−/− hepatocytes were stimulated with 10-ng/mL TNF-α or IL-6 for 10 and 30 minutes, respectively; 60 μL of cell lysates (1 μg/μL) was incubated with 10 U of recombinant PTP1B for 30 minutes at 37°C, and pJNK and pSTAT3 were analyzed by Western blot. As a loading control, membranes were blotted with total JNK or STAT3 antibodies. A representative Western blot is shown. Autoradiograms corresponding to three independent experiments were quantified by scanning densitometry. Results are expressed as arbitrary units of JNK and STAT3 phosphorylation and are the mean ± SEM. *P < 0.05 for PTP1B−/− hepatocytes reconstituted with active PTP1B versus nonreconstituted cells.
Increased EGFR and HGFR Tyrosine Phosphorylation in Livers of PTP1B−/− Mice after PH
After priming, hepatocyte proliferation is regulated by different mitogens, including HGF and ligands of the EGFR that are required for liver cell progression through the cell cycle. Both HGFR and EGFR are members of the PTP1B substrate family.3 Thus, we hypothesized that tyrosine phosphorylation of these receptors would be increased immediately after PH in the livers of PTP1B−/− mice compared with controls. To test this hypothesis, we performed Western blot analysis with anti–phospho-specific antibodies that recognize the activated forms of EGFR or HGFR. The early induction of EGFR tyrosine phosphorylation was prolonged more in regenerating livers lacking PTP1B (Figure 5A). Moreover, HGFR tyrosine phosphorylation peaked at 0.5 hours after PH in both genotypes of mice, but the response was enhanced in PTP1B−/− mice. Downstream from these activated receptors, Akt and ERK signaling cascades were subsequently activated. Although no differences in the course of ERK phosphorylation after PH were observed between both genotypes of mice, Akt phosphorylation was transiently activated at 0.5 hours in the PTP1B+/+ mice, but it was maintained up to 6 hours in regenerating livers from PTP1B−/− mice.
Figure 5.
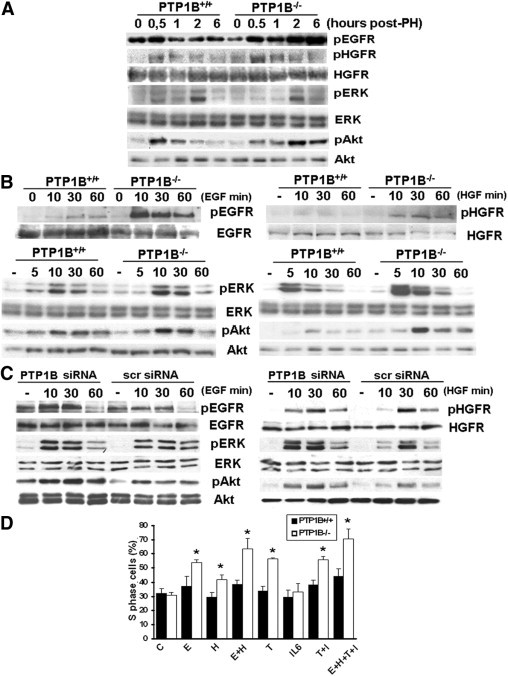
Increased EGFR- and HGFR-mediated signaling in hepatocytes and regenerating livers of PTP1B−/− mice. A: The PH was performed in PTP1B+/+ and PTP1B−/− mice, and regenerating livers were removed at the indicated periods. Total protein (50 μg) was analyzed by Western blot with the antibodies against phospho-EGFR (pEGFR) (Tyr1086), phospho-HGFR (pHGFR) (Tyr1234/1235), HGFR, phospho-Akt (Akt) (Ser473), Akt, phospho-ERK (pERK) (Thr202/Tyr204), and ERK. Representative autoradiograms of three independent series of PH in each genotype are shown. B: Primary hepatocytes (PTP1B+/+ and PTP1B−/−) were serum starved for 4 hours and then stimulated with 20-ng/mL EGF (left) or 30-ng/mL HGF (right) for the indicated periods. Control cells were maintained in the absence of growth factors. At the end of the culture time, cells were lysed and 50 μg of total protein was analyzed by Western blot with the indicated antibodies. Representative autoradiograms are shown. Similar results were obtained in two independent series of experiments. C: Immortalized PTP1B+/+ hepatocytes were transfected with PTP1B or scramble siRNA oligos. After 48 hours, cells were serum starved for 4 hours and stimulated with 20-ng/mL EGF (left) or 30-ng/mL HGF (right), as described in B, or left untreated. Total protein was analyzed by Western blot with the indicated antibodies. Results shown are representative of two independent experiments. D: Immortalized PTP1B+/+ and PTP1B−/− hepatocytes were grown to 60% to 70% confluence and serum deprived for 20 hours. Then, cells were stimulated with TNF-α (10 ng/mL) (T), IL-6 (10 ng/mL) (I), EGF (20 ng/mL) (E), or HGF (30 ng/mL) (H), either alone or in combination, for a further 24 hours (control: C). At the end of the culture time, the percentage of cells in the S phase of the cell cycle was analyzed by flow cytometry, as described in the respective section in Materials and Methods. Results are given as the mean ± SEM (n = 4–8). *P < 0.05 for PTP1B−/− versus PTP1B+/+ cells.
Increased EGFR- and HGFR-Mediated Akt and ERK Signaling and Proliferation in PTP1B−/− Hepatocytes
To substantiate the results obtained in vivo, we prepared primary hepatocytes from PTP1B+/+ and PTP1B−/− mice and analyzed the magnitude of EGFR- and HGFR-mediated signaling. Stimulation with EGF induced a rapid increase in EGFR tyrosine phosphorylation in primary hepatocytes from both genotypes of mice, with a more pronounced response in cells lacking PTP1B (Figure 5B). In PTP1B−/− hepatocytes, EGF-induced phosphorylation of EGFR was decreased dramatically in the presence of active PTP1B (see Supplemental Figure S1 at http://ajp.amjpathol.org). As expected, Akt phosphorylation was enhanced in EGF-stimulated PTP1B−/− hepatocytes, whereas only a slight increase in the activation of ERK was observed in the absence of the phosphatase. Similar results were found in immortalized hepatocytes transfected with the PTP1B siRNA oligonucleotide (Figure 5C). Regarding HGF-mediated signaling, the phosphorylation of HGFR was markedly increased in HGF-stimulated PTP1B−/− primary hepatocytes compared with controls (Figure 5B). Again, in PTP1B−/− hepatocytes, HGF-induced phosphorylation of HGFR was decreased dramatically in the presence of active PTP1B (see supplemental Figure S1 at http://ajp.amjpathol.org). Downstream HGFR Akt phosphorylation was higher in HGF-stimulated PTP1B−/− primary hepatocytes or immortalized hepatocytes transfected with PTP1B siRNA oligonucleotide than that of the corresponding controls (Figure 5, B and C). The ERK phosphorylation in response to HGF was pronounced more in cells lacking PTP1B by either genetic manipulation or RNA interference. Finally, we analyzed the effect of growth factors and cytokines on hepatocyte proliferation. For this goal, PTP1B+/+ and PTP1B−/− hepatocytes were stimulated with EGF or HGF alone or in combination with TNF-α and IL-6; the percentage of cells in the S phase of the cell cycle was analyzed. As shown in Figure 5D, the increase in the percentage of proliferating cells elicited by EGF or HGF was higher in hepatocytes lacking PTP1B than in the PTP1B+/+ control in either the absence or presence of priming cytokines.
Effect of PTP1B Deficiency on Hepatic Lipid Accumulation during Liver Regeneration
After PH, transient hepatic steatosis occurs during liver regeneration to provide energy fuel to support cell proliferation and tissue regrowth.32 Thus, we determined whether PTP1B deficiency affects the hepatocellular lipid accumulation in regenerating liver. Staining of liver sections with oil red O revealed that hepatic fat content was similar in the livers of both genotypes of mice before surgery (Figure 6A). However, PTP1B−/− mice accumulated more fat in the liver at an earlier period (12 hours) after PH compared with PTP1B+/+ controls; they accumulated lipids maximally at 24 hours. No differences in intrahepatic lipid accumulation were found between the two genotypes of mice at 36 and 48 hours after PH. Moreover, differential release of fatty acids from white adipose tissue deposits after PH, measured by serum free glycerol levels, was observed at 12 hours, with higher values in PTP1B−/− mice (Figure 6B). These results indicate that the lack of PTP1B has beneficial effects on hepatic lipid homeostasis during liver regeneration.
Figure 6.
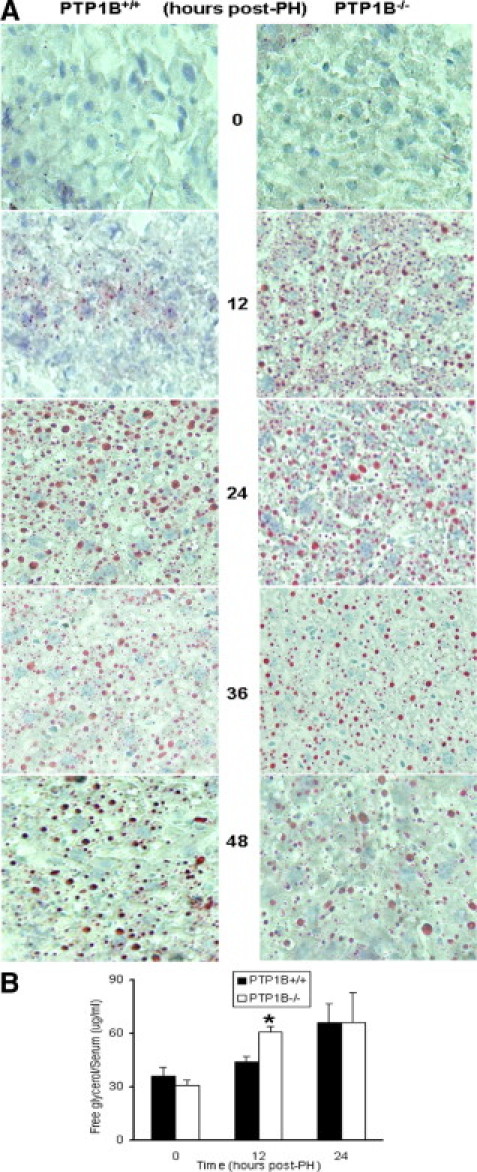
Effect of PTP1B deficiency on hepatic lipid accumulation during liver regeneration. Mice were subjected to PH and allowed to recover for 12 to 48 hours. A: Oil red O staining of liver sections from PTP1B−/− and PTP1B+/+ mice at the indicated periods after PH. Original magnification, ×10. B: Serum levels of free glycerol after PH. Results are given as the mean ± SEM. *P < 0.05 for PTP1B−/− versus PTP1B+/+ mice.
Decreased Pro-Inflammatory Signaling and Sustained Activation of Akt and ERK in the Late Phase of Liver Regeneration in PTP1B−/− Mice
Overactivation of JNK at late periods after PH impairs liver regeneration.33 Therefore, we analyzed JNK phosphorylation at 24 to 96 hours after surgery in both genotypes of mice. Phosphorylated JNK was detectable 24 hours after PH in PTP1B+/+ mice, coincident with a second phase of ERK activation (Figure 7A, left). However, JNK and c-Jun remained phosphorylated at 48 to 96 hours after PH, when ERK phosphorylation returned to basal levels. However, during this period, late JNK and c-Jun phosphorylations were almost undetectable in PTP1B−/− mice, despite the sustained ERK phosphorylation (72 to 96 hours). Moreover, activation of Akt was maximal at 36 hours after PH in PTP1B+/+ mice and declined thereafter, but activation was also maintained from 24 to 96 hours after PH in PTP1B−/− mice. Neither the presence of apoptotic cells quantified by terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling nor the activation of caspase-3 was found from 24 to 48 hours after PH in both genotypes of mice (results not shown). In addition, analysis of the serum alanine aminotransferase level revealed that, although the initial hepatocytolysis at 6 to 12 hours after PH was similar in both genotypes of mice, the serum transaminase level decreased more rapidly along the regeneration process in PTP1B−/− mice (Figure 7A, right).
Figure 7.
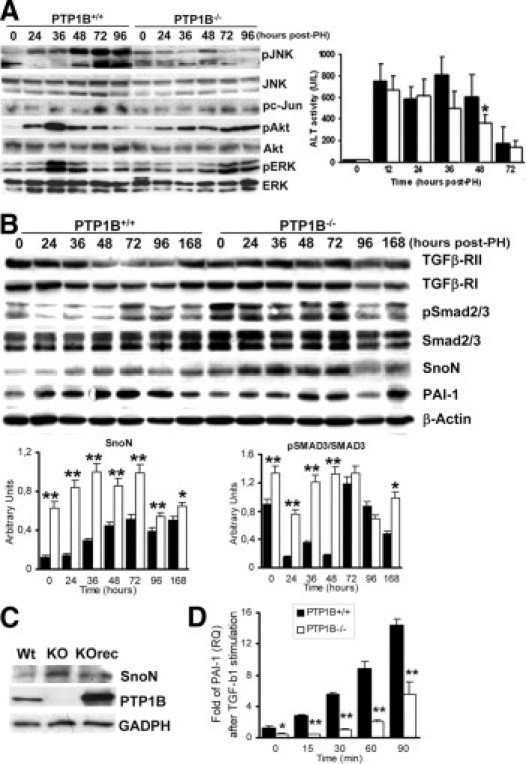
Decreased pro-inflammatory signaling and TGF-β–mediated effects during the late phase of liver regeneration in PTP1B−/− mice. A: Left: Western blot analysis of regenerating livers was performed in PTP1B+/+ and PTP1B−/− mice at the indicated periods with the antibodies against phospho-JNK (pJNK) (Thr183/Tyr185), JNK, phospho-c (pc)–Jun (Ser73), phospho-Akt (pAkt) (Ser473), Akt, phospho-ERK (pERK) (Thr202/Tyr204), and ERK. Representative autoradiograms of three independent series of PH in each genotype are shown. Right: Serum alanine aminotransferase activity at different periods after PH. Results are given as the mean ± SEM (n = 4–8). *P < 0.05 for PTP1B−/− versus PTP1B+/+ mice. B: Representative Western blot analysis of regenerating livers with antibodies against SnoN, phospho-Smad2 (Ser465/467)/Smad3 (Ser423/425) (pSmad2/3), Smad2/3, PAI-1, and β-actin as a loading control. Quantification of SnoN and pSmad2/3 from three independent animals in each period was normalized with β-actin or total Smad2/3, respectively. *P < 0.05; **P < 0.01 for PTP1B−/− versus PTP1B+/+ mice. C: Constitutive expression of SnoN in wild-type (Wt), PTP1B−/− (KO), and PTP1B−/−rec (KOrec) immortalized hepatocytes. GAPDH indicates glyceraldehyde-3-phosphate dehydrogenase. D: mRNA levels of PAI-1 were measured by real-time PCR in immortalized PTP1B+/+ and PTP1B−/− hepatocytes stimulated with TGF-β (5 ng/mL) for several periods. Results are given as the mean ± SEM (n = 6). **P < 0.001 for PTP1B−/− versus PTP1B+/+ cells.
PTP1B Deficiency Up-Regulates the TGF-β Inhibitor SnoN and Delays Termination of Liver Regeneration
Termination of liver regeneration is an important checkpoint that triggers feedback inhibition of growth factors and cytokine-mediated signaling pathways to regulate liver size. Transforming growth factor β via Smad is the main antiproliferative factor within the liver and, therefore, inhibitors of the TGF-β–Smad pathway, such as Ski-related novel gene (SnoN) and Ski, are up-regulated during regeneration.34 Because liver regeneration was increased at late periods after PH in PTP1B−/− mice compared with the PTP1B+/+ controls (Figure 1), we analyzed the expression of SnoN along this process. Figure 7B shows that increased SnoN levels were detected within 36 to 72 hours after PH in PTP1B+/+ mice, decreasing thereafter. The SnoN level was up-regulated in PTP1B−/− livers before surgery and remained highly elevated until 72 hours after PH. In agreement with this finding, the expression of PAI-1, a TGF-β target, increased after PH, reaching a maximum at 48 hours in PTP1B+/+ mice. However, it was decreased substantially and its induction was delayed in PTP1B−/− regenerating livers. In contrast, TGF-β–mediated upstream signaling, measured by Smad2/3 phosphorylation normalized with total Smad2/3 content, was unexpectedly up-regulated in PTP1B−/− livers before and during regeneration. Upstream of Smad, the expression of TGF-β receptors I and II followed the pattern described during liver regeneration in the PTP1B+/+ mice35; however, in mice lacking PTP1B, the expression of both receptors remained elevated up to 96 hours after PH. In vitro, the constitutive expression of SnoN was higher in PTP1B−/− immortalized hepatocytes compared with the controls and decreased further by stable reconstitution of PTP1B−/− cells with wild-type PTP1B (Figure 7C). To substantiate these results, we stimulated immortalized hepatocytes from both genotypes with TGF-β for several periods and the expression of PAI-1 was analyzed by real-time PCR. Figure 7D shows that mRNA levels of PAI-1 were decreased in TGF-β–stimulated PTP1B−/− hepatocytes, probably because of higher SnoN expression.
PTP1B Deficiency Protects from Diet-Induced Steatosis and Improves Hepatic Regeneration during HFD Conditions
Experiments performed in genetic-based and HFD-induced murine models have shown that pre-existing steatosis significantly impairs liver regeneration after PH.36–38 Because PTP1B−/− mice are protected from HFD-induced insulin resistance and obesity, the latter because of enhanced sensitivity to leptin actions in the brain,39 we investigated the presence of fat accumulation in the liver in both genotypes of mice fed an HFD for 12 weeks immediately after weaning. The protection of PTP1B deficiency against obesity and insulin resistance in our mice colony is shown in Supplemental Figure S2 (at http://ajp.amjpathol.org). As depicted in Figure 8, A and B, in PTP1B−/− mice fed an HFD, visceral fat mass and total body weight were significantly decreased when compared with PTP1B+/+ mice. More important, in PTP1B−/− mice fed an HFD, the histological analysis of liver sections revealed the absence of hepatic steatosis (Figure 8D, upper). Therefore, we studied the hepatic regeneration process under these experimental conditions. Analysis of the hepatosomal index revealed a significant increase at 48 to 72 hours after PH in PTP1B−/− mice fed an HFD compared with PTP1B+/+ mice (Figure 8C). Moreover, PCNA and cyclin E peaked at an early period in regenerating livers from HFD-fed PTP1B−/− mice (Figure 8E). Despite the absence of hepatic steatosis in the livers from PTP1B−/− mice fed an HFD, intrahepatic lipid accumulation was assessed by oil red O staining of liver sections from these mice at 48 hours after PH (Figure 8D, middle and lower). These results suggest that transient hepatic steatosis is a programmed adaptation response to PH that is maintained in PTP1B−/− mice fed an HFD. Regenerating livers from PTP1B+/+ mice showed additional lipid overload after PH during the same period. Altogether, these results indicate that PTP1B deficiency also improves hepatic regeneration during HFD conditions, in parallel to the protection against obesity and hepatic steatosis.
Figure 8.
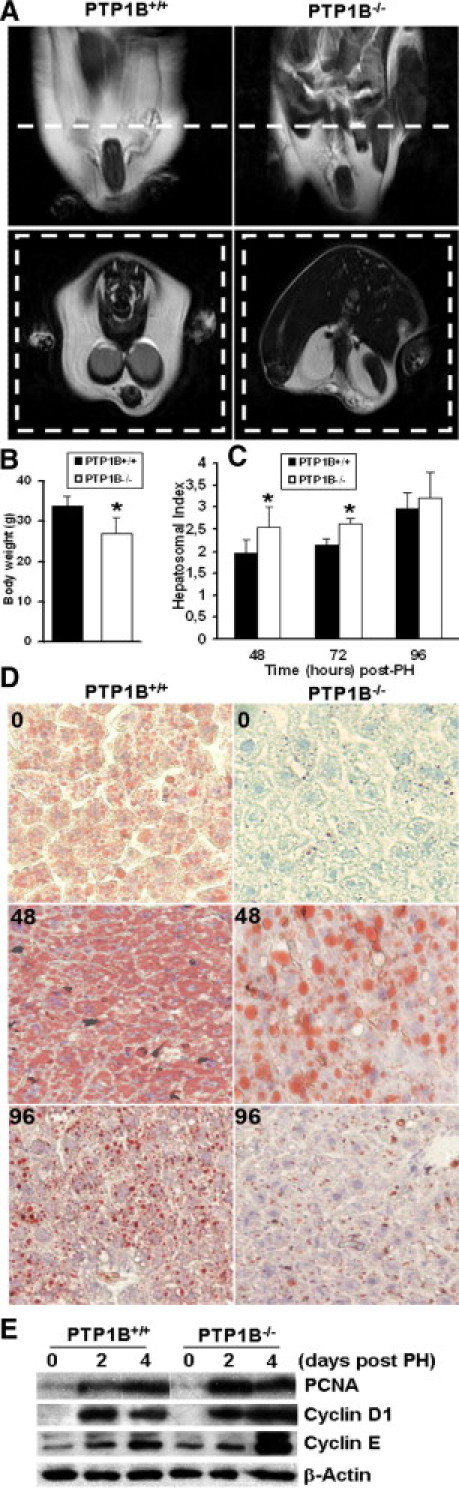
The PTP1B deficiency protects from diet-induced steatosis and improves hepatic regeneration under HFD conditions. A: Comparison of visceral fat mass in PTP1B+/+ and PTP1B−/− mice fed an HFD for 12 weeks after weaning. Animals were subjected to RMN scan analysis in fed conditions. The images were acquired in axial or coronal orientation. B: Comparison of total body weight in PTP1B+/+ and PTP1B−/− mice fed an HFD for 12 weeks after weaning. Results are given as the mean ± SEM (n = 8). *P < 0.05 for PTP1B−/− versus PTP1B+/+ mice. C: The PH was performed in PTP1B+/+ and PTP1B−/− mice after 12 weeks of an HFD, and regenerating livers were removed at the indicated periods. The liver regeneration index (liver/body weight ratio) is given after PH. Results are given as the mean ± SEM (n = 8). *P < 0.05 for PTP1B−/− versus PTP1B+/+ mice. D: Oil red O staining of liver sections from PTP1B−/− and PTP1B+/+ mice after 12 weeks of HFD and at 48 to 96 hours after PH. Original magnification, ×10. E: Representative Western blot analysis with the antibodies against PCNA, cyclins D1 and E, and β-actin as a loading control.
Discussion
Many intracellular pathways regulated by tyrosine phosphorylation play a role during liver regeneration.11,12,40 However, the function of negative modulators of tyrosine kinase–mediated signaling in this complex biological process remains largely unexplored. In this study, we investigated the role of PTP1B in liver regeneration after PH. We found that the early responses that initiate mitogenesis after surgery were enhanced in PTP1B−/− mice compared with PTP1B+/+ controls. These responses are mediated by cytokines and growth factors that stimulate tyrosine phosphorylation of membrane-bound receptors and/or intracellular proteins in hepatocytes. Regarding the priming signals that trigger the exit of G0 and transition to the G1 phase of the cell cycle, activation of JNK and STAT3 by the pro-inflammatory cytokines TNF-α and IL-6, respectively, was enhanced in regenerating livers from PTP1B−/− mice. This effect was mimicked by stimulation of PTP1B+/+ and PTP1B−/− primary hepatocytes with TNF-α or IL-6 or transfection of murine or HuH-7 human liver cells with PTP1B siRNA, indicating that hepatocytes lacking this phosphatase are hypersensitive to cytokine-mediated signaling. Although leptin-induced phosphorylation of the Tyr705 residue of STAT3 is dephosphorylated by PTP1B,17 in this study we reported, for the first time to our knowledge, a new substrate for PTP1B, the Tyr185 residue of JNK. We excluded the possibility of indirect effects of PTP1B deficiency, such as the up-regulation of dual-specificity phosphatase-9 (MKP-4/DUSP-9),41 because its expression in the liver does not differ between PTP1B+/+ and PTP1B−/− mice (results not shown). More important, the fact that JNK phosphorylation was severely decreased in PTP1B−/− hepatocytes reconstituted with active PTP1B indicates that this phosphatase directly modulates the effect of TNF-α on JNK phosphorylation in liver cells. Whether the same effect occurs in other TNF-α–responsive tissues or cell types remains to be investigated. Thus, the hyperactivation of TNF-α– and IL-6–mediated priming cascades in hepatocytes by PTP1B deficiency might trigger a rapid proliferative response after PH, as discussed later. On the other hand, circulating levels of TNF-α and IL-6 were rapidly elevated after PH, this effect being enhanced in the absence of PTP1B. These molecules are released by liver macrophages (Kupffer cells), suggesting an additional new role of PTP1B in Kupffer cells that modulate the secretion of early inducers of liver regeneration; this role deserves further investigation.
In the second phase of liver repair, growth factors are required for progression of hepatocytes through the cell cycle.30 In this step, PTP1B deficiency increased tyrosine autophosphorylation of EGFR and HGFR in hepatocytes and regenerating liver. This result agrees with the increased phosphorylation of EGFR reported in PTP1B−/− fibroblasts,3 whereas sustained HGFR tyrosine phosphorylation in response to an apoptotic trigger has been described in the livers of PTP1B−/− mice.42 Moreover, our results revealed that PTP1B modulates the early signaling pathways activated downstream of receptors of the tyrosine kinase superfamily in hepatocytes. Thus, phosphorylation of Akt and ERK was enhanced and/or prolonged in the absence of this phosphatase. However, the early responses induced in vivo after PH showed substantial differences between the two genotypes of mice because Akt, but not ERK, phosphorylation was prolonged in PTP1B−/− regenerating livers. Therefore, the absence of ERK hyperactivation during the early periods after PH in PTP1B−/− mice suggests the existence of compensatory mechanisms that probably avoid the dysregulation of the regenerative process. In fact, two recent articles43,44 noted that the loss of PTP1B in mouse models of breast cancer is accompanied by attenuation of the Ras/ERK pathway. Thus, during hepatic regeneration, the overall effect of cytokines and growth factors is a coordinated response that is enhanced in the absence of PTP1B, resulting in accelerated and/or increased expression of cell cycle markers and the re-expression of IRA.
One of the major hallmarks of liver regeneration after PH is the programmed adaptation response that triggers transient steatosis after surgery, with a peak before the initial wave of hepatocyte proliferation.45 The mechanisms underlying this accumulation of triglycerides include elevated fatty acid mobilization from white adipose tissue and delivery to the liver, coupled with increased hepatic lipogenesis.45,46 We found increased lipid accumulation in PTP1B−/− regenerating livers 12 hours after PH, in parallel with increased levels of serum free glycerol, an indicator of lipolysis in white adipose tissue. The PTP1B deficiency has tissue-specific effects on carbohydrate and lipid metabolism. In the liver, the lack of this phosphatase increased insulin signaling and, therefore, fatty acid synthesis. On the other hand, adipocytes from PTP1B−/− mice have attenuated insulin responses because of basal activation of the mammalian target of rapamycin complex 1, resulting in enhanced serine phosphorylation of insulin receptor substrate 1 by a feedback mechanism.47 Therefore, enhanced hepatic lipogenesis and elevated lipolysis in white adipose tissue induce a more rapid lipid load in the liver of PTP1B−/− mice immediately after PH. These metabolic adaptations might also play a role in the enhanced regenerative response in the liver of PTP1B−/− mice.
Notably, in hepatectomized PTP1B+/+ mice, the phosphorylation of both JNK and c-Jun was detected at later periods (ie, 24 hours and thereafter). In fact, the differential responses between PTP1B−/− and control mice became most evident at 24 to 96 hours after PH. At these later times, substantial differences were found between wild-type and Gadd45b−/− mice; the latter displayed overactivation of JNK and, therefore, defective hepatic regeneration.33 It is intriguing that PTP1B−/− mice lack the second peak of JNK activation during the 24 to 96 hours after PH (Figure 7A) because the opposite pattern was observed during earlier periods (Figure 2B). A temporal regulation of JNK activation seems critical during liver regeneration. Thus, JNK phosphorylation immediately after PH is required for the activation of nuclear factor κB, activator protein-1, and STAT3 transcription factors, resulting in G0 to G1 transition11,12,30; sustained phosphorylation at later phases might induce a pro-inflammatory–like milieu, with subsequent negative cross talk with the intracellular signaling pathways activated through regeneration. This molecular mechanism would orchestrate the later phases of liver regeneration to modulate the termination step. Thus, it is possible that low JNK activity, sustained Akt, and, to a lesser extent, ERK1/2 phosphorylation at 48 to 96 hours after PH would enhance and/or prolong hepatic regeneration in PTP1B−/− mice. In addition, alanine aminotransferase activity and liver regeneration indicate that the recovery of hepatocytolysis is improved in PTP1B−/− mice. Thus, the overall result is an improvement of the proliferative response after PH and in cultured hepatocytes in the absence of PTP1B.
Among the checkpoints that regulate the regenerative process, TGF-β and related family members are well-known antiproliferative factors within the liver.48 However, in the early periods after PH, hepatocytes become resistant to TGF-β and can proliferate despite the presence of this cytokine secreted by stellate cells.49 More important, levels of SnoN, an inhibitor of the TGF-β–Smad signaling pathway, are also increased during the main proliferative phase in regenerating liver.34 In light of these data, we show that PTP1B deletion concurs with an increase in SnoN expression and cell proliferation in both the in vivo model of liver regeneration and in vitro in cultured hepatocytes. In chronic kidney diseases, HGF abolished TGF-β signaling by inducing SnoN in tubular epithelial cells.50 Because PTP1B−/− hepatocytes are hypersensitive to HGF-induced ERK phosphorylation, this effect might explain the overexpression of SnoN in the liver of PTP1B−/− mice before surgery. In addition, after PH, SnoN expression is up-regulated by TGF-β/Smad2/3–mediated signaling.34 Therefore, the maintenance of TGF-β receptor expression up to 72 hours after PH and the high phospho-Smad2/3 content during this period likely contribute to the increased expression of SnoN in regenerating livers lacking PTP1B. These results are in agreement with the increased SnoN levels and improved liver regeneration in Cav-1−/− mice, as reported recently.51 More important, many human cancer cell lines that express high levels of SnoN are refractory to TGF-β–induced growth arrest.52 Altogether, these data suggest that PTP1B deficiency affects the early triggers of the regenerative response after PH and mediates gene expression related to the termination of this process. Therefore, the molecular mechanism by which PTP1B modulates SnoN expression deserves further investigation.
The role of PTP1B as a regulator of insulin sensitivity and body weight has been investigated extensively.19,20,53 In this study, we show protection against the development of hepatic steatosis together with improved regenerative responses in PTP1B−/− mice fed an HFD before PH. In addition to the protection against insulin resistance and obesity,20,54 an additional benefit of PTP1B deficiency may be the improvement of liver regeneration under conditions of increased lipid intake, as demonstrated herein by accelerated or increased induction of S-phase markers and an increased hepatosomal index compared with wild-type controls. In clinical situations, obese patients with fatty livers, who frequently develop insulin resistance, tend to have poor outcomes after resection or liver transplantation.55 Thus, our results suggest that, in addition to the amelioration of hepatic steatosis, PTP1B inhibitors could have additional therapeutic utility in these individuals with respect to the improvement of liver regeneration after PH.
In conclusion, the results presented herein have revealed an essential role of PTP1B in the response to cytokines/growth factors that initiate hepatic regeneration and in the control of its termination. More important, the overall process of liver regeneration in the absence of this phosphatase is accelerated but not deregulated. Therefore, PTP1B could be a novel drug target to improve liver regeneration in patients with acute or chronic liver injury.
Acknowledgments
We acknowledge Oscar Escribano, Ph.D., (Universidad Complutense, Madrid, Spain) for his expertise with PH in our initial experiments.
Footnotes
Supported by contracts from the Centro de Investigación Biomédica en Red de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM) (J.R.-C. and A.G.-R.) and by grants from CIBERDEM and the Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas, Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación (MICINN), Spain (SAF2007-60551 to P.M.S.; SAF2009-08114 and CIT-090100-2007-35 to A.M.V.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.12.020.
Supplementary data
Reconstitution of PTP1B−/− hepatocytes with purified PTP1B decreased EGFR and HGFR phosphorylation. Immortalized PTP1B−/− hepatocytes were stimulated with 20-ng/mL EGF (left) or 30-ng/mL HGF (right) for 10 minutes. A total of 60 μL of cell lysates (1 μg/μL) was incubated with 10 U of recombinant PTP1B for 30 minutes, and the phosphorylation of EGFR and HGFR was analyzed by Western blot against phospho-EGFR (Tyr1086), phospho-HGFR (Tyr1234/1235), and total HGFR and EGFR as loading controls. Representative autoradiograms of two independent experiments are shown.
The PTP1B−/− mice fed an HFD were protected against obesity, insulin resistance, and glucose intolerance. A: Body weight curves of age-matched PTP1B+/+ and PTP1B−/− mice fed an HFD for 12 weeks after weaning. Values are given as the mean ± SEM (n = 10-12). ***P < 0.001 for PTP1B−/− versus PTP1B+/+ mice. B: Glucose tolerance test (GTT) results (i.p. injection of 2 g d-glucose/kg). C: Insulin tolerance test (ITT) results (i.p. injection of 0.75 U/kg human regular insulin). Both tests were performed in PTP1B+/+ and PTP1B−/− mice fed an HFD. Animals fasted for 20 and 4 hours before the GTT and ITT, respectively. For GTTs, results are expressed as mean ± SEM blood glucose level (n = 8-10). For ITTs, results are expressed as mean ± SEM initial blood glucose level (n = 8-10). *P < 0.05, **P < 0.01, and ***P < 0.001 for PTP1B−/− versus PTP1B+/+ mice.
References
- 1.Frangioni J.V., Beahm P.H., Shifrin V., Jost C.A., Neel B.G. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell. 1992;68:545–560. doi: 10.1016/0092-8674(92)90190-n. [DOI] [PubMed] [Google Scholar]
- 2.Flint A.J., Tiganis T., Barford D., Tonks N.K. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haj F.G., Markova B., Klaman L.D., Bohmer F.D., Neel B.G. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatase-1B. J Biol Chem. 2003;278:739–744. doi: 10.1074/jbc.M210194200. [DOI] [PubMed] [Google Scholar]
- 4.Sangwan V., Paliouras G.N., Abella J.V., Dube N., Monast A., Tremblay M.L., Park M. Regulation of the Met receptor-tyrosine kinase by the protein-tyrosine phosphatase 1B and T-cell phosphatase. J Biol Chem. 2008;283:34374–34383. doi: 10.1074/jbc.M805916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salmeen A., Andersen J.N., Myers M.P., Tonks N.K., Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. 2000;6:1401–1412. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- 6.Buckley D.A., Cheng A., Kiely P.A., Tremblay M.L., O'Connor R. Regulation of insulin-like growth factor type I (IGF-I) receptor kinase activity by protein tyrosine phosphatase 1B (PTP-1B) and enhanced IGF-I-mediated suppression of apoptosis and motility in PTP-1B-deficient fibroblasts. Mol Cell Biol. 2002;22:1998–2010. doi: 10.1128/MCB.22.7.1998-2010.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herrera B., Fernandez M., Benito M., Fabregat I. cIAP-1, but not XIAP, is cleaved by caspases during the apoptosis induced by TGF-beta in fetal rat hepatocytes. FEBS Lett. 2002;520:93–96. doi: 10.1016/s0014-5793(02)02774-6. [DOI] [PubMed] [Google Scholar]
- 8.Valverde A.M., Fabregat I., Burks D.J., White M.F., Benito M. IRS-2 mediates the antiapoptotic effect of insulin in neonatal hepatocytes. Hepatology. 2004;40:1285–1294. doi: 10.1002/hep.20485. [DOI] [PubMed] [Google Scholar]
- 9.Mead J.E., Fausto N. Transforming growth factor alpha may be a physiological regulator of liver regeneration by means of an autocrine mechanism. Proc Natl Acad Sci U S A. 1989;86:1558–1562. doi: 10.1073/pnas.86.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyazaki M., Masaka T., Akiyama I., Nakashima E., Sakaguchi M., Huh N.H. Propagation of adult rat bone marrow-derived hepatocyte-like cells by serial passages in vitro. Cell Transplant. 2004;13:385–391. doi: 10.3727/000000004783983800. [DOI] [PubMed] [Google Scholar]
- 11.Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 12.Michalopoulos G.K. Liver regeneration. J Cell Physiol. 2007;213:286–300. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cressman D.E., Diamond R.H., Taub R. Rapid activation of the Stat3 transcription complex in liver regeneration. Hepatology. 1995;21:1443–1449. [PubMed] [Google Scholar]
- 14.FitzGerald M.J., Webber E.M., Donovan J.R., Fausto N. Rapid DNA binding by nuclear factor kappa B in hepatocytes at the start of liver regeneration. Cell Growth Differ. 1995;6:417–427. [PubMed] [Google Scholar]
- 15.Heim M.H., Gamboni G., Beglinger C., Gyr K. Specific activation of AP-1 but not Stat3 in regenerating liver in mice. Eur J Clin Invest. 1997;27:948–955. doi: 10.1046/j.1365-2362.1997.2100770.x. [DOI] [PubMed] [Google Scholar]
- 16.Fukada T., Hibi M., Yamanaka Y., Takahashi-Tezuka M., Fujitani Y., Yamaguchi T., Nakajima K., Hirano T. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity. 1996;5:449–460. doi: 10.1016/s1074-7613(00)80501-4. [DOI] [PubMed] [Google Scholar]
- 17.Zabolotny J.M., Bence-Hanulec K.K., Stricker-Krongrad A., Haj F., Wang Y., Minokoshi Y., Kim Y.B., Elmquist J.K., Tartaglia L.A., Kahn B.B., Neel B.G. PTP1B regulates leptin signal transduction in vivo. Dev Cell. 2002;2:489–495. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 18.Oe S., Lemmer E.R., Conner E.A., Factor V.M., Leveen P., Larsson J., Karlsson S., Thorgeirsson S.S. Intact signaling by transforming growth factor beta is not required for termination of liver regeneration in mice. Hepatology. 2004;40:1098–1105. doi: 10.1002/hep.20426. [DOI] [PubMed] [Google Scholar]
- 19.Elchebly M., Payette P., Michaliszyn E., Cromlish W., Collins S., Loy A.L., Normandin D., Cheng A., Himms-Hagen J., Chan C.C., Ramachandran C., Gresser M.J., Tremblay M.L., Kennedy B.P. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 20.Klaman L.D., Boss O., Peroni O.D., Kim J.K., Martino J.L., Zabolotny J.M., Moghal N., Lubkin M., Kim Y.B., Sharpe A.H., Stricker-Krongrad A., Shulman G.I., Neel B.G., Kahn B.B. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20:5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez-Rodriguez A., Clampit J.E., Escribano O., Benito M., Rondinone C.M., Valverde A.M. Developmental switch from prolonged insulin action to increased insulin sensitivity in protein tyrosine phosphatase 1B-deficient hepatocytes. Endocrinology. 2007;148:594–608. doi: 10.1210/en.2006-0644. [DOI] [PubMed] [Google Scholar]
- 22.Higgins G., Anderson R. Experimental pathology of the liver, I: restoration of the liver of the white rat following partial surgical removal. Arch Pathol Lab Med. 1931;12:186–202. [Google Scholar]
- 23.Martins P.N., Theruvath T.P., Neuhaus P. Rodent models of partial hepatectomies. Liver Int. 2008;28:3–11. doi: 10.1111/j.1478-3231.2007.01628.x. [DOI] [PubMed] [Google Scholar]
- 24.Mascrez B., Ghyselinck N.B., Watanabe M., Annicotte J.S., Chambon P., Auwerx J., Mark M. Ligand-dependent contribution of RXRbeta to cholesterol homeostasis in Sertoli cells. EMBO Rep. 2004;5:285–290. doi: 10.1038/sj.embor.7400094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benveniste R., Danoff T.M., Ilekis J., Craig H.R. Epidermal growth factor receptor numbers in male and female mouse primary hepatocyte cultures. Cell Biochem Funct. 1988;6:231–235. doi: 10.1002/cbf.290060403. [DOI] [PubMed] [Google Scholar]
- 26.Entingh A.J., Taniguchi C.M., Kahn C.R. Bi-directional regulation of brown fat adipogenesis by the insulin receptor. J Biol Chem. 2003;278:33377–33383. doi: 10.1074/jbc.M303056200. [DOI] [PubMed] [Google Scholar]
- 27.Assy N., Minuk G.Y. Liver regeneration: methods for monitoring and their applications. J Hepatol. 1997;26:945–952. doi: 10.1016/s0168-8278(97)80266-8. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez-Rodriguez A., Nevado C., Escriva F., Sesti G., Rondinone C.M., Benito M., Valverde A.M. PTP1B deficiency increases glucose uptake in neonatal hepatocytes: involvement of IRA/GLUT2 complexes. Am J Physiol Gastrointest Liver Physiol. 2008;295:G338–G347. doi: 10.1152/ajpgi.00514.2007. [DOI] [PubMed] [Google Scholar]
- 29.Frasca F., Pandini G., Scalia P., Sciacca L., Mineo R., Costantino A., Goldfine I.D., Belfiore A., Vigneri R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. 1999;19:3278–3288. doi: 10.1128/mcb.19.5.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fausto N. Liver regeneration. J Hepatol. 2000;32:19–31. doi: 10.1016/s0168-8278(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 31.Davis R.J. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 32.Shteyer E., Liao Y., Muglia L.J., Hruz P.W., Rudnick D.A. Disruption of hepatic adipogenesis is associated with impaired liver regeneration in mice. Hepatology. 2004;40:1322–1332. doi: 10.1002/hep.20462. [DOI] [PubMed] [Google Scholar]
- 33.Papa S., Zazzeroni F., Fu Y.X., Bubici C., Alvarez K., Dean K., Christiansen P.A., Anders R.A., Franzoso G. Gadd45beta promotes hepatocyte survival during liver regeneration in mice by modulating JNK signaling. J Clin Invest. 2008;118:1911–1923. doi: 10.1172/JCI33913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macias-Silva M., Li W., Leu J.I., Crissey M.A., Taub R. Up-regulated transcriptional repressors SnoN and Ski bind Smad proteins to antagonize transforming growth factor-beta signals during liver regeneration. J Biol Chem. 2002;277:28483–28490. doi: 10.1074/jbc.M202403200. [DOI] [PubMed] [Google Scholar]
- 35.Chari R.S., Price D.T., Sue S.R., Meyers W.C., Jirtle R.L. Down-regulation of transforming growth factor beta receptor type I, II, and III during liver regeneration. Am J Surg. 1995;169:126–131. doi: 10.1016/s0002-9610(99)80120-2. discussion, 131–132. [DOI] [PubMed] [Google Scholar]
- 36.Yang S.Q., Lin H.Z., Mandal A.K., Huang J., Diehl A.M. Disrupted signaling and inhibited regeneration in obese mice with fatty livers: implications for nonalcoholic fatty liver disease pathophysiology. Hepatology. 2001;34:694–706. doi: 10.1053/jhep.2001.28054. [DOI] [PubMed] [Google Scholar]
- 37.Yamauchi H., Uetsuka K., Okada T., Nakayama H., Doi K. Impaired liver regeneration after partial hepatectomy in db/db mice. Exp Toxicol Pathol. 2003;54:281–286. doi: 10.1078/0940-2993-00265. [DOI] [PubMed] [Google Scholar]
- 38.DeAngelis R.A., Markiewski M.M., Taub R., Lambris J.D. A high-fat diet impairs liver regeneration in C57BL/6 mice through overexpression of the NF-kappaB inhibitor, IkappaBalpha. Hepatology. 2005;42:1148–1157. doi: 10.1002/hep.20879. [DOI] [PubMed] [Google Scholar]
- 39.Bence K.K., Delibegovic M., Xue B., Gorgun C.Z., Hotamisligil G.S., Neel B.G., Kahn B.B. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 40.Fausto N., Campbell J.S., Riehle K.J. Liver regeneration. Hepatology. 2006;43:S45–S53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 41.Xu H., Dembski M., Yang Q., Yang D., Moriarty A., Tayber O., Chen H., Kapeller R., Tartaglia L.A. Dual specificity mitogen-activated protein (MAP) kinase phosphatase-4 plays a potential role in insulin resistance. J Biol Chem. 2003;278:30187–30192. doi: 10.1074/jbc.M302010200. [DOI] [PubMed] [Google Scholar]
- 42.Sangwan V., Paliouras G.N., Cheng A., Dube N., Tremblay M.L., Park M. Protein-tyrosine phosphatase 1B deficiency protects against Fas-induced hepatic failure. J Biol Chem. 2006;281:221–228. doi: 10.1074/jbc.M507858200. [DOI] [PubMed] [Google Scholar]
- 43.Bentires-Alj M., Neel B.G. Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Res. 2007;67:2420–2424. doi: 10.1158/0008-5472.CAN-06-4610. [DOI] [PubMed] [Google Scholar]
- 44.Julien S.G., Dube N., Read M., Penney J., Paquet M., Han Y., Kennedy B.P., Muller W.J., Tremblay M.L. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat Genet. 2007;39:338–346. doi: 10.1038/ng1963. [DOI] [PubMed] [Google Scholar]
- 45.Tijburg L.B., Nyathi C.B., Meijer G.W., Geelen M.J. Biosynthesis and secretion of triacylglycerol in rat liver after partial hepatectomy. Biochem J. 1991;277(pt 3):723–728. doi: 10.1042/bj2770723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Jongh K.S., Schofield P.J., Edwards M.R. Kinetic mechanism of sheep liver NADPH-dependent aldehyde reductase. Biochem J. 1987;242:143–150. doi: 10.1042/bj2420143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ruffolo S.C., Forsell P.K., Yuan X., Desmarais S., Himms-Hagen J., Cromlish W., Wong K.K., Kennedy B.P. Basal activation of p70S6K results in adipose-specific insulin resistance in protein-tyrosine phosphatase 1B−/− mice. J Biol Chem. 2007;282:30423–30433. doi: 10.1074/jbc.M700697200. [DOI] [PubMed] [Google Scholar]
- 48.Derynck R., Zhang Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 49.Koniaris L.G., McKillop I.H., Schwartz S.I., Zimmers T.A. Liver regeneration. J Am Coll Surg. 2003;197:634–659. doi: 10.1016/S1072-7515(03)00374-0. [DOI] [PubMed] [Google Scholar]
- 50.Tan R., Zhang X., Yang J., Li Y., Liu Y. Molecular basis for the cell type specific induction of SnoN expression by hepatocyte growth factor. J Am Soc Nephrol. 2007;18:2340–2349. doi: 10.1681/ASN.2007010128. [DOI] [PubMed] [Google Scholar]
- 51.Mayoral R., Valverde A.M., Llorente Izquierdo C., González-Rodríguez A., Boscá L., Martín-Sanz P. Impairment of transforming growth factor beta signaling in caveolin-1-deficient hepatocytes: role in liver regeneration. J Biol Chem. 2010;285:3633–3642. doi: 10.1074/jbc.M109.072900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sarker K.P., Wilson S.M., Bonni S. SnoN is a cell type-specific mediator of transforming growth factor-beta responses. J Biol Chem. 2005;280:13037–13046. doi: 10.1074/jbc.M409367200. [DOI] [PubMed] [Google Scholar]
- 53.González-Rodríguez A., Mas Gutierrez J.A., Sanz-González S., Ros M., Burks D.J., Valverde A.M. Inhibition of PTP1B restores IRS1-mediated hepatic insulin signaling in IRS2-deficient mice. Diabetes. 2010;59:588–599. doi: 10.2337/db09-0796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zabolotny J.M., Kim Y.B., Welsh L.A., Kershaw E.E., Neel B.G., Kahn B.B. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem. 2008;283:14230–14241. doi: 10.1074/jbc.M800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Behrns K.E., Tsiotos G.G., DeSouza N.F., Krishna M.K., Ludwig J., Nagorney D.M. Hepatic steatosis as a potential risk factor for major hepatic resection. J Gastrointest Surg. 1998;2:292–298. doi: 10.1016/s1091-255x(98)80025-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reconstitution of PTP1B−/− hepatocytes with purified PTP1B decreased EGFR and HGFR phosphorylation. Immortalized PTP1B−/− hepatocytes were stimulated with 20-ng/mL EGF (left) or 30-ng/mL HGF (right) for 10 minutes. A total of 60 μL of cell lysates (1 μg/μL) was incubated with 10 U of recombinant PTP1B for 30 minutes, and the phosphorylation of EGFR and HGFR was analyzed by Western blot against phospho-EGFR (Tyr1086), phospho-HGFR (Tyr1234/1235), and total HGFR and EGFR as loading controls. Representative autoradiograms of two independent experiments are shown.
The PTP1B−/− mice fed an HFD were protected against obesity, insulin resistance, and glucose intolerance. A: Body weight curves of age-matched PTP1B+/+ and PTP1B−/− mice fed an HFD for 12 weeks after weaning. Values are given as the mean ± SEM (n = 10-12). ***P < 0.001 for PTP1B−/− versus PTP1B+/+ mice. B: Glucose tolerance test (GTT) results (i.p. injection of 2 g d-glucose/kg). C: Insulin tolerance test (ITT) results (i.p. injection of 0.75 U/kg human regular insulin). Both tests were performed in PTP1B+/+ and PTP1B−/− mice fed an HFD. Animals fasted for 20 and 4 hours before the GTT and ITT, respectively. For GTTs, results are expressed as mean ± SEM blood glucose level (n = 8-10). For ITTs, results are expressed as mean ± SEM initial blood glucose level (n = 8-10). *P < 0.05, **P < 0.01, and ***P < 0.001 for PTP1B−/− versus PTP1B+/+ mice.