

Abstract
Ablation of mouse occipital cortex induces precisely timed and uniform p53-modulated and Bax-dependent apoptosis of thalamocortical projection neurons in the dorsal lateral geniculate nucleus (LGN) by 7 d after lesion. We tested the hypothesis that this neuronal apoptosis is initiated by oxidative stress and the mitochondrial permeability transition pore (mPTP). Preapoptotic LGN neurons accumulate mitochondria, Zn2+ and Ca2+, and generate higher levels of reactive oxygen species (ROS), including superoxide, nitric oxide (NO), and peroxynitrite, than LGN neurons with an intact cortical target. Preapoptosis of LGN neurons is associated with increased formation of protein carbonyls, protein nitration, and protein S-nitrosylation. Genetic deletion of nitric oxide synthase 1 (nos1) and inhibition of NOS1 with nitroindazole protected LGN neurons from apoptosis, revealing NO as a mediator. Putative components of the mPTP are expressed in mouse LGN, including the voltage-dependent anion channel (VDAC), adenine nucleotide translocator (ANT), and cyclophilin D (CyPD). Nitration of CyPD and ANT in LGN mitochondria occurs by 2 d after cortical injury. Chemical cross-linking showed that LGN neuron preapoptosis is associated with formation of CyPD and VDAC oligomers, consistent with mPTP formation. Mice without CyPD are rescued from neuron apoptosis as are mice treated with the mPTP inhibitors TRO-19622 (cholest-4-en-3-one oxime) and TAT-Bcl-XL-BH4. Manipulation of the mPTP markedly attenuated the early preapoptotic production of reactive oxygen/nitrogen species in target-deprived neurons. Our results demonstrate in adult mouse brain neurons that the mPTP functions to enhance ROS production and the mPTP and NO trigger apoptosis; thus, the mPTP is a target for neuroprotection in vivo.
Introduction
Apoptosis contributes to the pathophysiology of CNS damage in cerebral ischemia (Martin et al., 2000; Hou and MacManus, 2002; Moskowitz and Lo, 2003), trauma (Beattie et al., 2002), and chronic neurodegenerative disease, including Alzheimer's disease (Anderson et al., 1996; Martin, 2010), Parkinson's disease (Anglade et al., 1997; Hartmann et al., 2000; Martin, 2010), and amyotrophic lateral sclerosis (Martin, 1999). In rodent models of acute brain injury, specifically cerebral ischemia (Cheng et al., 1998; Nakajima et al., 2000; Gibson et al., 2001; Northington et al., 2001, 2005) and excitotoxicity (Lok and Martin, 2002; Pérez-Navarro et al., 2005), Bcl-2 family members and caspases have important roles in the evolution of neuropathology. However, cell death mechanisms in these models are confounded by a highly heterogeneous, asynchronous neuropathology and coactivation of several different cell death signaling pathways, including apoptosis, necrosis, and autophagy (Ferrer et al., 1995; van Lookeren Campagne et al., 1995; Portera-Cailliau et al., 1997a,b; Martin et al., 1998; Blomgren et al., 2001; Kawamura et al., 2005; Northington et al., 2007; Puyal et al., 2009; Martin, 2010).
To study the molecular regulation of neuronal apoptosis specifically within the CNS, divorced from other forms of cell death, and to identify potential targets for antiapoptosis neuroprotection, animal models featuring homogeneous and synchronous cellular pathology in discrete brain regions are desirable. Target ablation fulfills this need. Ablation of visual cortex induces selective retrograde neuronal degeneration in the dorsal lateral geniculate nucleus (dLGN) of thalamus (Lashley, 1941; Barron et al., 1973). The geniculocortical projection neurons die by a morphological process that is unequivocal apoptosis (Al-Abdulla and Martin, 1998; Al-Abdulla et al., 1998). Apoptosis of geniculocortical projection neurons requires the presence of the bax gene and is modulated by a functional p53 gene (Martin et al., 2001, 2003). This neuronal cell death emerges with apparent somal accumulation of mitochondria and oxidative damage to genomic DNA of vulnerable projection neurons (Al-Abdulla and Martin, 1998). Mitochondria are producers of toxic reactive oxygen species (ROS) that can damage cellular constituents and initiate many forms of cell death in mammalian cells (Hirsch et al., 1997; Wallace, 2005; Zorov et al., 2007). Mitochondria are sources of several apoptogenic proteins that on release execute the apoptotic process (Hirsch et al., 1997; Martin, 2010). Release of apoptogenic proteins from mitochondria can occur through mechanisms involving formation of membrane channels comprised of Bax (Antonsson et al., 1997), Bax and the adenine nucleotide translocator (ANT) (Marzo et al., 1998), the voltage-dependent anion channel (VDAC) (Shimizu et al., 2000), and the mitochondrial permeability transition pore (mPTP) (Hirsch et al., 1997; van Gurp et al., 2003). However, in non-neural cells, the role of the mPTP in regulating cell death is controversial because data show actions that are proapoptotic (Yasuda et al., 2006; Kang et al., 2007), antiapoptotic (Lin and Lechleiter, 2002; Schubert and Grimm, 2004; Machida et al., 2006), and pronecrotic (Baines et al., 2005; Nakagawa et al., 2005) in cell and animal models. It is not known whether mitochondria participate directly in mediating target deprivation-induced apoptosis of CNS neurons or whether they are bystanders. We tested the hypothesis mitochondria are effectors of apoptosis in adult brain neurons through the mPTP and that inactivation of mPTP protects neurons from apoptosis.
Materials and Methods
Mouse models of neuronal apoptosis in brain.
A unilateral occipital cortex ablation was the model for producing axotomy and target deprivation of dLGN neurons in mouse. Cortical ablations were done on adult (6–8 weeks of age) male mice. For experiments on wild-type mice, the C57BL/6J strain was used. For experiments on mice with NOS gene deletions, mice deficient in neuronal nitric oxide synthase (nNOS) (B6;129S4-Nos1tm1Plh/J; The Jackson Laboratory) and inducible nitric oxide synthase (iNOS) (B6;129P-Nos2tm1Lau) were used. B6129SF2/J mice were controls for nNOS−/− mice. B6129PF2/J mice were controls for iNOS−/− mice. Two different lines of cyclophilin D-null (ppif−/−) mice were used: one line on a SV129 genetic background (Baines et al., 2005; Martin et al., 2009b) and another line on a C57BL/6 genetic background (Basso et al., 2005). Mouse cohort sizes were 6–10 per genotype. The Institutional Animal Care and Use Committee approved the animal protocols. The validation and reproducibility of this model of neuronal apoptosis in mouse has been described previously (Martin et al., 2001, 2003; Natale et al., 2002). For corroboration of findings, an alternative model of pure target deprivation by excitotoxic lesioning was used (Portera-Cailliau et al., 1997b; Mueller et al., 2005). Unilateral visual cortical ablations were done on adult (6–8 weeks of age) male mice using direct cortical injection of the NMDA receptor agonist QA (quinolinic acid) (Sigma-Aldrich) at a concentration of 60 nmol (500 nl total volume at four sites) dissolved in PBS. Control mice received visual cortical injections of an equal volume of PBS.
Morphometric analyses.
Transmission electron microscopy (EM) was used for counting mitochondria. Mice received unilateral occipital cortex ablations and were killed for EM at 1, 2, 4, 5, and 6 d (n = 5 mice/group) after injury. The mice were deeply anesthetized and perfused through the heart with 2% glutaraldehyde/2% paraformaldehyde after which the brains were allowed to fix in situ overnight before removal from the cranium. Ipsilateral and contralateral dLGNs were viewed under a surgical microscope and microdissected from the brain, processed for EM, and serially thin sectioned as described previously (Martin et al., 1994; Al-Abdulla and Martin, 1998). Grids were viewed with JEOL 100S or Phillips CM12 electron microscopes and scanned for dLGN relay neuron profiles using previously defined ultrastructural characteristics (Al-Abdulla and Martin, 2002). Micrographs of serial dLGN neuron profiles were shot at a primary magnification of 4000×. Fifty cells were analyzed per postlesion time point in serial micrographs through the same cell. dLGN electron micrographs analyzed with an array that uses 1.2-cm-long line segments (each end defining one test point) and a total of 836 points (Weibel, 1979). The relative volume occupied by the dLGN neurons and the volume fraction occupied by mitochondria, Vv, were determined using the standard morphometric equation Vvi = Pi/Pt, where Pi and Pt are the sums of the number of points over mitochondria and over the reference space, respectively.
To control for cell body volume changes associated with degeneration, we measured the volume of dLGN neurons at different postlesion time points in sections stained with cresyl violet. Neuronal volumetric analyses using point counting and the Cavalieri principle were done on random neuron images throughout the dLGN.
Mitochondria and intracellular ROS, NO, Ca2+, and Zn2+ tracking.
In vivo tracking of mitochondria and intracellular ROS, NO, Ca2+, and Zn2+ in apoptotic dLGN corticopetal projection neurons was achieved using fluorescent tracers. dLGN cortical projection neuron mitochondria within the axons and synaptic terminals in occipital cortex were labeled using cell-permeable, fixable mitochondrian-selective probe MitoTracker Red CM-H2XRos (Invitrogen) as described in a different neuronal system (Martin et al., 2007). Stock solutions were prepared by dissolving the dye powder in anhydrous DMSO to a final concentration of 1 mm. Working solutions were diluted in Influx pinocytic cell-loading reagent (Invitrogen). Probe (100 nm/2 μl) was injected bilaterally into the occipital cortex to label mitochondria within geniculocortical projection neurons, and then after 1 d exposure the mice received a unilateral cortical ablation. The nonlesioned side served as control for each mouse. Controls for dye uptake were mice without injections of MitoTracker, which did not have any fluorescent labeling of mitochondria.
The productions of superoxide and general ROS in dLGN corticopetal neurons were visualized at incremental times after target deprivation. This was achieved by visual cortical injections of the cell-permeant superoxide indicator dihydroethidium (HE) (Invitrogen) and the ROS indicator 5-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA; DCF) (Invitrogen), which are retrogradely transported to dLGN neuron cell bodies as in other neuronal populations (Martin et al., 2005, 2007). DCF has properties that enhance cellular retention of the fluorescent product for improved localization and quantification. The probe is generic for ROS. DCF has been used to track peroxynitrite (ONOO−) in embryonic MN in vitro (Estévez et al., 1999), although it is not specific for only ONOO− as it can detect other oxidants. Stock solutions were prepared by dissolving the dye powder in anhydrous DMSO to a final concentration of 1 mm. Working solutions were diluted in pinocytic cell-loading solution. HE or DCF (100 nm/2 μl) was injected bilaterally into the occipital cortex to retrogradely label geniculocortical projection neurons, and then after 1 d exposure the mice received unilateral visual cortex ablation. The nonlesioned dLGN served as control for each mouse. Fluorescent probe controls were mice without injections of ROS indicators, which did not have any fluorescent labeling, viewing samples with excitation–emission filter combinations out of the appropriate spectral range, and using transgenic mice expressing human wild-type SOD1 (Martin et al., 2003) to attenuate signal.
NO production in dLGN neurons in vivo was tracked using 1,2-diaminoanthraquinone (DAA) (Invitrogen). As an aromatic vicinal diamine, DAA is nonfluorescent, but it reacts selectively with NO to yield a fluorescent product (Heiduschka and Thanos, 1998; Chen et al., 2001; von Bohlen und Halbach et al., 2002). DAA was prepared in Influx (Invitrogen) pinocytic cell-loading reagent. dLGN projections neurons were loaded in vivo by bilateral injection of DAA in visual cortex similar to that described using a different system of projection neurons (Martin et al., 2007). One day later, the mice received a unilateral occipital cortex ablation. The contralateral nonlesioned side served as control. As a negative control, l-NAME (NG-nitro-l-arginine methyl ester) was injected (50 mg/kg, i.p.) to inhibit all forms of NOS.
Neurons undergoing cell death show elevated intracellular Ca2+ and Zn2+, and these changes can be related mechanistically to the cytopathic process (Cheng and Reynolds, 1998; Li et al., 2009). Intracellular free Ca2+ and Zn2+ were detected fura-2 AM and FluoZin-3. Fura-2 AM-dextran (Invitrogen) was dissolved in saline and diluted in pinocytic uptake solution to a concentration of 25% (w/v). Dextran conjugates of fura-2 AM are transported retrogradely and remain in the cytoplasm without compartmentalization or leakage and can be used for long-term Ca2+ measurements (O'Donovan et al., 1994; Martin et al., 2007). However, most Ca2+-sensitive dyes can also bind Zn2+, so FluoZin-3 was used to measure intracellular Zn2+ distinct from Ca2+ determined by fura-2 AM (Devinney et al., 2005). FluoZin-3 (Invitrogen) was dissolved in DMSO for a 5 mm stock solution and then diluted in pinocytic uptake solution to a concentration of 100 μm. Mice received bilateral parenchymal injections of fura-2 AM-dextran into the occipital cortex or intracerebroventricular injections of FluoZin-3 to load dLGN projection neurons, and 1 d later a unilateral occipital cortex ablation was performed. As a negative control, a pledget of BAPTA-AM-saturated Gelfoam with was placed in the cortical lesion site.
Mice that received fluorescent tracer injections and unilateral occipital cortex ablations were killed at 1, 6, and 12 h and at 1, 2, 4, 5, 6, and 7 d (n = 6 mice/group) after injury by anesthetic overdose and intracardiac perfusion with PBS followed by 4% paraformaldehyde. All fluorescent probes used were fixable. The brains were removed 2 h later and postfixed overnight followed by cryoprotection in 20% glycerol. Sections were cut serially at 40 μm in thickness using a freezing sliding microtome. Every 10th section through the dLGN was counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) and analyzed.
Imaging and single-cell densitometry.
Brain sections were viewed on Zeiss Axiophot epifluorescent microscope equipped with a Spot digital camera. Filter combinations for fluorophore detections were as follows: 580/590 (MitoTracker and DAA) and 494/517 (DCF, fura-2 AM-dextran, and FluoZin-3). We estimated dLGN neuron perikaryal mitochondrial “mass” (an indirect measure based on total pixel intensity of MitoTracker), ROS index (based on total pixel intensity of DCF fluorescence), NO index (based on total pixel intensity of DAA fluorescence), intracellular Ca2+ index (based on total pixel intensity of fura-2 AM-dextran fluorescence), and intracellular Zn2+ index (based on total pixel intensity of FluoZin-3 fluorescence). Fluorescent signals in dLGN neurons were analyzed on a single-cell basis (Martin et al., 2005, 2007) by an observer unaware of sample history. For each mouse, digital images from ∼50 neurons, cut through the approximate cell center as judged by the DAPI-stained nucleus, were acquired from the target-deprived and target-intact dLGN. For each neuron, measurements of cell size and fluorescence intensity were obtained by delineating the cell of interest using ImageJ densitometry software and necessary plug-ins (Chang and Martin, 2009). An alternative approach was used for corroboration of ImageJ analysis. Images of dLGN neurons were captured photographically on black-and-white film at 1000× magnification. The films of individual dLGN neurons were scanned and saved as TIFF files. For each neuron, measurements were obtained by delineating the cell of interest using densitometry software (Inquiry Software; Loats Associates). Relative fluorescence intensity is reflected by the average-integrated intensity of film emulsion grain density.
In vivo chemical cross-linking.
We determined whether the mPTP was formed in target-deprived dLGN using a novel approach to chemical cross-linking in vivo. Wild-type and ppif−/− mice received unilateral occipital cortex ablations, and at 1, 2, 3, and 4 d after lesioning mice (n = 4/time point) received intracerebroventricular injection of disuccinimidyl suberate (DSS) (Pierce) at a concentration of 5 mm. Mice were survived for 1 h after DSS and killed by decapitation. The brain from each mouse was removed quickly from the cranium and placed in ice-cold Hanks buffer. Under a stereomicroscope, the cerebrum was removed to reveal the brainstem and the LGN was carefully microdissected from each side (ipsilateral and contralateral to the occipital cortex lesion). Samples were rinsed briefly in ice-cold 50 mm Tris-glycine and then lysed and homogenized and evaluated for CyPD, ANT, and VDAC oligomerization by immunoblotting. Equivalent protein loading was assessed by Ponceau S staining of nitrocellulose membranes.
Protein oxidation assay.
Oxidative modification of proteins in mouse LGN after target deprivation was determined by OxyBlot (Millipore Bioscience Research Reagents) analysis of carbonyl groups (aldehydes and ketones). Wild-type mice received unilateral occipital cortex ablations and were killed 1, 2, 3, 4, 5, and 6 d later by anesthesia overdose and decapitation. The brain for each mouse (n = 6 per time point) was removed quickly from the cranium and placed in ice-cold Hanks buffer. Microdissected LGN samples were lysed and homogenized to prepare soluble protein and mitochondrial-enriched membrane protein fractions by subcellular fractionation using a confirmed protocol (Martin et al., 2003). Aliquots of equal amounts of protein (5 μg) from mitochondrial-enriched and soluble fractions were subjected to derivatization with 2–4 dinitrophenylhydrazine, SDS-PAGE, and Western blotting for proteins with hydrazone moieties as described previously (Martin et al., 2009b).
Protein S-nitrosylation assay.
Endogenously produced NO can exert a variety of effects in neurons. One effect of NO that can initiate apoptosis in cultured neurons is S-nitrosylation (a direct covalent modification of cysteine residues) to form nitrosothiols (Hara et al., 2005). The biotin switch method (Jaffrey and Snyder, 2001) was used to detect S-nitrosylated proteins in the ipsilateral and contralateral LGN at 1, 2, 3, and 4 d after target deprivation. Fresh brains (n = 3–4 mice/time point) were removed quickly from the cranium and placed in ice-cold Hanks buffer. Left and right LGN regions were carefully microdissected from the dorsolateral brainstem under a stereomicroscope. Microdissected LGN samples were lysed and homogenized to prepare crude protein fractions. Aliquots of equal amounts of protein extracts (0.2–3 μg) were subjected to nitrosothiol derivatization with maleimide-biotin (Cayman Chemical) followed by SDS-PAGE, and Western blotting for biotinylated proteins developed with ECL and exposed to x-ray film. The negative control was LGN extract without nitrosothiol derivatization to detect endogenously biotinylated proteins. Positive controls were LGN extracts treated in vitro with sodium nitroprusside and biotinylated IgG.
Western blotting.
The relative levels of nNOS, nitrated proteins, CyPD, ANT, and VDAC in mouse LGN after cortical target deprivation were evaluated by immunoblotting. Wild-type mice received unilateral occipital cortex ablations and were killed 1, 2, 3, 4, 5, and 6 d later by anesthesia overdose and decapitation. Precise microdissections of LGN samples were used to prepare subcellular fractions or a crude homogenate. Protein fractions were subjected to SDS-PAGE and immunoblotting using enhanced chemiluminescence (ECL) detection (Pierce) as described previously (Martin et al., 2003). nNOS was detected with four different affinity-purified rabbit polyclonal antibodies (Transduction Laboratories; Millipore; Millipore Bioscience Research Reagents; ImmunoStar) that have been well characterized (Martin et al., 2005). Nitrated proteins were detected with two different affinity-purified mouse monoclonal antibodies, clone 2A12 (Abcam) and clone 1A6 (Millipore), to 3-nitrotyrosine. Antibodies to mPTP proteins were purchased from commercial sources. For CyPD, a mouse monoclonal antibody (Mitoscience; clone E11AE12BD4) was used. For ANT, a mouse monoclonal antibody (Mitoscience; clone 5F51BB5AG7) and a rabbit polyclonal antibody (Santa Cruz; H-188) were used. For VDAC, two mouse monoclonal antibodies (Mitoscience, clone 20B12AF2; Calbiochem, clone 89-173/016) were used. ANT and VDAC antibodies were not isoform specific. The antibodies were used at concentrations for visualizing protein immunoreactivity within the linear range (Lok and Martin, 2002).
Immunoprecipitation and detection of nitrated proteins.
Immunoprecipitation and Western blot analysis of control and target-deprived LGN was used to identify peroxynitrite-mediated nitration of CyPD, ANT, and VDAC. LGN protein (100 μg) was immunoprecipitated using 5 μg of monoclonal nitrotyrosine antibody (Millipore) as described previously (Martin et al., 2007, 2009b). After immunocapture, the samples were subjected to SDS-PAGE and Western blot detection of CyPD, ANT, and VDAC using ECL. Equivalent input was assessed by Western blot detection of CyPD in equal amounts of LGN extract.
Pharmacologic interventions.
We examined the participation of NOS and the mPTP in the mechanisms of neuronal apoptosis in adult mouse brain by determining whether an nNOS inhibitor and mPTP antagonists influence the amount of apoptosis of LGN neurons after target deprivation. Wild-type male C57BL/6 were injected intraperitoneally with 3-bromo-7-nitroindazole (Sigma-Aldrich) at a dosage of 25 mg/kg body weight (dissolved in peanut oil vehicle) or vehicle alone 2 h before cortical ablation and every day after ablation until killing at day 7. For mPTP inhibitors, wild-type male mice were implanted with intracerebroventricular cannulas and then received unilateral occipital cortex ablations. Mice were treated daily on day 0 (day of lesion) and on postlesion days 1–6 with the olesoxime cholest-4-en-3-one oxime (TRO-19622) (Sigma-Aldrich), at a concentration of 10 μm (dissolved in cyclodextrin/ethanol) in 4 μl or a cell-permeable peptide composed of the conserved N-terminal Bcl-2 homology domain (BH4) of Bcl-XL (Bcl-XL-BH4) linked to a 10 aa HIV-TAT48-57 carrier peptide (Shimizu et al., 2000) at a concentration of 10 μm (dissolved in cyclodextrin/DMSO) in 4 μl. TRO-19622 is a cholesterol-like compound with robust neuroprotective properties for neurotrophin-deprived motor neurons in cell culture and rodent models (Bordet et al., 2007). TRO-19622 can rescue cultured motor neurons from apoptosis induced by trophic factor deprivation and can protect motor neurons from axotomy-induced degeneration in neonatal rat (Bordet et al., 2007). TRO-19622 acts on the mPTP by binding to VDAC and TSPO (translocator protein of 18 kDa) (Bordet et al., 2007). Bcl-XL-BH4 directly binds to VDAC to block its activity and to inhibit loss of mitochondrial membrane potential and cytochrome c release (Shimizu et al., 2000) and has strong antiapoptotic actions in cultured mouse cortical neurons (Martin et al., 2009a). The control for TRO-19622 was vehicle. The control for Bcl-XL-BH4 was TAT dissolved in vehicle. Each treatment group had eight mice.
Cell counting.
For neuroprotection experiments, mice were killed at 7 d after cortical injury. Animals were anesthetized with an overdose of sodium pentobarbital and perfused intracardially with ice-cold PBS (100 mm), pH 7.4, followed by ice-cold 4% paraformaldehyde in PBS. Transverse serial symmetrical sections (40 μm) through the thalamus were cut, and sections were subsampled from each mouse brain and stained with cresyl violet for neuronal counting. Neuronal counts in the ipsilateral and contralateral dLGN were made at 1000× magnification using the stereological optical disector method as described previously (Al-Abdulla and Martin, 1998; Martin et al., 1999). dLGN neurons without apoptotic structural changes were counted using strict morphological criteria. These criteria included a round, open, pale nucleus (not condensed and darkly stained), globular Nissl staining of the cytoplasm, and a diameter of ∼10–15 μm. With these criteria, astrocytes, oligodendrocytes, and microglia were excluded from the counts.
Data analysis.
Neuronal counts and fluorescence intensities were used to determine group means and variances, and comparisons among groups were analyzed using a one-way ANOVA and a Newman–Keuls post hoc test. The experiments were controlled at two levels. Neuronal counts in the contralateral dLGN always served as controls for the ipsilateral dLGN in lesioned NOS and CyPD-null mice. In addition, neuron counts in wild-type mice served as strain controls.
Results
Mitochondria from distal axons accumulate in dLGN neurons after target ablation
EM was used to evaluate directly the number of mitochondria in dLGN neuron perikarya during their apoptosis (Fig. 1A–C). dLGN relay neuron profiles were identified by ultrastructural characteristics (Fig. 1A,B), including nuclear eccentricity and chromatolysis in response to distal axotomy (Al-Abdulla and Martin, 2002). Electron micrographs were taken of dLGN neurons represented in serial ultrathin sections. Morphologically intact mitochondria, easily identifiable by EM, were counted in ∼50 neurons per mouse per time point (Fig. 1A,B). We found that mitochondria accumulate progressively in preapoptotic neurons (Fig. 1A–C). By 4–5 d after lesion, there is a threefold increase in perikaryal mitochondria (Fig. 1C). Target-deprived dLGN neurons often contained mitochondria in contact with cleavage furrows (Fig. 1B, inset, arrows) indicative of mitochondrial fission (Martin et al., 2009b). At 6 d after lesion, there was a sharp decline in intact mitochondria, coinciding precisely with apoptotic nuclear condensation (Fig. 1C).
Figure 1.
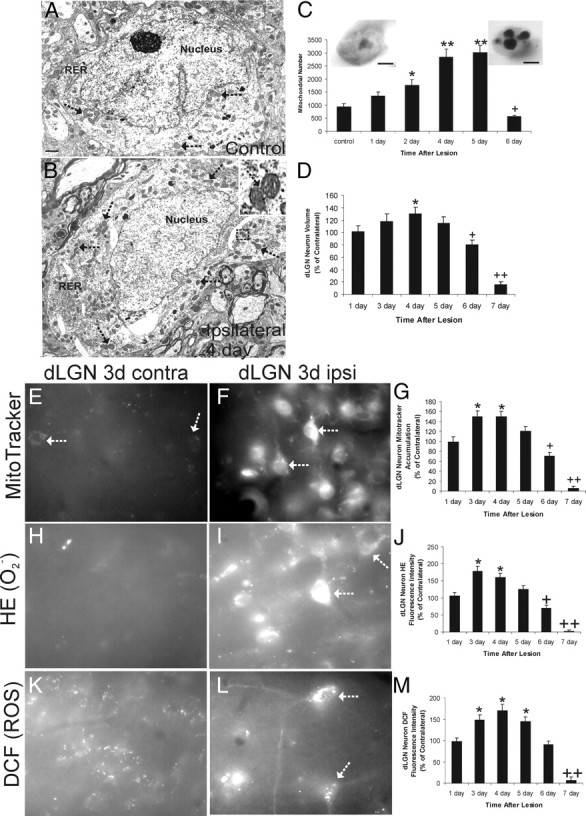
Target-deprived dLGN neurons accumulate mitochondria and elevate production of ROS preapoptotically. A, B, Representative electron micrographs of dLGN neuron cell body profiles in cross-section showing the perikaryal distribution of mitochondria in a control target-intact neuron (A) and a target-deprived neuron (B) at 4 d after cortical ablation. Mitochondria (arrows), nucleus, and rough endoplasmic reticulum (RER) are identified. The target-deprived neuron has accumulated mitochondria (arrows), some of which are undergoing fission (B, top right inset). Scale bar: A, bottom left, 1.7 μm. C, Counts of mitochondrial numbers derived from serial electron micrographs of dLGN neuron perikarya with intact cortex (control) and ablated occipital cortex at 1, 2,4, 5, and 6 d after lesion. Values (n = 5 mice per group) are mean ± SD of 50 cells analyzed for each time point with significant increases seen at 2 d (*p < 0.05) and at 4 and 5 d (**p < 0.001) and a significant decrease seen at 7 d (+p < 0.05) compared with control. Insets show a control dLGN neuron (left) and a near end-stage apoptotic dLGN neuron (right) on the sixth day after target deprivation. Scale bars, 4.5 μm. D, Graph showing the volume of dLGN neurons cell bodies (represented as percentage of control) at 1, 3, 4, 5, 6, and 7 d after target deprivation. Values are mean ± SD of 50 cells analyzed for each time point with a significant increase seen at 4 d (*p < 0.05) and significant decreases seen at 6 d (+p < 0.05) and 7 d (++p < 0.001) compared with control. E, F, MitoTracker Red CM-H2XRos labeling of control contralateral (contra) and target-deprived ipsilateral (ipsi) dLGN neurons at 3 d after occipital cortex ablation (white arrows identify selected neurons with fluorescent labeling of mitochondria). G, Graph showing MitoTracker Red fluorescence intensity of dLGN neuron cell bodies (represented as percentage of control) at 1, 3, 4, 5, 6, and 7 d after target deprivation. Values (n = 6 mice per group) are mean ± SD of 50 cells analyzed for each time point with significant increases seen at 3 and 4 d (*p < 0.05) and significant decreases seen at 6 d (+p < 0.05) and 7 d (++p < 0.001) compared with control. H, I, HE labeling of control contralateral (contra) and target-deprived ipsilateral (ipsi) dLGN neurons at 3 d after occipital cortex ablation (white arrows identify selected neurons with fluorescent labeling). J, Graph showing HE fluorescence intensity of dLGN neuron cell bodies (represented as percentage of control) at 1, 3, 4, 5, 6, and 7 d after target deprivation. Values (n = 6 mice per group) are mean ± SD of 50 cells analyzed for each time point. Significant increases were seen at 3 and 4 d (*p < 0.05), and significant decreases were seen at 6 d (+p < 0.05) and 7 d (++p < 0.001) compared with control. K, L, DCF labeling of control contralateral (contra) and target-deprived ipsilateral (ipsi) dLGN neurons at 3 d after occipital cortex ablation (white arrows identify selected neurons with fluorescent labeling of mitochondria). M, Graph showing DCF fluorescence intensity of dLGN neuron cell bodies (represented as percentage of control) at 1, 3, 4, 5, 6, and 7 d after target deprivation. Values (n = 6 mice per group) are mean ± SD of 50 cells analyzed for each time point. Significant increases were seen at 3, 4, and 5 d (*p < 0.05), and a significant decrease was seen at 7 d (+p < 0.001) compared with control.
Mitochondrial accumulation appeared to be occurring in dLGN neurons before apoptotic shrinkage of cells, not as a morphological consequence of apoptosis. To confirm this impression, dLGN neuron cell volume was measured morphometrically. Neuronal cell body volume in target-deprived dLGN increased significantly at 4 d, consistent with an axonal injury-induced chromatolytic response (Lieberman, 1971; Martin et al., 1999). The mitochondrial accumulation occurred at a time when cell body volume increased transiently (Fig. 1D).
We tested the hypothesis that mitochondria return from the distal axons and synapses to accumulate in the cell body of dLGN neurons after target ablation. Distal axon- and terminal-derived mitochondria of dLGN neuron terminal fields in occipital cortex were visualized using MitoTracker Red. In dLGN neurons with an intact target, subsets of MitoTracker-labeled distal axon-derived mitochondria are trafficked back to the dLGN neuron cell body where they are visualized by 3 d after labeling (Fig. 1E). The MitoTracker labeling is distinct from general retrograde transport of dyes (Al-Abdulla and Martin, 1998) and has an appearance (discrete granules or puncta) and distribution typical of mitochondria (Fig. 1E). Mice without injections of MitoTracker did not have any fluorescent labeling of mitochondria. MitoTracker fluorescence signal in target-deprived dLGN neurons was much greater than in the nonlesioned contralateral dLGN neurons at 3, 4, and 5 d after cortical ablation (Fig. 1F,G), suggesting that retrograde transport of mitochondria is encouraged in target-deprived neurons. Similar results were found using an excitotoxic ablation of visual cortex (data not shown). These data corroborate by a different approach the finding by EM that mitochondria accumulate during preapoptosis in dLGN neurons.
Preapoptotic dLGN neurons have increased production of ROS after target deprivation
To determine whether the mitochondrial accumulation in axotomized/target-deprived dLGN neurons has consequences, in vivo dLGN prelabeling experiments were done using the superoxide indicator HE and the general ROS indicator carboxy-H2DCFDA (DCF) to determine whether oxidative stress is enhanced in preapoptotic dLGN neurons (Fig. 1H–M). HE and DCF signals were increased significantly in target-deprived dLGN neurons (Fig. 1J,M). Superoxide productions were increased at 3 and 4 d after lesion, whereas total ROS production was increased at days 3 through 5 after target deprivation. HE signal was seen mostly within the perikaryon (Fig. 1I), whereas DCF signal was seen throughout the somatodendritic compartment and concentrated in discrete particles/aggregates at perinuclear locations (Fig. 1L).
Oxidative damage in the form of carbonyl modification of mitochondrial proteins occurs in dLGN after target deprivation
Oxidative stress can trigger neuronal apoptosis (Ratan et al., 1994). The presence of protein carbonyl groups, a hallmark of oxidative damage to proteins, was identified by oxyblot analysis of ipsilateral and contralateral dLGN after target deprivation. Protein carbonyl formation in mitochondrial fractions was elevated moderately in ipsilateral dLGN at 3 d after lesion (Fig. 2A, top). Protein carbonyl signal in the ipsilateral dLGN at 4 d after lesion was much greater than in the contralateral dLGN and was greater compared with ipsilateral dLGN at 3 d. The proteins damaged ranged broadly in sizes (from <25 to 200 kDa) (Fig. 2A, top). The high level of protein oxidation was surprisingly transient, because oxidatively damaged proteins were cleared within 1 d (Fig. 2A, top), probably because the damaged cellular constituents are degraded and not because the process is reversible. At the same times, carbonyl levels in soluble protein fractions were unaltered in ipsilateral dLGN at 3 and 4 d after lesion (Fig. 2A, bottom).
Figure 2.
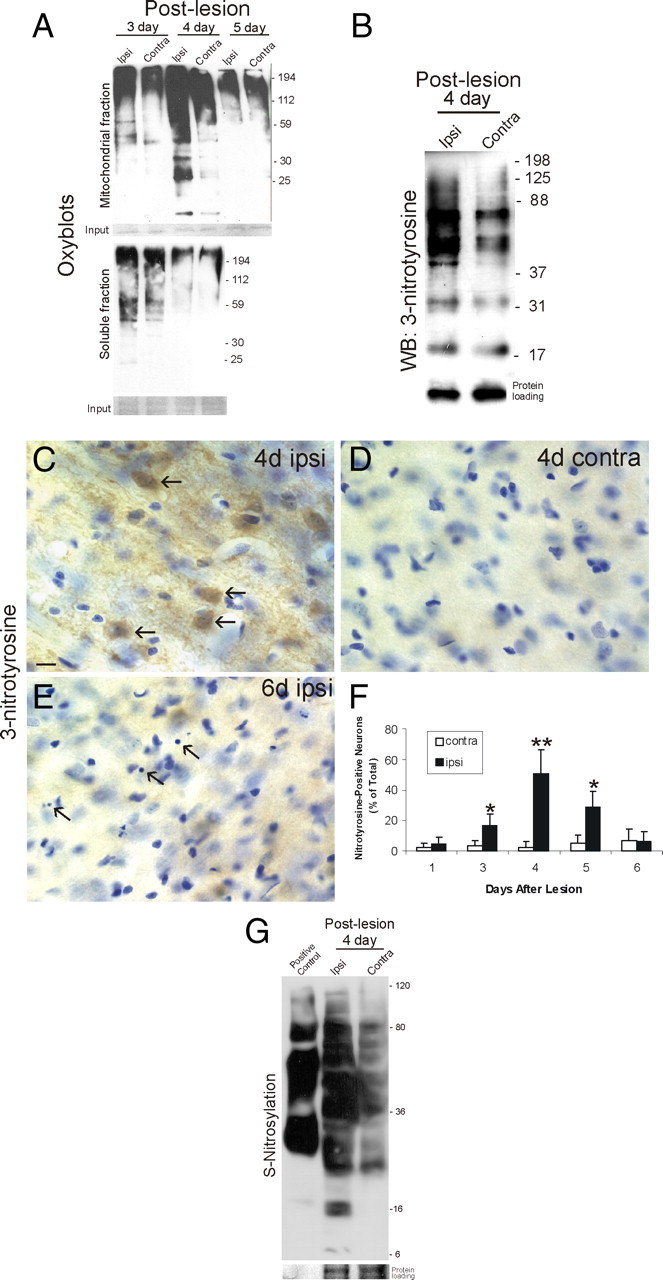
Preapoptotic dLGN neurons accumulate oxidative damage, nitrated proteins, and S-nitrosylated proteins. A, Oxyblot analysis of microdissected ipsilateral target-deprived and contralateral target-intact LGN after fractionation into mitochondrial (top blot) and soluble (bottom blot) protein fractions as described previously (Martin et al., 2003). Mitochondrial proteins (5 μg/lane) from ipsilateral LGN showed a modest increase in protein carbonyls at 3 d and then an abrupt robust transient accumulation of protein carbonyls at 4 d. Molecular weight standards (in kilodaltons) are shown at right. Soluble protein extracts from the same LGN samples used for mitochondrial extractions did not show differences between ipsilateral and contralateral. The result was reproduced in four different mice in each group. Equivalent inputs are shown by Ponceau S staining of membranes (shown at the bottom of each oxyblot). B, Western blot for 3-nitrotyrosine-immunoreactive proteins in ipsilateral and contralateral LGN total extracts at 4 d after occipital cortex ablation. The ipsilateral LGN showed a marked accumulation of nitrated proteins in the 40–125 kDa range. Molecular weight standards (in kilodaltons) are shown at right. Protein loading control is show at bottom. The result was reproduced in four different mice. C–E, Immunohistochemical localization of 3-nitrotyrosine immunoreactivity in the dLGN. Immunoperoxidase was used to visualize nitrated proteins using monoclonal antibody to 3-nitrotyrosine and DAB (brown) and then sections were counterstained with cresyl violet. A marked accumulation was seen in neurons in the ipsilateral dLGN at 4 d after target deprivation (C, arrows). The contralateral dLGN on the same section showed only faint labeling (D). At 6 d after target deprivation, end-stage apoptotic profiles became discernible (E, arrows), whereas 3-nitrotyrosine immunoreactivity dissipated. Scale bar: (in C) C–E, 12 μm. F, Graph showing number of 3-nitrotyrosine-positive dLGN neuron cell bodies at 1, 3, 4, 5, and 6 d after target deprivation. Values (n = 6 mice per group) are mean ± SD. Significant increases were seen at 3 d (*p < 0.05), 4 d (**p < 0.01), and 5 d (*p < 0.05) compared with contralateral control. G, S-Nitrosylation of proteins in LGN of mouse brain at 4 d after target deprivation. Protein S-nitrosylation was detected using the biotin switch method (Jaffrey and Snyder, 2001). The ipsilateral LGN showed a marked accumulation of nitrosylated proteins (3.5 μg of protein loaded in each lane). Molecular weight standards (in kilodaltons) are shown at right. Protein input is shown at bottom. The positive control for protein biotinylation was biotin-labeled IgG.
Protein nitration is elevated in dLGN neurons after target deprivation
Reactive nitrogen species can mediate apoptosis of cultured neurons triggered by trophic factor withdrawal (Estévez et al., 1999), and peroxynitrite is thought to be involved in the mechanisms of motor neuron apoptosis or necrosis in vivo induced by axotomy and mutant SOD1, respectively (Martin et al., 2005, 2007). Western blotting for 3-nitrotyrosine-modified proteins, a signature of peroxynitrite, showed elevated protein nitration in ipsilateral dLGN at 4 d after cortical lesion (Fig. 2B). Western blotting for 3-nitrotyrosine was done on crude homogenates of LGN because there was no reason to suspect that peroxynitrite is generated within mitochondria, unlike ROS causing the protein carbonyl formation. Immunohistochemical localization of 3-nitrotyrosine revealed numerous positive neurons in ipsilateral dLGN at 4 d after lesion (Fig. 2C,D,F), but, as neuronal apoptosis achieved near completion at 6 d, 3-nitrotyrosine immunoreactivity dissipated (Fig. 2E,F). Thus, protein nitration specifically occurs at preapoptotic and early apoptotic stages and is not a secondary consequence of the death process.
Protein S-nitrosylation is elevated in LGN after target deprivation
Neuronal cell death can be caused by NO reacting with superoxide to form peroxynitrite (Beckman et al., 1993; Estévez et al., 1999) and by NO reacting with cysteine residues to form S-nitrosothiols (Hara et al., 2005). We assessed the participation of protein S-nitrosylation in the possible mechanisms of dLGN neuron apoptosis using the biotin switch method (Jaffrey and Snyder, 2001). Substantial baseline endogenous S-nitrosylation of proteins was detected in the mouse LGN with modified proteins ranging broadly in sizes (from ∼25 to ∼100 kDa) (Fig. 2G, contra). Background biotinylation of proteins was negligible as assessed with this assay (data not shown). The target-deprived LGN shows a dramatic elevation in S-nitrosylated proteins at 3 and 4 d (Fig. 2G). The patterns seen in the apoptotic LGN reflected an enhancement of baseline nitrosylated proteins and nitrosylation of new proteins not seen in target-intact LGN (Fig. 2G).
dLGN neurons after target ablation accumulate intracellular Ca2+ and generate NO
The preapoptotic elevations in superoxide (Fig. 1H–J) and signatures for peroxynitrite (Fig. 2B–F) and S-nitrosylation (Fig. 2G) led to the suspicion that NO is involved in the mechanisms of dLGN neuron apoptosis. Moreover the elevated DCF signals in ipsilateral dLGN neurons (Fig. 1K–M) could be attributable to the formation of NO or ONOO− (Estévez et al., 1999). Intracellular Ca2+ itself does not cause the oxidative stress, but it fosters the development of oxidative stress through its availability to activate enzymes that can generate prooxidants (Mattson, 2007). Ca2+ is required for nNOS activity. We therefore examined intracellular Ca2+ and NO production directly in dLGN neurons.
Mouse dLGN neurons were loaded bilaterally by retrograde labeling with dextran-conjugated fura-2 AM, an indicator dye that increases its fluorescence when intracellular Ca2+ levels increase (O'Donovan et al., 1994), before unilateral target ablation. This approach has been developed by others for retrograde preloading of neurons (O'Donovan et al., 1994). Fluorescent signal for intracellular Ca2+ was elevated significantly in target-deprived dLGN neurons (compared with the contralateral nonlesioned dLGN as control) at 1 d after lesion, with the accumulation peaking at 2–3 d after ablation (Fig. 3A–C). As a negative control, Gelfoam saturated with BAPTA-AM was placed in the cortical lesion site. Fura-2-dextran signals were attenuated (data not shown). These data show that intracellular Ca2+ is increased in dLGN neurons within 12–24 h after disconnection from their target.
Figure 3.

Early dysregulation of intracellular Ca2+, Zn2+, and NO in target-deprived dLGN neurons. A, B, Fura-2 AM-dextran labeling of control contralateral (contra) and target-deprived ipsilateral (ipsi) dLGN neurons at 2 d after occipital cortex ablation. Very faint fluorescent signal is seen in control neurons, whereas intense intracellular Ca2+ signal is seen in target-deprived neurons (arrows). Scale bar: (in A) A–E, 12 μm. C, Graph showing fura-2 AM-dextran fluorescence intensity of dLGN neuron cell bodies (represented as percentage of control) at 1 and 12 h (hr) and 1, 2, 3, 4, and 5 d after target deprivation. Values (n = 6 mice per group) are mean ± SD of 50 cells analyzed for each time point with indicated significant increases (*p < 0.05 or **p < 0.01) compared with control. D, E, DAA labeling of control contralateral (contra) and target-deprived ipsilateral (ipsi) dLGN neurons at 2 d after occipital cortex ablation. No fluorescent signal is seen in control neurons, whereas intense intracellular NO signal is seen in target-deprived neurons (arrows). F, Graph showing DAA fluorescence intensity of dLGN neuron cell bodies (represented as percentage of control) at 1 and 12 h (hr) and 1, 2, 3, 4, and 5 d after target deprivation. Values (n = 6 mice per group) are mean ± SD of 50 cells analyzed for each time point with indicated significant increases (*p < 0.05 or **p < 0.01) compared with control. G, H, FluoZin-3 labeling of control contralateral (contra) and target-deprived ipsilateral (ipsi) dLGN neurons at 6 h after occipital cortex ablation. No fluorescent signal is seen in control neurons, whereas intense intracellular Zn2+ signal is seen in target-deprived neurons (arrows). Scale bar: (in G) G, H, 12 μm. I, Graph showing FluoZin-3 fluorescence intensity of dLGN neuron cell bodies (represented as percentage of control) at 1, 6, and 12 h (hr) and 1, 2, 3, 4, and 5 d after target deprivation. Values (n = 3 mice per group) are mean ± SD of 50 cells analyzed for each time point with indicated significant increases (*p < 0.05 or **p < 0.01) compared with control.
Because of concern about the possibility of other divalent cations (Zn2+) contributing the intracellular Ca2+ signal in dLGN neurons as determined by dextran-conjugated fura-2 AM, we used a highly specific fluorescent probe (FluoZin-3) to investigate the role of Zn2+ in neuron apoptosis induced by target deprivation. Two important observations were made with this experiment. First, intracellular Zn2+ increased significantly in dLGN neurons undergoing apoptosis (Fig. 3G–I), consistent with observations made on non-neuronal cells (Kimura et al., 2004). Second, to our surprise the temporal pattern of the Zn2+ accumulation in apoptotic dLGN neurons was very different from the intracellular Ca2+ pattern, particularly early in the apoptotic process. At 12 h after target deprivation of dLGN neurons, intracellular Zn2+ was increased significantly (Fig. 3I), whereas intracellular Ca2+ was unchanged (Fig. 3C). Because of this early increase in intracellular Zn2+, we analyzed another time point and found that Zn2+ was increased significantly in target-deprived dLGN neurons already at 6 h, but not at 1 h (Fig. 3I).
In a parallel experiment, NO production in dLGN neurons was tracked using DAA. NO production was significantly elevated in target-deprived dLGN neurons compared with contralateral neurons (Fig. 3D–F). The onset and peak in NO production coincided with the elevations in intracellular Ca2+ (Fig. 3C,F). DAA signal was abolished in l-NAME-treated mice (94 ± 12% of control) with images being similar to contralateral sides (Fig. 3F).
nNOS is upregulated in the dLGN after target ablation and nNOS gene deletion and enzyme inhibition rescue dLGN neurons from apoptosis
We examined the involvement in nNOS in the mechanisms of dLGN neuron apoptosis. Immunolocalization of nNOS in mouse brain sections revealed an upregulation of nNOS in dLGN after target ablation (Fig. 4A,B). Very low levels of nNOS immunoreactivity were detected in the nonlesioned contralateral dLGN (Fig. 4B). Western blots confirmed the low basal levels of nNOS in dLGN and the upregulation of nNOS protein in dLGN after target ablation, occurring as early as 2 d after cortical ablation with an effect sustained through 5 d (Fig. 4C).
Figure 4.

nNOS mediates apoptosis of dLGN neurons induced by target deprivation. A, B, Immunohistochemistry showing the nNOS upregulation in ipsilateral (ipsi) dLGN at 2 d (2d) after target deprivation (A). Scale bar: (in A) A, B, 30 μm. Immunoreactivity for nNOS is low in the dLGN with an intact cortical target at 2 d (B). C, Western blot showing the nNOS upregulation in the ipsilateral versus contralateral LGN at 2, 4, and 5 d after target deprivation. Western blot reprobed for β-tubulin is shown for protein loading. D, Graph showing the number dLGN neurons in nNOS−/− and iNOS−/− mice at 7 d after occipital cortex ablation. Values are mean ± SD (n = 8 mice per genotype). The asterisk indicates significantly different from wild type (p < 0.01). E, Graph showing the number dLGN neurons at 7 d after occipital cortex ablation in wild-type mice treated daily (25 mg/kg, i.p.) with the nNOS inhibitor 3-bromo-7-nitroindazole (7-NI) or vehicle (peanut oil). Values are mean ± SD (n = 8 mice per group). The asterisk indicates significantly different from wild type (p < 0.01).
To determine directly whether nNOS contributes to the mediation of apoptosis of dLGN neurons after target ablation, lesions were done on mice without nNOS (n = 8). Target deprivation-induced apoptosis of dLGN neurons was significantly mitigated in nNOS−/− mice (Fig. 4D). Nos−/− mice had only ∼40% loss of dLGN neurons compared with the 90% loss in wild types at 7 d after ablation (Fig. 4D). The specificity of the nNOS isoform effect was determined by lesioning mice with homozygous deletion of iNOS (n = 8). dLGN neuron apoptosis after target ablation in iNOS−/− mice was similar to wild type (Fig. 4D). To follow up on results based of genetic manipulation, pharmacologic experiments were done using the specific nNOS inhibitor 3-bromo-7-nitroindazole, which rescued dLGN neurons from apoptosis (Fig. 4E).
The mPTP mediates target deprivation-induced apoptosis of dLGN neurons
Peroxynitrite and NO can induce apoptosis in non-neural cells by triggering mitochondrial permeability transition (Hortelano et al., 1997). We therefore determined whether putative core components of the mPTP (Crompton et al., 1998) are present in mouse dLGN and whether inactivation of the mPTP blocks dLGN neuron apoptosis after target deprivation. Western blotting showed that VDAC, ANT, and CyPD are present in adult mouse dLGN at the expected monomeric sizes (Martin et al., 2009b), but the levels of each in target-deprived dLGN were not different from contralateral dLGN at 3–6 d after target deprivation (Fig. 5A). Complete deletion of CyPD in ppif−/− mice on two different genetic backgrounds protected, nearly 100%, dLGN neurons from apoptosis induced by target deprivation (Fig. 5B). The effects of pharmacological inactivation of the mPTP on dLGN neuron apoptosis were assessed by intraventricular injection of TRO-19622 and TAT-Bcl-XL-BH4 (Fig. 5C,D). Mice with these treatments had significant protection from apoptosis (Fig. 5C,D). To examine the formation of the mPTP, the chemical cross-linker DSS was injected into the lateral ventricle of mice with cortical ablations. Western blotting of cross-linked dLGN extracts for CyPD revealed the expected monomer in ipsilateral and contralateral dLGN and the presence of proteins containing CyPD at higher molecular weights only in the ipsilateral dLGN (Fig. 5E). Western blotting of cross-linked dLGN extracts for VDAC revealed in contralateral dLGN the predominant monomeric from at ∼33 kDa and a less abundant higher molecular weight band containing VDAC (Fig. 5E), whereas in ipsilateral dLGN this pattern was reversed (Fig. 5E). To determine whether the mPTP is a target of peroxynitrite during apoptosis, dLGN proteins were immunoprecipitated with 3-nitrotyrosine monoclonal antibody and then immunoblotted for CyPD, ANT, and VDAC. CyPD and ANT became nitrated at preapoptotic stages of apoptosis (Fig. 5F). To examine the upstream consequences of mPTP inactivation, superoxide and NO production were measured in target-deprived dLGN neurons in ppif−/− mice by in vivo tracking of HE and DAA, respectively. CyPD deletion occluded the preapoptotic burst of superoxide and NO in dLGN neurons at 2 and 3 d after target deprivation (Fig. 5G,H).
Figure 5.

The mPTP drives apoptosis of dLGN neurons induced by target deprivation through increased ROS production and is associated with the formation of mPTP protein assemblies and nitration of core components. A, Western blot showing three core components of the mPTP (VDAC, ANT, and CyPD) in the ipsilateral versus contralateral LGN at 3, 4, 5, and 6 d after target deprivation. No major changes in the levels of monomeric proteins are evident under reducing conditions. B, Graph showing the number dLGN neurons at 7 d after occipital cortex ablation in CyPD−/− mice on two different genetic backgrounds. Values are mean ± SD (n = 10 mice per genotype). The asterisks indicate significantly different from wild type (p < 0.001). C, Graph showing the number dLGN neurons at 7 d after occipital cortex ablation in wild-type mice treated daily (10 μm, i.c.v.) with TRO-19622 or vehicle. Values are mean ± SD (n = 8 mice per group). The asterisk indicates significantly different from wild type (p < 0.01). D, Graph showing the number dLGN neurons at 7 d after occipital cortex ablation in wild-type mice treated daily (10 μm, i.c.v.) with TAT-Bcl-XL:BH4 or vehicle. Values are mean ± SD (n = 8 mice per group). The asterisk indicates significantly different from wild type (p < 0.01). E, Western blots for CyPD and VDAC in wild-type mouse LGN treated with chemical cross-linker at 3 d after unilateral target deprivation. Ipsilateral and contralateral LGN extracts of the same mouse are shown. Monomeric CyPD, detected at ∼20 kDa in comparable amounts, is seen in both contralateral and ipsilateral LGN, but higher molecular weight forms of CyPD (asterisks) are seen only in ipsilateral LGN. Protein loading in each lane is shown by Ponceau S staining of membrane (bottom). Monomeric VDAC is detected at ∼33 kDa. Monomeric VDAC predominates in the control contralateral LGN with a less abundant band immunoreactive for VDAC at ∼45 kDa. This higher molecular form of VDAC (arrow) predominates in the target-deprived LGN, whereas the monomer dissipates under cross-linking conditions. Protein loading in each lane is shown by Ponceau S staining of membrane (bottom). Equivalent lysate input is shown by CyPD immunoblot on ∼25% of the crude fraction used for immunoprecipitation. Negative control for immunoprecipitation of LGN extracts was normal preimmune IgG. F, Immunoprecipitation of nitrated proteins followed by Western blotting shows that CyPD and ANT are nitrated in the LGN by 2 d after target deprivation. Ipsilateral and contralateral LGN samples (250 μg protein) were immunoprecipitated with 3-nitrotyrosine monoclonal antibody (1 μg) and subjected to SDS-PAGE and sequential Western blotting for CyPD, ANT, and VDAC, respectively. G, Graph showing HE fluorescence intensity of dLGN neurons in wild-type and CyPD−/− mice at 3 d after target deprivation. Values (n = 4 mice per group) are mean ± SD (represented as percentage of contralateral) of 100 cells analyzed for each mouse. H, Graph showing DAA fluorescence intensity of dLGN neurons in wild-type and CyPD−/− mice at 3 d after target deprivation. Values (n = 4 mice per group) are mean ± SD (represented as percentage of contralateral) of 100 cells analyzed for each mouse.
Discussion
We used a mouse model of neuronal apoptosis to uniquely examine in vivo aspects of mitochondrial biology and mechanisms of neuronal cell death. Our model features neuronal apoptosis induced in a remote brain nucleus by ablation of visual cortex. The neuronal apoptosis occurs discretely in dLGN and is homogeneous, precisely timed, and synchronous; thus, this model can be used to delineate cell death pathways in brain neurons destined to undergo unequivocal apoptosis (Al-Abdulla and Martin, 1998; Martin et al., 2003). We show that preapoptotic dLGN neurons accumulate mitochondria, intracellular Ca2+ and Zn2+, and have enhanced ROS production, including superoxide and NO, with associated increased formation protein carbonyls, nitration, and S-nitrosylation. The ROS bursts are transient and causal to the mechanisms, not merely secondary aspects of cell death, because genetic and pharmacological inactivation of NOS1 blocks the apoptosis and because the ROS bursts are generated by formation of the mPTP, the inactivation of which occludes the ROS overproduction and provides robust protection of neurons. These observations demonstrate in an in vivo setting of neuronal apoptosis in brain that (1) mitochondria redistribute to the perikaryon preapoptotically, (2) transiently enhanced ROS production precedes morphologically evident apoptosis, (3) aberrant upregulation of NOS1 and NO production in neurons is proapoptotic, and (4) activation of the mPTP mediates the preapoptotic ROS bursts and the subsequent apoptosis of neurons.
We found that mitochondria accumulate in neurons destined for apoptosis. The accumulation of mitochondria in target-deprived dLGN neurons occurs acutely before morphological evidence for apoptosis (Al-Abdulla et al., 1998; Martin et al., 2001), when cell body volume increases during the chromatolytic reaction after axonal injury (Lieberman, 1971); thus, the accumulation is not an artifact of somal shrinkage. A redistribution of mitochondria also occurs in hippocampal pyramidal neurons in human Alzheimer's disease brain (Wang et al., 2009). Possible explanations for increased number of perikaryal mitochondria in preapoptotic neurons are enhanced retrograde transport of mitochondria from dendrites and axon, decreased anterograde export of mitochondria to neuronal processes, or increased mitochondrial fission. Our fluorescent mitochondria tracking experiments reveal that preapoptotic perikaryal mitochondrial burden has considerable contribution from axon/terminal-derived mitochondria. Our ultrastructural analysis suggests that mitochondrial fission is a coinciding mechanism responsible for mitochondrial accumulation. Our finding of increased mitochondrial number coinciding with increased NO production in preapoptotic neurons is consistent with work showing that mitochondrial fission is induced by NO in non-neural cells (Nisoli et al., 2003). Increased mitochondrial fission has been described in cultured primary cortical neurons challenged by NO (Barsoum et al., 2006) and ROS (Cheung et al., 2007). The accumulation of mitochondria specifically in neurons destined for apoptosis suggests a novel messenger role of mitochondria located at distal parts of neurons involving NO signaling in the mechanisms of cell death.
A dilemma in understanding mechanisms of neurodegeneration in vivo is whether oxidative stress causes the neurodegeneration or is secondary to the process (Balaban et al., 2005; Souza et al., 2008). We determined by tracking ROS whether preapoptotic perikaryal accumulation of mitochondria has important consequences related to cell death signaling. Two indicators for ROS showed that preapoptotic dLGN neurons overproduce ROS compared with neurons with an intact target. Enhanced production of superoxide is causal, not secondary, to the mechanisms of apoptosis in target-deprived dLGN neurons because mice expressing human wild-type SOD1 are resistant to this cell death (Martin et al., 2003). The pattern of superoxide fluorescence in dLGN neurons was discrete within the cytoplasm and reminiscent of superoxide flashes produced by the mPTP in apoptotic cardiac cells (Wang et al., 2008). We also found that protein carbonyl formation in mitochondrial fractions of target-deprived LGN is a robust signature of impending apoptosis, whereas oxidative damage to soluble proteins is unchanged preapoptotically. Production of ROS in target-deprived dLGN neurons is blocked in mice without CyPD, suggesting mPTP involvement in the mechanisms of enhanced ROS production in neurons. In vitro studies of liver mitochondria show that mitochondrial permeability transition generates superoxide anion (Han et al., 2003). Thus, mitochondrial permeability transition driven ROS production, nonstochastic oxidative stress, and aberrant modification of mitochondrial proteins contribute to the mechanisms of target deprivation-induced neuron apoptosis in vivo.
Intracellular Ca2+ and Zn2+ overloads are believed to be mechanisms in neurodegeneration in vitro (Lee et al., 1999; Devinney et al., 2005; Mattson, 2007). We show that dLGN neurons accumulate intracellular Ca2+ and Zn2+ early after target ablation. Zn2+ accumulation occurred before Ca2+ accumulation. A preapoptotic increase in intracellular Ca2+ in dLGN neurons is consistent with work on motor neuron axotomy in chick embryo (O'Donovan et al., 1994). Our experiments do not reveal the mechanisms of how Ca2+ and Zn2+ accumulate in apoptotic neurons. Our current results suggest links between intracellular Ca2+ accumulation in dLGN neurons and aberrant NO production through NOS activation as well as activation of the mPTP, which is known to be driven by elevated intracellular Ca2+ (Zorov et al., 2007).
Our results demonstrate a role for NO in the in vivo mechanisms of adult brain neuron apoptosis resulting from target deprivation. NO has roles in adult spinal motor neuron apoptosis after nerve avulsion (Martin et al., 2005) and is involved in the mechanisms of neurodegeneration induced by cerebral ischemia (Huang et al., 1994; Hara et al., 1996), and NMDA receptor excitotoxicity (Ayata et al., 1997), but its contributions specifically to neuronal apoptosis in these latter models is unclear because of the inhomogeneity of cell death mechanisms (Portera-Cailliau et al., 1997b; Martin et al., 2002). Work in cell culture has shown that overproduction of NO can compromise neuronal bioenergetics, open the mPTP, and cause mitochondrial fission (Kindler et al., 2003; Moncada and Bolaños, 2006; Cho et al., 2009). The mechanisms are in part related to S-nitrosylation (Cho et al., 2009) and to ONOO−, which is formed by a diffusion-limited reaction between NO and superoxide anion (Beckman et al., 1993). We show a robust increase in S-nitrosylation of proteins in target-deprived LGN and that NO and superoxide levels and 3-nitrotyrosine immunoreactivity accumulate transiently in preapoptotic dLGN neurons, but then dissipate as degeneration advances. We also identify mPTP proteins as targets of nitration in preapoptotic neurons and found that target-deprived dLGN neurons in mice without NOS1 or inhibited NOS1 have considerable protection against apoptosis. However, it is still not definite that ONOO− is the killer of dLGN neurons. ROS exist in many different interconvertible forms. ONOO− can homolyze after protonation to generate ·OH (Coddington et al., 1998). Increased 8-hydroxy-deoxyguanosine (8OHdG) staining in preapoptotic dLGN neurons is consistent with formation of ·OH (Martin et al., 2003). It is noteworthy that 3-nitrotyrosine and OHdG immunoreactivities are prominent features of neurodegeneration in human Alzheimer's disease brain (Smith et al., 1997; Nunomura et al., 2004) and may be indicative of oxidative damage as a disease mechanism (Nunomura et al., 2001).
Oxidative stress and intramitochondrial Ca2+ overload can induce apoptosis by triggering mitochondrial permeability transition (Hortelano et al., 1997). Mitochondrial permeability transition is a mitochondrial state in which the proton-motive force is disrupted reversibly or irreversibly (Crompton, 1999; Leung and Halestrap, 2008). This altered state of mitochondria involves the mPTP that functions as a voltage, thiol, and Ca2+ sensor (Crompton, 1999; Leung and Halestrap, 2008). The mPTP is a poly-protein transmembrane channel formed at contact sites between the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM). The components of the mPTP are controversial, but VDAC in the OMM, ANT in the IMM, and CyPD in the matrix are involved (Crompton, 1999; Bernardi et al., 2006). Permeability transition is activated when ANT changes its conformation from its native state into a nonselective pore (Crompton, 1999). This process is catalyzed by CyPD, which is a peptidylprolyl isomerase that functions in protein cis-trans isomerization and chaperoning (Waldmeier, 2003) and as a redox sensor at cysteine-203 (Linard et al., 2009). The ANT and CyPD interact directly (Woodfield et al., 1998). Oxidative stress can enhance CyPD binding to ANT (McStay et al., 2002), and thiol modification of ANT can cause mPTP opening (Costantini et al., 2000; Vieira et al., 2001; García et al., 2007). When this occurs, small ions and metabolites permeate freely across the IMM and oxidation of metabolites by O2 proceeds with electron flux not coupled to proton pumping, resulting in collapse of ΔP, dissipation of ATP production, and production of ROS (Zoratti and Szabò, 1995).
CyPD is a regulator of the mPTP (Baines et al., 2005). We found in CyPD-null mice a near-complete protection of target-deprived dLGN neurons against apoptosis. This effect was robust in two different null lines on completely different genetic backgrounds. Pharmacological approaches using small molecule and peptide antagonists to the mPTP also blocked apoptosis of dLGN neurons.
We conclude that mitochondria mediate the apoptotic process in adult brain neurons by mPTP-triggered ROS and NO production after their accumulation and priming instigated by Zn2+ and Ca2+ accumulation (Fig. 6). Mitochondrial targeted drugs such as TRO-19622 and Bcl-XL:BH4 peptides can block apoptosis of neurons within the adult mouse CNS. Thus, the mPTP is an important molecular target for the design of drugs and small molecules as antiapoptotic neuroprotectants with in vivo CNS efficacy.
Figure 6.

Schematic diagram illustrating a possible sequence of events leading to neuronal apoptosis in our animal model. This neuronal cell death is highly synchronous and uniformly apoptotic. This scheme has been generated based on data from this study and other studies (Al-Abdulla and Martin, 1998; Martin et al., 2001, 2003). So far, the earliest preapoptotic event that we have detected is the intracellular accumulation of Zn2+, consistent with a previous finding (Land and Aizenman, 2005).
Footnotes
This work was supported by United States Public Health Service–National Institutes of Health–National Institute on Aging Grant AG016282. We are grateful to Frank Barksdale for his excellent work processing the specimens for electron microscopy. We thank Dr. Michael Forte at the Vollum Institute and Dr. Jeffery Molkentin at the Cincinnati Children's Medical Center for providing the original breeder pairs of homozygous cyclophilin D-null mice.
References
- Al-Abdulla NA, Martin LJ. Apoptosis of retrogradely degenerating neurons occurs in association with the accumulation of perikaryal mitochondria and oxidative damage to the nucleus. Am J Pathol. 1998;153:447–456. doi: 10.1016/S0002-9440(10)65588-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Abdulla NA, Martin LJ. Projection neurons and interneurons in the lateral geniculate nucleus undergo distinct forms of degeneration ranging from retrograde and transsynaptic apoptosis to transient atrophy after cortical ablation in rat. Neuroscience. 2002;115:7–14. doi: 10.1016/s0306-4522(02)00363-9. [DOI] [PubMed] [Google Scholar]
- Al-Abdulla NA, Portera-Cailliau C, Martin LJ. Occipital cortex ablation in adult rat causes retrograde neuronal death in the lateral geniculate nucleus that resembles apoptosis. Neuroscience. 1998;86:191–209. doi: 10.1016/s0306-4522(98)00014-1. [DOI] [PubMed] [Google Scholar]
- Anderson AJ, Su JH, Cotman CW. DNA damage and apoptosis in Alzheimer's disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J Neurosci. 1996;16:1710–1719. doi: 10.1523/JNEUROSCI.16-05-01710.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou JC. Inhibition of bax channel-forming activity by bcl-2. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- Ayata C, Ayata G, Hara H, Matthews RT, Beal MF, Ferrante RJ, Endres M, Kim A, Christie RH, Waeber C, Huang PL, Hyman BT, Moskowitz MA. Mechanisms of reduced striatal MNDA excitotoxicity in type I nitric oxide synthase knock-out mice. J Neurosci. 1997;17:6908–6917. doi: 10.1523/JNEUROSCI.17-18-06908.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Barron KD, Means ED, Larsen E. Ultrastructure of retrograde degeneration in thalamus of rat. I. Neuronal somata and dendrites. J Neuropathol Exp Neurol. 1973;32:218–244. doi: 10.1097/00005072-197304000-00004. [DOI] [PubMed] [Google Scholar]
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Gräber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Hermann GE, Rogers RC, Bresnahan JC. Cell death in models of spinal cord injury. Prog Brain Res. 2002;137:37–47. doi: 10.1016/s0079-6123(02)37006-7. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD and peroxynitrite. Nature. 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, Mallard C, Hagberg H. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis?”. J Biol Chem. 2001;276:10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- Bordet T, Buisson B, Michaud M, Drouot C, Galéa P, Delaage P, Akentieva NP, Evers AS, Covey DF, Ostuni MA, Lacapère JJ, Massaad C, Schumacher M, Steidl EM, Maux D, Delaage M, Henderson CE, Pruss RM. Identification and characterization of cholest-4-en-3-one, Oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J Pharmacol Exp Ther. 2007;322:709–720. doi: 10.1124/jpet.107.123000. [DOI] [PubMed] [Google Scholar]
- Chang Q, Martin LJ. Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: a confocal quantitative analysis. Am J Pathol. 2009;174:574–585. doi: 10.2353/ajpath.2009.080557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Sheng C, Zheng X. Direct nitric oxide imaging in cultured hippocampal neurons with diaminoanthraquione and confocal microscopy. Cell Biol Int. 2001;25:593–598. doi: 10.1006/cbir.2001.0734. [DOI] [PubMed] [Google Scholar]
- Cheng C, Reynolds IJ. Calcium-sensitive fluorescent dyes can report increases in intracellular free zinc concentration in cultured forebrain neurons. J Neurochem. 1998;71:2401–2410. doi: 10.1046/j.1471-4159.1998.71062401.x. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Deshmukh M, D'Costa A, Demaro JA, Gidday JM, Shah A, Sun Y, Jacquin MF, Johnson EM, Holtzman DM. Caspase inhibitor affords neuroprotection with delayed adminstration in a rat model of neonatal hypoxic-ischemic brain injury. J Clin Invest. 1998;101:1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, McBride HM, Slack RS. Mitochondrial dynamics in the regulation of neuronal cell death. Apoptosis. 2007;12:979–992. doi: 10.1007/s10495-007-0745-5. [DOI] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-Nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coddington JW, Hurst JK, Lymar SV. Hydroxyl radical formation during peroxynitrous acid decomposition. J Am Chem Soc. 1998;121:2438–2443. [Google Scholar]
- Costantini P, Belzacq AS, Vieira HL, Larochette N, de Pablo MA, Zamzami N, Susin SA, Brenner C, Kroemer G. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene. 2000;19:307–314. doi: 10.1038/sj.onc.1203299. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- Devinney MJ, 2nd, Reynolds IJ, Dineley KE. Simultaneous detection of intracellular free calcium and zinc using fura-2FF and FluoZin-3. Cell Calcium. 2005;37:225–232. doi: 10.1016/j.ceca.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Estévez AG, Crow JP, Sampson JB, Reiter C, Zhuang Y, Richardson GJ, Tarpey MM, Barbeito L, Beckman JS. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Martin F, Serrano T, Reiriz J, Pérez-Navarro E, Alberch J, Macaya A, Planas AM. Both apoptosis and necrosis occur following intrastriatal administration of excitotoxins. Acta Neuropathol. 1995;90:504–510. doi: 10.1007/BF00294812. [DOI] [PubMed] [Google Scholar]
- García N, Martínez-Abundis E, Pavón N, Correa F, Chávez E. Copper induces permeability transition through its interaction with the adenine nucleotide translocase. Cell Biol Int. 2007;31:893–899. doi: 10.1016/j.cellbi.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Gibson ME, Han BH, Choi J, Knudson CM, Korsmeyer SJ, Parsadanian M, Holtzman DM. Bax contributes to apoptotic-like death following neonatal hypoxia-ischemia: evidence for distinct apoptosis pathways. Mol Med. 2001;7:644–655. [PMC free article] [PubMed] [Google Scholar]
- Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem. 2003;278:5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- Hara H, Huang PL, Panahian N, Fishman MC, Moskowitz MA. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J Cereb Blood Flow Metab. 1996;16:605–611. doi: 10.1097/00004647-199607000-00010. [DOI] [PubMed] [Google Scholar]
- Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. S-Nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- Hartmann A, Hunot S, Michel PP, Muriel M-P, Vyas S, Faucheux BA, Mouatt-Prignet A, Turmel H, Srinivasan A, Ruberg M, Evan GI, Agid Y, Hirsch EC. Caspase-3: a vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson's disease. Proc Natl Acad Sci U S A. 2000;97:2875–2880. doi: 10.1073/pnas.040556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiduschka P, Thanos S. NO production during neuronal cell death can be directly assessed by a chemical reaction in vivo. Neuroreport. 1998;9:4051–4057. [PubMed] [Google Scholar]
- Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, Geuskens M, Kroemer G. The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene. 1997;15:1573–1581. doi: 10.1038/sj.onc.1201324. [DOI] [PubMed] [Google Scholar]
- Hortelano S, Dallaporta B, Zamzami N, Hirsch T, Susin SA, Marzo I, Boscá L, Kroemer G. Nitric oxide induces apoptosis via triggering mitochondrial permeability transition. FEBS Lett. 1997;410:373–377. doi: 10.1016/s0014-5793(97)00623-6. [DOI] [PubMed] [Google Scholar]
- Hou ST, MacManus JP. Molecular mechanisms of cerebral ischemia-induced neuronal death. Int Rev Cytol. 2002;221:93–148. doi: 10.1016/s0074-7696(02)21011-6. [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;86:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- Kawamura M, Nakajima W, Ishida A, Ohmura A, Miura S, Takada G. Calpain inhibitor MDL 28170 protects hypoxic-ischemic brain injury in neonatal rats by inhibition of both apoptosis and necrosis. Brain Res. 2005;1037:59–69. doi: 10.1016/j.brainres.2004.12.050. [DOI] [PubMed] [Google Scholar]
- Kimura E, Takasawa R, Tanuma S, Aoki S. Monitoring apoptosis with fluorescent Zn2+-indicators. Sci STKE. 2004;223:pl7. doi: 10.1126/stke.2232004pl7. [DOI] [PubMed] [Google Scholar]
- Kindler DD, Thiffault C, Solenski NJ, Dennis J, Kostecki V, Jenkins R, Keeney PM, Bennett JP., Jr Neurotoxic nitric oxide rapidly depolarizes and permeabilizes mitochondria by dynamically opening the mitochondrial transition pore. Mol Cell Neurosci. 2003;23:559–573. doi: 10.1016/s1044-7431(03)00074-5. [DOI] [PubMed] [Google Scholar]
- Land PW, Aizenman E. Zinc accumulation after target loss: an early event in retrograde degeneration of thalamic neurons. Eur J Neurosci. 2005;21:647–657. doi: 10.1111/j.1460-9568.2005.03903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashley KS. Thalamo-cortical connections of the rat's brain. J Comp Neurol. 1941;75:67–121. [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399(6738 Suppl):A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Leung AW, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2008;1777:946–952. doi: 10.1016/j.bbabio.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Li YM, Shi J, Wu X, Luo ZF, Wang FL, Guo QX. Tracing of intracellular zinc (II) fluorescence flux to monitor cell apoptosis by using FluoZon-3AM. Cell Biochem Funct. 2009;27:417–423. doi: 10.1002/cbf.1598. [DOI] [PubMed] [Google Scholar]
- Lieberman AR. The axon reaction: a review of the principal features of perikaryal responses to axon injury. Int Rev Neurobiol. 1971;14:49–124. doi: 10.1016/s0074-7742(08)60183-x. [DOI] [PubMed] [Google Scholar]
- Lin DT, Lechleiter JD. Mitochondrial targeted cyclophilin D protected cells from cell death by peptidyl prolyl isomerization. J Biol Chem. 2002;277:31134–31141. doi: 10.1074/jbc.M112035200. [DOI] [PubMed] [Google Scholar]
- Linard D, Kandlbinder A, Degand H, Morsomme P, Dietz KJ, Knoops B. Redox characterization of human cyclophilin D: identification of a new mammalian mitochondrial redox sensor? Arch Biochem Biophys. 2009;491:39–45. doi: 10.1016/j.abb.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Lok J, Martin LJ. Rapid subcellular redistribution of Bax precedes caspase-3 and endonuclease activation during excitotoxic neuronal apoptosis in rat brain. J Neurotrauma. 2002;19:815–828. doi: 10.1089/08977150260190410. [DOI] [PubMed] [Google Scholar]
- Machida K, Ohta Y, Osada H. Suppression of apoptosis by cyclophilin D via stabilization of hexokinase II mitochondrial binding in cancer cells. J Biol Chem. 2006;281:14314–14320. doi: 10.1074/jbc.M513297200. [DOI] [PubMed] [Google Scholar]
- Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. 1999;58:459–471. doi: 10.1097/00005072-199905000-00005. [DOI] [PubMed] [Google Scholar]
- Martin LJ. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals. 2010;3:839–915. doi: 10.3390/ph3040839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Pardo CA, Cork LC, Price DL. Synaptic pathology and glial responses to neuronal injury precede the formation of senile plaques and amyloid deposits in the aging cerebral cortex. Am J Pathol. 1994;145:1358–1381. [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res Bull. 1998;46:281–309. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Kaiser A, Price AC. Motor neuron degeneration after sciatic nerve avulsion in adult rat evolves with oxidative stress and is apoptosis. J Neurobiol. 1999;40:185–201. [PubMed] [Google Scholar]
- Martin LJ, Sieber FE, Traystman RJ. Apoptosis and necrosis occur in separate neuronal populations in hippocampus and cerebellum after ischemia and are associated with alterations in metabotropic glutamate receptor signaling pathways. J Cereb Blood Flow Metab. 2000;20:153–167. doi: 10.1097/00004647-200001000-00020. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Kaiser A, Yu JW, Natale JE, Al-Abdulla NA. Injury-induced apoptosis of neurons in adult brain is mediated by p53-dependent and p53-independent pathways and requires Bax. J Comp Neurol. 2001;433:299–311. doi: 10.1002/cne.1141. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Price AC, McClendon KB, Al-Abdulla NA, Subramaniam JR, Wong PC, Liu Z. Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J Neurochem. 2003;85:234–247. doi: 10.1046/j.1471-4159.2003.01659.x. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Chen K, Liu Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J Neurosci. 2005;25:6449–6459. doi: 10.1523/JNEUROSCI.0911-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Liu Z, Chen K, Price AC, Pan Y, Swaby JA, Golden WC. Motor neuron degeneration in amyotrophoc lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J Comp Neurol. 2007;500:20–46. doi: 10.1002/cne.21160. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Liu Z, Pipino J, Chestnut B, Landek MA. Molecular regulation of DNA damage-induced apoptosis in neurons of cerebral cortex. Cereb Cortex. 2009a;19:1273–1293. doi: 10.1093/cercor/bhn167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD, Chang Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp Neurol. 2009b;218:333–346. doi: 10.1016/j.expneurol.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Jürgensmeier JM, Susin SA, Vieira HL, Prévost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–350. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- McStay GP, Clarke SJ, Halestrap AP. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem J. 2002;367:541–548. doi: 10.1042/BJ20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Bolaños JP. Nitric oxide, cell bioenergetics and neurodegeneration. J Neurochem. 2006;97:1676–1689. doi: 10.1111/j.1471-4159.2006.03988.x. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH. Neurogenesis and apoptosis cell death. Stroke. 2003;34:324–326. doi: 10.1161/01.str.0000054047.14853.ad. [DOI] [PubMed] [Google Scholar]
- Mueller D, Shamblott MJ, Fox HE, Gearhart JD, Martin LJ. Transplanted human embryonic germ cell-derived neural stem cells replace neurons and oligidendrocytes in the forebrain of neonatal mice with excitotoxic brain damage. J Neurosci Res. 2005;82:592–608. doi: 10.1002/jnr.20673. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulated some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Nakajima W, Ishida A, Lange MS, Gabrielson KL, Wilson MA, Martin LJ, Blue ME, Johnston MV. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci. 2000;20:7994–8004. doi: 10.1523/JNEUROSCI.20-21-07994.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natale JE, Cheng Y, Martin LJ. Thalamic neuron apoptosis emerges rapidly after cortical damage in immature mice. Neuroscience. 2002;112:665–676. doi: 10.1016/s0306-4522(02)00098-2. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol Dis. 2001;8:207–219. doi: 10.1006/nbdi.2000.0371. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Graham EM, Martin LJ. Apoptosis in perinatal hypoxic-ischemic brain injury: how important is it and should it be inhibited? Brain Res Brain Res Rev. 2005;50:244–257. doi: 10.1016/j.brainresrev.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Zelaya ME, O'Riordan DP, Blomgren K, Flock DL, Hagberg H, Ferriero DM, Martin LJ. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience. 2007;149:822–833. doi: 10.1016/j.neuroscience.2007.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer's disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Chiba S, Lippa CF, Cras P, Kalaria RN, Takeda A, Honda K, Smith MA, Perry G. Neuronal RNA oxidation is a prominent feature of familial Alzheimer's disease. Neurobiol Dis. 2004;17:108–113. doi: 10.1016/j.nbd.2004.06.003. [DOI] [PubMed] [Google Scholar]
- O'Donovan M, Ho S, Yee W. Calcium imaging of rhythmic network activity in the developing spinal cord of the chick embryo. J Neurosci. 1994;14:6354–6369. doi: 10.1523/JNEUROSCI.14-11-06354.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Navarro E, Gavaldà N, Gratacòs E, Alberch J. Brain-derived neurotrophic factor prevents changes in Bcl-2 family members and caspase-3 activation induced by excitotoxicity in the striatum. J Neurochem. 2005;92:678–691. doi: 10.1111/j.1471-4159.2004.02904.x. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Price DL, Martin LJ. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J Comp Neurol. 1997a;378:70–87. [PubMed] [Google Scholar]
- Portera-Cailliau C, Price DL, Martin LJ. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evidence for an apoptosis-necrosis continuum. J Comp Neurol. 1997b;378:88–104. [PubMed] [Google Scholar]
- Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–389. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical cultures. J Neurochem. 1994;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- Schubert A, Grimm S. Cyclophilin D, a component of the permeability transition-pore, is an apoptosis repressor. Cancer Res. 2004;64:85–93. doi: 10.1158/0008-5472.can-03-0476. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Konishi A, Kodama T, Tsujimoto Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci U S A. 2000;97:3100–3105. doi: 10.1073/pnas.97.7.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza JM, Peluffo G, Radi R. Protein tyrosine nitration—functional alteration or just a biomarker? Free Radic Biol Med. 2008;45:357–366. doi: 10.1016/j.freeradbiomed.2008.04.010. [DOI] [PubMed] [Google Scholar]
- van Gurp M, Festjens N, van Loo G, Saelens X, Vandenabeele P. Mitochondrial intermembrane proteins in cell death. Biochem Biophys Res Comm. 2003;304:487–497. doi: 10.1016/s0006-291x(03)00621-1. [DOI] [PubMed] [Google Scholar]
- van Lookeren Campagne M, Lucassen PJ, Vermeulen JP, Balázs R. NMDA and kainate induced internucleosomal DNA cleavage associated with both apoptotic and necrotic cell death in the neonatal rat brain. Eur J Neurosci. 1995;7:1627–1640. doi: 10.1111/j.1460-9568.1995.tb01158.x. [DOI] [PubMed] [Google Scholar]
- Vieira HL, Belzacq AS, Haouzi D, Bernassola F, Cohen I, Jacotot E, Ferri KF, Hamel CE, Bartle LM, Melino G, Brenner C, Goldmacher V, Kroemer G. The adenine nucleotide translocator: a target of nitric oxide, peroxynitrite, and 4-hydroxynonenal. Oncogene. 2001;20:4305–4316. doi: 10.1038/sj.onc.1204575. [DOI] [PubMed] [Google Scholar]
- von Bohlen und Halbach O, Albrecht D, Heinemann U, Schuchmann S. Spatial nitric oxide imaging using 1,2-diaminoanthraquione to investigate the involvement of nitric oxide in long-term potentiation in rat brain slices. Neuroimage. 2002;15:633–639. doi: 10.1006/nimg.2001.1045. [DOI] [PubMed] [Google Scholar]
- Waldmeier PC. Prospects for antiapoptotic drug therapy of neurodegenerative diseases. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:303–321. doi: 10.1016/S0278-5846(03)00025-3. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn of evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel ER. Practical methods for biological morphometry. New York: Academic; 1979. Stereological methods. [Google Scholar]
- Woodfield K, Rück A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J. 1998;336:287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda O, Fukuo K, Sun X, Nishitani M, Yotsui T, Higuchi M, Suzuki T, Rakugi H, Smithies O, Maeda N, Ogihara T. Apop-1, a novel protein inducing cyclophilin D-dependent but Bax/Bak-related channel-independent apoptosis. J Biol Chem. 2006;281:23899–23907. doi: 10.1074/jbc.M512610200. [DOI] [PubMed] [Google Scholar]
- Zoratti M, Szabò I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Isave NK, Plotnikov EY, Zorova LD, Stelmashook EV, Vasileva AK, Arkhagelskaya AA, Khrjapenkova TG. The mitochondrion as janus bifrons. Biochemistry (Mosc) 2007;72:1115–1126. doi: 10.1134/s0006297907100094. [DOI] [PubMed] [Google Scholar]