

Abstract
CA1 pyramidal neurons are the final integrators of information flow leaving the hippocampus, yet are singularly vulnerable to activity-dependent cell death. Zinc (Zn) entry into cells may add to this vulnerability. Here, we find that Slc39a1 and Slc39a3, members of the Zip (Zrt/Irt-like protein) plasmalemmal Zn transporter family, are predominantly expressed in the hippocampus. We examined Zip-1,3-deficient mice to investigate their role in neurodegeneration following intense synaptic activation. When isolated by blockade of NMDA receptors and voltage-gated calcium channels, the absence of both transporters slowed passive Zn uptake into CA1 neurons measured with intracellular fluorescent Zn dyes. In vivo CA1 cell damage following kainic acid exposure was greatly attenuated. Consistent with the hypothesis that Zn entry contributes to neurodegeneration, Znt-3-deficient mice lacking synaptic Zn also show less hippocampal cell damage following kainic acid injection. Zip transporters may provide selective therapeutic targets to protect these neurons from early Zn-induced neurodegeneration following injury.
Introduction
Hippocampal pyramidal neurons play key roles in learning and memory, yet are especially vulnerable to brain injury. Early neurodegeneration following seizures and ischemia is largely a consequence of glutamate excitotoxicity that activates NMDA receptors and Ca-permeable AMPA/kainate receptors, leading to elevated intracellular Ca2+ and eventual cell death (Kwak and Weiss, 2006; Liu and Zukin, 2007). However, attempts to block these entry routes have been therapeutically ineffective (Villmann and Becker, 2007; Besancon et al., 2008). Excessive zinc (Zn) translocation has also been proposed as a molecular trigger of the cellular apoptotic cascade (Weiss et al., 2000; Frederickson et al., 2005; Kwak and Weiss, 2006; Hershfinkel et al., 2009). Several possible routes of transmembrane Zn entry have been identified. When externally applied, Zn permeates cultured neurons through NMDA receptors, Ca-permeable AMPA/Kainate receptors, voltage-gated calcium channels, and neuronal plasma membrane Zn transporters (Freund and Reddig, 1994; Sensi et al., 1997, 1999; Colvin et al., 2003). Of the latter, members of the solute carrier 39a (Slc39a) Zn transporter family called Zips (Zrt/Irt-like protein) are interesting candidates, since they mediate inward Zn transport and are expressed in brain (Dufner-Beattie et al., 2003, Belloni-Olivi et al., 2009, Emmetsberger et al., 2010).
The contribution of Zn accumulation to neurodegeneration following brain injury has been extensively explored. Chelation of extracellular Zn is the most widely used technique. While in some studies, Zn chelation successfully protects neurons (Koh et al., 1996), in others Zn removal increases cell death (Domínguez et al., 2003; Côté et al., 2005; Lavoie et al., 2007). Lowering brain Zn content by a low Zn diet or genetic ablation of presynaptic vesicular Zn transporters also fails to prevent neurodegeneration (Lee at al, 2000; Takeda et al., 2005). In these studies, however, it is unclear to what extent the reduction of neuronal Zn pools actually affected postsynaptic Zn entry. Since extracellular Zn can modulate membrane excitability by targeting receptors for NMDA, GABAA, glycine, voltage-gated Ca2+ channels, and ATP-K+ channels (Westbrook and Mayer, 1987; Bancila et al., 2004; Hirzel et al., 2006; Cataldi et al., 2007; Traboulsie et al., 2007), any experimental removal of Zn (by dietary deficiency, by reduction in the quantal size of presynaptic vesicular Zn pools, or particularly by chelation) before it enters postsynaptic neurons may mask the actual amount of Zn movement into cells. For example, endogenous Zn inhibits NMDA receptors (Vogt et al., 2000; Molnár and Nadler, 2001), but chelation of extracellular Zn greatly magnifies the effects of NMDA receptor excitotoxicity (Domínguez et al., 2006), allowing more Ca entry, thereby obscuring the ionic mechanisms contributing to cell death.
To isolate the contribution of extracellular Zn entry to neurodegeneration following brain injury while sparing other receptors and channels essential for hippocampal signaling, a means of directly reducing Zn entry into postsynaptic neurons without perturbing upstream Zn availability is required. In the present study, we combined fluorescent Zn dye imaging, electrophysiology, and histochemistry methods to demonstrate that two members of the plasma membrane inward Zn transporter family, Slc39a1 (Zip-1) and Slc39a3 (Zip-3) are strongly expressed in hippocampal pyramidal neurons and contribute to Zn homeostasis. When normalized for the amount of electrographic seizure activity within a 24 h period following kainate injection, we found a significant reduction of cell damage in CA1 principal cells in mice lacking both Zip-1 and Zip-3 transporters compared with wild-type littermates. Our results identify Zip-1,3 transporters as novel therapeutic targets to protect CA1 neurons from Zn entry-induced neuronal degeneration following seizure or ischemia attacks.
Materials and Methods
Generation of Zip-1 and Zip-3 double-knock-out mice.
The targeted disruption of both Zip-1 and Zip-3 gene expression in mouse has been previously described in detail by Dufner-Beattie et al. (2006). Briefly, the targeting construct fused the initiator methionine codon of the targeted Zip gene with the open reading frame of the enhanced green fluorescent protein (EGFP) reporter. This disrupts the protein coding sequence of the targeted Zip gene and inserts a stop codon and a polyadenylation signal into exon 2 of the gene. This allows for EGFP expression that is driven by the targeted Zip promoter and disruption of the targeted Zip gene expression. The loxP flanked MC1-Neo cassette was removed from the knock-out insertion in targeted Zip-1 and Zip-3 mice by mating with Cre-expressing transgenic mice (Jackson Laboratories). Both strains of knock-out mice were generated by inbreeding the neo-negative Zip-1 or 3 −/− offspring from the breeding with Cre-expressing transgenic mice. Homozygous (Zip-1 −/− and Zip-3 −/−) mice were crossbred, and then offspring were inbred to generate homozygous Zip-1, Zip-3 double-knock-out mice.
Whole-cell patching and optical recording.
Hippocampal slices were prepared from Zip-1,3 double-knock-out mice and homozygous wild-type littermates of the same strain using a vibratome (350 μm thickness) as described in a previous study (Qian and Noebels, 2005). Brain slices were incubated in artificial CSF (ACSF) at 32°C for 1 h and then transferred into a submerged recording chamber mounted on a microscope (Axioskop 2, Zeiss) for dye loading and electrophysiology recording. Brain slices were constantly perfused with ACSF contained the following (in mm): 125 NaCl, 2.5 KCl, 2.5 CaCl2, 25 NaHCO3, 2 MgSO4, 1.25 NaH2PO4, and 11 d-glucose, gassed with 95% O2/5% CO2 to maintain a constant pH of 7.4. The temperature of the recording chamber was controlled at 30°C. The microscope was fitted with a 40×/0.8 numerical aperture Zeiss water-immersion objective and differential interference contrast optics to visualize the hippocampal slices. CA1 pyramidal neurons were identified based on their location in the stratum pyramidale and characteristic morphology (pyramidal shape of the cell body and the distinct apical dendritic trunk). Whole-cell recording pipettes (4–6 MΩ) were pulled from borosilicate glass and filled with the following (in mm): 107.5 Cs-gluconate, 10 KCl, 20 HEPES, 8 NaCl, 6 phosphocreatine, 5 QX-314, 4 Mg-ATP, 0.3 Na-GTP, and 0.2 EGTA, pH 7.3. The membrane-impermeable fluorescent dyes Alexa 586 (4 μm) and FluoZin-3 (40 μm) were added to the pipette solution. The superficial layer of cells damaged during cutting was carefully removed over the CA1 area by gentle suction through a polished pipette, and whole-cell patch-clamp recordings were then made from the visually identified CA1 pyramidal somata with an Molecular Devices Axoclamp-1D amplifier. The membrane potential was clamped at −70 mV. The measured input membrane resistance was in the range of 60–100 MΩ. Optical recordings were made 30 min after membrane break-in. To assay passive Zn uptake, NaH2PO4 was removed from the ACSF solution during perfusion of 10 μm Zn2+ to avoid precipitation of Zn (Frederickson et al., 2006), which may reduce the actual Zn2+ concentration applied.
Fluorescence images were obtained with a NeuroCCD-SMQ (80 × 80) imaging system (RedshirtImaging). For Alexa 586, the excitation filter was 580/20 nm and the emission filter was 630/60 nm. For FluoZin-3, the excitation filter was 488/20 nm and the emission filter was 535/25 nm. The GFP image was obtained using the same optical setting as for FluoZin-3. Each sample consisted of paired Alexa 586 and FluoZin-3 fluorescence images. Autofluorescence was corrected by subtracting the fluorescence intensity measured from an area within 50 μm devoid of neuronal processes of the dye-loaded cell. The frame rate of the CCD camera was set at 40 Hz and an average of 50 frames per image was used for data analysis.
Kainic acid injection and detection of cell injury.
Adult mice from mutant and wild-type genotypes (30–35 d of age) were injected intraperitoneally with kainic acid (KA) (either 15 mg/kg or a higher dose of 30 mg/kg consisting of two injections of 15 mg/kg separated by 1 h). Early neurodegeneration following KA injection was assayed by the Fluoro-Jade (FJ) staining technique. Briefly, 24 h after the KA injection, mice were killed, and the brains were quickly removed and placed into PBS containing 4% paraformaldehyde at 4°C for overnight fixation. Brains were then infiltrated with a sucrose tissue-freezing solution and then frozen at −80°C. Transverse sections (20 μm thick) were cut in a cryostat from the midsection of hippocampi (the same portion of hippocampi as used in electrophysiology experiments) and mounted onto glass coverslips for Fluoro-Jade staining. The staining procedures were followed according to the instructions for the Fluoro-Jade detection kit (Millipore Bioscience Research Reagents).
The areal intensity of Fluoro-Jade staining in the hippocampal formation was measured by calculating an FJ staining index (FJI) for each section, determined by the number of Fluoro-Jade-positive pixels in the pyramidal cell layer region of interest (CA1, CA3). This value was normalized by the length of the whole pyramidal cell layer in each section, and compared across genotypes.
Video/electroencephalographic recording.
Mice ranging from 30 to 35 d of age were surgically implanted under avertin anesthesia with a microminiature connector attached to silver wire electrodes (0.005 inch diameter). EEG electrodes were positioned through cranial burr holes overlying the cortical surface in the subdural space over the brain. After ≥1 d of postsurgical recovery, the cortical EEG activity of freely moving mice was monitored before and after intraperitoneal KA injection. Electrographic seizure activity was visually identified and quantified by measuring the time of onset and the total aggregate time spent in synchronized epileptiform EEG discharge activity. A computer program was developed to plot the cumulative EEG discharge data as shown in Figure 4. The synchronized EEG discharges were classified into two categories: typical seizure-like synchronized EEG discharges that exhibit a burst frequency of >1 Hz; and discharges with a burst frequency <1 Hz.
Figure 4.

Higher seizure susceptibility in Zip-1,3 −/− mouse brain. Summary data quantifying shorter latency to seizure onset and prolonged time spent in active seizure activity in mutant compared with controls injected with kainic acid. EEG seizure activity recorded from mice (−/− n = 8; +/+ n = 6) injected with KA (15 mg/kg). Before injection, Zip1,3 mice show normal EEG patterns (right, gray sample trace) without any spontaneous cortical discharge activity similar to wild-type mice. After injection of KA, behavioral and electrographic seizure activity gradually developed over 30 min (blue and red sample traces). As shown at left bottom, the average latency for the initial onset of high-frequency EEG spiking was 11 ± 9 min in the −/− group, which is less than one half that in the +/+ group (26 ± 17 min, p = 0.05). Blue bar marks the time during which the EEG shows low-frequency (<1 Hz) aberrant synchronized spike discharges (blue sample trace) Red bar indicates the period of high-frequency synchronized EEG seizure discharges (red sample trace). By comparison of measures of latency to seizure onset and total time spent in seizure activity, Zip-1,3 −/− mutants exhibit higher seizure susceptibility to the KA injection than their +/+ littermates.
Western immunoblotting.
Cortex and hippocampi were dissected from freshly isolated whole brain of Zip +/+ and −/− mice, flash frozen in liquid nitrogen, and subsequently homogenized on ice with a Tissue Tearor in lysis buffer containing the following (in mm): 20 Tris, pH 7.5, 138 NaCl, 3 KCl, 1% Triton X-100, 1 EGTA, 2 EDTA, 1 benzamidine, 1 phenylmethylsulfonylfluoride, 1 dithiothreitol, and 5 μg/ml each of aprotinin, leupeptin, and pepstatin A. Total protein concentration of the brain and heart tissue lysates were determined using the Bio-Rad Protein Assay. One hundred micrograms of cortex or hippocampal protein lysate were separated on 8% Tris-HEPES-SDS polyacrylamide gels, analyzed by Western immunoblot using rabbit polyclonal anti-NMDA receptor 1 (NMDAR1) antibody (1:300 in vehicle; catalog #4204, Cell Signaling Technology) and HRP-tagged goat anti-rabbit IgG secondary antibody (1:5000 dilution in vehicle, Santa Cruz Biotechnology), and subsequently detected using a commercial chemiluminescent substrate (SuperSignal, Pierce Chemical).
Data analysis.
Data from mice in each genotype were pooled and represented with mean/SDs. The Student's t test was used for significance analysis.
Results
Expression of Zn transporter subfamily II members Zip-1 and Zip-3 in the hippocampus
Zip-1- and Zip-3-containing neurons were visualized in the CNS by detecting the expression of an EGFP reporter engineered into mice doubly homozygous for targeted deletions of these genes (Dufner-Beattie et al., 2006). These transporters are expressed at low levels throughout the forebrain, brainstem, and cerebellum; however, they show a predominant localization within the hippocampal formation. Figure 1A shows the intense EGFP signal found within the hippocampal formation after immunostaining with anti-GFP-Alexa 594 (Texas Red) in a coronal section from Zip-1,3 double-knock-out mouse brain. In comparison, anti-GFP-Alexa 594 staining in +/+ brain shows no specific signal such as a highlighted staining in the pyramidal cell layers, as observed in the −/− mouse brain (data not shown). The fluorescence intensity of EGFP antibody staining in the pyramidal cell layer is ∼50% higher than that in the nearby entorhinal cortex after being normalized by the cell density in each region (obtained from densitometry of adjacent Nissl-stained sections). Figure 1B,C demonstrates that the Zip-1 and Zip-3 expression in the hippocampal pyramidal cell layer arises from staining of the principal cells. To verify that the strong expression within the cell layer represented intrinsic transporter expression in individual pyramidal cells, a patch pipette was used to load visually identified CA1 pyramidal cells in Zip-1,3 mutant (Fig. 1B) and wild-type (Fig. 1C) mouse brain slices with the fluorescent tracer Alexa 568. In Zip-1,3-null mutants, but not in control wild-type mice, significant GFP fluorescence could be detected in pyramidal cells as shown in the corresponding EGFP image in Figure 1B. EGPF staining was also detected in cells at very low intensity similar to neocortex in other regions of brain including, thalamus, pons, and cerebellum (data not shown).
Figure 1.

Expression of Zip-1,3 Zn transporters in the hippocampus. A, Immunofluorescence staining of anti-GFP-Alexa 594 (Texas Red) in a coronal section of Zip-1,3-null mice labeled with targeted EGFP reporter. Intense signal indicates strong expression of Zip-1 and Zip-3 in the hippocampal pyramidal (CA1, CA3) and dentate granule cell layers with low levels in overlying neocortex. B, C, High-magnification EGFP fluorescence images taken from Zip-1,3 −/− (B) and +/+ (C) brain slices. Scale bar, 10 μm. To verify EGFP staining in identified CA1 neurons, Alexa dye was injected through a patch-clamp electrode and visualized with filtering for Alexa (B, left), then EGFP (B, right). In the mutant (B), fluorescent Alexa pyramidal cell (dotted outline) colabels EGFP-filled cells. In the wild-type cell (C), no EGFP is present (C, right). To highlight the somatic GFP fluorescence, the EGFP fluorescence intensity in the dendritic region was subtracted from the raw image data. To compare the EGFP fluorescence intensity between two genotypes, the EGFP image from the +/+ group is normalized to the one taken from the −/− group.
Ablation of Zip-1 and Zip-3 transporters slows Zn uptake into CA1 pyramidal cells
We used whole-cell patch-clamp recording combined with fluorescent imaging to examine the potential contribution of Zip-1,3 transporters to Zn uptake into CA1 pyramidal neurons in vitro. We isolated the inward transporter-mediated fraction by adding the NMDA receptor antagonist D-APV and the AMPA receptor antagonist DNQX to the perfusate of hippocampal slices to eliminate possible Zn entry through glutamatergic receptors (GluRs). To prevent Zn entry through open voltage-gated Ca2+ channels in the target cell, the intracellular solution contained 5 mm QX-314 to block voltage-gated Na+ channels, and the somatic membrane potential was constantly held at −70 mV. To monitor intracellular Zn levels, single CA1 pyramidal neurons were loaded with the membrane-impermeable fluorescent tracer Alexa 568 and the Zn-sensitive dye FluoZin-3. A pair of Alexa 568 and FluoZin-3 fluorescence images, providing a baseline reference of intracellular Zn activity, was obtained 30 min after membrane break-in. We determined that the ratio F-FluoZin-3/F-Alexa is homogeneous across the cell surface from soma to the proximal one third of the apical dendrite, as demonstrated by a linear relationship between FluoZin-3 fluorescence (F-FluoZin-3) and Alexa fluorescence (F-Alexa) sampled along the line shown in the Alexa image (Fig. 2A, left), indicating that cytoplasmic free Zn levels are buffered to a uniform level. To minimize the sampling variance, we measured the ratio F-FluoZin-3/F-Alexa, instead of F-FluoZin-3 alone, before (Fig. 2A, center) and after (Fig. 2A, right) exposure of the slice to extracellular Zn2+ to calculate Zn uptake. In wild-type brain slices, perfusion of 10 μm Zn2+ for 30 min increased the F-FluoZin-3/F-Alexa ratio from 6.7 ± 0.9% to 23.1 ± 8.1% (n = 11) (Fig. 2A–C, top). In Figure 2A–C (bottom), the corresponding result for neurons in Zip-1/3 −/− slices was 4.8 ± 0.9% and 12.1 ± 2.2% (n = 9), which represents a significant reduction of ∼50% (p < 0.01) compared with wild-type neurons. This sharp reduction is consistent with the transporter localization obtained with EGFP staining (Fig. 1) and supports the notion that Zip-1,3 transporters expressed in hippocampal CA1 pyramidal cells regulate passive Zn homeostasis.
Figure 2.

Slowed Zn uptake in mice lacking Zip-1,3 transporters. A, Fluorescence images taken from a representative pyramidal neuron filled with both Alexa and the Zn dye FluoZin-3 in wild-type (top) and mutant (bottom) neurons. Top left, Alexa image. scale bar, 10 μm. Top center, FluoZin-3 image taken in control solution. Top right, FluoZin-3 image taken after the exposure of 10 μm Zn2+ to slice for 30 min. B, Linear relationship between FluoZin-3 fluorescence (F-FluoZin-3) and Alexa fluorescence (F-Alexa) sampled across the cell soma/proximal dendrite surface in the sample cell as marked by the line in the Alexa image in A. The F-FluoZin-3/F-Alexa ratio is quite homogeneous across the cell surface. Therefore, the ratio of F-FluoZin-3/F-Alexa before and after Zn2+ exposure was measured to minimize the sampling variance in Zn uptake. C, Summary data for passive Zn uptake assay for +/+ and −/− groups. A 30 min perfusion of 10 μm Zn2+ increases the ratio of F-FluoZin-3/F-Alexa from 6.7 ± 0.9% to 23.1 ± 8.1% (n = 11). The corresponding result for the −/− group is 4.8 ± 0.9% and 12.1 ± 2.2% (n = 9), a ∼50% reduction (p < 0.01), indicating that lack of Zip-1,3 Zn transporters in CA1 pyramidal neurons reduces Zn uptake in response to elevation of extracellular Zn concentration.
Protection from seizure-induced cell injury in the CA1 area of Zip-1,3 −/− mutants
To determine whether this twofold reduction in the contribution of transporter-mediated Zn entry during intense synaptic activation could influence cell survival following injury in vivo, we used a widely used hippocampal convulsant that generates extensive excitotoxic pyramidal cell damage while allowing survival of the animal. Zip-1,3 mutants and wild-type littermates were injected intraperitoneally with KA (15 mg/kg), monitored by video EEG to quantify seizure activity, and early hippocampal cell injury was evaluated 24 h later with FJ staining. Figure 3A shows representative images of FJ staining in the hippocampus of Zip-1/3 −/− and +/+ brains analyzed 1 d after KA injection. The Fluoro-Jade staining is largely confined to the pyramidal cell layers except for the CA2 area. The dentate granule layer was not stained at all. This pattern of Fluoro-Jade staining is consistent with the cell death pattern in the hippocampus following excitotoxic insults.
Figure 3.

Attenuated neurodegeneration in the hippocampal CA1 area in Zip-1,3 −/− mutants after KA-induced seizures. A, Representative Fluoro-Jade-stained sections taken from transverse mid-hippocampal sections from +/+ and −/− mutant mice show less CA1 injury in Zip1,3-null mutants following KA seizure activity. B, Fluoro-Jade staining patterns in +/+ and −/− mutant mice reveal region-specific patterns of cell injury. For each mouse, the CA1 FJI was plotted against its CA3 FJI. In wild-type mice (left), the upper left quadrant represents mice with selective and extensive CA1 cell injury but little CA3. This pattern is strikingly reduced in Zip-1,3 −/− mutant mice (right). C, Summary data for the extent of CA1 and CA3 neurodegeneration 24 h after administration of KA (15 mg/kg).
In a large sample of mice from both genotypes, disruption of Zip-1,3 transporters produced clear protection in the hippocampal CA1 region. We found positive cellular FJ staining in 21 of 41 injected +/+ mice in the CA1 area; in contrast, 11 of 43 injected −/− mutants showed positive FJ staining in the same area. We calculated an FJI for hippocampal pyramidal subregions in each section to quantify cell injury in the mutant and littermate control genotypes. We found evidence that the two hippocampal subregions exhibit different vulnerability profiles to kainate-induced injury. The average FJI in CA1 for the −/− group was 9 ± 19% (n = 43) of its maximal value, which is significantly smaller than the value for the +/+ group (23 ± 32%, n = 41, p = 0.01). Only 5 of 43 Zip-1,3 −/− mice fell in the upper 60 percentile of FJIs compared with 13 of 41 from the +/+ group. These data demonstrate a striking reduction in early CA1 injury in the −/− group after KA injection compared with wild-type mice. The pattern of selective protection of CA1 neurons in −/− mice was even more evident when cell damage from both subregions is directly compared, as shown in Figure 3B. In wild-type mice, a group of mice (8/41) exhibits a characteristic pattern of vulnerability exclusively confined to the CA1 pyramidal cell region (Fig. 3B, left). These mice display extensive CA1 FJ staining (>40% of maximum FJI), while the CA3 region is mostly spared (<10% of maximum FJI). In the −/− group (Fig. 3B, right), the number of mice with exclusive CA1 cell damage was greatly diminished. In contrast, due to significant neuroprotection in the CA1 region, Zip-1,3-null mutants show damage largely confined to the CA3 region, with minor amounts of CA1 injury. Only 2 of 43 mutants showed evidence of isolated CA1 damage. The FJI for the CA3 area was positive in a similar proportion of Zip-1,3-null mice (26/43) compared with wild-type mice (26/41); however, the average FJI for CA3 in Zip 1,3-null mice was 29 ± 27% (n = 43) of its maximal value, which is significantly larger than the average value from the +/+ group (19 ± 23%, n = 41, p = 0.04), indicating more CA3 cell damage occurs in the −/− group. Figure 3C summarizes cell injury in each subregion of hippocampus.
Zip-1,3-null mutants exhibit a lower seizure threshold to kainate
The extent of hippocampal cell injury in experimental seizure models is strongly dependent on the severity of seizure activity. It is therefore possible that if developmental absence of Zip-1,3 transporters lowered the underlying network excitability, weaker seizures, rather than reduced vulnerability to Zn entry, might explain the differences in cellular damage between the mutant and wild-type brains. To compare seizure severity between the genotypes, both behavioral and EEG seizure monitoring was continuously performed in mice from both groups during the period of KA exposure and accurately quantified by frequency analysis of the digitized EEG trace. Zip-1,3-null mutants display normal patterns of EEG activity during routine monitoring, but reveal a latent hyperexcitability upon administration of the glutamate agonist. Figure 4 shows EEG activity recorded from mice (−/−, n = 8; +/+, n = 6) before and during the first 4 h after KA injection. Zip-1,3 mutants exhibited more synchronized high-frequency spike bursting (Fig. 4, marked in red) than wild-type mice, and 3 of 8 mutant mice showed essentially constant EEG seizure activity, lasting even beyond the 4 h experimental monitoring window. Among the six +/+ mice recorded for EEG activity, none displayed seizures beyond 4 h, and the EEG seizure discharges were discontinuous. The average latency for the initial onset of high-frequency EEG spiking was 11 ± 9 min (n = 8) in the −/− group, which is less than one half that in the +/+ group (26 ± 17 min, n = 6, p = 0.05). Seizure-induced lethality also differed between genotypes; 17% of the tested mutants died during a prolonged seizure, compared with only 4% in the +/+ group after injection of 15 mg/kg KA. At a higher dose (30 mg/kg), the difference in mortality was even greater (mortality rate was 66% and 22%, respectively, for −/− and +/+ mice). These results demonstrate that less seizure-induced neurodegeneration occurs in the CA1 area of Zip-1,3 −/− mice even though they experience far more severe seizure activity after KA injection than the +/+ group.
During seizures, glutamate excitotoxicity, particularly mediated by NMDA receptors, also contributes to hippocampal pyramidal cell injury. If Zip −/− mice express a lower NMDA receptor density, then less neuronal cell damage in the −/− group could be explained by a reduction in glutamate excitotoxicity. To evaluate this possibility, we compared the expression levels of NMDA receptors by Western blot between +/+ and −/− mice. Figure 5 shows NMDA receptor expression in the cortex and hippocampus of +/+ and Zip-1,3 −/− mouse brains. Comparable immunoreactive bands corresponding to the 120 kDa NMDAR1 subunit were detected in cortex and hippocampus, indicating unaltered global NMDA receptor expression in Zip-1,3 −/− mouse. This result also suggests that the increased seizure severity in Zip-1,3 −/− brain is unlikely to be explained by secondary changes in NMDA receptor density.
Figure 5.

Unaltered NMDA receptor expression in Zip-1,3 −/− mouse brain. NMDA receptor expression, a major alternative pathway for neurodegeneration following seizures was examined by Western blot with antibody against NMDAR1 subunit. Similar immunoreactive bands corresponding to the 120 kDa NMDAR1 subunit were detected in cortex and hippocampus of +/+ and Zip-1,3 −/− mouse brain, indicating unaltered global NMDA receptor expression in both cortex and hippocampus of the −/− group.
Less neuronal cell damage in Znt-3 −/− mouse with KA injection
If postsynaptic Zn uptake contributes to pyramidal cell vulnerability, then reduction of extracellular Zn content during seizure activity might resemble the effect of Zip-1,3 transporter deletion. We examined early neurodegeneration in mice lacking synaptic Zn by injecting Znt-3 −/− mice with KA (15 mg/kg) in parallel studies. The same dose of KA, which usually evokes a full body convulsion in either the Zip-1,3 −/− or +/+ group within the first 30 min, only caused forelimb tremors in Znt-3 −/− mice within the first hour of the injection. After a second dose of 15 mg/Kg KA injection 1 h later, Znt-3 −/− mice started to develop full body convulsions and were killed for Fluoro Jade staining 24 h later. Figure 6 summarizes the result for the Znt-3 −/− group. There is much less cell damage in both the CA3 and CA1 areas after two injections of 15 mg/kg KA compared with the +/+ group with one injection of 15 mg/kg KA. The overwhelming majority of mice did not show any cell damage in the CA1 area. Only 3 of 34 Znt-3 −/− mice fell in the upper 60 percentile of FJIs. The mean FJI in the CA1 area was 8 ± 17% (n = 34), which is significantly less than that in the +/+ group (p = 0.01). In the CA3 area, the average FJI was 9 ± 12% (n = 34), which is also significantly smaller than the average value from the +/+ group (p = 0.03). As shown in Figure 6B, cell injury for both CA1 and CA3 remained in a smaller range compared with the +/+ group. Interestingly, EEG monitoring of Znt-3 −/− mice after only one injection of KA at 15 mg/kg showed more seizure activity than the +/+ group even though their behavioral seizure activity scores were low. Although their average latency for the initial onset of high-frequency EEG spiking was not significantly different from mice in the +/+ group, Znt-3 −/− mice displayed longer, on average, seizure episodes of high-frequency EEG spiking. Our results indicate that reduction of Zn uptake, either by knockout of Zip-1,3 transporters or abolishment of synaptic Zn, can protect neurons from neurodegeneration following prolonged seizures.
Figure 6.
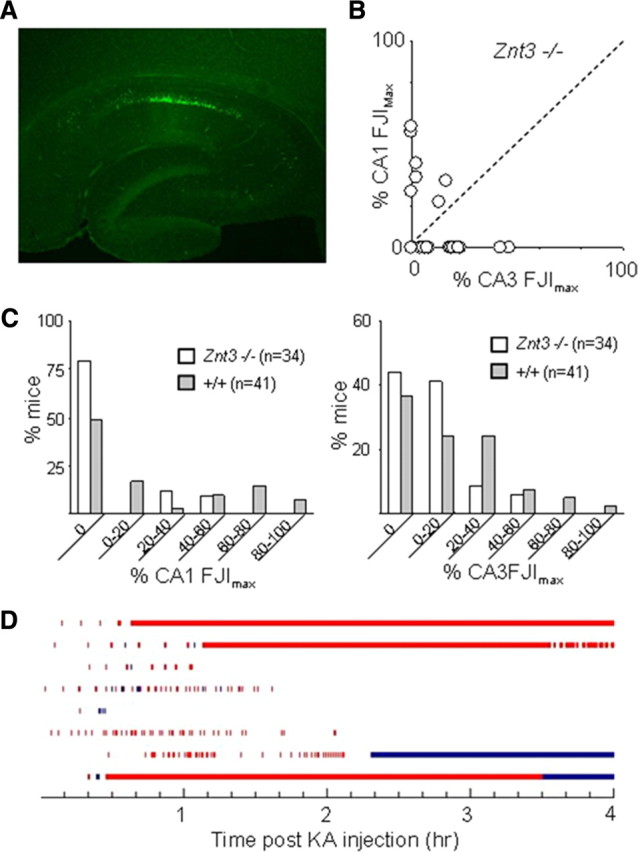
Less hippocampal cell damage in Znt3 −/− mouse brain following KA injection. A, A representative Fluoro-Jade-stained section taken from transverse mid-hippocampal sections from Znt-3 −/− mutant mice shows less cell injury compared with +/+ group. B, In both CA1 and CA3 areas, cell injury is confined to a smaller region of the pyramidal cell layer. C, Direct comparison of cell damage between Znt-3 −/− and +/+. About 75% of Znt3 −/− mice tested showed no CA1 damage, compared with 50% in the +/+ control group. More mice in the +/+ group exhibited severe cell damage than in the Znt-3 −/− group within both CA1 and CA3 regions. D, Quantitative plot of EEG seizure activity (red bar indicates high-frequency spiking activity, blue bar indicates low-frequency intermittent bursts) recorded in 8 individual Znt-3 −/− mice injected with KA (15 mg/kg). Although the average latency for the initial onset of high-frequency EEG spiking was not significantly different from that in the +/+ group, on average the −/− group displayed high EEG seizure activity for a much longer period.
Discussion
The ZIP Slc39a superfamily of metal transporters exists in all eukaryotes and imports Zn ions into the cytoplasm. Another family, Slc30a, is involved in Zn export (Kambe et al., 2004). ZIP proteins include eight transmembrane domains, the fourth of which contains fully conserved histidyl and glycyl residues in a putative amphipathic α helix. In mouse and humans, 14 genes encoding ZIP proteins have been identified with widely differing patterns of tissue expression; of these, 7 (Zip 1, 3, 4, 5, 10, 13, and 14) are variably expressed in brain (Dufner-Beattie et al., 2003; Belloni-Olivi et al., 2009; Emmetsberger et al., 2010) (Allen Brain Atlas). Although no specific antibodies for mammalian Zip proteins expressed in brain have been reported, our analysis is the first to identify the in vivo functional presence of these transporters in CNS neurons.
Zip-1,3 transporters regulate Zn homeostasis in hippocampal neurons
Loading high-affinity Zn-sensitive fluorescent dye into patch-clamped CA1 pyramidal cells bathed with D-APV and DNQX allowed us to demonstrate the passive uptake of Zn not mediated by voltage- or glutamate receptor-dependent channels in mouse brain slices. The relative reduction of passive Zn uptake in mutant neurons with targeted deletions of the plasma membrane Zn transporters Zip-1 and Zip-3 provides the first direct evidence for the involvement of members of the Zip transporter superfamily in the regulation of Zn homeostasis in hippocampal CA1 neurons. We found that Zip-1 and Zip-3 transporters account for ∼50% of the passive Zn uptake in these cells when extracellular Zn concentration reaches the low micrometer range (10 μm), a concentration that is in line with their Michaelis–Menten kinetics. When assayed in cultured HEK293 cells, both Zip-1 and Zip-3 have an apparent Km of ∼1.7 μm (Dufner-Beattie et al., 2003). We used the Allen Brain Atlas to determine that two other genes of the Slc39A family, Zip-5 and Zip-10, show particularly strong hippocampal mRNA transcript expression and presumably contribute to the remaining passive influx. To localize the putative Zip-1 and Zip-3 transporters in neuronal dendritic and somatic compartments, we tried three Zip-1 and Zip-3 antibodies made available by other laboratories; however, none proved to be specific in brain tissue. The exact cellular and compartmental location of Zip transporter proteins in central neurons remains to be further characterized.
Slower Zn uptake reduces neurodegeneration in the CA1 area but not CA3 neurons
The reduction of CA1 pyramidal cell injury in mouse mutants with targeted deletion of Zip-1 and Zip-3 also provides the first direct evidence that reduced postsynaptic Zn entry is neuroprotective in these cells. In addition to Zn transporters, Zn can also enter cells via Ca2+-permeable AMPA receptors lacking the GluR2 subunit. CA1 pyramidal cells in the mature hippocampus abundantly express AMPA receptors containing GluR2 subunit (Wenthold et al., 1996). Under pathological situations such as seizures and ischemia, these AMPA receptors can undergo subunit reorganization (Grooms et al., 2000; Noh et al., 2005; Kwak and Weiss, 2006; Liu and Zukin, 2007), and the resulting GluR2-deficient AMPA receptors are permeable to both Ca2+ and Zn2+. While these receptors contribute to cell death after ischemia (Yin et al., 2002), it is not clear how much they contribute to Zn entry. Despite their activation during KA-induced seizures and potential Zn entry through NMDA receptor and voltage-gated Ca channels at synaptic sites, our results indicate that tonic Zn uptake by Zn transporters contributes to a significant additional fraction of early hippocampal cell injury. Interestingly, the pattern of cell damage due to loss of Zip-1,3 was not uniform in the hippocampus. As shown by EEG recordings and the increased mortality rate following KA injection, even though Zip-1,3-null mutants were more susceptible to KA-induced seizures, they showed less neuronal cell injury in the CA1 pyramidal region. In contrast, in the CA3 area more pyramidal cell injury was observed in mutants than wild-type control mice. Several factors may contribute to the disparity in different hippocampal regions, including a difference in activity-induced release of Zn at the two sites. Due to the extensive innervation by Zn-rich mossy fibers and severer seizure activity, massive Zn release and Zn uptake in CA3 of the −/− group during kainate seizures may surpass that seen in CA1 even though Zip-1,3 transporters are not present in those CA3 cells. Less cell death in CA1 but more in CA3 following acute brain injury has also been reported in mice deficient in metallothionein, a major intracellular Zn binding protein (Lee et al., 2003). Intrinsic activity-dependent mechanisms other than Zn release, entry and synaptic connectivity may also contribute to region-specific variation in excitability and neurodegeneration levels, as confirmed by our Znt-3 −/− experiments that showed minor cell injury after seizures even in a hippocampus devoid of synaptic Zn.
Synaptic Zn contributes to neurodegeneration of pyramidal cells in the hippocampus
In the present study, we also show that lack of synaptic Zn leads to less pyramidal cell neurodegeneration in both CA1 and CA3 areas following KA injection of Znt-3 −/− mice. A previous study using the same Znt-3 −/− mutant mice reported that although the CA3 area exhibited less cell damage than +/+ wild-type mice, little difference was found in the CA1 area (Lee et al., 2000). Our results generally confirm these findings, and several factors may contribute to the disparity that CA1 neurons were severely damaged in their study, while the same region was relatively spared in ours. First, a higher KA dose was given to induce status epilepticus in their study then used here. Therefore, more severe seizure activity and a higher KA peak concentration in the brain, along with the loss of Zn-mediated inhibition of NMDA receptors in Znt-3 −/− mice might amplify overall glutamate excitotoxicity in that study. Consequently, the contribution of Zn entry-mediated cell injury may be masked by the greater amount of NMDA receptor mediated, Ca-induced neurodegeneration in the CA1 area, a region well known for its vulnerability to glutamate excitotoxicity. Second, prolonged seizures were a selection criterion for analysis in their study, whereas we studied mice following more moderate seizure activity. To control for the relatively high interanimal variation in cell degeneration patterns found in convulsant models, we investigated cell injury in a very large experimental cohort. Extensive CA1 pyramidal cell damage, as identified in the top 60th percentile, could still be detected in mice lacking synaptic Zn but with a much lower incidence (3/34) compared with their wild-type littermates (13/41). The results obtained in both studies of Znt-3 −/− mice are consistent with our demonstration that entry of Zn into postsynaptic cells contributes to neurodegeneration in the early stages of brain injury.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS 29709 (J.L.N.) and the Blue Bird Circle Pediatric Neurology Research Foundation. We are grateful to Ed Glasscock for generous assistance.
References
- Bancila V, Nikonenko I, Dunant Y, Bloc A. Zinc inhibits glutamate release via activation of pre-synaptic K channels and reduces ischaemic damage in rat hippocampus. J Neurochem. 2004;90:1243–1250. doi: 10.1111/j.1471-4159.2004.02587.x. [DOI] [PubMed] [Google Scholar]
- Belloni-Olivi L, Marshall C, Laal B, Andrews GK, Bressler J. Localization of zip1 and zip4 mRNA in the adult rat brain. J Neurosci Res. 2009;87:3221–3230. doi: 10.1002/jnr.22144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besancon E, Guo S, Lok J, Tymianski M, Lo EH. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci. 2008;29:268–275. doi: 10.1016/j.tips.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Cataldi M, Lariccia V, Marzaioli V, Cavaccini A, Curia G, Viggiano D, Canzoniero LM, di Renzo G, Avoli M, Annunziato L. Zn(2+) slows down Ca(V)3.3 gating kinetics: implications for thalamocortical activity. J Neurophysiol. 2007;98:2274–2284. doi: 10.1152/jn.00889.2006. [DOI] [PubMed] [Google Scholar]
- Colvin RA, Fontaine CP, Laskowski M, Thomas D. Zn2+ transporters and Zn2+ homeostasis in neurons. Eur J Pharmacol. 2003;479:171–185. doi: 10.1016/j.ejphar.2003.08.067. [DOI] [PubMed] [Google Scholar]
- Côté A, Chiasson M, Peralta MR, 3rd, Lafortune K, Pellegrini L, Tóth K. Cell type-specific action of seizure-induced intracellular zinc accumulation in the rat hippocampus. J Physiol. 2005;566:821–837. doi: 10.1113/jphysiol.2005.089458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez MI, Blasco-Ibáñez JM, Crespo C, Marqués-Marí AI, Martínez-Guijarro FJ. Zinc chelation during nonlesioning overexcitation results in neuronal death in the mouse hippocampus. Neuroscience. 2003;116:791–806. doi: 10.1016/s0306-4522(02)00731-5. [DOI] [PubMed] [Google Scholar]
- Domínguez MI, Blasco-Ibáñez JM, Crespo C, Nacher J, Marqués-Marí AI, Martínez-Guijarro FJ. Neural overexcitation and implication of NMDA and AMPA receptors in a mouse model of temporal lobe epilepsy implying zinc chelation. Epilepsia. 2006;47:887–899. doi: 10.1111/j.1528-1167.2006.00501.x. [DOI] [PubMed] [Google Scholar]
- Dufner-Beattie J, Langmade SJ, Wang F, Eide D, Andrews GK. Structure, function, and regulation of a subfamily of mouse zinc transporter genes. J Biol Chem. 2003;278:50142–50150. doi: 10.1074/jbc.M304163200. [DOI] [PubMed] [Google Scholar]
- Dufner-Beattie J, Huang ZL, Geiser J, Xu W, Andrews GK. Mouse ZIP1 and ZIP3 genes together are essential for adaptation to dietary zinc deficiency during pregnancy. Genesis. 2006;44:239–251. doi: 10.1002/dvg.20211. [DOI] [PubMed] [Google Scholar]
- Emmetsberger J, Mirrione MM, Zhou C, Fernandez-Monreal M, Siddiq MM, Ji K, Tsirka SE. Tissue plasminogen activator alters intracellular sequestration of zinc through interaction with the transporter ZIP4. J Neurosci. 2010;30:6538–6547. doi: 10.1523/JNEUROSCI.6250-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Giblin LJ, 3rd, Balaji RV, Rengarajan B, Masalha R, Frederickson CJ, Zeng Y, Lopez EV, Koh JY, Chorin U, Besser L, Hershfinkel M, Li Y, Thompson RB, Krezel A. Synaptic release of zinc from brain slices: factors governing release, imaging, and accurate calculation of concentration. J Neurosci Methods. 2006;154:19–29. doi: 10.1016/j.jneumeth.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Freund WD, Reddig S. AMPA/Zn(2+)-induced neurotoxicity in rat primary cortical cultures: involvement of L-type calcium channels. Brain Res. 1994;654:257–264. doi: 10.1016/0006-8993(94)90487-1. [DOI] [PubMed] [Google Scholar]
- Grooms SY, Opitz T, Bennett MV, Zukin RS. Status epilepticus decreases glutamate receptor 2 mRNA and protein expression in hippocampal pyramidal cells before neuronal death. Proc Natl Acad Sci U S A. 2000;97:3631–3636. doi: 10.1073/pnas.050586497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershfinkel M, Kandler K, Knoch ME, Dagan-Rabin M, Aras MA, Abramovitch-Dahan C, Sekler I, Aizenman E. Intracellular zinc inhibits KCC2 transporter activity. Nat Neurosci. 2009;12:725–727. doi: 10.1038/nn.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirzel K, Müller U, Latal AT, Hülsmann S, Grudzinska J, Seeliger MW, Betz H, Laube B. Hyperekplexia phenotype of glycine receptor alpha1 subunit mutant mice identifies Zn(2+) as an essential endogenous modulator of glycinergic neurotransmission. Neuron. 2006;52:679–690. doi: 10.1016/j.neuron.2006.09.035. [DOI] [PubMed] [Google Scholar]
- Kambe T, Yamaguchi-Iwai Y, Sasaki R, Nagao M. Overview of mammalian zinc transporters. Cell Mol Life Sci. 2004;61:49–68. doi: 10.1007/s00018-003-3148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- Kwak S, Weiss JH. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr Opin Neurobiol. 2006;16:281–287. doi: 10.1016/j.conb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Lavoie N, Peralta MR, 3rd, Chiasson M, Lafortune K, Pellegrini L, Seress L, Tóth K. Extracellular chelation of zinc does not affect hippocampal excitability and seizure-induced cell death in rats. J Physiol. 2007;578:275–289. doi: 10.1113/jphysiol.2006.121848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Cole TB, Palmiter RD, Koh JY. Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: evidence against synaptic vesicle origin. J Neurosci. 2000;20:RC79. doi: 10.1523/JNEUROSCI.20-11-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kim JH, Palmiter RD, Koh JY. Zinc released from metallothionein-iii may contribute to hippocampal CA1 and thalamic neuronal death following acute brain injury. Exp Neurol. 2003;184:337–347. doi: 10.1016/s0014-4886(03)00382-0. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Molnár P, Nadler JV. Synaptically-released zinc inhibits N-methyl-D-aspartate receptor activation at recurrent mossy fiber synapses. Brain Res. 2001;910:205–207. doi: 10.1016/s0006-8993(01)02720-2. [DOI] [PubMed] [Google Scholar]
- Noh KM, Yokota H, Mashiko T, Castillo PE, Zukin RS, Bennett MV. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc Natl Acad Sci U S A. 2005;102:12230–12235. doi: 10.1073/pnas.0505408102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Noebels JL. Visualization of transmitter release with zinc fluorescence detection at the mouse hippocampal mossy fibre synapse. J Physiol. 2005;566:747–758. doi: 10.1113/jphysiol.2005.089276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Canzoniero LM, Yu SP, Ying HS, Koh JY, Kerchner GA, Choi DW. Measurement of intracellular free zinc in living cortical neurons: routes of entry. J Neurosci. 1997;17:9554–9564. doi: 10.1523/JNEUROSCI.17-24-09554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci U S A. 1999;96:2414–2419. doi: 10.1073/pnas.96.5.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda A, Tamano H, Nagayoshi A, Yamada K, Oku N. Increase in hippocampal cell death after treatment with kainate in zinc deficiency. Neurochem Int. 2005;47:539–544. doi: 10.1016/j.neuint.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Traboulsie A, Chemin J, Chevalier M, Quignard JF, Nargeot J, Lory P. Subunit-specific modulation of T-type calcium channels by zinc. J Physiol. 2007;578:159–171. doi: 10.1113/jphysiol.2006.114496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villmann C, Becker CM. On the hypes and falls in neuroprotection: targeting the NMDA receptor. Neuroscientist. 2007;13:594–615. doi: 10.1177/1073858406296259. [DOI] [PubMed] [Google Scholar]
- Vogt K, Mellor J, Tong G, Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- Weiss JH, Sensi SL, Koh JY. Zn(2+): a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci. 2000;21:395–401. doi: 10.1016/s0165-6147(00)01541-8. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook GL, Mayer ML. Micromolar concentrations of Zn2+ antagonize NMDA and GABA responses of hippocampal neurons. Nature. 1987;328:640–643. doi: 10.1038/328640a0. [DOI] [PubMed] [Google Scholar]
- Yin HZ, Sensi SL, Ogoshi F, Weiss JH. Blockade of Ca2+-permeable AMPA/kainate channels decreases oxygen-glucose deprivation-induced Zn2+ accumulation and neuronal loss in hippocampal pyramidal neurons. J Neurosci. 2002;22:1273–1279. doi: 10.1523/JNEUROSCI.22-04-01273.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]