

Abstract
Foldamers present a particularly difficult challenge for accurate computational design compared to the case for conventional peptide and protein design due to the lack of a large body of structural data to allow parameterization of rotamer libraries and energies. We therefore explored the use of molecular mechanics for constructing rotamer libraries for non-natural foldamer backbones. We first evaluated the accuracy of molecular mechanics (MM) for the prediction of rotamer probability distributions in the crystal structures of proteins is explored. The van der Waals radius, dielectric constant and effective Boltzmann temperature were systematically varied to maximize agreement with experimental data. Boltzmann-weighted probabilities from these molecular mechanics energies compare well with database-derived probabilities for both an idealized α-helix (R = 0.95) as well as β-strand conformations (R = 0.92). Based on these parameters, de novo rotamer probabilities for secondary structures of peptides built from β-amino acids were determined. To limit computational complexity, it is useful to establish a residue-specific criterion for excluding rare, high-energy rotamers from the library. This is accomplished by including only those rotamers with probability greater than a given threshold (e.g. 10%) of the random value, defined as 1/n where n is the number of potential rotamers for each residue type.
Introduction
In the past decade the field of computational protein design has rapidly expanded, providing an approach to test the structural basis for function as well as a tool for designing useful molecules.1-3 In parallel, chemists have demonstrated the ability to design and synthesize foldamers, short sequence-specific oligomers of non-natural building blocks that adopt secondary and tertiary structures.4 Recent studies from the labs of Dill,5-7 Zuckermann,8-10 Seebach,11-14 Gellman,15-21 and Schepartz22-26 have demonstrated the feasibility of designing protein-like tertiary structures, which has set the stage for application of computational methods akin to those used in protein design to the problem of foldamer design.
Protein design depends strongly on rotamer libraries that define available conformations for side chains and their relative frequencies.27 Rotamers reflect the low-energy conformations of amino acid side chains and a rotamer library consists of those rotamers that are energetically reasonable for structure prediction or design. While molecular mechanics28-31 (MM) and quantum mechanics (QM) approaches have been successful at defining the geometry of amino acid rotamers, they have been less successful at capturing their relative energetics. Handel and coworkers have shown both the challenges and limitations of using physics-based force fields.32 Therefore, protein design typically uses rotamer libraries obtained from analysis of the very large database of high-resolution crystallographic structures of proteins.27,33,34
Many computational approaches to protein design35-39 and comparative modeling40-42 combine these statistically defined rotamer libraries with energy functions that are carefully parameterized to best reproduce the native rotamers of proteins of known three-dimensional structure. As the number of available structures has increased, rotamer definitions have been refined,27,34,43,44 energy equations optimized,45-48 and force-field parameters improved.47,49-51 Recent efforts have resulted in libraries and energy functions that have been able to achieve high recovery rates of χ1 (∼90%) and χ1+2 (∼80%).52 These systematic improvements have been a result of both the growing database of protein structures as well as the increased stringency for inclusion into a library. For example, recent work has shown that electron-density maps generated by X-ray crystallography experiments leave some room for interpretation.53 The Penultimate Rotamer library34 and more recently the backbone-dependent rotamer library (R. Dunbrack and M. Shapovalov, unpublished) have demonstrated substantial improvement in rotamer library construction by careful selection and refinement of the underlying database of rotamers used to generate the respective libraries. The use of such rotamer libraries in protein design has substantially decreased the possible search space to a large, yet tractable problem.
The challenge becomes substantially greater when a non-natural backbone is employed and there is no database to guide selection of low-energy rotamers as well as the avoidance of improbable, high-energy rotamers. In this paper, we therefore explore the scope and limitations of using molecular mechanics for developing rotamer libraries, initially calibrating the approach using conventional proteins (α-amino acids, αAA) and then extending this approach to develop libraries for β3 substituted β-amino acid peptides (βAA).
βAA peptides are similar to their α-amino acid counterparts, the difference being an additional methylene group in the backbone between Cα and the carboxylic acid group, as shown in Figure 1. This backbone modification has several interesting effects on βAA peptides specifically: protease resistance,54-59 an additional branch-point (C2) for R-group substitution of each residue,4 and additional degrees of backbone freedom. Each of these features makes this field of growing interest for applications in medicinal chemistry and material science.
Figure 1.
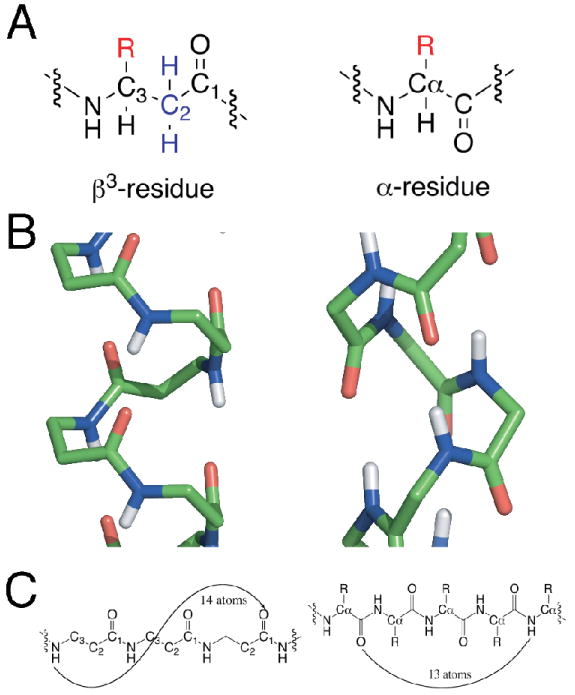
A. backbone units for a β3AA (left) and αAA (right) peptides. B. Wireframe of β314 helix (left) and α-helix (right, non-polar hydrogens omitted for clarity, atoms colored by elemental type: carbon (green), oxygen (red), nitrogen (blue), hydrogen (white). C. Hydrogen-bonding patterns for each structural motif. Note the direction of the carbonyl group in each subunit. The carbonyl points in the opposite direction in some β3AA peptide helices from that in the α-helix.
The additional degree of backbone freedom of βAA peptides contributes to their ability to adopt a family of stable helical backbone conformations, each forming a unique hydrogen-bonding pattern. The most studied backbone conformations include the 314, 312, 310/12 helices, each of which has a different number of heavy atoms enclosed within a hydrogen bonded ring.4,60 The traditional α-helix has 13 atoms encompassing four residues within a hydrogen bond, while the βAA β314 helix spans three residues and 14 atoms (Figures 1B and 1C).
The β314 helix has been extensively studied because this is a preferred conformation for water soluble peptides composed of β3 amino acids, which are easily obtained by homologation of the corresponding αAAs.61 Sequence-directed approaches have been successfully employed to design secondary and tertiary architectures.23,24,62-64 In several cases the β-peptides were designed by exchanging residues from known αAA peptides63 as well as charge patterning along the βAA helical wheel.22,25 These studies have not only demonstrated the feasibility of β-peptide foldamer design, but also the need for more computationally intensive approaches that can combine the advantages of traditional computational protein design with the structural versatility of foldamer scaffolds.
We have therefore developed a program to facilitate foldamer design that allows the input of a variety of backbones with a suite of options including the construction of secondary and tertiary structures, side-chain repacking using either Monte Carlo65 or Dead End Elimination,66 flexible and rigid-body docking, coiled-coil generation67 together with a customizable energy equation that draws upon parameters from either AMBER68 or CHARMM.69 An essential component of the repacking algorithm is an accurate rotamer library with associated energies. Here we explore the accuracy of molecular mechanics (MM) computed rotamer library probabilities in the context of αAAs and extend this approach to βAAs, and evaluate the library for repacking of a tertiary structure.
Methods
Rotamer energetics and repacking experiments were calculated using a custom C++ implementation of the CHARMM software package (TIGER). All atom, bonded and non-bonded parameters used to generate structures were taken from the CHARMM27 force field.45,69 Hydrogen atoms were placed according to atom type and bonding patterns, using bond lengths and angles specified in the CHARMM27 parameter file.
Calibration of α-amino acid rotamer calculations against experimental data
An idealized poly-glycine 20 residue α-helix as well as a three residue β-sheet scaffold were generated using bond lengths and angles from the CHARMM27 force field, followed by backbone dihedral angle modifications to arrive at the desired homogeneous secondary structure. The central amino acid of the secondary structure was mutated to each residue type in turn.
Each side chain torsional angle for 17 of the 20 natural amino acids (glycine and alanine as well as proline were excluded) was rotated in 10° steps to generate candidate rotamers. The energetics for each candidate rotamer were then evaluated. Energetic evaluation included: (1) the Lennard-Jones van der Waals potential with atomic radii scaled at 90%, 95%, or 100% with a 12 Å distance cutoff and no switching function; (2) Coulombic electrostatics with a distance-dependent dielectric constant set to either 10 or 20, also with a 12 Å distance cutoff and no switching function; and (3) dihedral angle terms. Polar hydrogen positions for serine, threonine and tyrosine were optimized by selecting the minimum energy as a function of polar hydrogen dihedral angle searched in 10° steps.
Rotamer energetics for a particular amino acid in the α-helix were then converted into their equivalent Boltzmann probabilities, using Equation 1 (see below). Individual probabilities were then summed within angular bins used to define each rotamer (e.g., 0°≤χ<120° for g+; 120°≤χ<240° for trans; 240°≤χ<360° for g-) to arrive at a final probability for a given rotamer. The temperature used for the Boltzmann conversion was optimized to obtain the highest correlation with a statistical database of rotameric probabilities for a subset of the amino acids (ILVFYSTV).
β-amino acid rotamer library generation
Various idealized β-amino acid helices of length 40 were generated using bond lengths and angles from the CHARMM27 force field, followed by backbone dihedral angle modifications to arrive at a homogeneous atomic structure. All side chains were set to β-homoglycine for each candidate scaffold. Side chains were placed at the C3 position of the central residue on each scaffold to create an L-β-amino acid.
Each side chain torsional angle for 17 of the 20 amino acids (non-rotameric glycine and alanine as well as proline were excluded) was rotated in 10° steps to generate candidate rotamers. The energetics for each candidate rotamer were evaluated using the process optimized during the α-helix calibration.
Rotamer energetics for a particular amino acid were then converted into their equivalent Boltzmann probabilities using the α-helix optimized conversion temperature of 450K, in a manner similar to the α amino acids. It is of interest to note that the minimum energy within a given rotameric state is not necessarily the center of the rotameric bin, however there was never more than a single distinct minimum within each bin (some bins did not have any minimum due to poor energetics).
This process was repeated for each of the candidate β-peptide helix scaffolds, including the use of 3-residue and 20-residue helices. The correlation coefficient between the β314 3.0 helices of these lengths is 0.857, indicating that 26.6% (1-R2) of the variance in the data is independent of the neighboring atoms.
Backbone-dependent rotamer libraries for each secondary structure type
A backbone (ϕ,ψ) dependent rotamer library was created for both α-helices and β-sheets from a set of 2028 protein chains of resolution ≤ 1.7 Å, R-factor ≤0.20, and less than 50% mutual sequence identity using the PISCES server70,71. Secondary structure of chains was determined with the program Stride72. For α-helices, only helices of length 12 or longer were used and the first four and last four residues of each helix were excluded from the data set for the rotamer library. For β-sheets, only strands of length 8 or longer were used and the first two and last two residues of each strand were excluded from the data set for the rotamer library. The resulting data sets consisted of 51,290 α-helix residues and 22,853 β-strand residues.
Rotamer frequencies were determined using kernel density estimates of the ϕ,ψ probability distribution function (pdf) for each rotamer, p(ϕ,ψ|r), and Bayes' rule to determine p(r|ϕ,ψ) on a grid every 10° from these pdfs (R. Dunbrack and M. Shapovalov, unpublished). The kernels used were von Mises functions with the bandwidth parameter κ set to 50.
β-peptide scaffold repacking
Using in-house foldamer repacking software we conducted several different experiments using the crystallographic representation (five octameric bundles, each a dimer of tetramers generated using symmetry information from the structure) of the Zwit1f bundle. For each exercise, native sidechains were removed and candidate sidechains were evaluated using the CHARMM27 molecular mechanics force-field. Candidate sidechains were drawn from the generated rotameric preferences for idealized β-314 helix presented in this work. Each candidate sidechain was evaluated using the same energetic form used in the calibration of α-amino acid sidechain energetics as described above. Experiments included the preservation of sequence identity allowing rotameric freedom, conservation of position specific charge (positive: KOR, negative: DE, neutral: ILVAFW) and finally a general sequence recovery experiment (core: ILVFA, polar exposed: DENQSTKOR, neutral exposed: ILVFYW). Symmetric constraints were applied, linking identity and rotamer selections between related chains. A Monte Carlo search algorithm was applied to the available rotameric search space and concluded after 50,000 steps.
Graphics & Regressions
The PyMol 1.1beta3 graphics package, MatLab (R2008a), Microsoft Excel 2004, Gnuplot 4.0 and ChemBioDraw Ultra 2008 were used to generate figures. Microsoft Excel 2004 was used to perform regression analysis.
Results
Prediction of αAA rotamer probability distributions
To develop reliable and accurate rotamer libraries and associated energies and probabilities for βAA libraries, we first asked how well the CHARMM2745,69 force field, as implemented in our general-purpose program for foldamer design, predicts αAA rotamer libraries. Specifically we examined two parameters that are typically varied in computational protein design. To compensate for the rigid backbone employed in protein/foldamer design, one often dampens the Lennard Jones potential69 by scaling the van der Waals (vdw) radii for the atoms. Furthermore, the effective dielectric constant in the electrostatic term is often scaled in a distance-dependent manner to simulate screening of electrostatic effects. We apply the Boltzmann equation:
| (1) |
to compute rotamer probability distributions from computed MM energies. The Boltzmann equation relates the energy (E) of a given state (i) to its probability at a given temperature (T) in the context of all n possible states. kB is the Boltzmann constant. An important parameter in this equation is the temperature, which is not necessarily a physically meaningful quantity. This parameter relates the energy difference between distinct rotamers to their probabilities in the overall population; for a given energy difference, a very low temperature would give rise to a single rotamer, while at limiting high temperature all rotamers would be equally populated. Therefore, we systematically varied this parameter to determine the temperature range that yields the best agreement with observed populations.
We computed the rotameric energies considering different vdw radii scaling factors (90%-100%) and distance-dependent electrostatic treatments (10 < ε < 20) in conjunction with torsional potentials. We then used these calculated energies to compute the Boltzmann-weighted probabilities for comparison with experimental frequency distributions. An initial analysis focused on the short, neutral αAA (ILVFYST) on an α-helical backbone with surrounding residues set to glycine. The experimental frequencies came from the most recent backbone-dependent rotamer library27 with backbone dihedrals ϕ and ψ near the standard values of -60° and -50° respectively. We observe an excellent correlation over a range of temperatures (300-500 K) with an optimum correlation coefficient R=0.946 at 450K with vdw scaling of 100% and distance-dependent dielectric constant of ε = 10, as shown in Figure 2.
Figure 2.
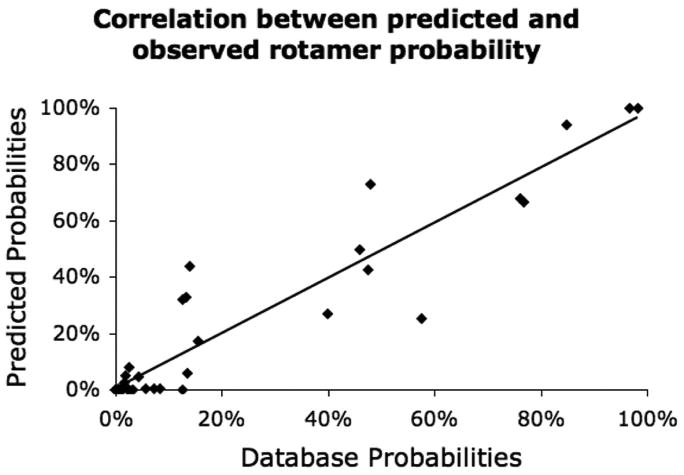
Comparison between predicted and observed rotamer probabilities of α-helical rotamers (ILVFYST), R=0.946. Optimized Boltzmann temperature of 450 K shown. Correlation of remaining α-helical rotamers (CMWDENQHKR) which include extended sidechains, R = 0.568.
The excellent correlation with experiment for the α-helix bodes well for prediction of rotamers for the helical conformations of βAA. However, we also wished to explore the ability of this approach to predict the distributions within the β-sheet conformation of conventional proteins, which has relevance to the general problem of αAA protein design. Rotameric probabilities were calculated using an idealized three residue β-strand (ϕ=-120°, ψ=110°), which provides enough side-chain/backbone interactions for all intra-strand rotameric interactions. The results using MM together with a ϕ,ψ based backbone-dependent rotamer library27 as a benchmark resulted in an R of 0.900 (VDW scaling 95%, ε = 20, T=450K). However, many residues in a ϕ,ψ defined library are in extended conformations but not necessarily in β-sheets. To overcome this limitation, we developed a backbone-dependent rotamer library based only on residues in by β-sheet strands of at least 8 consecutive residues, excluding the first two and last two residues. The ϕ,ψ-dependent rotamer library frequencies were then determined from the filtered data using kernel density estimates (R. Dunbrack and M. Shapovalov, unpublished). The R for the β-sheet motif improved to 0.916 (VDW scaling 90%, ε = 20, T= 450K, ϕ=-120°, ψ=110°).
Criteria for constructing a rotamer library for protein design
In the context of computational protein design, we considered the effect of a minimum rotamer probability inclusion cutoff of 5% on the various amino acid types. For short side chains with one degree of freedom, this results in a reasonable representation of the rotameric possibilities. For valine, for instance, the unfavorable g+ rotamer occurs at a frequency of about 5% in α-helices. However, for long side chains such as lysine, the 5% criteria excludes a large portion of the available rotamers. For example, lysine has potentially 81 rotameric states (3 states for each of four degrees of freedom); thus if each were equally populated they would occur at 1/81 or 1.2% abundance. In fact, three of these 81 rotamers occur with greater than 5% probability in the crystallographic database in the α-helix. Moreover, less than half (47.8%) of the lysine residues in the crystallographic database adopt one of these three rotameric states. Thus, a majority of observed α-helical lysine states are excluded in design projects that use a simple 5% cutoff criteria. Clearly the use of a constant probability inclusion cutoff eliminates a very large number of possible rotamers given that the random expectation for this identity would be 1.2% (1/81). We therefore adopted the ratio of the predicted probability, pi, or observed frequency, fi, of a given rotamer i and the frequency expected if rotamers were equally distributed (Ratio to the null hypothesis, RN) as our inclusion criterion:
| (2) |
| (3) |
where n is the number of rotamers available to a specific residue type, generally 3d where d is the number of degrees of freedom of the side chain. Using the RN approach, with a cutoff of 0.1 (log RN) would include 9 additional lysine rotamers (12 total, additional 34.7% of observed states, 82.5% of total), more accurately representing the diversity of this larger amino acid. In contrast, including 95% of all observed rotamers from the structural database would require the inclusion of 25 lysine rotamers in a given design project, placing undue demands on conformational search and including relatively rare (high energy) rotamers.
In constructing a rotamer library, it is particularly important to exclude highly unfavorable states. In Figure 3, we show predicted versus observed RN for residues in α helices for the residues ILFVSTD. Here we define a bad rotamer as one with an RNobs(i) < 0.1 (log value -1). Of 48 possible rotamers for ILFVSTD, 22 are “bad” rotamers based on their value of RNobs(i). All 22 bad rotamers were correctly predicted by MM equations based on their having a value of RNpred(i) < 0.1. A second consideration is to capture as many highly favorable rotamers as possible. Along these lines, again we find that the MM force field correctly predicts all 15 favorable rotamers whose values of RNobs(i) are greater than 0.5 (log value -0.30). Furthermore, there is a good linear correlation between log(RNobs(i)) and log(RNpred(i)) for this set of favorable rotamers (R = 0.67), which are most suited for protein design. The borderline cases, consisting of moderately unfavorable rotamers whose values of RNobs(i) lie between 0.1 and 0.5 (log values -1 and -0.30), are more difficult to predict (highlighted in yellow in figure 3). Using the best combination of vdw radii-scaling, dielectric constant and Boltzmann temperature, we were able to capture only two of the eleven rotamers in this regime. Thus, for the purposes of protein design, we would lose some of these moderately unfavorable rotamers based on a molecular mechanics rather than a structural bioinformatics approach. These borderline rotamers in total make up 5.2% (1868) of the total residues in the structural database, while the ‘good’ rotamers encompass 93.4% (33,443) of the residues, with the ‘bad’ rotamers covering the remaining 1.4% (505) of residues. These data are summarized for α-helices in Table 1 for different combinations of MM parameters.
Figure 3.
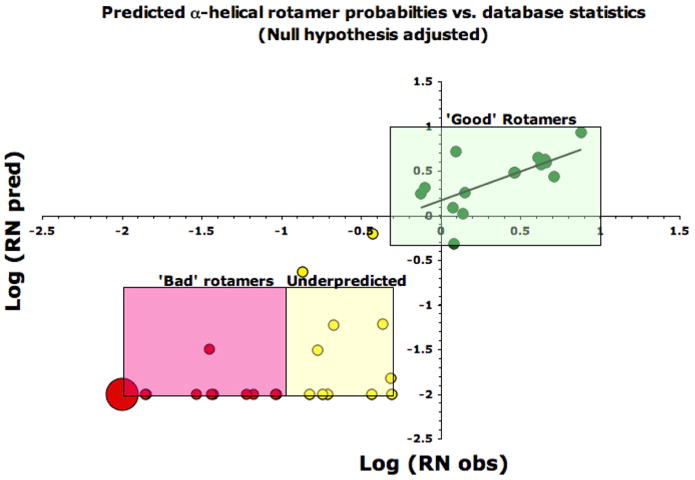
Log (Predicted probabilities / Null probability) vs. Log (Database probabilities / Null probability) for the α-helix (αAA residues VILSTFY, Boltzmann temperature 450° for predicted probabilities). Green points are ‘good’ predicted rotamers, RNobs > 0.5 (the majority of which are over-represented when compared to the null hypothesis expectations); yellow points are ‘underpredicted’ rotamers, RNobs = 0.1 - 0.5; red points are rotamers that are ranked poorly, RNobs <0.1 (the point at -2, -2 represents 11 unique rotamer evaluations and is sized accordingly). Predicted probabilities with RNobs < 0.01 are plotted at log(RNobs)= -2.
Table 1. Performance of RN (odds ratio) metric in rotamer library design for αAA residues.
| VDW=90% ε = 20 |
VDW=90% ε = 10 |
VDW=95% ε = 20 |
VDW=95% ε = 10 |
VDW=100% ε = 20 |
VDW=100% ε = 10 |
|
|---|---|---|---|---|---|---|
| False Negatives | ||||||
| α-Helix | 3 | 6 | 5 | 7 | 5 | 8 |
| β-Sheet | 2 | 3 | 4 | 4 | 5 | 5 |
| False Positives | ||||||
| α-Helix | 0 | 0 | 0 | 0 | 0 | 0 |
| β-Sheet | 5 | 5 | 1 | 1 | 1 | 1 |
VDW is the vdw radii-scaling factor, ε is the distance-dependent dielectric constant. False positives and false negatives are listed when compared to knowledge-based αAA rotamer library of 48 rotameric states. ‘Good’ (positive) and ‘bad’ (negative) rotamers have odds ratio RNobs of >0.1 and <0.01 respectively.
Construction of secondary structure dependent rotamer library for βAA
There have been several definitions of the minimum energy conformations of the β314 helix, which vary slightly in their backbone torsional angles. We examine three such sets of β314 backbone angles, one derived from a recent crystal structure,22 one the result of quantum mechanics optimization,73 and one derived from an energy-optimized backbone that maintains a precise 3.0 periodicity (Figure 4). Using these backbones we computed rotamer energies and the associated probabilities. In 10° steps, all possible side-chain χ angles for the natural residue types (proline was excluded) were evaluated on each scaffold, which resulted in a set of allowed rotameric states for each identity and backbone combination. The energetics of these states were then converted into Boltzmann weighted probabilities using the predetermined optimal conversion temperature (Equation 1) and vdw and electrostatic parameters optimized for α-helices and rank ordered to enable more effective rotamer searches in typical design programs. These files can be downloaded from http://degrado.med.upenn.edu/rotamers.
Figure 4.
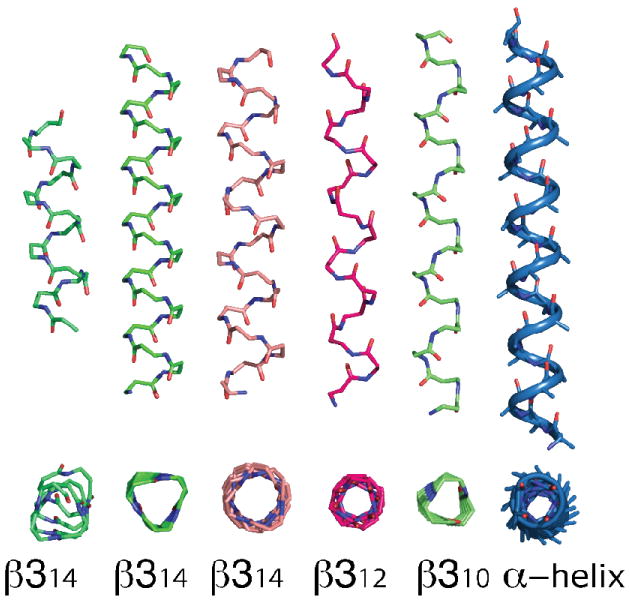
Various β-peptide helix motifs. From left to right: β314 helix from crystal structure22, energy minimized β314 3.0 helix, β314 QM minimized β314 helix73, β312 helix77, β310 helix76. Note the direction of the carbonyl groups in the β312 helix. Side-chain and hydrogen atoms omitted for clarity. α-helix shown for comparison at right.
Comparison of the isoleucine side-chain χ1 rotameric preferences across backbone scaffolds and motifs provides further insight into the differences between the secondary structures of αAA and βAA scaffolds. The α-helix and β314 helix demonstrate a strong preference for the gauche- rotamer (g- = -60°), while the β-sheet preferences are predominantly spread between the g- and trans (t = 180°) rotamers, with a small population for the g+ (+60°) rotamer. However, Ile in the β312 helix favors the g- rotamer with a 58% preference for the g+ rotamer, as shown in Figure 5.
Figure 5.
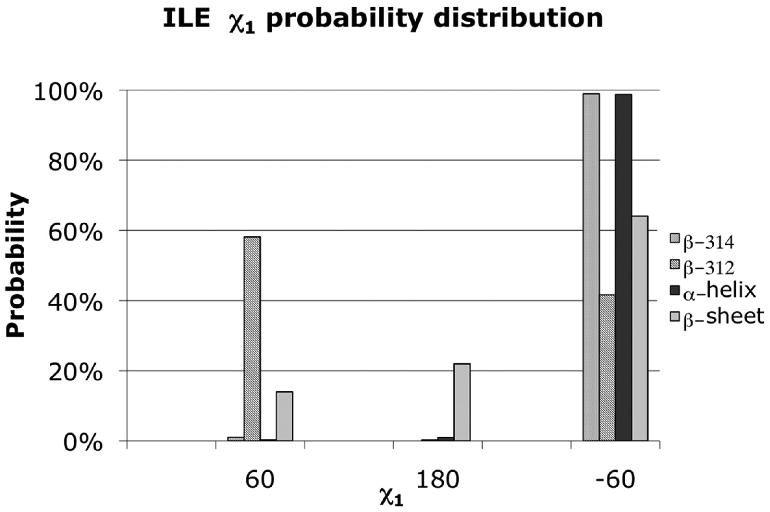
χ1 rotamer distributions for the isoleucine side chain in the context of the α-helix, β-sheet, β314 helix, and the β312 helix. α-helix and β-sheet rotameric preference derived from a binned backbone dependent rotamer library for each secondary structure.
Using rotamer probabilities calculated using a crystal structure of a protein-like β-peptide22 backbone as a reference, we find the best agreement with the minimized 3.0 helix amongst the computed log probabilities for each of the idealized candidate backbones. In each case, there is reasonable agreement with correlation coefficients (R) in the range of 0.8-0.9 (Table 2). Nevertheless this exercise demonstrates the subtle interdependence of backbone and rotamer preferences. Even greater variation in rotamer probabilities is observed if one compares different secondary structures of αAA and βAAs. Again taking the rotamer probabilities calculated on the β314 crystallographically observed helix backbone as a reference we see correlations of 0.6-0.9 when comparing rotamer probabilities of the β310, β312, the α-helix and β-sheet motifs (Table 3). These data further demonstrate the importance of computing backbone dependent rotamer libraries in the context of the correct secondary structure.
Table 2. The dependence of rotamer probabilities on the backbone conformation of the β314 helix.
| Helix | ϕ† | ψ† | θ† | β314 Crystal (R) †† | β314 Literature (R) †† | β314 3.0 Idealized (R) †† |
|---|---|---|---|---|---|---|
| β314 Crystal22 | 54.2 | -135.1 | -132.3 | 1 | ||
| β314 Literature73 | 60.0 | -134.3 | -139.9 | 0.889 | 1 | |
| β314 3.0 Idealized | 55.0 | -123.4 | -139.3 | 0.929 | 0.824 | 1 |
Backbone torsional angles in degrees.
The correlation coefficients in the three rightmost columns are based on the linear regression of the log of the calculated rotamer probabilities of residues ILVFYSTD between the helical backbones under consideration.
Table 3. Rotamer library similarity for different scaffolds.
| Helix | ϕ† | ψ† | θ† | Crystal (R) †† | Available Rotameric States | Polarity | Ref |
|---|---|---|---|---|---|---|---|
| 310 | 64 | 59 | 75 | 0.771 | 14 | N | 76 |
| 312 | -90 | 90 | -110 | 0.932 | 19 | C | 24 |
| α Helix (αAA) | -57 | -47 | - | 0.634 | 19 | C | 33 |
| β-sheet (αAA) | -119 | 113 | - | 0.667 | 18 | C |
Available rotameric states reflect the total size of the rotamer library for amino acid side chains (ILVFYSTD) after excluding rotamers with less than a 5% probability.
Backbone torsional angles in degrees.
Correlation between the log of the null hypothesis adjusted probabilities of the library based on the average β314 crystal backbone and libraries for the same amino acids derived from the respective scaffolds.22 The α-helix and β-sheet data are derived from a backbone dependent rotamer library database determined using the indicated backbone dihedrals angles.
Finally, to confirm that the rotamer probabilities were accurately predicted for the β314 helix, we compared predicted Leu and Phe energy surface with the dihedral angles of residues observed in the crystal structure. These observed data points fall in minimum energy wells that are associated with the highest probability rotameric states, as shown in Figure 6.
Figure 6.
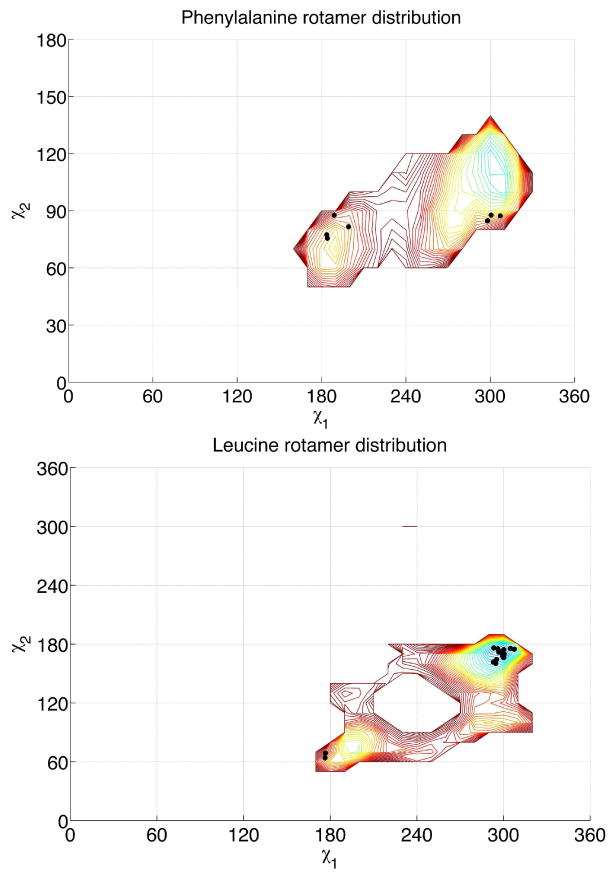
Contour plot of χ1/χ2 dependent rotamer probabilities for β3homoleucine and β3homophenylalanine in the β314 3.0 rotamer library. The negative logs of rotamer probabilities are indicated via the contour map, in 10°×10° bins. Experimental rotamers from the crystal structure22 are overlaid (black dots).
To evaluate the β-amino acid rotamer library for repacking of a complex tertiary structure, we repacked the crystallographic structure of the 8-helix bundle formed by the β-peptide Zwit1f22. The CHARMM27 force field was used to evaluate energies, and the computation conducted as described in methods. We first evaluated the recovery of amino acid rotamers while keeping the sequence held fixed; the χ1 and χ1/χ2 recoveries from this treatment were to 95% and 65% respectively. This compares quite favorably with the accuracy achieved using the most advanced repacking algorithms for natural proteins52. This is a significant accomplishment because repacking of natural proteins employs sidechain rotamer libraries defined using crystallographic structures rather than computed energetics. We next addressed the extent to which the native sequence would be recapitulated when the amino acids identities were allowed to vary.
Figure 8 compares the sequence and structures of the starting Zwit1f sequence with that of the computed sequence. Because of asymmetry in the crystallographic structure, the sidechain repacking was conducted using a dimer as the asymmetric unit. Of eight Leu residues in the starting sequence, six were recovered as leucine in the crystallographically observed χ1/χ2 rotamers. The other two residues were converted to Ala, due to small clashes with other residues when these positions were held Leu. The four Phe residues were converted to three Tyr residues, whose hydroxyl groups were on the surface of the protein. This finding is consistent with the observation that para-idodo-phenylalanine can substitute for Phe in Zwit1f. The remaining starting hPhe was converted to hLeu, which shares a large hydrophobic sidechain. The remaining solvent-exposed residues were more variable, because the positions of amino acids in salt-bridging positions can interchange with retention of energetics. Nevertheless, the retention of χ1 in the repacked structure was 75%. A further repacking in which the charge of the sidechains was retained from the original sequence, the χ1 recovery increased to 83.3% and sequence recovery was 33.3% for the entire structure, in-line with the other modern repacking algorithms74.
Figure 8.
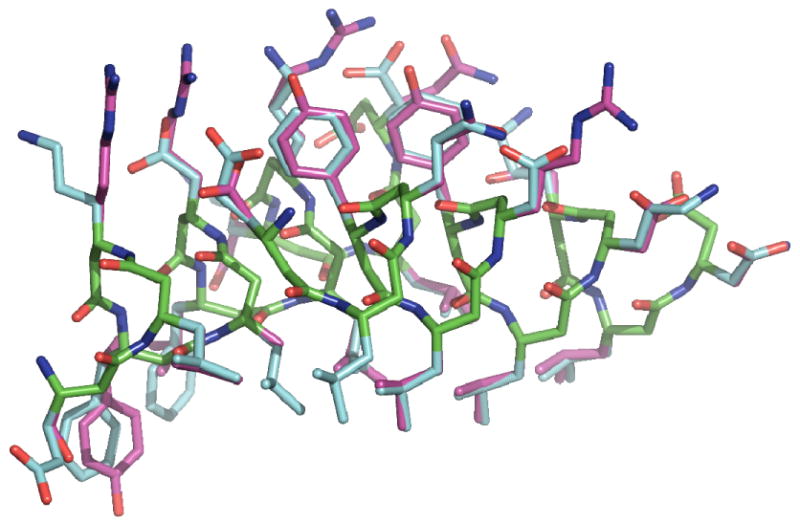
Comparison between experimental and computationally selected rotamers for the Zwit1f β-peptide scaffold using denovo β-peptide rotamer library. β314 backbone carbon atoms are shown in green, crystallographic sidechain carbon atoms shown in blue and computationally selected rotamers shown in magenta. Oxygen and nitrogen atoms are shown in red and blue respectively. Note the good identity and rotameric agreement between 6 of the 8 core β3-homoLeucines and 3 of 4 aromatic residues.
We note that the structure of zwit1f was not designed to form the observed octameric structure. Thus the resulting sequence of zwit1f is not necessarily optimal to maximize stability, and some of the changes to polar residues might in fact improve stability of the structure, although it is unlikely that the above-mentioned replacement of hleu for hala would be favorable.
Discussion
In the area of protein design, Handel and coworkers have shown both the challenges and limitations of using physics-based force fields.32 The challenge becomes substantially greater when a non-natural backbone is employed and there is no database to guide selection of low-energy rotamers as well as the avoidance of improbable high-energy rotamers. There is also the issue of setting energetic limits on the inclusion of rotamers in a library for non-natural backbones. For example, including high-energy rotamers will significantly slow convergence in any repacking algorithm, while the exclusion of too many rotamers decreases the complexity of the library. Furthermore, if the energy of a rotamer is overestimated, there is a danger of excluding this state, while if it is underestimated, a physically unreasonable structure would be obtained.
Therefore, before computing rotamer libraries for non-natural backbones, we began by asking what set of parameters in the CHARMM force-field would provide the best prediction of the relative abundance of rotamers as seen in the crystallographic structures of natural proteins. We computed the rotameric energies considering different van der Waals radii scaling factors and electrostatic treatments in conjunction with torsional interactions. The resulting energies were used to compute the Boltzmann-weighted probabilities for comparison with experimental frequency distributions. A good overall correlation was obtained, particularly in the α-helix.
Using the α-helix parameters, we developed rotameric distributions for several βAA scaffolds. In the context of β314 helices, our analysis suggests that the backbone details of the modeled β314 3.0 helix correlate most closely with experimental evidence. Computed rotameric probabilities generated from the β314 scaffold are most similar to the β312 helix followed by the β310 helix and are dramatically different from their αAA counterparts.
Using this technique we have addressed how the additional methylene in the backbone influences the rotamer distributions. Rotamer frequencies are predominantly determined by interactions of atoms separate by three and four atomic bonds75. In αAAs, the γ heavy atom of a side chain has ϕ-dependent steric interactions with the carbonyl carbon of preceding residue, placing restrictions on g+ and g- rotamers. Also, in αAAs, the γ heavy atom may have ψ-dependent unfavorable interactions with the adjacent backbone amide, affecting the g+ and t rotamers.
The β314 helix g+ rotamer has steric interactions with its own amide NH and carbonyl group which interacts with the C2 group. The g- rotamer has no unfavorable interactions with the local backbone in β314 helices and is therefore expected to be the most probable rotamer, followed by trans at moderate frequency, and g+ at very low frequency.
This analysis is in good accord with the rotamer frequencies predicted by our molecular mechanics calculations. For instance, Met has χ1 predicted frequencies of 77%, 22%, and 1% for the g-, trans, and g+ rotamers respectively. The numbers for Phe are 86%, 13%, and <1% and for Cys they are 70%, 30%, and 0%. As with αAAs, Val and Ile have only one viable rotamer since there are two γ heavy atoms to interact with the local backbone. This analysis predicts that Val (CG1 at χ1 and CG2 at χ1+120°) should exist only in the trans rotamer in β314 helices, which is what is predicted by the MM calculations. For Ile (CG1 at χ1 and CG2 at χ1-120°), only the g- rotamer is expected and predicted to be observable. To confirm our hypothesis, probabilities were computed using a three-residue β314 scaffold. As expected, the trans and g- rotamers are both accessible to valine (56.6% vs. 43.3%), while the g+ rotamer is highly disfavored (0.09%).
There are additional interactions with the helical structure, outside of the local ϕ and ψ dependent distances. Good examples of this are the aromatic ring side chains of β3homophenylalanine, β3homotyrosine and β3homotryptophan. These rings demonstrate a preference for a single rotamer (χ1,χ2 =g-, g+), which affords them significant side-chain-backbone interaction against the β314 helix. If they were to occupy the same rotameric state in the α-helix, they would extend away from the backbone and forego favorable intra-helical interactions. However, these extended rotamers could prove fruitful in higher-order structures where side chains participate in side-chain-side-chain interactions.
In addition, since the backbone carbonyl points in the opposite direction in β314 helices compared to α-helices, they have a different geometry for the hydrogen bonding side chains Ser and Thr. This causes a significant increase in the g+ rotamer of Ser relative to other side chain types, and a change in the rotameric preferences for threonine relative to Ile. Thr has a predicted frequency of 78% and 22% for the g+ and g- rotamers respectively, due to the favorable hydrogen bond for the g+ rotamer (Figure 7). By contrast, in the α-helix, Thr and Ser make an inter-residue hydrogen bond by assuming the -60° (g-) rotamer.
Figure 7.
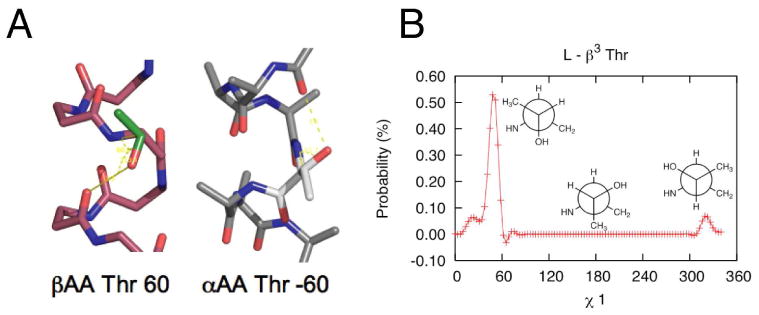
A. β314 helix and α-helix each with the threonine side chain. The preferred rotamer for each scaffold is shown, g+ for β314 helix and g- for the α-helix. B. Rotameric probabilities for β3 substituted threonine. (Newman projections shown in inset)
While local atomic interactions are useful for development of rotamer libraries, critical evaluation of more complex sequence-dependent and tertiary structure-dependent effects require a full forcefield, as exemplified in the repacking of a protein-like β-peptide crystal structure22. The results presented here show the first attempt to overcome this challenging problem through the rigorous generation of a rotameric library, for use in the computational design of β-peptides.
Supplementary Material
Acknowledgments
The authors would like to thank the anonymous reviewers for their useful input. We thank NIH (GM54616 to W.F.D, P20 GM76222 and R01 GM84453 to R.L.D.) and DOE (DE-FG-02-04ER46156-A006 to W.F.D) for financial support.
Footnotes
Supporting Information: Computer readable versions of the β314 and β312 rotamer libraries for C3 and C2, along with file format information can be found at http://degrado.med.upenn.edu/rotamers. Complete references can be found in the supporting information.
References
- 1.Jiang L, Althoff EA, Clemente FR, Doyle L, Röthlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, Hilvert D, Houk KN, Stoddard BL, Baker D. Science. 2008;319:1387–91. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yin H, Slusky JS, Berger BW, Walters RS, Vilaire G, Litvinov RI, Lear JD, Caputo GA, Bennett JS, Degrado WF. Science. 2007;315:1817–22. doi: 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
- 3.Slovic AM, Kono H, Lear JD, Saven JG, Degrado WF. Proc Natl Acad Sci USA. 2004;101:1828–33. doi: 10.1073/pnas.0306417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodman CM, Choi S, Shandler S, Degrado WF. Nat Chem Biol. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee BC, Zuckermann RN, Dill KA. J Am Chem Soc. 2005;127:10999–1009. doi: 10.1021/ja0514904. [DOI] [PubMed] [Google Scholar]
- 6.Armand P, Kirshenbaum K, Falicov A, Dunbrack RL, Jr, Dill KA, Zuckermann RN, Cohen FE. Fold Des. 1997;2:369–75. doi: 10.1016/S1359-0278(97)00051-5. [DOI] [PubMed] [Google Scholar]
- 7.Kirshenbaum K, Zuckermann RN, Dill KA. Curr Opin Struct Biol. 1999;9:530–5. doi: 10.1016/S0959-440X(99)80075-X. [DOI] [PubMed] [Google Scholar]
- 8.Kirshenbaum K, Barron AE, Goldsmith RA, Armand P, Bradley EK, Truong KT, Dill KA, Cohen FE, Zuckermann RN. Proc Natl Acad Sci U S A. 1998;95:4303–8. doi: 10.1073/pnas.95.8.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horn T, Lee BC, Dill KA, Zuckermann RN. Bioconjugate Chemistry. 2004;15:428–435. doi: 10.1021/bc0341831. [DOI] [PubMed] [Google Scholar]
- 10.Lee BC, Chu TK, Dill KA, Zuckermann RN. Journal of the American Chemical Society. 2008;130:8847–8855. doi: 10.1021/ja802125x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seebach D, Beck AK, Bierbaum DJ. Chem Biodivers. 2006;1:1111–239. doi: 10.1002/cbdv.200490087. [DOI] [PubMed] [Google Scholar]
- 12.Rueping M, Mahajan YR, Jaun B, Seebach D. Chemistry (Weinheim an der Bergstrasse, Germany) 2004;10:1607–15. doi: 10.1002/chem.200305571. [DOI] [PubMed] [Google Scholar]
- 13.Gademann K, Häne A, Rueping M, Jaun B, Seebach D. Angew Chem Int Ed Engl. 2003;42:1534–7. doi: 10.1002/anie.200250290. [DOI] [PubMed] [Google Scholar]
- 14.Rueping M, Schreiber JV, Lelais G, Jaun B, Seebach D. Helv Chim Acta. 2002;85:2577–2593. [Google Scholar]
- 15.Vaz E, Pomerantz WC, Geyer M, Gellman SH, Brunsveld L. Chembiochem. 2008;9:2254–2259. doi: 10.1002/cbic.200800355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horne WS, Price JL, Gellman SH. Proc Natl Acad Sci USA. 2008;105:9151–6. doi: 10.1073/pnas.0801135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadowsky JD, Murray JK, Tomita Y, Gellman SH. Chembiochem. 2007 doi: 10.1002/cbic.200600546. [DOI] [PubMed] [Google Scholar]
- 18.Lee MR, Raguse TL, Schinnerl M, Pomerantz WC, Wang X, Wipf P, Gellman SH. Org Lett. 2007;9:1801–4. doi: 10.1021/ol070511r. [DOI] [PubMed] [Google Scholar]
- 19.Choi SH, Guzei IA, Gellman SH. J Am Chem Soc. 2007;129:13780–1. doi: 10.1021/ja0753344. [DOI] [PubMed] [Google Scholar]
- 20.Porter EA, Wang X, Schmitt MA, Gellman SH. Org Lett. 2002;4:3317–9. doi: 10.1021/ol0266370. [DOI] [PubMed] [Google Scholar]
- 21.Cheng RP, Gellman SH, Degrado WF. Proc Natl Acad Sci USA. 2001;101:3219–32. doi: 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]
- 22.Daniels DS, Petersson EJ, Qiu JX, Schepartz A. J Am Chem Soc. 2007;129:1532–3. doi: 10.1021/ja068678n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hart SA, Bahadoor AB, Matthews EE, Qiu XJ, Schepartz A. J Am Chem Soc. 2003;125:4022–3. doi: 10.1021/ja029868a. [DOI] [PubMed] [Google Scholar]
- 24.Kritzer JA, Tirado-Rives J, Hart SA, Lear JD, Jorgensen WL, Schepartz A. J Am Chem Soc. 2005;127:167–78. doi: 10.1021/ja0459375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petersson EJ, Schepartz A. J Am Chem Soc. 2008;130:821–3. doi: 10.1021/ja077245x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiu JX, Petersson EJ, Matthews EE, Schepartz A. J Am Chem Soc. 2006;128:11338–9. doi: 10.1021/ja063164+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunbrack RL. Curr Opin Struct Biol. 2002;12:431–40. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- 28.Brooks BR, Bruccoleri RE, Olafson DJ, States DJ, Swaminathan S, Karplus M. Journal of Computational Chemistry. 1983;4:187–217. [Google Scholar]
- 29.Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, Profeta S, Weiner P. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 30.Jorgensen WL, Tiradorives J. Journal of the American Chemical Society. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 31.Renfrew PD, Butterfoss GL, Kuhlman B. Proteins. 2008;71:1637–46. doi: 10.1002/prot.21845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pokala N, Handel TM. J Mol Biol. 2005;347:203–27. doi: 10.1016/j.jmb.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 33.Dunbrack RL, Cohen FE. Protein Sci. 1997;6:1661–81. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lovell SC, Word JM, Richardson JS, Richardson DC. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- 35.Baker D, Degrado WF. Curr Opin Struct Biol. 1999;9:485–6. doi: 10.1016/s0959-440x(99)80068-2. [DOI] [PubMed] [Google Scholar]
- 36.Kuhlman B, Baker D. Proc Natl Acad Sci USA. 2000;97:10383–8. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dahiyat BI, Mayo SL. Protein Sci. 1996;5:895–903. doi: 10.1002/pro.5560050511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voigt CA, Gordon DB, Mayo SL. J Mol Biol. 2000;299:789–803. doi: 10.1006/jmbi.2000.3758. [DOI] [PubMed] [Google Scholar]
- 39.Gordon DB, Hom GK, Mayo SL, Pierce NA. Journal of computational chemistry. 2003;24:232–43. doi: 10.1002/jcc.10121. [DOI] [PubMed] [Google Scholar]
- 40.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Science. 2003;302:1364–8. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 41.Levitt M. J Mol Biol. 1992;226:507–33. doi: 10.1016/0022-2836(92)90964-l. [DOI] [PubMed] [Google Scholar]
- 42.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 43.Ponder JW, Richards FM. J Mol Biol. 1987;193:775–91. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 44.Jiang L, Kuhlman B, Kortemme T, Baker D. Proteins. 2005;58:893–904. doi: 10.1002/prot.20347. [DOI] [PubMed] [Google Scholar]
- 45.Mackerell AD. Journal of computational chemistry. 2004;25:1584–604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 46.Kortemme T, Baker D. Proc Natl Acad Sci USA. 2002;99:14116–21. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lazaridis T, Karplus M. Proteins. 1999;35:133–52. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 48.Lazaridis T, Karplus M. Curr Opin Struct Biol. 2000;10:139–45. doi: 10.1016/s0959-440x(00)00063-4. [DOI] [PubMed] [Google Scholar]
- 49.Senes A, Chadi DC, Law PB, Walters RF, Nanda V, Degrado WF. J Mol Biol. 2006;366:436–48. doi: 10.1016/j.jmb.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 50.Summa CM, Levitt M, Degrado WF. J Mol Biol. 2005;352:986–1001. doi: 10.1016/j.jmb.2005.07.054. [DOI] [PubMed] [Google Scholar]
- 51.Zhou H, Zhou Y. Protein Sci. 2002;11:2714–26. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krivov GG, Shapovalov MV, Dunbrack RL., Jr Proteins. 2009 doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shapovalov MV, Dunbrack RL. Proteins. 2007;66:279–303. doi: 10.1002/prot.21150. [DOI] [PubMed] [Google Scholar]
- 54.Gopi HN, Ravindra G, Pal PP, Pattanaik P, Balaram H, Balaram P. FEBS Lett. 2003;535:175–8. doi: 10.1016/s0014-5793(02)03885-1. [DOI] [PubMed] [Google Scholar]
- 55.Saikumari YK, Ravindra G, Balaram P. Protein Pept Lett. 2006;13:471–6. doi: 10.2174/092986606776819501. [DOI] [PubMed] [Google Scholar]
- 56.Hook DF, Bindschädler P, Mahajan YR, Sebesta R, Kast P, Seebach D. Chem Biodivers. 2006;2:591–632. doi: 10.1002/cbdv.200590039. [DOI] [PubMed] [Google Scholar]
- 57.Hook DF, Gessier F, Noti C, Kast P, Seebach D. Chembiochem. 2004;5:691–706. doi: 10.1002/cbic.200300827. [DOI] [PubMed] [Google Scholar]
- 58.Wiegand H, Wirz B, Schweitzer A, Camenisch GP, Rodriguez Perez MI, Gross G, Woessner R, Voges R, Arvidsson PI, Frackenpohl J, Seebach D. Biopharmaceutics & drug disposition. 2002;23:251–62. doi: 10.1002/bdd.334. [DOI] [PubMed] [Google Scholar]
- 59.Frackenpohl J, Arvidsson PI, Schreiber JV, Seebach D. Chembiochem. 2002;2:445–55. doi: 10.1002/1439-7633(20010601)2:6<445::aid-cbic445>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 60.Chatterjee S, Roy RS, Balaram P. J Nat Prod. 2007 [Google Scholar]
- 61.Seebach D, Ciceri PE, Overhand M, Jaun B, Rigo D, Oberer L, Hommel U, Amstutz R, Widmer H. Helvetica Chimica Acta. 1996;79:2043–2066. [Google Scholar]
- 62.Guarracino DA, Chiang HR, Banks TN, Lear JD, Hodsdon ME, Schepartz A. Org Lett. 2006;8:807–10. doi: 10.1021/ol0527532. [DOI] [PubMed] [Google Scholar]
- 63.Kritzer JA, Lear JD, Hodsdon ME, Schepartz A. J Am Chem Soc. 2004;126:9468–9. doi: 10.1021/ja031625a. [DOI] [PubMed] [Google Scholar]
- 64.Petersson EJ, Craig CJ, Daniels DS, Qiu JX, Schepartz A. J Am Chem Soc. 2007;129:5344–5. doi: 10.1021/ja070567g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Metropolis N, Ulam S. Journal Of The American Statistical Association. 1949;44:335–341. doi: 10.1080/01621459.1949.10483310. [DOI] [PubMed] [Google Scholar]
- 66.Pierce NA, Spriet JA, Desmet J, Mayo SL. Journal of computational chemistry. 2000;21:999–1009. [Google Scholar]
- 67.Dieckmann GR, Degrado WF. Curr Opin Struct Biol. 1997;7:486–94. doi: 10.1016/s0959-440x(97)80111-x. [DOI] [PubMed] [Google Scholar]
- 68.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. J Comput Chem. 2005;26:1668–88. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.MacKerell AD, Jr, et al. J Phys Chem. 1998;B102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 70.Wang G, Dunbrack RL., Jr Bioinformatics. 2003;19:1589–91. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- 71.Wang G, Dunbrack RL., Jr Nucleic Acids Res. 2005;33:W94–8. doi: 10.1093/nar/gki402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frishman D, Argos P. Proteins. 1995;23:566–79. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 73.Bode KA, Applequist J. Macromolecules. 1997;30:2144–2150. [Google Scholar]
- 74.Barth P, Schonbrun J, Baker D. Proc Natl Acad Sci USA. 2007 [Google Scholar]
- 75.Dunbrack RL, Karplus M. Nature Structural Biology. 1994 doi: 10.1038/nsb0594-334. [DOI] [PubMed] [Google Scholar]
- 76.Gunther R, Hofmann HJ, Kuczera K. The Journal of Physical Chemistry B. 2001;105:5559–5567. [Google Scholar]
- 77.Applequist J, Bode KA, Appella DH, Christianson LA, Gellman SH. J Am Chem Soc. 1998;120:4891–4892. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.