

Abstract
We report a full account of our work towards the development of an eight-step synthesis of anti-influenza drug (−)-oseltamivir (Tamiflu®) from commercially available starting material. The final synthetic route proceeds with an overall yield of 30 %. Key transformations include a novel palladium-catalyzed asymmetric allylic alkylation reaction (Pd-AAA) as well as a rhodium-catalyzed chemo-, regio-, and stereoselective aziridination reaction.
Keywords: Tamiflu, palladium catalysis, deracemization, rhodium catalysis, aziridination
Introduction
Tamiflu® (oseltamivir phosphate, 1), developed by Gilead Sciences, is an orally active neuraminidase inhibitor, which had been widely used for the treatment of H5N1 influenza as well as a recent outbreak of H1N1 flu.1 In order to be effective in treating infected patients, tamiflu must be administered to influenza patients no later than 36–48 h after the manifestation of the symptoms. With a daily dose of 150 mg per patient, sufficient stockpiles of tamiflu are necessary in times of a flu pandemic, as it is both a therapy and a preventive agent.
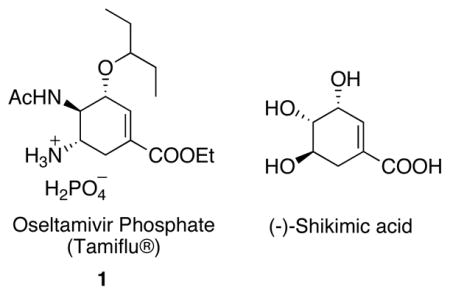
Such high public demands raise the challenge of providing large quantities of tamiflu to meet the worldwide need.2 Currently, tamiflu is marketed by Roche and is prepared via a semisynthetic approach starting from (−)-shikimic acid, which is still a limited resource given the massive demand.3 Therefore, the development of alternative synthetic approaches that start from simple materials has drawn extensive attention from the chemical community.4 Our group, among many others, have contributed to the development of alternative chemical synthetic routes and completed an eight-step azide-free synthesis of (−)-oseltamivir in 2008, which represents the shortest synthesis to date.4i This article elaborates in detail our development of the aforementioned synthetic route.
Results and Discussion
1. Retrosynthetic Analysis
In our first-generation retrosynthetic analysis, the alkyl chain of (−)-oseltamivir 2 would be installed by etherification of the allylic alchohol 3, which could come from isomerization of epoxide 4. The expoxide would in turn result from an allyl amine 5. To install the second amino group, aziridination was proposed starting from alkene intermediate 7. Finally, a novel Pd-AAA reaction is envisioned, where a nitrogen-centered nucleophile is to be used to directly open racemic cis-lactone 8 and to set the requisite stereochemistry for the entire synthesis. (Scheme 1)
Scheme 1.

Retrosynthetic analysis. Pg = protecting group.
2. Pd-AAA Opening of Lactone
Commercially available lactone 8 was chosen as the starting material because of its six-membered carbon backbone, the cis-disubstituted stereochemistry, and its potential to undergo a deracemization.5 Scheme 2 illustrates how a Pd-AAA deracemization process differs from the normal nucleophilic opening of lactone 8: Instead of a normal nucleophilic attack at the carbonyl carbon C4 to generate ring-opened product 9, a Pd-AAA process results in nucleophilic attack at the ether carbon C1, generating an alternative ring-opened product 10, forming a new bond on the six-membered ring.
Scheme 2.

Different product outcome between normal nucleophilic opening of lactone and Pd-AAA deracemizing lactone.
When a chiral ligand is used, this Pd-AAA opening of lactone process also generates enantiomeric excess. Thus, lactone 8 is not only opened by the incoming nucleophile, but also deracemized. The deracemization mechanism is shown in Scheme 3. A chiral palladium complex reacts with a racemic mixture of 8 to form a pseudo-meso π-allyl intermediate with the attached carboxylate as a leaving group. Chiral ligand biased nucleophilic attack yields an enantiomerically enriched product 10.
Scheme 3.

Mechanism of Pd-AAA deracemizing lactone.
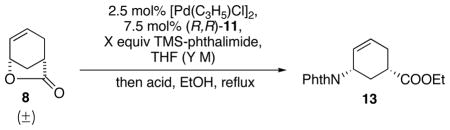
Our group had demonstrated that stabilized carbon-nucleophiles such as nitromethane and malonate can deracemize the lactone with high enantioselectivity (Table 1, entry 1 and 2).6,7 Using similar strategies, several stabilized nitrogen nucleophiles that were successfully used in previous Pd-AAA reactions were examined to deracemize lactone 8 and install the first amino group at C1. Literature known compound HN(Boc)28,9 was examined as the initial nucleophile. Using standard Pd-AAA conditions, no reaction was observed (Table 1, entry 3). We hypothesized that the HN(Boc)2 was not sufficiently nucleophilic, therefore, a variety of bases (Table 1, entry 4, 5 and 6) were tested, however, still no reaction was observed. Then more reactive Pd-source, [Pd(C3H5)Cl]2, was examined, both with and without base, showing no improved reactivities (Table 1, entry 7 and 8). HN(Cbz)2 has also shown promising results in related Pd-AAA reactions.10,11 However, as shown in Table 1, no reaction was observed in the Pd-AAA opening lactone reaction at various temperatures (Table 1, entry 9 10, and 11). Another known Pd-AAA nucleophile, NaN(CHO)212, also failed to proceed with or without phase-transfer reagent THAB (Table 1, entry 12 and 13). Phthalimide, either in salt form13 or neutral form14, has shown to function as good nucleophiles in AAA reactions. First, sodium phthalimide was tested but showed zero reactivity in Pd-AAA reaction with 8. Considering the charge repulsion between the negatively-charged carboxylate leaving group and the incoming sodium phthalimide may have prevented the addition (Table 1, entry 14),15 neutral phthalimide was tested as the nucleophile with base as an additive to increase the nucleophilicity. However, no reaction occurred with either buffered system (CsOAc/HOAc) or with the addition of excess base (Table 1, entry 15 and 16).
Table 1.
Opening of lactone with imide nucleophiles.
 | |||
|---|---|---|---|
| Entry | Nuc | Conditions | Results[a] |
| 1 | CH3NO2[6] | 2 mol% Pd2(dba)3•CHCl3, 6 mol% (S,S)-11, (C4H9)4NCl, BSA, DCM, rt | 74% yield, 99%ee |
| 2 | malonate[7] | 2.5 mol% [Pd(C3H5)Cl]2, 7.5 mol% (S,S)-11, 1 equiv NaH, DCM, 1eq THAB[b], rt | 57%, 85%ee |
| 3 | HN(Boc)2[8,9] | 2.5 mol% Pd2(dba)3•CHCl3, 7.5 mol% (S,S)-11, DCM, rt-40 °C | NR |
| 4 | HN(Boc)2 | 2.5 mol% Pd2(dba)3•CHCl3, 7.5 mol% (S,S)-11, 1.1 equiv Cs2CO3, DCM, rt-40 °C | NR |
| 5 | HN(Boc)2 | 2.5 mol% Pd2(dba)3•CHCl3, 7.5 mol% (S,S)-11, 1 equiv NaH, DCM, rt-40 °C | NR |
| 6 | HN(Boc)2 | 2.5 mol% Pd2(dba)3•CHCl3, 7.5 mol% (S,S)-11, 1 equiv BSA, DCM, rt-40 °C | NR |
| 7 | HN(Boc)2 | 2.5 mol% [Pd(C3H5)Cl]2, 7.5 mol% (S,S)-11, 1.1 equiv Cs2CO3, DCM, rt-40 °C | NR |
| 8 | HN(Boc)2 | 2.5 mol% [Pd(C3H5)Cl]2, 7.5 mol% (S,S)-11, DCM, rt-40 °C | NR |
| 9 | HN(Cbz)2[10,11] | 2.5 mol% [Pd(C3H5)Cl]2, 7.5 mol% (S,S)-11, 1.1eq Cs2CO3, DCM, rt-40°C | NR |
| 10 | HN(Cbz)2 | 2.5 mol% [Pd(C3H5)Cl]2, 7.5 mol% (S,S)-11, DCM, rt-40°C | NR |
| 11 | HN(Cbz)2 | 2.5 mol% [Pd(C3H5)Cl]2, 7.5 mol% (S,S)-11, 1.2equiv Cs2CO3, THF, 50°C | NR |
| 12 | NaN(CHO)2[12] | 2 mol% [Pd(C3H5)Cl]2, 6 mol% (S,S)-11, CH3CN, rt-50 °C | NR |
| 13 | NaN(CHO)2 | 2 mol% [Pd(C3H5)Cl]2, 6 mol% (S,S)-11, CH3CN, 1equiv THAB[b], rt-60 °C | NR |
| 14 | NaPhth[13][c] | 2.5 mol% [Pd(C3H5)Cl]2, 7.5 mol% (S,S)-11, THAB, THF, rt | NR |
| 15 | HPhth[14] | 2.5 mol% [Pd(C3H5)Cl]2, 7.5 mol% (S,S)-11, 20 mol% CsOAc, 10 mol% THF, rt-60 °C | NR |
| 16 | HPhth | 2 mol% [Pd(C3H5)Cl]2, 6 mol% (S,S)-11, 3 eq Cs2CO3, THF, 50 °C | NR |
NR = no reaction.
THAB = tetrahexylammonium bromide.
NaPhth = sodium phthalimide.
As nitrogen-centered nucleophiles were not effective for the desired Pd-AAA reaction for lactone 8, a subsequent strategy involving ring-opening of the lactone prior to the Pd-AAA was envisioned. Lactone 8 was transformed in two steps into the ring-opened 15. Interestingly, in contrast to the reactions with lactone 8, the Pd-AAA reaction with 15 proceeded excellently with phthalimide as the nucleophile in the presence of Cs2CO3, affording 13 in high yield and enantiomeric excess (Scheme 4).
Scheme 4.

Pd-AAA with phthalimide.
A rationale for the different reactivities between 8 and 15 is provided in Scheme 4. Upon oxidative insertion of the Pd(0) catalyst into substrate 15, an intermediate involving a pseudo-meso Pd(II) π-allyl complex bearing an attached carboxylate ester is formed (16). The chiral ligand biases the nucleophilic attack by the deprotonated phthalimide and leads to enantiomerically enriched 13. However, when using lactone 8 as the substrate, it generates an intermediate 12 with an attached carboxylate anion. Presumably, the approach of an anionic phthalimide towards the Pd π-allyl complex is disfavored by repulsion between the negative charges on the nitrogen atom and the carboxylate group. In addition, it is also possible that the intramolecular reaction with the carboxylate is favored over the intermolecular reaction with the phthalimide, therefore the reverse reaction of 12 back to 8 is faster than the formation of 13. We hypothesized that, to overcome the repulsion and the intramolecular trapping of 12, the carboxylate anion of 12 must be trapped prior to the attack of the phthalimide without diminishing the nucleophilicity of the phthalimide.
Based upon the above analysis, we proposed using a trimethylsilyl (TMS) group, whose oxophilicity would result in a selective trapping of the carboxylate oxygen over the nitrogen (Scheme 5). Considering the simple addition of external silylating reagent did not facilitate the reaction (Table 1, entry 6), we considered bringing in the TMS group attached to the nitrogen nucleophile, wherein silyl transfer should capture the carboxylate and simultaneously reveal the imide nucleophile (17). Therefore, we chose commercially available TMS-phthalimide. Utilizing another advantage of the TMS group, that is, its susceptibility to acid hydrolysis, esterification could be carried out in the same pot, forming the carboxylic ethyl ester.
Scheme 5.

Opening of lactone with TMS-phthalimide.
To our delight and in stark contrast to phthalimide itself, with [Pd(C3H5)Cl]2 and ligand (R,R)-11 as the catalyst, and TMS-phthalimide as the nucleophile, the racemic lactone 8 was opened and desymmetrized, and the resulted TMS-carboxylate was in situ converted to the ethyl ester affording the desired compound 13. The initial conditions afforded 13 in 24% yield and 96% ee (Scheme 6). This approach installed both the amino group and the ester functionality in one step and established the desired stereochemistry for the entire synthesis. (R,R) Ligand was randomly chosen at this point and was later confirmed to lead to the desired absolute stereochemistry.
Scheme 6.

Initial results of the opening of lactone with TMS-phthalimide.
Further optimization of this reaction is shown in Table 2. Our goal was to improve the yield without using large excess of reagents. Therefore, higher reaction concentration, temperature, and various esterification conditions were examined (Table 2, entry 1–3). The enantioselectivity was optimal at 0.2 M. Unfortunately, the yield was still unacceptable. TMS-phthalimide was stored and handled in the glove box. Despite this precaution and the use of rigorous drying techniques, the TMS-phthalimide was found to undergo competitive hydrolysis to the unreactive phthalimide during the AAA reaction. This undesired side reaction explained the low conversion. To rectify this observation, we first decided to add 1 equiv external silylating reagent BSA to maintain the effective concentration of TMS-phthalimide in the reaction. Satisfyingly, such method led to full conversion in the Pd-AAA step, however, the following esterification became problematic with BSA present (Table 2, entry 4). Finally, we decided to use excess amount of TMS-phthalimide. As little as 1.5 equiv of the nucleophile was sufficient enough to lead to full conversion to 13 with an isolated yield of 80% (Table 5, entry 5 and 6). The yield improved further when scaled up (Table 5, entry 7).
Table 2.
Optmization of opening of lactone with TMS-phthalimide.
 | ||||
|---|---|---|---|---|
| Entry | X | Y | Acid | Results |
| 1 | 1.1 | 0.1 | conc. H2SO4, rt | 24%, 96% ee |
| 2 | 1 | 0.2 | conc H2SO4, 40 °C | 32%, 97% ee |
| 3 | 1 | 0.3 | 10 mol% TsOH•H2O, 40 °C | 47%, 94% ee |
| 4 | 1 | 0.2 | 1 equiv BSA, 10 mol% TsOH•H2O, 40 °C | Full conv. in Pd-AAA; low conv. in hydrolysis step |
| 5 | 2 | 0.2 | 10 mol% TsOH•H2O, 40 °C | 80%, 97% ee (0.3 mmmol) |
| 6 | 1.5 | 0.2 | 10 mol% TsOH•H2O, 40 °C | 80%, 98% ee (3 mmol) |
| 7 | 1.5 | 0.2 | 10 mol% TsOH•H2O, 40 °C | 84%, 98% ee (20 mmol) |
Table 5.
Attempted 1,6-conjugate addition on model compound.[a]
 | ||
|---|---|---|
| Entry | Conditions | Results |
| 1 | AcNHOAc, Cs2CO3, LA[b], CH3CN, rt-80 °C | NR[c] |
| 2 | nBuLi, AcNHOAc, THF, −40 °C-rt, | NR |
| 3 | (AcNOAc)2CuLi-LiCN, THF, -78-0 °C | NR |
| 4 | AcNHOAc, NaH, DMF, rt-90 °C | NR |
| 5 | BocNHOP(O)(OEt)2, NaH, DMF, rt −70 °C | NR |
Pg = protecting group.
LA= Pd(CH3CN)2Cl2, Sc(OTf)3, Yb(OTf)3, In(OTf)3, or Cu(OTf)3.
NR = no reaction.
In summary, the first amino group was successfully installed via a novel Pd-AAA deracemizing lactone reaction with a unique N-centered nucleophile in high yield and ee, and in the same pot the desired ethyl ester was formed. This reaction set up the absolute stereochemistry for the entire synthesis.
3. Installation of the Second Amino Group
With 13 in hand, we turned to install the second amino group onto the ring. Before following our synthetic plan of utilizing aziridination reaction to form aziridine 6, we made an attempt of accessing allylic amine 5 directly from 13. Initially, ene reaction was examined. It is known that an ene type reaction between an alkene and a nitroso compound affords an allylic amine.16 It is proposed that the reaction occurs via a six-membered transition state (Scheme 7). Nitroso compound 17 is highly reactive and thus cannot be isolated. Therefore, it is generated in situ and subsequently trapped by the olefin species via an ene reaction.17 However, under literature conditions the ene reaction with 13 failed to proceed, even when heated to 80 °C (Scheme 7). Starting material 13 was recovered in both cases. We hypothesized that the alkene of 13 is not electron-rich enough to react with 17.
Scheme 7.

Attempted second amination via ene reaction.
Alternatively, the second amino group could be installed via a reaction between olefin 13 and a highly reactive AcNH+ equivalent.18 A straightforward way to generate AcNH+ equivalence is reacting N-haloacetamide such as 19 with a silver salt, such as AgBF4. A non-nucleophilic base, 2,6-di-tert-butylpyridine, was added to deprotonate the allylic proton of intermediate 21 thus leading to the formation of allylic amine 20. Unfortunately, although the AgBr precipitation was observed under the reaction conditions, none of the desired amination product was detected (Scheme 8).
Scheme 8.

Attempted amination with acyl cation equivalent.
Acknowledging the challenge of installing the second amino group on an electron deficient double bond via ene or acyl cation strategy, we decided to investigate aziridination reactions as originally planned, as they are more developed amination reactions for electron deficient double bonds.19 Gratifyingly, under standard aziridination conditions, the desired aziridine 22 was formed in 69–74% yield as a single diastereoisomer (Scheme 9). One major drawback of the reaction was the requirement of large excesses of PhI=NTs and high catalyst loading. Previously, our group had reported that Cu(IPr)Cl is an effective aziridination catalyst.20 However, in this case, it led to no reaction for substrate 13.
Scheme 9.

Copper-catalyzed aziridination.
In summary, the second amine was installed via a copper-catalyzed diastereoselective aziridination in good yield. The next step in the total synthesis of oseltamivir was regioselective ring opening of the aziridine 22.
4. Further Conversions
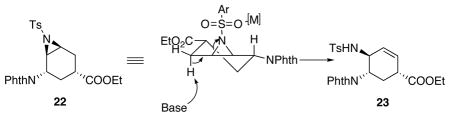
In order to obtain the desired allylic amine 23 from aziridine 22, an elimination reaction was proposed (Table 3). In the literature, allyl amines can be accessed from aziridines utilizing either R2NLi or RLi to promote the desired eliminative rearrangement.21,22 However, in our case, it would lead to the undesired regioisomer due to chelating effects of such bases. Nonetheless, when we did perform the reaction with LDA, it simply led to decomposition even at −78 °C (Table 3, entry 1). Additionally, nucleophilic bases would attack the ester functionality, which should also be avoided. Therefore, our initial studies focused on non-nucleophilic, non-coordinating bases and E2 type elimination conditions to promote the formation of 23. When 1 equiv of potassium tert-butoxide (KOtBu) was used, no desired product was observed (Table 3, entry 2). Adding excess amount of potassium tert-butoxide resulted in messy results, where attack of the ester group by the base was observed (Table 3, entry 3). Superbase LIDAKOR21,23 was used with the intention to minimize the nucleophilic attack at the ester. However, either no reaction was observed in hexanes or decomposition occurred in THF (Table 3, entry 4 and 5). Changing to organic base DBU gave no reaction (Table 3, entry 6). Next, instead of only using base to promote the elimination, Lewis acid–non-nucleophilic base combinations were used to generate a “push-pull” effect. However, still no reaction was observed (Table 3, entry 7 and 8). In order to force the reaction under the “push-pull” conditions to occur, chlorobenzene was used as the solvent in order to increase the substrate solubility as well as the reaction temperature (Table 3, entry 9). Oxophilic lanthanide holmium triflate (Ho(OTf)3) was examined as a Lewis acid to activate the sulfonyl group on the aziridine. However, no reaction occurred even at 100 °C. As lanthanides did not assist the aziridine opening, main group Lewis acids were studied. Gratifyingly, Al(OTf)3 gave nearly quantative yield of aziridine opening product 24, which could potentially lead to allylic amine 23 via a dehydration reaction (Table 3, entry 10). Delightfully, by switching to less Lewis acidic indium triflate, at an elevated temperature, elimination product 23 was observed for the first time, although irreproducible, in 23% yield (Table 3, entry 11). The desired product 23 coeluted with another product, which is believed to be 25. In addition to 23 and 25, the reaction also afforded 30% yield of 24. We hypothesized that water was acting as the nucleophile for the opening of aziridine 22 to form 24, in which case, addition of molecular sieves might prevent this undesired reaction. However, when 3Å MS was added, no reaction was observed after 24 h (Table 3, entry 12).
Table 3.
Attempted elimination conditions.
 | ||
|---|---|---|
| Entry | Conditions | Notes |
| 1 | 2 equiv. LDA, THF, −78 °C | Decomposition to baseline by TLC |
| 2 | 1 equiv. KOtBu, THF, 0 –40 °C | No desired product |
| 3 | 5 equiv. KOtBu, THF, rt | Messy, t-BuO− attacked ester |
| 4 | 1 eq LIDAKOR[a], hexanes, 0 °C-rt | NR (substrate not dissolving) |
| 5 | 1 eq LIDAKOR, THF, −78 °C | Decomposition to baseline by TLC |
| 6 | 1.5 eq DBU, toluene, reflux | NR |
| 7 | 50 mol% In(OTf)3, 2,6-di-tert-butylpyridine, THF, 50 °C | NR |
| 8 | 20 mol% In(OTf)3, proton-sponge, THF, reflux | No desired reaction. In(OTf)3 reacted with proton-sponge to form insoluble solids in the reaction. |
| 9 | Ho(OTf)3, 2,6-di-tert-butylpyridine, chlorobenzen, 100 °C | NR, recovered starting material |
| 10 | Al(OTf)3, 2,6-di-tert-butylpyridine, DCM, 0 °C-rt | 100% NMR yield[c] of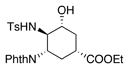 24 |
| 11 | In(OTf)3, 2,6-di-tert-butylpyridine, chlorobenzene, 100 °C | NMR yields: 23% 23, 30% 24,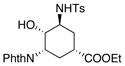 25 30% |
| 12 | In(OTf)3, 2,6-di-tert-butylpyridine, chlorobenzene, 3Å MS, 100 °C | NR, recovered starting material |
LIDAKOR=LDA + tBuOK.
NR = no reaction
NMR yields were determined by using 1,3,5-trimethoxybenzene as an internal standard.
Taking the good result from Table 3, entry 10, we tried to convert 24 into 23 (Scheme 10). Various one-step dehydration conditions were investigated, however, no desired product was observed. Therefore, a step-wise procedure was pursued. Interestingly, after transforming 24 to its corresponding triflate, DBU induced elimination afforded aziridine 22 instead of the desired allylic amine 23, presumably due to adjacent group participation.
Scheme 10.

Attempted dehydration of alcohol 24.
At the same time we were studying the one-step E2 rearrangement, we also started investigating a two-step sequence to access 23 as a backup plan (Scheme 11). Because of the observed adjacent group participation indicated in Scheme 10, we proposed to open the aziridine utilizing a nucleophile that would not participate in the intramolecular SN2 reactions. Therefore, PhSe− was chosen to generate intermediate 26, followed by oxidative thermal elimination to afford the allylic amine 23. One major challenge of this strategy was the regioselectivity of the aziridine-opening step. Electronically, the equatorial attack to afford desired product 26 is favored (Scheme 11, path a; similar result has been observed in Table 3, entry 10). However, stereoelectronically, the axial attack of the selenide to afford the undesired regioisomer 27 is favored (Scheme 11, path b), leading to allylic amine 28. A mixture of regioisomers may also be generated due to the competition between the two pathways (as already shown in Table 3, entry 11). With this in mind, we started to look into the reactivity and the selectivity of the aziridine opening reaction.
Scheme 11.

Planned two-step sequence to form allylic amine 23.
As shown in Scheme 12, the aziridine opening reaction was carried out in ethanol, however, only 10% conversion was observed; this was presumably due to the low solubility of 22 in ethanol. The conversion increased to 62% when THF was added as a co-solvent to dissolve the starting material. When TFA was added to activate the Ts-aziridine, full conversion was achieved with a 90% isolated yield for the opening of aziridine. Unfortunately, the subsequent elimination did not occur under the standard oxidative conditions. Later it was discovered, the reaction yielded 27 rather than the desired 26, which explained the difficulty of elimination due to lack of aligned proton. This intriguing result indicates that the axial attack (Scheme 11, path b) predominates under those reaction conditions, suggesting that the torsional strain in the transition state is dictating the regioselectivity when a nucleophile is used, leading to the formation of undesired 27 (Scheme 11 and 12).
Scheme 12.

Opening of aziridine 22 with selenide.
5. Revised Synthetic Plan
In observation of the difficulties in the direct E2 type elimination from 22, we decided to revise our synthetic plan. As shown in Table 3, Al(OTf)3 and In(OTf)3 assisted the ring-opening of aziridine 22 with small nucleophiles such as water to afford the desired regioisomer 24. We decided to take advantage of this result. With a careful choice of the nucleophile employed to open 22 and a slight alteration of our synthetic plan, the target molecule 2 may be accessed with equal efficiency as the first generation synthetic plan. As shown in Scheme 13, oxidative opening of aziridine 22 with DMSO may be followed by further oxidation to the enone 30. Subsequent selective reduction would afford allylic alcohol 31. Finally, an etherification reaction would complete the synthesis of oseltamivir.
Scheme 13.

Revised Synthetic Plan.
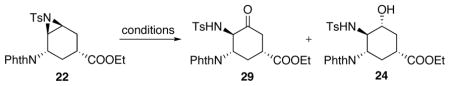
Oxidative opening of aziridine 22 was examined and the results are summarized in Table 4. At 80 °C with In(OTf)3 and DMSO, no reaction occurred (Table 4, entry 1). Increasing the reaction temperature to 200 °C and applying a microwave reactor resulted in aromatization of the starting material (Table 4, entry 2). Further screening showed that at 120 °C the desired ketone 29 could be obtained in moderate yields together with a significant amount of alcohol 24 (Table 4, entry 3). Water was considered to be the source of the formation of 24. Therefore, several methods were applied in order to minimize water’s presence in the reaction. Utilization of ultra dry In(OTf)3 as well as freshly distilled DMSO led to no improvement in the yield of 29 (Table 4, entry 4). Addition of desiccant Na2SO4 into the reaction increased the yield of 29 to 58% (Table 4, entry 5). Finally, while keeping the catalyst concentration unchanged, increasing the substrate concentration, and therefore lowering the water concentration in the reaction resulted in 54% yield of 29 (Table 4, entry 6).
Table 4.
Oxidative opening of aziridine.
 | ||
|---|---|---|
| Entry | Conditions | Results |
| 1 | 50 mol% In(OTf)3, 0.1 M DMSO, 80 °C | No reaction |
| 2 | 50 mol% In(OTf)3, 0.1 M DMSO, MW, 200 °C, 30 min | aromatization |
| 3 | 50 mol% In(OTf)3 (wet), 0.1 M DMSO, 120 °C | 46% 29, 50% 24 |
| 4 | 50 mol% In(OTf)3 (dry), 0.1 M DMSO (distilled), 120 °C | 43% 29, 42% 24 |
| 5 | 50 mol% In(OTf)3 (dry), 0.1 M DMSO, 1 equiv Na2SO4, 120 °C | 58% 29, 34% 24 |
| 6 | 5 mol% In(OTf)3 (dry), 1M in DMSO, 120 °C | 54% 29, 22% 24 |
Having ketone 29 in hand, enone 30 was successfully formed by IBX oxidation (Scheme 14).24 Fortunately, by doubling the equivalence of IBX, alcohol 24 could be directly oxidized to enone 30, albeit in a modest yield.
Scheme 14.

Further oxidation to enone.
Since the ring opening of aziridine 22 and subsequent oxidation both took place in DMSO, we investigated a one-pot procedure for the two steps (Scheme 15). By adding all reagents at once, enone 30 was formed in a modest 30% yield and was accompanied by 49% yield of the unoxidized alcohol 24. When IBX was added after the opening of aziridine but still in the same pot, the yield of 30 can be improved to 52%.
Scheme 15.

One-step procedure for transforming aziridine to enone.
Moving forward in the synthesis, enone 30 was reduced to allylic alcohol 31 as a single stereoisomer in 76% yield (Scheme 16). The relative stereochemistry was determined by comparison of the NMR spectra with the other isomer 35 obtained later in the synthesis.
Scheme 16.
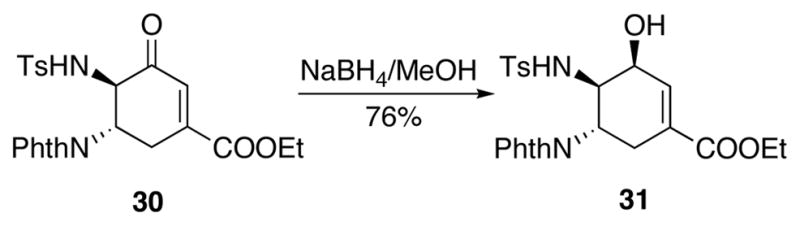
Reduction of enone 30 to allylic alcohol 31.
Further transformation of 31 to 2 showed interesting results (Scheme 17): The planned mesylation of the allylic alcohol 31 in order to install the alkyl chain via an SN2 displacement led to the formation of aziridine 34 rather than the mesylate, despite the cis-configuration of the sulfonamide and hydroxy groups.
Scheme 17.

Attempted installation of alkyl fragment.
At this point, we decided to take advantage of the formation of 34 and install the 3-pentoxy side chain via a Lewis acid catalyzed opening of vinyl aziridine reaction with 3-pentanol as the nucleophile. Such method should lead to 2 in the same number of steps as the planned mesylation-displacement protocol. Encouragingly, in the presence of BF3.Et2O, reaction between 34 and 3-pentanol gave a 26% yield of the desired product 32, along with a substantial amount of hydrated product 35 (Scheme 18). Attempted reductive etherification of 35 to form 32 gave only 9% yield.25
Scheme 18.

Opening of vinyl aziridine with 3-pentanol.
In addition to the low yield in the alkylation step, later it was found the removal of the tosyl group on 32 was rather difficult. Both direct deprotection of tosyl26 and the acylation27 of tosylamine to facilitate the tosyl removal failed. In addition to the deprotection problem, this synthetic route suffered several low yielding steps after the Pd-AAA step (Scheme 19). Therefore, we decided to investigate an alternative synthetic plan to address these problems.
Scheme 19.

Overview of the early synthetic route.
6. Final Synthetic Plan
As shown in Scheme 19, from the aziridine 22 to the vinyl aziridine 34, many oxidation stage changes were made only to install one double bond as a net result. It was against our intial goal of developing an efficient synthesis. Therefore, a major revision of the previous synthetic route is needed to access the vinyl aziridine 34 from the amino ester intermediate 13 more efficiently.
To avoid the multiple oxidation stage changes involved in installing the C-C double bond in 34, we proposed to first take advantage of the existing double bond in 13 to introduce a second double bond in conjugation (38), and then install the aziridine by a selective aziridination reaction. In the mean time, we sought alternative protecting groups, in the hope of improving the yields of the aziridination and opening of the aziridine, as well as easy deprotection. Accordingly, a final revision of our synthetic plan is shown in Scheme 20.
Scheme 20.

Final synthetic plan. Pg = protecting group.
First, we attempted to oxidatively form 38 directly from 13. Similar strategy to form α,β-unsaturated system from carbonyl compound has been reported for ketones and was also demonstrated in our synthetic study in Scheme 14.24 However, under similar or more rigorous conditions, the formation of 38 was not observed (Scheme 21).
Scheme 21.

Attempted oxidative installation of conjugation.
Hence, a stepwise strategy was undertaken. Starting with 22, α–selenation followed by oxidative thermal elimination should afford diene 38. However, the α–selenation only afforded about 10% NMR yield of 37 (Scheme 22).
Scheme 22.

α–Selenation.
Alternatively, a highly reactive sulfenylating reagent phenyl benzenethiosulfonate (PhSSO2Ph) discovered by our group has been successfully utilized in α-sulfenylation of ketones and esters.28 Compared with PhSeCl, the reactions with it are cleaner, less odoriferous and much less toxic. In this case, it led to satisfying sulfenylation of 13 (Scheme 23). KHMDS was found to be the optimal base after careful screening. Sulfenylation of 13 with PhSSO2Ph and KHMDS yielded an approximately 1:1 diastereomeric mixture of β-ketosulfide 39 in 94% yield (Scheme 23). The sulfide was oxidized to the sulfoxide and a subsequent thermal elimination occurred upon refluxing in toluene. The elimination afforded diene 3829 and 40 in 5.8:1 ratio. Lower temperature gave higher ratio of 38 with a lower conversion. Further investigation revealed that one diastereomer of 39 spontaneously eliminated after oxidation to give a 11:1 regioselectivity favoring 38 (Scheme 24). Unfortunately, the other diastereomer of 39, upon oxidation, was stable up to room temperature. When heated to 60 °C, a 2.7:1 ratio of 38 : 40 was obtained. After optimization, DBU was found effective in increasing the regioselectivity in the elimination step (Scheme 25). After chromatography, 38 and 40 was obtained as a 10:1 regioisomeric mixture in 85% yield (Scheme 25). This mixture was carried on to the next step.
Scheme 23.

Sulfenylation-elimination.
Scheme 24.

Difference in elimination of the two α–thioester isomers.
Scheme 25.

Optimized elimiation conditions.
With diene 38 in hand, we would like to install the second amino group via a selective aziridination reaction. Considering the difficulty of removing the tosyl group in the previous synthetic route, this time we wanted to carefully choose a protecting group on the nitrogen.
Ideally, we wanted to bring in the acyl-protected amine directly as it is present in the final product. But considering the difficulties of performing a metal catalyzed aziridination of acyl nitrene, a 1,6-conjugate addition-elimination process was proposed instead to install an acyl aziridine on the double bond distal to the ester.30,31 Unfortunately, utilizing O-acetyl acetamide as nucleophile, and copper thiophenecarboxylate (CuTC) as the catalyst, no reaction occurred with reaction temperatures up to 40 °C (Scheme 26).
Scheme 26.

Planned 1,6-conjugate addition.
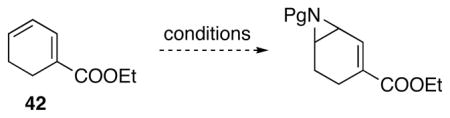
In order to develop conditions for the conjugate addition, a model compound 42 was studied under a variety of Lewis Acid catalyzed 1,4-conjugate addition reaction conditions (Table 5, entry 1). No reaction was observed under all conditions. Since Lewis acidic conjugate addition didn’t work, we turned our attention to basic conditions. Commonly used conjugate addition reagents lithium anion and cuprates gave no reaction with 42 (Table 5, entry 2 and 3). Using strongly basic sodium hydride along with with heating still led to no reaction (Table 5, entry 4). Lastly, suspecting the α-protons of the acyl group may be deprotonated under basic conditions, we changed the acyl amine to a Boc protected amine, and switched the acetate leaving group to a more active phosphate leaving group. However, such changes did not give improved results (Table 5, entry 5).
Delightfully, diphenylsulfilimine, a good nucleophile for 1,4-conjugate addition,32 reacted with model compound 42 via 1,6-conjugate addition to yield the desired aziridine product 43 as a single regioisomer (Scheme 27). Unfortunately, the same conditions on real system 38 led to exclusive addition to the carbonyl group of the phthalimide functionality.
Scheme 27.

1,6-Conjugate addition on model compound. TMG = 1,1,3,3-tetramethylguanidine
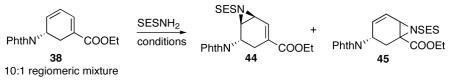
Considering the difficulties in conjugate 1,6-addition, a selective aziridination was proposed using copper as catalyst.33 First, we tried to find a good protecting group on the aziridine. 4-Nitrobenzenesulfonyl group is known for its ease of removal. However, it led to decomposition in the aziridination step. A more stabilized, yet relatively easy-to-remove protecting group, benzenesulfonyl group, was then quickly examined. Unfortunately, standard benzenesulfonyl group removal condition led to decomposition. Therefore, finding a sulfonyl group which deprotected under mild conditions was necessary for the synthesis. 2-(Trimethylsilyl)ethanesulfonyl (SES) group was known for its ease of removal by fluoride sources and thus was chosen to be the protecting group on the aziridine. A variety of conditions and catalysts were examined for the aziridination of 38 with SESNH2 (Table 6).
Table 6.
Selected conditions for the aziridination of diene 38.
 | |||
|---|---|---|---|
| Entry | Catalyst | Conditions | Result[b] |
| 1 | 10 mol% Cu(CH3CN)4PF6 | PhI=O; 3 A MS; CH3CN; rt | 84%; 44 : 45 = 1.7 : 1 |
| 2 | 10 mol% Cu(IPr)Cl[c] + AgBF4 | PhI=O; 3 A MS; CH3CN; 0 °C | (80%); 44 : 45 = 1.6 : 1 |
| 3 | 10 mol% Ag2(tBu3tPy)2(NO3)2[d] | PhI=O; 3 A MS; CH3CN; 0 °C-rt | (< 48%);[e] 44 only |
| 4 | 10 mol% Au(tBu3tPy)OTf[d] | PhI(OAc)2; 4 A MS; CH3CN; 80-50 °C | (< 53%);[e] 44 only |
| 5 | 2 mol% Rh2(CF3CONH)4 | PhI(OAc)2; MgO; C6H5Cl; 0 °C-rt | (18%); 44 only |
| 6 | 5 mol% Rh2(OCOCPh3)4 | PhI(OAc)2; MgO; C6H5Cl; 0 °C-rt | (35%); 44 only |
| 7 | 5 mol% Rh2(OCOCMe3)4 | PhI(OAc)2; MgO; C6H5Cl; 0 °C-rt | (56%); 44 only |
| 8 | 5 mol% Rh2(esp)2 | PhI(OAc)2; MgO; C6H5Cl; 0 °C-rt | (71%); 44 only |
| 9 | 2 mol% Rh2(esp)2 | PhI(OAc)2; MgO; C6H5Cl; 0 °C-rt | (72%); 44 only |
| 10 | 2 mol% Rh2(esp)2 | PhI(OPiv)2; MgO; C6H5Cl; 0 °C-rt | 86% (91%); 44 only |
| 11 | 2 mol% Rh2(esp)2 | PhI(OPiv)2; MgO; isopropyl acetate; 0 °C-rt | (91%);[f] 44 only |
The stereochemistry of 45 is undetermined. Compound 44 and 45 are inseparable on column.
Yields in parentheses were determined by 1H NMR with 1,3,5-trimethoxybezene as an internal standard.
IPr = 1,3-bis(2,6-di-i-propylphenyl)imidazol-2-ylidene.
tBu3tPy = 4,4′,4″-tri-tert-butyl-2,2′;6′,2″-terpyridine.
Conversion was accompanied by decomposition, resulting in irreproducible results.
Dry isopropyl acetate (IPAC), a non-halogenated solvent, gave the same result as dry chlorobenzene.
An unexpected difficulty arose in the regio- and stereoselective aziridination of 38. Extensive conditions were screened; selected examples are shown in Table 6. Copper catalysts, which were commonly used in aziridination reactions, showed virtually no selectivity between the α,β- and the γ,δ-double bonds of 38 and formed an inseparable mixture of 44 and 45. This lack of selectivity was seen throughout extensive screening of ligands (Selected results: Table 6, entry 1 and 2). Additionally, changing copper for larger metals such as Ag or Au, resulted in low reactivity though excellent regioselectivity of the desired γ,δ-aziridine 44 was obtatined (Selected results: Table 6, entry 3 and 4).34 Guthikonda and Du Bois reported a rhodium-catalyzed intermolecular aziridination reaction, highlighting in situ generation of a nitrene species. In these reactions the olefin is the limiting reagent.35 Screening of Rh-catalysts revealed Du Bois’s C–H insertion catalyst, Rh2(esp)2 (bis[rhodium(α,α,α′,α′-tetramethyl-1,3-benzenedipropionic acid)]),36 gave higher conversions than Rh2(O2CCPh3)4, Rh2(O2CCMe3)4 or Rh2(CF3CONH)4 (Table 6, entry 5–8). The catalyst loading could be lowered to 2 mol% without decay in yield (Table 6, entry 9). Moreover, changing the oxidant from PhI(OAc)2 to PhI(O2CCMe3)2 afforded even better conversion to 44 (Table 11, entry 10).37 The environmentally benign solvent isopropyl acetate could be used, replacing chlorobenzene, and leading to similar yield and selectivity, however, with a slightly inconvenient workup. Most satisfyingly, γ,δ-aziridine 44 was observed as the exclusive product in all Rh-catalyzed cases. Finally, in chlorobenzene, using PhI(O2CCMe3)2 and SESNH2, 2 mol% Rh2(esp)2, and MgO as the base and dessicant, an 86% isolated yield of a single isomer 44 was obtained from the diene mixture 38 (Scheme 28). Replacing chlorobenzene with isopropyl acetate gave the same result (entry 11).
Scheme 28.

Optimized aziridination condition.
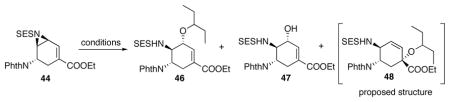
With 44 in hand, similar opening of aziridine as shown in Scheme 18 was performed with good regio- and diastereoselectivity (Table 7). Similar to tosyl aziridine, the alcohol 47 was a major byproduct in this ring-opening reaction (Table 7, entry 1). At lower temperatures, 46 was formed more slowly than 47, resulting in less 46 formation (Table 7, entry 2). Running the reaction at higher temperature accelerated the formation of 46 relative to 47 (Table 7, entry 3). Weaker Lewis acids, such as In(OTf)3 or Cu(OTf)2 also slowed down the formation of 46, and thus increased the yield of the undesired 47 (Table 7, entry 4 and 5). Entry 1–5 in Table 6 were performed when 44 still contained 45 as an impurity and yields were calculated based on the assay of 44. Later, with pure 44 in hand, a third product with an unconfirmed structure 48 was isolated (Table 7, entry 6–12). Therefore 46, 47 and 48 nicely account for all the mass balance of this reaction. Lowering the substrate concentration in order to increase the concentration of the nucleophile 3-pentanol did not improve the yield (Table 7, entry 6 and 7). Adding molecular sieves to minimize water’s presence also did not reduce the formation of 47 (Table 7, entry 8). Preformation of the 3-pentyl borate and subjection of it to the reaction conditions led to no conversion (Table 7, entry 9). However, thoroughly drying 44 slightly improved the yield of 46 (Table 7, entry 10). Adding 1 equiv N,O-bis[trimethylsilyl]acetamide (BSA) to chemically remove any residual or newly generated water afforded a lower yield of 46 (Table 7, entry 11). Eventually, after premixing BSA with dry 44 followed by full removal of all volatiles, the aziridine-opening gave a 65% isolated yield of 46 (Table 7, entry 12).
Table 7.
Opening of vinyl aziridine.
 | ||
|---|---|---|
| Entry | Conditions | Yields |
| 1 | 1.5 equiv BF3•Et2O, 0.15 M in 3-pentanol, rt | 31% 46, 54% 47 |
| 2 | 1.2 equiv BF3•Et2O, 0.15 M in 3-pentanol, −20 °C | 20% 46, 41% 47 |
| 3 | 1.5 equiv BF3•Et2O, 0.15 M in 3-pentanol, 75 °C | 60% 46, 22% 47 |
| 4 | 1.5 equiv In(OTf)3, 0.15 M in 3-pentanol, 75 °C | 17% 46, 71% 47 |
| 5 | 10 mol% Cu(OTf)2, 0.1 M in 3-pentanol, 0-rt-100 °C | 87% 47 |
| 6 | 1.5 equiv BF3•Et2O, 0.15 M in 3-pentanol, 75 °C | 54% 46, 36% 47, 12% 48 |
| 7 | 1.5 equiv BF3•Et2O, 0.05 M in 3-pentanol, 70 °C | 54% 46, 34% 47, 12% 48 |
| 8 | 1.5 equiv BF3•Et2O, 0.05 M in 3-pentanol, 3Å MS, 70 °C | 50% 46, 34% 47, 16% 48 |
| 9 | 1.5 equiv B(OCHEt2)3, 0.15 M in 3-pentanol, 75 °C | No reaction |
| 10 | Azeotropically dired with toluene, then 1.5 equiv BF3•Et2O, 0.15 M in 3-pentanol, 75 °C | 60% 46, 26% 47, 13% 48 |
| 11 | 1.5 equiv BF3•Et2O, 1 equiv BSA in rxn, 0.15 M in 3-pentanol, rt | 25% 46, 57% 47, 7% 48, 11% starting material |
| 12 | Pre-mixed with BSA, volatile fully removed; then 1.5 equiv BF3•Et2O, 0.15 M in 3-pentanol, 75 °C | 65% 46, 20% 47, 15% 48 |
As the formation of 47 could not be avoided, conditions to convert 47 to 46 were studied. Unfortunately, various attempts to directly convert 47 to 46 failed. However, under Mitsunobu conditions, we were able to convert compound 47 back to 44 which would allow the recycling of 44 (Scheme 29).
Scheme 29.

Attempted alkylation of allylic alcohol 47.
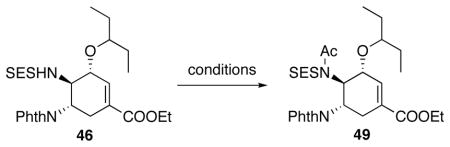
The next step in the synthesis of 2 was to deprotect the SES group. However, direct removal of the SES group was unsuccessful under a variety of literature conditions. Therefore, we decided to acylate the SES-amino group prior to the deprotection. Optimization of the acylation step is summarized in Table 8. Under standard acylation conditions, no reaction occurred at room temperature even with large excesses of reagents (Table 8, entry 1 and 2). At elevated temperature as high as 120 °C with acetic anhydride as solvent, conversion was observed (Table 8, entry 3). Addition of pyridine improved the yield from to 64% after 3 hrs and 74% after overnight heating (Table 8, entry 4 and 5). Eventually, a microwave reactor was used and 84% yield was obtained at 150 °C after 1 hr (Table 8, entry 6).
Table 8.
Acylation of 46.
 | ||
|---|---|---|
| Entry | Conditions | results |
| 1 | 10 equiv. AcCl, 10 equiv. DMAP, DMF, rt | sm[a]+ elimination |
| 2 | 10 equiv Ac2O, 10 equiv DMAP, Py, rt | sm |
| 3 | 3 equiv. DMAP, 0.1 M in Ac2O, 120 °C, 4 hr | 54% product+sm |
| 4 | 1 equiv. DMAP, 10 equiv. Py, 0.1 M in Ac2O, 120 °C, 3 h | 64% product + 36% sm |
| 5 | 1 equiv. DMAP, 10 equiv. Py, 0.1 M in Ac2O, 120 °C, overnight | 74% product |
| 6 | 2 equiv. DMAP, 20 equiv. Py, 0.1 M in Ac2O, MW 150 °C, 1 h | 84% product |
sm = starting material.
The optimized acylation conditions and the final steps in the synthesis are summarized in Scheme 30. After acylation, the SES group was easily removed by TBAF. Finally, removal of the phthaloyl with hydrazine finished the synthesis of 2.
Scheme 30.

Completion of the synthesis.
Conclusion
In summary, we have accomplished an azide-free eight-step synthesis of (−)-oseltamivir from commercially available materials with 30% overall yield. Key transformations involve a novel Pd-catalyzed AAA deracemization of a lactone with a nitrogen nucleophile and an unprecedented Rh-catalyzed chemo-, regio- and stereoselective direct aziridination reaction on an electron deficient conjugated diene system. The final synthetic route for (−)-oseltamivir is shown in Scheme 31.4i
Scheme 31.

Final synthetic scheme.
Experimental Section
Selected experimental procedures for the two key steps, Pd-AAA deracemization of lactone and Rh-catalyzed selective azirdination, are shown below. Full experimental details for all new compounds and (−)-oseltamivir are given in the Supporting Information.
Pd-AAA of deracemization Lactone 8: (1S,5S)-ethyl 5-(1,3-dioxoisoindolin-2-yl)cyclohex-3-enecarboxylate (13)
All glassware was flame-dried under high vacuum. All chemicals were flushed with N2 by evacuating under high-vacuum then refilling with N2 for 5 cycles prior to the reaction. THF was degassed using a freeze-thaw technique under N2 for 5 cycles. Lactone 8 was redistilled before use and TMS-phthalimide was stored and weighed in a glove box. To a 100 ml pear shaped flask was loaded [Pd(C3H5)Cl]2 (183 mg, 0.50 mmol), (R,R)-11 (1.04 g, 1.50 mmol) and a stir bar. Degassed dry THF (70 ml) was injected into the flask and the mixture was stirred for 20 min at 40 °C to give a yellow solution. At 40 °C under N2, lactone 5 (2.48 g, 20 mmol) in 5 ml THF was cannulated into the solution. THF (2 × 5 ml) was used to rinse the flask containing 8 and was also cannulated into the yellow solution. After 5 min, TMS-phthalimide (6.58 g, 30 mmol) in 15 ml THF was cannulated into the yellow solution under N2. The reaction was then stirred at 40 °C under N2 for 8 h at which time the TLC showed the disappearance of 8. THF was removed in vacuo. To the residue was added TsOH•H2O (380 mg, 2.0 mmol) and 67 ml absolute ethanol. The mixture was heated at reflux for 10 h monitored by TLC. Ethanol was removed in vacuo, and the resulting solid mixture was mixed evenly with 33 g silica gel by dissolving in DCM and concentrating in vacuo. The silica gel containing crude product was loaded on top of a silica gel column. Eluting with 15:1 petroleum ether : ethyl acetate yielded 5.0 g 13 as a white solid. Rf = 0.76 (1:1 petroleum ether : ethyl acetate). HPLC OD 220 nm 90:10 heptane : 2-propanol 0.8 ml/min, tmajor= 11.44 min, tminor= 15.54 min. [α]D = −93.8 ° (CHCl3, c=1.05) mp 108–112 °C. IR (neat): 3033, 2984, 1770, 1731, 1702, 1613, 1464, 1387, 1179, 1105 cm−1. 1H NMR (CDCl3, 500 MHz): δ 7.75 (dd, 5.59, 3.07, 2H), 7.65 (dd, 5.4, 3.1, 2H), 5.85 (m, 1H), 5.52 (d, 10.2, 1H), 4.92 (m, 1H), 4.07 (q, 7.0, 2H), 2.72 (m, 1H), 2.34 (m, 2H), 2.27 (q, 12.4, 1H), 2.13 (m, 1H), 1.18 (t, 7.6, 3H). 13C NMR (CDCl3, 125 MHz) δ 174.05, 167.8, 133.9, 131.9, 127.9, 126.6, 123.1, 60.5, 47.3, 39.4, 29.0, 27.0, 14.1. Anal. Calc’d for C17H17NO4: C, 68.21; H, 5.72. Found: C, 67.88; H, 6.00. HRMS Calc’d for C17H17NO4(M+): 299.1158. Found: 299.1171.
Rh-catalyzed selective azirdination: (1S,5S,6R)-ethyl 5-(1,3-dioxoisoindolin-2-yl)-7-(2-(trimethylsilyl)ethylsulfonyl)-7-aza-bicyclo[4.1.0]hept-2-ene-3-carboxylate (44)
All glassware was flamed dried under high vacuum. MgO was activated by flame-dry under high vacuum before use. Chlorobenzene was distilled over P2O5 and stored under inert atomasphere over 4 Å MS beads. Under a positive pressure of N2, to SESNH2 in 1 ml C6H5Cl was added sequentially mixture 38 (149 mg, 0.5 mmol), MgO (46 mg, 1.15 mmol), Rh2(esp)2 (7.6 mg, 0.01 mmol). The reaction was cooled to 0 °C. PhI(OPiv)2 (264 mg, 0.65 mmol) was added in one portion. The reaction was slowly warmed to rt and stirred for 4 hrs till the disappearance of 38 by TLC. The MgO was filtered and washed thoroughly with DCM. The combined filtrate was concentrated in vacuo with gentle heating and loaded to a silica gel column. Eluting with 10:1 petroleum ether : ethyl acetate yielded 206 mg 44 as a white foam. Rf = 0.50 (2.5:1 petroleum ether : ethyl acetate). MP = 55~58 °C. IR (film): 2955, 1775, 1715, 1468, 1388, 1327, 1263, 1144, 1109, 1005 cm−1. [α]D = + 42.1 ° (CHCl3, c = 1.08). 1H NMR (CDCl3, 500 MHz): δ 7.75(dd, 5.4, 3.1, 2H), 7.67(dd, 5.5, 3.1, 2H), 7.15(dd, 4.5, 2.4, 1H), 4.90(ddd, 7.6, 3.5, 2.2, 1H), 4.14-4.07(m, 2H), 3.47(dd, 6.6, 4.5, 1H), 3.40(dt, 6.6, 1.7, 1H), 3.06-3.02(m, 2H), 2.78(dd 17.9, 3.3, 1H), 2.53(ddd, 17.7, 7.6, 2.6, 1H), 1.20(t, 7.1, 3H), 1.04-1.00(m, 2H), 0.005(s, 9H). 13C NMR (CDCl3, 125 MHz) δ 167.5, 165.4, 134.3, 132.4, 131.4, 130.9, 123.3, 61.0, 49.2, 42.9, 40.3, 36.5, 26.1, 14.1, 9.5, −2.1. HRMS Calc’d for C22H29N2O6SSi (M+ + H): 477.1516 Found: 477.1494.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM 033049) and the National Science Foundation for their generous support of our programs. T.Z. thanks Novartis and Roche for graduate fellowships. Mass spectra were provided, in part, by the Mass Spectrometry Regional Center of the University of California, San Francisco. Palladium salts were generously supplied by Johnson-Matthey. We thank Gilead Sciences for a reference sample of Tamiflu.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.For information on H5N1 avian flu and H5N1 virus, visit: http://www.nature.com/avianflu/index.html. For information on H1N1 flu and H1N1 virus, visit: http://www.cdc.gov/h1n1flu/
- 2.For reviews on Tamiflu synthesis, see: Farina V, Brown JD. Angew Chem Int Ed. 2006;45:2. doi: 10.1002/anie.200602623.Shibasaki M, Kanai M. Eur J Org Chem. 2008;11:1839.Magano J. Chem Rev. 2009;109:4398. doi: 10.1021/cr800449m.
- 3.Contributions from Gilead Sciences: Kim CU, Lew W, Williams MA, Liu H, Zhang L, Swaminathan S, Bischofberger N, Chen MS, Mendel DB, Tai CY, Laver G, Stevens RC. J Am Chem Soc. 1997;119:681. doi: 10.1021/ja963036t.Rohloff JC, Kent KM, Postich MJ, Becker MW, Chapman HH, Kelly DE, Lew W, Louie MS, McGee LR, Prisbe EJ, Schultze LM, Yu RH, Zhang L. J Org Chem. 1998;63:4545.Bischofberger NW, Kim CU, Lew W, Liu H, Williams MA. 5763483. US Patent. 1998Extensive studies at Roche: Abrecht S, Harrington P, Iding H, Karpf M, Trussardi R, Wirz B, Zutter U. Chimia. 2004;58:621.Karpf M, Trussardi R. Angew Chem Int Ed. 2009;48:5760. doi: 10.1002/anie.200901561.
- 4.a) Yeung YY, Hong S, Corey EJ. J Am Chem Soc. 2006;128:6310. doi: 10.1021/ja0616433. [DOI] [PubMed] [Google Scholar]; b) Fukuta Y, Mita T, Fukuda N, Kanai M, Shibasaki M. J Am Chem Soc. 2006;128:6312. doi: 10.1021/ja061696k. [DOI] [PubMed] [Google Scholar]; c) Cong X, Yao ZJ. J Org Chem. 2006;71:5365. doi: 10.1021/jo060633h. [DOI] [PubMed] [Google Scholar]; d) Mita T, Fukuda N, Roca FX, Kanai M, Shibasaki M. Org Lett. 2007;9:259. doi: 10.1021/ol062663c. [DOI] [PubMed] [Google Scholar]; e) Yamatsugu K, Kamijo S, Suto Y, Kanai M, Shibasaki M. Tetrahedron Lett. 2007;48:1403. [Google Scholar]; f) Bromfield KM, Graden H, Hagberg DP, Olsson T, Kann N. Chem Commun. 2007:3183. doi: 10.1039/b703295a. [DOI] [PubMed] [Google Scholar]; g) Satoh N, Akiba T, Yokoshima S, Fukuyama T. Angew Chem. 2007;119:5836. doi: 10.1002/anie.200701754. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:5734. [Google Scholar]; Angew Chem Int Ed. 2007;46:5734. [Google Scholar]; h) Shie JJ, Fang JM, Wang SY, Tsai KC, Cheng YSE, Yang AS, Hsiao SC, Su CY, Wong CH. J Am Chem Soc. 2007;129:11892. doi: 10.1021/ja073992i. [DOI] [PubMed] [Google Scholar]; i) Trost BM, Zhang T. Angew Chem Int Ed. 2008;47:3759. doi: 10.1002/anie.200800282. [DOI] [PubMed] [Google Scholar]; j) Ishikawa H, Suzuki T, Hayashi Y. Angew Chem Int Ed. 2009;48:1304. doi: 10.1002/anie.200804883. [DOI] [PubMed] [Google Scholar]; k) Mandai T, Oshitari T. Synlett. 2009:787. [Google Scholar]; l) Nie LD, Shi XX. Tetrahedron: Asymmetry. 2009;20:124. [Google Scholar]; m) Matveenko M, Willis AC, Banwell MG. Tetrahedron Lett. 2008;49:7018. [Google Scholar]; n) Yamatsugu K, Yin L, Kamijo S, Kimura Y, Kanai M, Shibasaki M. Angew Chem Int Ed. 2009;48:1070. doi: 10.1002/anie.200804777. [DOI] [PubMed] [Google Scholar]; o) Satoh N, Akiba T, Yokoshima S, Fukuyama T. Tetrahedron. 2009;65:3239. [Google Scholar]; p) Sullivan B, Carrera I, Drouin M, Hudlicky T. Angew Chem Int Ed. 2009;48:4229. doi: 10.1002/anie.200901345. [DOI] [PubMed] [Google Scholar]; q) Yamatsugu K, Kanai M, Shibasaki M. Tetrahedron. 2009;65:6017. [Google Scholar]; r) Sun H, Lin YJ, Wu YL, Wu Y. Synlett. 2009:2473. [Google Scholar]; s) Osato H, Jones IL, Chen A, Chai CLL. Org Lett. 2010;12:60. doi: 10.1021/ol9024716. [DOI] [PubMed] [Google Scholar]; t) Ma J, Zhao Y, Ng S, Zhang J, Zeng J, Than A, Chen P, Liu XW. Chem Eur J. 2010;16:4533. doi: 10.1002/chem.200902048. [DOI] [PubMed] [Google Scholar]; u) Kamimura A, Nakano T. J Org Chem. 2010;75:3133. doi: 10.1021/jo1002856. [DOI] [PubMed] [Google Scholar]; v) Zhu S, Yu S, Wang Y, Ma D. Angew Chem Int Ed. 2010;49:4656. doi: 10.1002/anie.201001644. [DOI] [PubMed] [Google Scholar]
- 5.The lactone can also be synthesized in two steps: Barlett PA, McQuaid LA. J Am Chem Soc. 1984;106:7854.
- 6.Trost BM, Surivet J-P. Angew Chem Int Ed. 2000;39:3122. doi: 10.1002/1521-3773(20000901)39:17<3122::aid-anie3122>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 7.Trost BM, Bunt RC. J Am Chem Soc. 1996;118:235. [Google Scholar]
- 8.Grehn L, Ragnarsson U. Synthesis. 1987:275. [Google Scholar]
- 9.Pd-catalyzed AAA with HN(Boc)2: Evans DA, Campos KR, Tedrow JS, Michael FE, Gagne MR. J Am Chem Soc. 2000;122:7905.
- 10.Pd-catalyzed stereospecific allylic alkylation with HN(Cbz)2: Fairhurst RA, Taylor RJ. 2008006563. WO Patent. 2008
- 11.Pd-catalyzed AAA with HN(Cbz)2: Fandrick DR. PhD thesis. Stanford University; 2006.
- 12.Pd-catalyzed AAA with NaN(CHO)2: Zhang R, Yu L, Xu L, Wang Z, Ding K. Tetrahedron Lett. 2001;42:7659.
- 13.Pd-catalyzed AAA with sodium phthalimide: Trost BM, Radinov R. J Am Chem Soc. 1997;119:5962.
- 14.Pd-catalyzed AAA with phthalimide: Trost BM, Bunt RC. J Am Chem Soc. 1994;116:4089.Trost BM, Bunt RC, Lemoine RC, Calkins TL. J Am Chem Soc. 2000;122:5968.Trost BM, Krueger AC, Bunt RC, Zambrano J. J Am Chem Soc. 1996;118:6520.Trost BM, Jiang C, Hammer K. Synthesis. 2005:3335.Trost BM, Horne DB, Woltering MJ. Angew Chem, Int Ed. 2003;42:5987. doi: 10.1002/anie.200352857.Trost BM, Horne DB, Woltering MJ. Chem–Eur J. 2006;12:6607. doi: 10.1002/chem.200600202.Trost BM, Aponick A. J Am Chem Soc. 2006;128:3931. doi: 10.1021/ja0578348.Trost BM, Aponick A, Stanzl BN. Chem–Eur J. 2007;13:9547. doi: 10.1002/chem.200700832.Trost BM, O’Boyle BM. Org Lett. 2008;10:1369. doi: 10.1021/ol800127a.
- 15.Bunt RC. PhD thesis. Stanford University; 1996. [Google Scholar]
- 16.Adam W, Bottke N, Krebs O, Sahao-Moller CR. Eur J Org Chem. 1999:1963. [Google Scholar]
- 17.Keck GE, Webb RR, Yates JB. Tetrahedron. 1981;37:4007. [Google Scholar]
- 18.a) Lessard J, Mondon M, Touchard D. Can J Chem. 1981;59:431. [Google Scholar]; b) Lessard J, Tuaillon J. J Org Chem. 1982;47:5013. [Google Scholar]; c) Koser GF, Kokil PB, Shah M. Tetrahedron Lett. 1987;28:5431. [Google Scholar]; d) Glover SA, Goosen A, McCleland CW, Schoonraad JL. Tetrahedron. 1987;43:2577. [Google Scholar]; e) Glover SA, Rowbottom CA, Scott AP. Tetrahedron. 1990;46:7247. [Google Scholar]
- 19.For aziridination reactions, see a recent review and references therein: Watson IDG, Yu L, Yudin AK. Acc Chem Res. 2006;39:194. doi: 10.1021/ar050038m.
- 20.Trost BM, Dong G. J Am Chem Soc. 2006;128:6054. doi: 10.1021/ja061105q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mordini A, Russo F, Valacchi M, Zani L, Degl’Innocenti A, Reginato G. Tetrahedron. 2002;58:7153. [Google Scholar]
- 22.O’Brien P, Rosser CM, Caine D. Tetrahedron. 2003;59:9779. [Google Scholar]
- 23.Mordini A, Sbaragli L, Valacchi M, Russo F, Reginato G. Chem Commun. 2002:778. doi: 10.1039/b200708h. [DOI] [PubMed] [Google Scholar]
- 24.Nicolaou KC, Montagnon T, Baran PS, Zhong YL. J Am Chem Soc. 2002;124:2245. doi: 10.1021/ja012127+. [DOI] [PubMed] [Google Scholar]
- 25.a) Kato J-i, Iwasawa N, Mukaiyama T. Chem Lett. 1985:743. [Google Scholar]; b) Hatakeyama S, Mori H, Kitano K, Yamada H, Nishizawa M. Tetrahedron Lett. 1994;35:4367. [Google Scholar]; c) Lee SH, Park YJ, Yoon CM. Tetrahedron Lett. 1999;40:6049. [Google Scholar]; d) Iwanami K, Seo H, Tobita Y, Oriyama T. Synthesis. 2005:183. [Google Scholar]; e) Iwanami K, Yano K, Oriyama T. Chem Lett. 2007;36:38. [Google Scholar]
- 26.Hill DC, Flugge LA, Petillo PA. J Org Chem. 1997;62:4864. [Google Scholar]
- 27.Kumar V, Ramesh NG. Chem Commun. 2006:4952. doi: 10.1039/b612151a. [DOI] [PubMed] [Google Scholar]
- 28.Trost BM, Massiot GS. J Am Chem Soc. 1977;99:4405. [Google Scholar]
- 29.A similar diene intermediate bearing an NH-Boc group rather than an N-phthaloyl group was reported in Corey’s synthesis ( Yeung YY, Hong S, Corey EJ. J Am Chem Soc. 2006;128:6310. doi: 10.1021/ja0616433.). In their approach, the synthesis of the diene required 8 steps, whereas in our approach, compound 38 took only 3 steps. From the conjugate diene, they applied a two-step haloamination-SN2 displacement sequence to form the desired aziridine while we developed a direct aziridination process.
- 30.Examples for 1,4-conjuate addition to form aziridines: Fioravanti S, Colantoni D, Pellacani L, Tardella PA. J Org Chem. 2005;70:3296. doi: 10.1021/jo050044w.Colantoni D, Fioravanti S, Pellacani L, Tardella PA. Org Lett. 2004;6:197. doi: 10.1021/ol0361554.Fioravanti S, Morreale A, Pellacani L, Tardella PA. Synlett. 2004;6:1083.Vyas R, Gao GY, Harden JD, Zhang XP. Org Lett. 2004;6:1907. doi: 10.1021/ol049691k.Fioravanti S, Morreale A, Pellacani L, Tardella PA. Eur J Org Chem. 2003:4549. doi: 10.1021/jo025670x.Jain SL, Sain B. Tetrahedron Lett. 2003;44:575.Gasperi T, Loreto MA, Tardella PA, Gambacorta A. Tetrahedron Lett. 2002;43:3017.Aires-de-Sousa J, Prabhakar S, Lobo AM, Rosa AM, Gomes MJS, Corvo MC, Williams DJ, White AJP. 2001;12:3349.Fioravanti S, Morreale A, Pellacani L, Tardella PA. J Org Chem. 2002;67:4972. doi: 10.1021/jo025670x.Ishikawa T, Kudoh T, Yoshida J, Yasuhara A, Manabe S, Saito S. Org Lett. 2002;4:1907. doi: 10.1021/ol025906j.
- 31.Example of 1,6-addition onto diene esters: Bernardi L, Lopez-Cantarero J, Niess B, Jorgensen KA. J Am Chem Soc. 2007;129:5772. doi: 10.1021/ja0707097.
- 32.a) Schirmeister T. Leibigs Ann. 1997:1895. [Google Scholar]; b) Clark DA, De Riccardis F, Nicolaou KC. Tetrahedron. 1994;50:11391. [Google Scholar]
- 33.For a recent review on metal-catalyzed aziridination reactions, see: Halfen JA. Current Organic Chemistry. 2005;9:657.
- 34.For a recent report on Ag catalyzed aziridination reactions, see: Cui Y, He C. J Am Chem Soc. 2003;125:16202. doi: 10.1021/ja038668b.For a recent report on Au catalyzed aziridination reactions, see: Li Z, Ding X, He C. J Org Chem. 2006;71:5876. doi: 10.1021/jo060016t.
- 35.a) Guthikonda K, Du Bois J. J Am Chem Soc. 2002;124:13672. doi: 10.1021/ja028253a. [DOI] [PubMed] [Google Scholar]; b) Guthikonda K, Wehn PM, Caliando BJ, Du Bois J. Tetrahedron. 2006;62:11331. [Google Scholar]
- 36.a) Espino CG, Fiori KW, Kim M, Du Bois J. J Am Chem Soc. 2004;126:15378–15379. doi: 10.1021/ja0446294. [DOI] [PubMed] [Google Scholar]; b) Fiori KW, Du Bois J. J Am Chem Soc. 2007;129:562–568. doi: 10.1021/ja0650450. [DOI] [PubMed] [Google Scholar]
- 37.a) Chan J, Baucom KD, Murry JA. J Am Chem Soc. 2007;129:14106. doi: 10.1021/ja073872a. [DOI] [PubMed] [Google Scholar]; b) Zalatan DN, Du Bois J. J Am Chem Soc. 2009;131:7558. doi: 10.1021/ja902893u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.